
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shell-isolated nanoparticle-enhanced Raman spectroscopy study of the adsorption behaviour of DNA bases on Au(111) electrode surfaces†
Bao-Ying
Wen
a,
Xi
Jin
a,
Yue
Li
a,
Ya-Hao
Wang
a,
Chao-Yu
Li
a,
Miao-Miao
Liang
b,
Rajapandiyan
Panneerselvam
a,
Qing-Chi
Xu
*a,
De-Yin
Wu
a,
Zhi-Lin
Yang
b,
Jian-Feng
Li
*a and
Zhong-Qun
Tian
a
aMOE Key Laboratory of Spectrochemical Analysis and Instrumentation, State Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: li@xmu.edu.cn; xuqingchi@xmu.edu.cn
bDepartment of Physics, Xiamen University, Xiamen 361005, China
First published on 14th March 2016
Abstract
For the first time, we used the electrochemical shell-isolated nanoparticle-enhanced Raman spectroscopy (EC-SHINERS) technique to in situ characterize the adsorption behaviour of four DNA bases (adenine, guanine, thymine, and cytosine) on atomically flat Au(111) electrode surfaces. The spectroscopic results of the various molecules reveal similar features, such as the adsorption-induced reconstruction of the Au(111) surface and the drastic Raman intensity reduction of the ring breathing modes after the lifting reconstruction. As a preliminary study of the photo-induced charge transfer (PICT) mechanism, the in situ spectroscopic results obtained on single crystal surfaces are excellently illustrated with electrochemical data.
1. Introduction
DNA and RNA play the most significant roles in inheritance and the gene expression.1–3 Particularly, the four DNA bases, adenine, guanine, thymine, and cytosine, are essential components of ATP, NADH and some co-enzymes.4 To deeply understand life processes, the nucleobases have been extensively investigated in the past few decades. Over the last decade, the electrochemical behaviour of the DNA bases was examined using scanning tunneling microscopy (STM) and cyclic voltammetry (CV) on Au(111),5–10 Au(100),11 Au(110),12 Cu(110),13 and Cu(111)14 surfaces. In addition, other techniques such as normalized interfacial Fourier-transform infrared spectroscopy (SNIFTIRS),7 surface-enhanced infrared reflection-absorption spectroscopy with the attenuated total reflection technique (ATR-SEIRAS),15,16 surface-enhanced Raman spectroscopy (SERS)17, surface X-ray scattering (SXS)18 and X-ray photoelectron spectroscopy (XPS)19 have been used for nucleobase investigation.For instance, Tao et al. combined the STM and CV method for the investigation of the electrochemical behaviour of DNA bases on Au(111) electrodes; the polymeric aggregates were formed by adenine/guanine, but the oblique lattice had been observed with cytosine/thymine.5 Maleki and coworkers used STM and molecular dynamics in their work; adenine, guanine and thymine formed hydrogen-bonded chains on Au(111), however, cytosine was clustered into an island at low coverage. And at high coverage, adenine was formed as a ordered two-dimensional hexagonal structure, while guanine, cytosine and thymine aggregated into small random clusters.9 In another work, Salvatore and coworkers evidenced that DNA bases chemisorbed on the Au(110) surface, and this study revealed that the chemisorption was accompanied by lifting reconstruction of Au(110).12,20 Although the structural transformation of DNA bases on the low-index single crystal gold electrodes could be imaged by STM, it is highly necessary to obtain the chemical information of these vital molecules on metal substrates (Fig. 1).
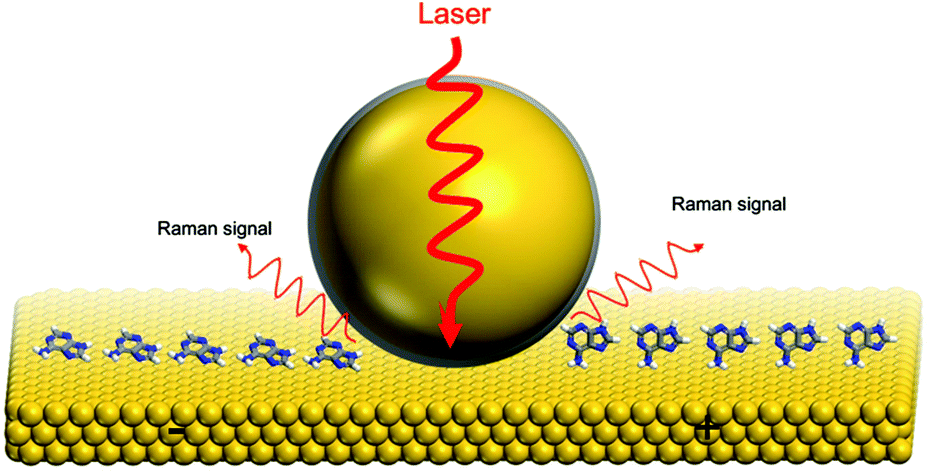 | ||
| Fig. 1 Schematic diagram of the EC-SHINERS measurement on a gold single crystal electrode surface. The left side of the electrode surface is more negative and the right side is more positive. | ||
To examine the interaction between DNA bases and metal substrates, the Raman spectroscopy technique can be used to accrue chemical information of these processes. Uniquely, Raman scattering has been widely applied in biological molecule characterization because of its non-destructive analysis, simplicity, and highly sensitive detection.21–24 Meanwhile, the influence of water and carbon dioxide is significantly low in Raman measurements when compared with infrared spectroscopy.23 Along with the nanostructured or roughened metal substrate morphologies, the Raman signals of probe molecules will be amplified to a great extent.25 This phenomenon is called surface-enhanced Raman scattering (SERS). SERS has gained great attention in the field of electrochemistry and biology due to its sensitivity and molecular specificity.26 However, there are still two main generality limitations regarding the applications of SERS. First, only a few noble metals (e.g. Au, Ag, and Cu) could provide the large enhancement of SERS. Second, the substrate should be roughened or nanostructured.27 In 2010, our group invented a novel method called shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS)28 which had been successfully applied to acquire high quality Raman signals on various well-defined substrates. The key advantage of the shell-isolated nanoparticles is their uniform and ultrathin SiO2 shell, which permits the Au core to generate strong electromagnetic near-fields and simultaneously protects it from the chemical environment. For instance, this technique was used to in situ monitor the electro-oxidation processes at low-index Au(hkl) surfaces.29
In this work, we utilize the in situ electrochemical SHINERS method to study the adsorption behaviours of the four DNA bases at single crystal Au(111) electrode surfaces for the first time. The adsorption-induced lifting reconstruction of the Au(111) surface has been evidenced by the cycle voltammetry studies with the four bases. The Raman signal intensity attenuation of the ring breathing mode has been observed along with the lifting reconstruction process, and the photo-induced charge transfer (PICT) mechanism was used to explain these spectroscopic results. As an emerging vibrational spectroscopic method, electrochemical SHINERS has great potential for the in situ analysis of biological molecules at electrochemical interfaces with a well-defined substrate.
2. Experimental
2.1 Materials
The nucleic acid bases adenine (A, 99%), thymine (T, 97%), cytosine (C, 98%+), and guanine (G, 98%), sodium citrate (99.0%), chloroauric acid (HAuCl4·3H2O, 99.99%), sodium perchlorate (NaClO4, 98%–102%), and (3-aminopropyl)trimethoxysilane (97%) were purchased from Alfa Aesar. Sodium silicate solution (27% SiO2) was purchased from Sigma-Aldrich. All the chemicals were used directly without purification. Deionized Milli-Q water (∼18.2 MΩ cm) was used throughout the study.2.2 Apparatus
Scanning electron microscopy (SEM) images were recorded with a HITACHI S-4800, and transmission electron microscopy (TEM) images were recorded with a JEM 2100. CV was recorded with a CHI-630E potentiostat. All the electrochemical measurements were carried out using a standard glass three-room cell. A saturated calomel electrode (SCE) and a Pt wire served as the reference and counter electrodes, respectively. In the case of EC-SHINERS measurements, the electrode potential was controlled with an Autolab PGSTAT30 (Metrohm). The EC-SHINERS measurements were carried out with a confocal Raman system Xplora (Jobin-Yvon Horiba). The excitation wavelength was 638 nm. The microscope objective was 50× magnification with a numerical aperture of 0.55. All the EC-SHINERS measurements were carried out using a Teflon three-electrode cell. SCE and Pt wire were used as the reference and counter electrodes, respectively. In this work, all potentials given were referred to the SCE potential scale.2.3 Preparation of shell-isolated Au nanoparticles
First, 55 nm Au core nanoparticles were prepared according to the literature:30 1.4 mL of 1 wt% sodium citrate solution was added into 200 mL of 0.01 wt% boiling HAuCl4 solution. The mixture was refluxed for 1 h and then cooled down to room temperature for the following silica coating. The Au@SiO2 nanoparticles were produced according to the previous publications of our group:28 0.4 mL of 1 mM APTMS solution was added into 30 mL of the as-prepared Au sol, and the mixture was stirred for 15 min at room temperature. Next, 3 mL of 0.54 wt% sodium silicate solution with pH ∼10.3 was added to the sol. Finally, the sol was then transferred to a 95 °C bath and stirred for 30 min for the coating of the silica shell with a thickness of about 4 nm. The hot sol was transferred to a 1.5 mL test tube and cooled in an ice bath. The SHINs were centrifuged at 5500 rpm twice and washed with Milli-Q water. Then the concentrated SHINs were diluted with Milli-Q water to about 200 μL for the in situ EC-SHINERS measurements (Fig. 2).31 | ||
| Fig. 2 (a) SEM image of a Au(111) bead electrode. (b) High-resolution TEM image of a single Au SHINERS particle with a 4 nm SiO2 shell. (c) SEM image of Au SHINERS particles on a Au(111) single crystal electrode. | ||
3. Results and discussion
3.1 The adsorption behaviour of adenine on a Au(111) surface
The adsorption behaviour of adenine (1 mM) on a Au(111) electrode in 0.1 M NaClO4 solution was monitored by CV at a scan rate of 50 mV s−1. From Fig. 3a, we can distinguish the potential dependence of current and spectral characteristics in three regions: (I) within a potential range of −0.7 to −0.45 V, adenine molecules formed disordered adsorption on Au(111), and the planar adenine molecules were oriented nearly parallel to the electrode surface; (II) from −0.45 to 0 V, two couples of broad peaks were observed. Similarly, Camargo et al.8 and Xiao et al.32 reported the dissolution and formation of the charge transfer complex in this region. Besides, the formation of metal–adenine complexes could be also confirmed by the shifting of the bands compared with the Raman spectra in solution (ESI Fig. S2†).32 According to the work of Martins et al.,33 adsorbed adenine molecules undergo deprotonation with lifting of the reconstruction (Fig. 3a), simultaneously. At the potential of −0.45 V, adenine formed an ordered physisorbed film on the non-reconstructed gold surface; (III) at positive electric potential, adenine molecules were adsorbed in a vertical orientation with the anchors of the N7 atom and NH2 group. The positively charged surface stabilized the coordination of lone-pair orbitals to the metal surface. At more positive potential, oxidation of the Au surface leads to adenine desorption at positive potentials. To avoid the oxidation, the maximum potential of the CV scan is set to be 0.4 V.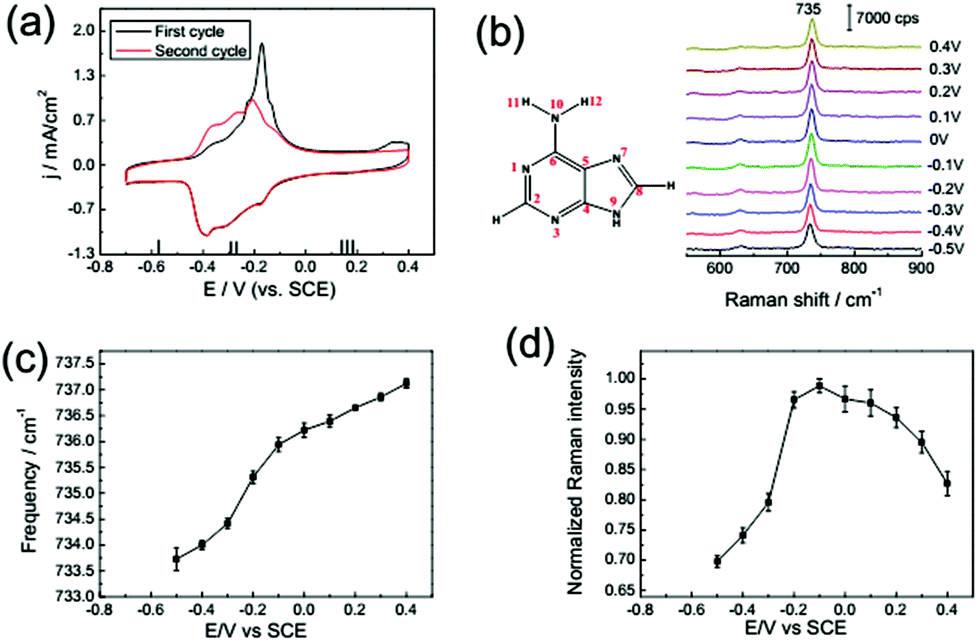 | ||
| Fig. 3 (a) CV from a Au(111) electrode in 0.1 M NaClO4 solution with 1 mM adenine: first cycle (black line), second cycle (red line). (b) SHINER spectra of adenine adsorption on a Au(111) electrode in 0.1 M NaClO4 + 1 mM adenine. The dependence of the Raman frequency (c) and normalized Raman intensity (d) on applied potentials for the adenine ring breathing mode. Error bars represent standard deviations obtained by measuring at different locations on the samples. | ||
The in situ SHINER spectra of adenine adsorption on a Au(111) single crystal electrode from 1 mM solution are shown in Fig. 3b (the spectral range from 550 to 1700 cm−1 contains more fingerprint information, as shown in the ESI Fig. S3,† and the spectra of applied potential are reversible, as shown in the ESI Fig. S4†). The potential increment of the SHINER spectra sampling was 0.1 V, with about 60 s at each step for stabilization of the current. The strong peaks appearing between 600 and 800 cm−1 for all DNA bases are attributed to the ring breathing vibrational modes.23,34
The vibrational frequency shift of molecules at electrochemical interfaces is a sensitive probe of molecular orientation on electrode surfaces, because the bonding interaction between DNA bases is highly correlated with the adsorption geometries on noble metal surfaces.35,36 The hybridization of occupied orbitals of nucleobases and the d-band of the Au surface dominate the bonding interaction for both the vertical and flat orientations, while the delocalized occupied π orbital coupled more strongly with the d-band in the vertically orientated adsorption.35 Such enhancement of the intramolecular π bonding orbital results in the blue-shift of the ring breathing modes. Consequently, the vibrational frequency of the ring breathing modes experiences significant blue-shift during the orientation variation of the lifting reconstruction process, as shown in Fig. 3a and c.
The spectral intensity of the ring breathing mode is plotted against potential in Fig. 3d. The intensity of the ring breathing mode for adenine was firstly enhanced with the increase of coverage, but drastically decreased immediately after the lifting reconstruction process, while the adsorption kept full monolayer coverage. This intensity variation profile could be explained by the photo-induced charge transfer (PICT) mechanism.37 The electronic excitation gap for the nucleobases in the gas phase is far from resonance with the energy of the excitation laser (about 2 eV). Nevertheless, the Fermi level of Au lying between the HOMO and LUMO of the molecular orbitals narrowed the off-resonance intramolecular excitation gap to near-resonance matching with the excitation laser,36 which provided the possible metal-to-molecule PICT mechanism. As the electrochemical potential shifts positively, the chemical potential of interfacial electrons gets closer to the HOMO level of the nucleobases, thus the near-resonance excitation switched to off-resonance excitation. We also performed a SHINERS experiment with a 785 nm excitation laser (ESI Fig. S5†). The results showed that the potential of the maximum Raman intensity experienced negative shifts to −0.3 V with the increase of excitation wavelength. Consequently, at the potential range before the reconstruction, the intensities of the ring breathing mode of adenine were dominated by the coverage, while after the reconstruction, the PICT became the main contribution.
3.2 The adsorption behaviour of thymine on a Au(111) surface
The CV curve of 1 mM thymine on a Au(111) electrode in the electrolyte containing 0.1 M NaClO4 is shown in Fig. 4a; it was recorded at a rate of 50 mV s−1. The CV of thymine is also distinguished into three potential ranges. The first adsorption configuration (E < −0.2 V) refers to a disordered adsorption of thymine molecules on the surface with a negative charge, and the plane of the thymine molecules was oriented nearly parallel to the electrode surface. The second adsorption configuration was formed in the potential range from around −0.20 up to 0.35 V. In this region, a couple of broad peaks emerged, indicating the transformation from a reconstructed surface into an unreconstructed surface. Correspondingly, the physisorbed monolayer was transformed into a chemisorbed configuration.19,38 In the third region, at potential E > 0.35 V, the surface configuration was completely transformed into a chemisorbed state, the N3 and O8 atoms of thymine were anchored to metal atoms,39 and the molecules prefer a vertical orientation on the single crystal surface.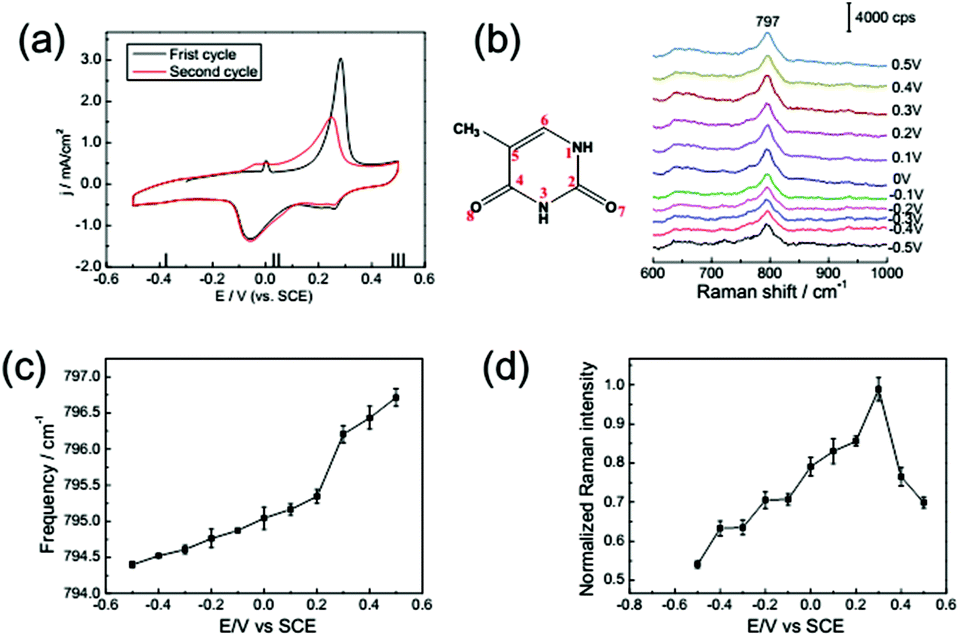 | ||
| Fig. 4 (a) CV from a Au(111) electrode in 0.1 M NaClO4 solution with 1 mM thymine: first cycle (black line), second cycle (red line). (b) SHINER spectra of thymine adsorption on a Au(111) electrode in 0.1 M NaClO4 + 1 mM thymine. The dependence of the Raman frequency (c) and normalized Raman intensity (d) on applied potentials for the thymine ring breathing mode. Error bars represent standard deviations obtained by measuring at different locations on the samples. | ||
The frequency shift and intensity variation of the ring breathing mode of thymine are qualitatively similar to those of adenine, as shown in Fig. 4c and d. Meanwhile the potential range of the lifting reconstruction was shifted, as shown in Fig. 4a. The frequency shift is dominated by the surface charge-dependent molecular reorientation, as noted in the discussion of adenine adsorption. The potential of zero charge (PZC) is changed for different adsorption species. Consequently, the CV peak of the lifting reconstruction, accompanied by the potential of the maximum slope in the frequency shifting curve, is displaced towards higher electric potential. The potential of the extremely enhanced SHINERS intensity is also displaced for the different bases’ molecular adsorption, because both the chemical potential of the surface electrons and alignment of molecular energy levels are changed for different adsorption species.
3.3 The adsorption behaviour of cytosine on a Au(111) surface
The cyclic voltammogram of 1 mM cytosine on a Au(111) electrode in the electrolyte containing 0.1 M NaClO4 is shown in Fig. 5a; it was recorded at a rate of 50 mV s−1. Similar to the two bases discussed before, the CV is distinguished into three electric potential ranges. For the potential range before −0.20 V, cytosine molecules were oriented nearly parallel to the electrode surface. In the second region, from −0.2 to 0.2 V, the broad peaks indicate the reorientation of the cytosine molecules on the electrode surface, and the accompanied lifting of the reconstruction on the single crystal surface.6 In this section, the intensity of the ring breathing mode was increased with higher coverage. The slope of the frequency shift is even higher than the other potential ranges, because the coupling strength between molecular orbitals and the d-band of the surface experience significant change during the reorientation. The third region can be assigned to a chemisorbed phase of the cytosine molecules, which shows great stability even at very positive potentials due to the strong coordination of N3 and NH2 lone-pair orbitals. The intensity of the ring breathing mode is sharply decreased due to the mismatching of electronic levels and photonic energy within the PICT mechanism. Both the frequency shift and the intensity variation profiles of cytosine are similar to those of the previously discussed nucleobases.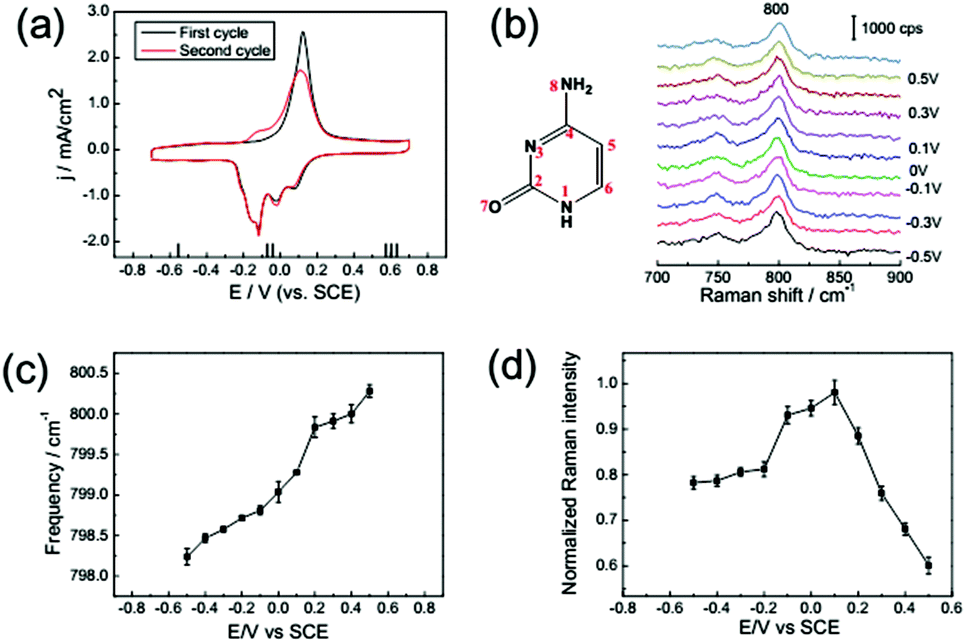 | ||
| Fig. 5 (a) CV from a Au(111) electrode in 0.1 M NaClO4 solution with 1 mM cytosine: first cycle (black line), second cycle (red line). (b) SHINER spectra of cytosine adsorption on a Au(111) electrode in 0.1 M NaClO4 + 1 mM cytosine. The dependence of the Raman frequency (c) and normalized Raman intensity (d) on applied potentials for the cytosine ring breathing mode. Error bars represent standard deviations obtained by measuring at different locations on the samples. | ||
3.4 The adsorption behaviour of guanine on a Au(111) surface
As shown in Fig. 6a, the CV of guanine is qualitatively similar with the CV of the other bases, as discussed in detail previously. The most characteristic peaks between −0.45 to −0.2 V indicate the process of molecular reorientation-induced lifting reconstruction of single crystal surfaces. Besides, after the lifting reconstruction, a chemisorbed stacked monolayer of guanine formed at Au(111),40 and the N7 and O13 lone-pairs of the guanine molecule are the anchors for vertical adsorption.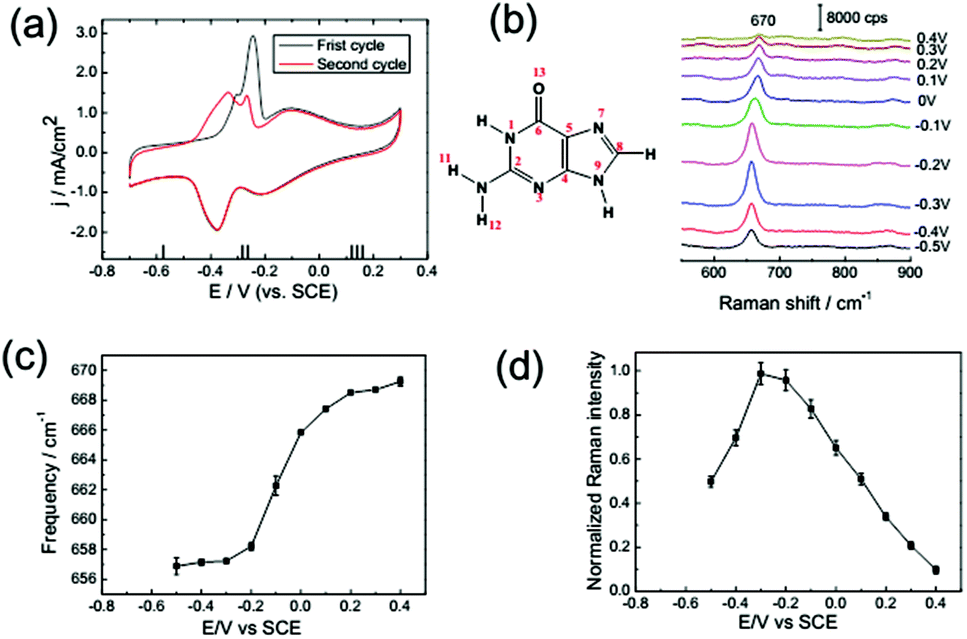 | ||
| Fig. 6 (a) CV from a Au(111) electrode in 0.1 M NaClO4 solution with 1 mM guanine: first cycle (black line), second cycle (red line). (b) SHINER spectra of guanine adsorption on a Au(111) electrode in 0.1 M NaClO4 + 1 mM guanine. The dependence of the Raman frequency (c) and normalized Raman intensity (d) on applied potential for the guanine ring breathing mode. Error bars represent standard deviations obtained by measuring at different locations on the samples. | ||
As shown in Fig. 6b, the frequency shift and intensity variation could be attributed to the same mechanism as that of adenine. But the ring breathing frequency shift is more significant than that for the other bases, the intensity of guanine is even stronger than that of the other nucleobases, and the variation of intensity with potential tuning is more significant. Because for the guanine molecule, the highest energy levels of delocalized occupied π orbitals hybridize with metal bands which are closer to the Fermi level.36 Consequently, the PICT profile of the spectral intensity of guanine is more significantly enhanced.
4. Conclusions
In this work, we employed electrochemical SHINERS to investigate the adsorption behaviours of adenine, guanine, thymine, and cytosine molecules on Au(111) single crystal electrodes. Based on CV data and SHINER spectra, we found that all four of the nucleobases resulted in the reconstruction of single crystal Au(111) electrodes and have similar behaviours. Concurrently, after the lifting of reconstruction, with a positive potential, the Raman intensity of the ring breathing modes of the four nucleobases drastically decreased immediately due to the PICT mechanism. Due to the strong interaction between the nucleobases and the metal electrode, the Raman frequency of the ring breathing modes increased continuously after the potential of lifting reconstruction. Overall, this in situ SHINERS study at the electrochemical interface reveals remarkable significance, and evidently it promotes the applications of in situ SHINERS on well-defined single crystal surfaces.Acknowledgements
This research was supported by the NSFC (21522508, 21427813, and 21503175), Thousand Youth Talents Plan of China, the Fundamental Research Funds for the Central Universities (no. 20720150039), and NFFTBS (no. J1310024).Notes and references
- B. Commoner, Am. Sci., 1964, 52, 365–388 CAS.
- T. Kouzarides, Cell, 2007, 128, 693–705 CrossRef CAS PubMed.
- X. Feng and S. Guang, J. Genet. Genomics, 2013, 40, 153–160 CrossRef CAS PubMed.
- R. N. Goyal, A. Kumar and A. Mittal, J. Chem. Soc., Perkin Trans., 1991, 2, 1369–1375 RSC.
- N. J. Tao, J. A. DeRose and S. M. Lindsay, J. Phys. Chem., 1993, 97, 910–919 CrossRef CAS.
- T. Wandlowski, D. Lampner and S. M. Lindsay, J. Electroanal. Chem., 1996, 404, 215–226 CrossRef.
- W. Haiss, B. Roelfs, S. N. Port, E. Bunge, H. Baumgärtel and R. J. Nichols, J. Electroanal. Chem., 1998, 454, 107–113 CrossRef CAS.
- A. P. M. Camargo, H. Baumgärtel and C. Donner, Phys. Chem. Chem. Phys., 2003, 5, 1657–1664 RSC.
- A. Maleki, S. Alavi and B. Najafi, J. Phys. Chem. C, 2011, 115, 22484–22494 CAS.
- C. Vaz-Domínguez, M. Escudero-Escribano, A. Cuesta, F. Prieto-Dapena, C. Cerrillos and M. Rueda, Electrochem. Commun., 2013, 35, 61–64 CrossRef.
- D. Bogdan and C. Morari, J. Phys. Chem. C, 2012, 116, 7351–7359 CAS.
- P. Salvatore, R. R. Nazmutdinov, J. Ulstrup and J. Zhang, J. Phys. Chem. B, 2015, 119, 3123–3134 CrossRef CAS PubMed.
- Q. Chen, D. J. Frankel and N. V. Richardson, Langmuir, 2002, 18, 3219–3225 CrossRef CAS.
- H. Tanaka, T. Nakagawa and T. Kawai, Surf. Sci., 1996, 364, L575–L579 CrossRef CAS.
- K. Ataka and M. Osawa, J. Electroanal. Chem., 1999, 460, 188–196 CrossRef CAS.
- S. Pronkin and T. Wandlowski, J. Electroanal. Chem., 2003, 550, 131–147 CrossRef.
- B. Giese and D. McNaughton, J. Phys. Chem. B, 2002, 106, 101–112 CrossRef CAS.
- T. Wandlowski, B. M. Ocko, O. M. Magnussen, S. Wu and J. Lipkowski, J. Electroanal. Chem., 1996, 409, 155–164 CrossRef.
- B. Roelfs, E. Bunge, C. Schröter, T. Solomun, H. Meyer, R. J. Nichols and H. Baumgärtel, J. Phys. Chem. B, 1997, 101, 754–765 CrossRef CAS.
- P. Maksymovych, D. C. Sorescu and J. T. Yates, Phys. Rev. Lett., 2006, 97, 146103 CrossRef PubMed.
- Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2007, 42, 493–541 CrossRef CAS.
- A. M. Robinson, S. G. Harroun, J. Bergman and C. L. Brosseau, Anal. Chem., 2012, 84, 1760–1764 CrossRef CAS PubMed.
- R. A. Karaballi, A. Nel, S. Krishnan, J. Blackburn and C. L. Brosseau, Phys. Chem. Chem. Phys., 2015, 17, 21356–21363 RSC.
- N. Rajendiran and J. Thulasidhasan, Spectrochim. Acta, Part A, 2015, 144, 183–191 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- Z. Q. Tian, B. Ren and D. Y. Wu, J. Phys. Chem. B, 2002, 106, 9463–9483 CrossRef CAS.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- C. Y. Li, J. C. Dong, X. Jin, S. Chen, R. Panneerselvam, A. V. Rudnev, Z. L. Yang, J. F. Li, T. Wandlowski and Z. Q. Tian, J. Am. Chem. Soc., 2015, 137, 7648–7651 CrossRef CAS PubMed.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- J. F. Li, Y. J. Zhang, A. V. Rudnev, J. R. Anema, S. B. Li, W. J. Hong, P. Rajapandiyan, J. Lipkowski, T. Wandlowski and Z. Q. Tian, J. Am. Chem. Soc., 2015, 137, 2400–2408 CrossRef CAS PubMed.
- Y. J. Xiao, Y. F. Chen and X. X. Gao, Spectrochim. Acta, Part A, 1999, 55, 1209–1218 CrossRef.
- A. Martins, A. Queirós and F. Silva, ChemPhysChem, 2005, 6, 1056–1060 CrossRef CAS PubMed.
- J. De Gelder, K. De Gussem, P. Vandenabeele and L. Moens, J. Raman Spectrosc., 2007, 38, 1133–1147 CrossRef CAS.
- S. Kilina, S. Tretiak, D. A. Yarotski, J.-X. Zhu, N. Modine, A. Taylor and A. V. Balatsky, J. Phys. Chem. C, 2007, 111, 14541–14551 CAS.
- M. Rosa, S. Corni and R. Di Felice, J. Chem. Theory Comput., 2013, 9, 4552–4561 CrossRef CAS PubMed.
- J. R. Lombardi and R. L. Birke, Acc. Chem. Res., 2009, 42, 734–742 CrossRef CAS PubMed.
- E. Avci, Ph.D. thesis, Freie University, Berlin, 2007 Search PubMed.
- K. H. Cho, J. Choo and S. W. Joo, J. Mol. Struct., 2005, 738, 9–14 CrossRef CAS.
- A. P. Martins Camargo, Ph.D. thesis, Freie University, Berlin, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an00180g |
| This journal is © The Royal Society of Chemistry 2016 |