
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Raman spectroscopy: an evolving technique for live cell studies
Rachael
Smith
ab,
Karen L.
Wright
b and
Lorna
Ashton
*a
aDepartment of Chemistry, Lancaster University, LA1 4YG, UK. E-mail: l.ashton@lancaster.ac.uk
bFaculty Health and Medicine, Division of Biomedical and Life Sciences, Lancaster University, Lancaster LA1 4YG, UK
First published on 5th April 2016
Abstract
One of the most exciting developments in Raman spectroscopy in the last decade has been its application to cells and tissues for diagnostic and pharmaceutical applications, and in particular its use in the analysis of cellular dynamics. Raman spectroscopy is rapidly advancing as a cell imaging method that overcomes many of the limitations of current techniques and is earning its place as a routine tool in cell biology. In this review we focus on important developments in Raman spectroscopy that have evolved into the exciting technique of live-cell Raman microscopy and highlight some of the most recent and significant applications to cell biology.
Introduction
The cell is the basic biological unit of all living organisms, and the understanding of cellular dynamics and processes is vital for biological research as most diseases occur as a result of cellular abnormalities due to intracellular biochemical changes.1 Cell imaging allows these intracellular biochemical changes, along with normal behaviour, to be observed. Raman spectroscopy is an advancing cell imaging method that overcomes some of the limitations of current techniques.Raman spectroscopy is a measure of the Raman effect, or inelastic scattering, first discovered by C. V. Raman in 1928.2 He and K. S. Krishnan described the observation of a ‘new type of secondary radiation’ after experiments that involved illuminating samples with sunlight focused through a lens. They observed scattered light with a different wavelength to the original, incident wavelength; this scattering is now known as Raman scattering. This is induced by monochromatic light, usually from a laser, directed onto the cell; photons interact with this sample and energy can either be lost (Stokes) or gained (anti-Stokes). This difference in energy between the incident and scattered photon corresponds to the energy required to excite a particular molecular vibration; detection of these scattered photons produces a Raman spectrum, with different bands corresponding to the vibrational frequencies of different functional groups. Each molecule therefore has a unique fingerprint, or spectrum, according to the chemical bonds within it.3
While for cells these spectra can be complex, containing information about many different molecules and often requiring complex data interpretation, the potential of Raman spectroscopy in live cell imaging cannot be ignored. One of the key advantages of Raman spectroscopy is the ability to determine the underlying chemical structure of a cell: proteins, lipids and DNA can be visualised according to their vibrational spectra and as a result cells do not need to be labelled or stained prior to imaging.4 Furthermore, as water has a weak Raman signal, cells can be imaged within aqueous environments, meaning that live cell imaging is possible and making Raman spectroscopy an exciting alternative to existing imaging methods, allowing for the observation of living cells under normal physiological conditions.
Numerous other imaging techniques are currently used today in order to visualise cells. Historically, cell imaging was carried out only on fixed cells, primarily by electron microscopy. While providing valuable structural and biochemical information, these techniques only provide a ‘snapshot’ of the cells current state.5 In addition, cells often have to be stained prior to imaging, which has the potential to introduce artefacts and give an inaccurate representation of the natural intracellular state. The development of brightfield microscopy based techniques such as phase contrast and differential interference contrast microscopy meant that cells could be observed without staining, and live cell imaging became a possibility.6 Images obtained via these techniques can typically be collected more quickly than those acquired via Raman spectroscopy, but there is a lack of chemical specificity and there is the potential for artefacts to be introduced due to the halo affect.7
More recently, live cell imaging has been carried out using fluorescence microscopy, which allows for the detection of single molecules within a cell. Fluorescence microscopy is used to visualise the pattern of fluorescence within cells that have been stained with fluorescent molecules. The resolution of images achieved via fluorescence microscopy can be improved by techniques like STORM (stochastic optical reconstruction microscopy) or PALM (photo-activated localization microscopy) which sequentially selectively activate fluorophores throughout a single cell (for more detail on these techniques see ref. 8). Both of these techniques have been used in a number of live cell studies.9–11 FRET (fluorescence resonance energy transfer) is another fluorescence-related technique that can be used to detect and image protein–protein interactions within cells to give insight into cellular events like signal transduction and gene transcription.12 Whilst generally, these techniques have the advantage of giving a better resolution than can typically be achieved with Raman spectroscopy (down to ∼10 nm (ref. 13)), Raman spectroscopy has the advantage of increased chemical specificity and a lack of labels. Fluorescent microscopy relies on the expression of fluorescent molecules within cells, often by introducing external genes,6 which can directly affect cell physiology14,15 and as such, fluorescence imaging may not reflect the normal cellular environment in comparison to Raman-related techniques.
Infrared spectroscopy (IR) is another vibrational spectroscopy technique that can be utilised for live cell imaging. IR spectroscopy shares many of the advantages of Raman spectroscopy, in that it is non-invasive, non-destructive, and identifies the underlying chemical structure and as a result does not require labels. However, as water is a strong absorber of infrared radiation3 this can make the analysis of substances in an aqueous environment difficult; Raman spectroscopy is therefore usually preferred over IR spectroscopy as cells can be maintained under physiological conditions. Additionally, Raman spectroscopy is typically able to achieve a better resolution.16
Due to its advantages over current techniques, there is a huge interest in developing Raman spectroscopy and imaging in biomedicine, with a particular emphasis on its potential for non-invasive in vivo disease diagnosis.
Single cell Raman spectroscopy
Currently, despite its advantages and potential, Raman spectroscopy is not widely used in clinical practice. This is primarily due to the fact that spontaneous Raman scattering is an inherently weak process, with only approximately 1 × 108 of photons inelastically scattered.3 Traditionally, this has resulted in long acquisition times and high laser intensity which is detrimental in examining biological samples and results in degradation.31 However, a number of new developments, techniques and instrumentation are starting to overcome these limitations (summarised in Fig. 1), leading to vast improvements and allowing for cell imaging via Raman spectroscopy to become a viable alternative to traditional methods. One of the most important step-changes in single cell Raman spectroscopy was the combining of a Raman spectrometer with a standard confocal microscope, the typical set-up of which can be seen in Fig. 2 below. In this technique, the laser beam is focused onto the cell by a microscope lens in order to give a much better resolution than can be achieved with traditional Raman spectroscopy.26 By coupling the Raman spectrometer to a camera, usually a charged-coupled device (CCD), confocal Raman spectroscopy is able to image or map samples in addition to collecting their Raman spectra, allowing for the visualisation of intracellular components.32 Wide-field Raman imaging is a direct approach that illuminates the entire sample with a laser, and a narrowband filter is used to measure a specific wavenumber range. Raman mapping is more commonly used in cell studies, and traditionally involves raster-scanning a confocal laser spot and collecting the full Raman spectra sequentially at each location across the sample after dispersion onto the spectrometer CCD.23,33 A Raman map can them be computationally constructed to produce a pseudo-coloured image shaded according to the relative intensity of the Raman spectra at a given wavelength at each pixel.34 The first confocal Raman microscope was described in 1990, and was used to visualise and study single cells and chromosomes with a high spatial resolution.26 Since 1990, many different studies have been carried out using confocal Raman spectroscopy to map cells.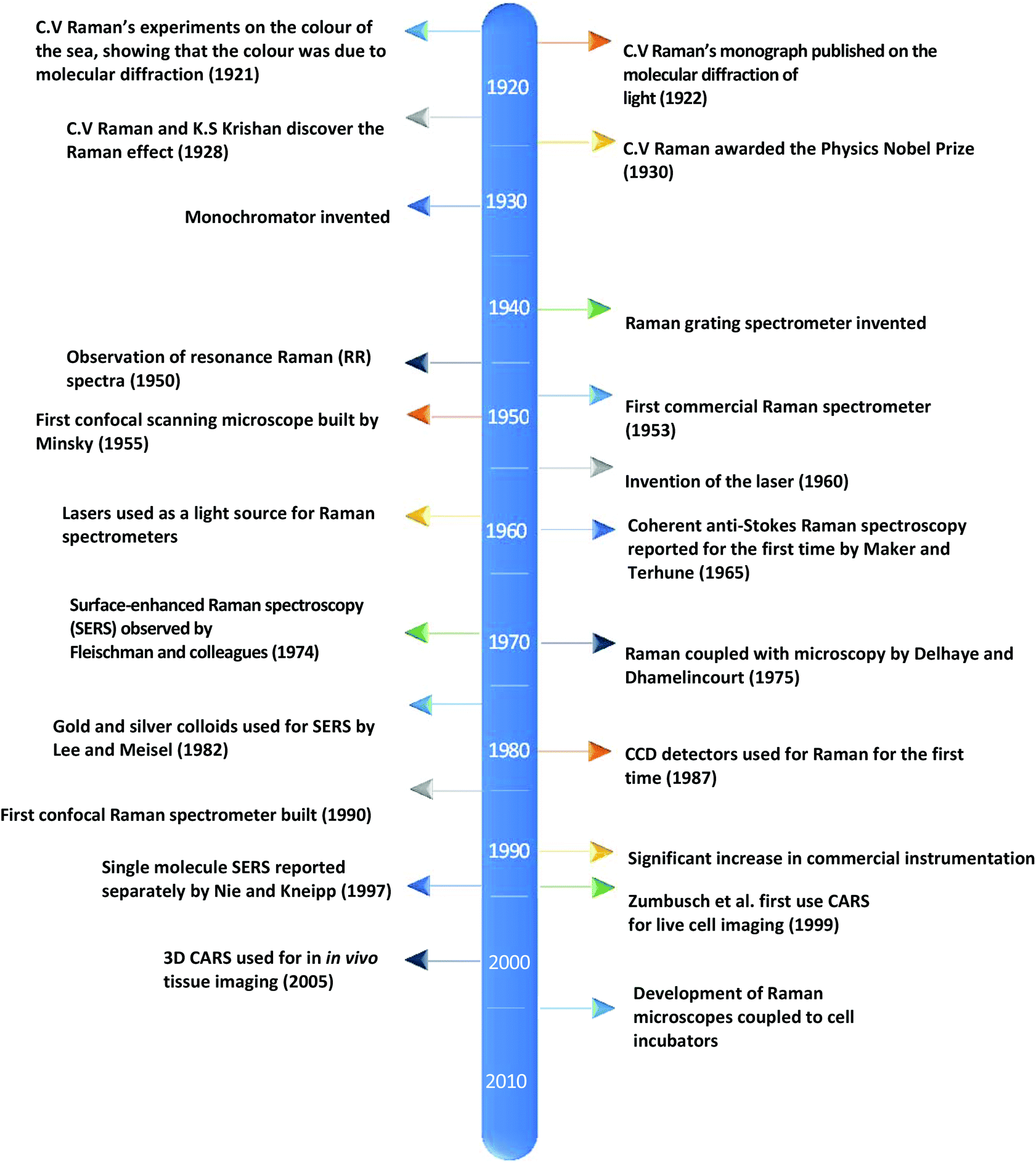 | ||
| Fig. 1 A timeline of the key events in the history of Raman spectroscopy that have led to significant advancements in the field.2,17–30 | ||
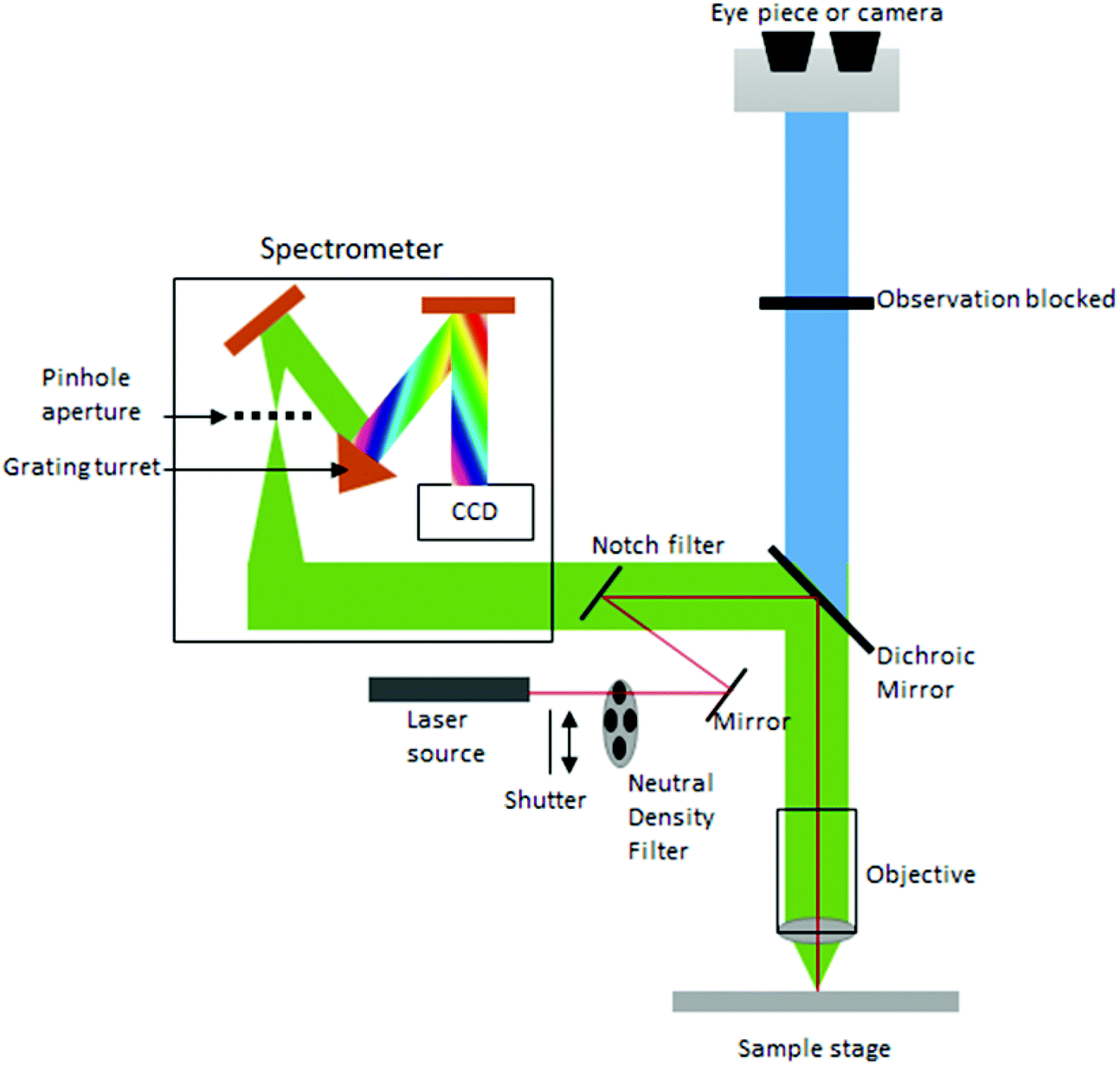 | ||
| Fig. 2 Standard configuration of a confocal Raman microscope. | ||
As with many traditional cell imaging techniques, cells were initially fixed for these Raman spectroscopy experiments. Fixatives are used to preserve and to prevent the degradation of biological samples before they can be observed microscopically. The main fixatives used in Raman microscopy are alcohols (which fix samples by disrupting the hydrophobic bonds within proteins to denature them) and aldehydes (crosslinking fixatives that work by inducing covalent bonds between proteins).35 In fixed cells, Raman microspectroscopy has been used to map and visualise cell organelles, including: DNA by detecting the intensity of the 788 cm−1 band, associated with nucleic acids;36 proteins by measuring the bands for alkane groups at 2850 cm−1, 2885 cm−1 and 2935 cm−1;37 lipid bodies by measuring the intensity of bands at 1658 cm−1, 1267 cm−1 and 1411 cm−1;38 and mitochondria by using bands at 750 cm−1, indicative of cytochrome c distribution.39 In addition to this, studies on fixed cells have also been used to image cells at different stages of the cell cycle,40 to show subcellular changes that occur as a result of the stress-induced response,41 and to identify differences between benign and malignant breast tissue.42
However, whilst all of these above studies have provided valuable biochemical information, there is evidence that fixing procedures may alter the Raman spectra of cells in comparison to unfixed cells treated in the same way. Chan et al. reported changes in the intensity of bands associated with DNA, RNA, protein and lipid vibrations between fixed and unfixed cells, with methanol fixation showing greater changes than paraformaldehyde.43 A more recent study supported this, with ethanol inducing a greater change in the Raman spectra of nucleolar components than formaldehyde,44 and it has also been shown that formalin fixation induces changes in the Raman spectra of nucleic acids and proteins.45,46 Together, this data suggests that studies on fixed cells may not give accurate Raman spectra and has the potential to lead to data being misinterpreted, and studies on live cells are necessary to overcome this and to give more accurate results.
Live-cell studies
Raman microscopy is ideal for live cell studies, because it does not require the use of labels that may harm cells, and because it can be carried out in aqueous conditions due to the weak Raman intensity of water, cells can be imaged in either phosphate buffer solution (PBS) or medium, so do not need to be fixed prior to imaging. Many live cell studies use a laser with a wavelength of 785 nm, just outside of the visible range. This is because there is evidence that visible wavelengths can cause degradation and photodamage to biological samples even at a low power and with short acquisition times.31,47 In contrast, it has been shown in several studies that a 785 nm laser does not cause this degradation, even with a high power and longer acquisition times,3,48 making it the preferred wavelength for the study of living cells.Since the development of live-cell Raman imaging, many studies using living cells have been carried out and Raman microscopy has proved to be an important imaging technique in a number of different research areas. Initial Raman spectroscopy studies on live cells acquired spectra of cells immersed in PBS. One of the earliest studies occurred in 2005, when Wood et al. used Raman spectroscopy in order to image the oxygenation process of haemoglobin in human erythrocytes. Red blood cells were isolated from healthy volunteers and suspended in PBS before being transferred to a Petri dish for Raman measurements. Cells were imaged in the 1210–1250 cm−1 region over a period of 90 minutes; during the first 50 minutes, cells were deoxygenated (by the addition of nitrogen), before being reoxygenated by atmospheric oxygen. Changes in the intensity of the measured band meant that the process could be followed and demonstrated the potential of Raman spectroscopy for single cell analysis of erythrocytes.47 Confocal Raman spectroscopy has also been applied to other cell types, and has been used to construct maps of lung carcinoma cells,49 and glioma cells,50 showing the distribution of nucleic acids, cell membrane lipids and proteins in both of these cell types. Both of these studies had an acquisition time of between 45–60 minutes, and while there was little loss of cell viability,49 this acquisition time is still long for biological samples and could be improved. More recent studies, including the investigation of hemozoin uptake in macrophages51 demonstrate that collection times can be reduced to 5–12 minutes depending on the cell size. The resonance effect can also be exploited in order to reduce acquisition times. In this technique, the frequency of the laser is adjusted so that it coincides with an electronic transition of the molecule of interest in order to enhance the Raman scatter and allow for high contrast spatial distribution imaging.3 This has been used in a number of Raman experiments to map the intracellular distribution of a number of different molecules in fixed cells,52,53 and more recently in living cells to visualise changes in cytochrome c distribution during apoptosis.39 Studies such as these highlight the potential of Raman spectroscopy for not only determining different cell types, but also as an analytical tool to monitor biochemical changes in single cells in response to changes in the medium and cell culture conditions.
While studies in PBS have proved useful for studying biochemical processes, they do not give an accurate representation of normal physiological conditions, or as close as is possible in vitro. The need for in vitro models is great, especially in the field of drug development and cellular response to specific compounds, but also in terms of studying differences between normal and diseased or malignant cells and tissues in order to potentially develop Raman spectroscopy as a diagnostic tool. This has led to the development of Raman spectrometers that can be coupled to a cell incubator, allowing cells to be maintained under normal physiological conditions (37 °C and 5% carbon dioxide).32 In ‘true’ live cell studies, cells are usually seeded directly onto the Raman substrate, held within sample holders or chambers – these are then immersed in medium and placed inside the incubator within the Raman microscope (Fig. 3). Several commercial microscope incubators are now available that include the incubator and the facilities to control the humidity, temperature and carbon dioxide and oxygen levels enabling cells to be kept alive for days at a time. Single cells have been mapped in this way by confocal microscopy to visualise the nucleus and cytoplasm of human breast cancer cells over a period of several hours, showing the potential of Raman spectroscopy in time-course studies.32
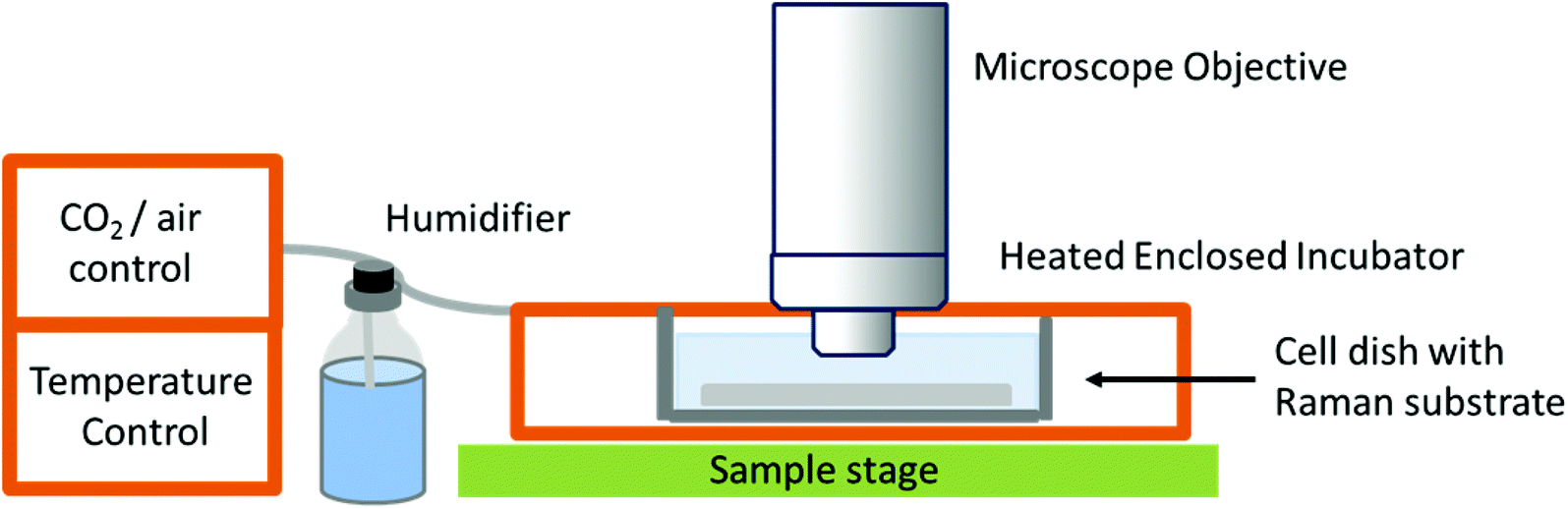 | ||
| Fig. 3 Schematic of a typical live cell incubator set-up for combining with Raman spectroscopy. While this is the typical set up by commercial manufacturers, some live cell experiments for long-term studies use an inverted microscope as unlike this set-up, the microscope objective does not need to be immersed in media.32 | ||
At present there are two major limitations in the translation of live cell Raman spectroscopy from the laboratory into the clinic: difficulties in analysing the data and as previously mentioned slow acquisition times. There is often a misconception outside of the field of Raman spectroscopy that data analysis is difficult and time consuming for non-specialists. Although there is no standardized approach for how to do this, with different groups using different methods,34 rapid improvements in data analysis software supplied with Raman spectrometers is making it easier and quicker to convert the Raman maps into meaningful pictures. However, there is still a need for those carrying out single cell Raman spectroscopy to work together to establish robust and reliable protocols that are user-friendly and applicable to cell biology. As previously discussed, progress is being made with the second limitation of slow acquisition times.
One method to increase imaging speed as well as gaining increased chemical specificity is stable-isotope labelling (SILAC), primarily using amino acid isotopes. These can be easily incorporated into all newly synthesised proteins within cells by being added to the culture medium, and are an exciting labelling method as, unlike the fluorophores mentioned earlier in this review, there is little chemical difference between these isotopes and the natural amino acid, meaning that cellular behaviour will not be altered.54 The incorporation of stable isotopes within cells will shift the Raman band at a particular frequency which can be observed and used for labelling purposes. This technique has been used in a number of Raman-based studies to further investigate cell physiology and behaviour beyond the limitations of traditional Raman microscopic techniques. SILAC has been combined with Raman spectroscopy to show the previously unobserved localisation of newly synthesised proteins to lipid droplets in live yeast cells,55 to visualise the uptake and distribution of labelled lipids and the subsequent formation of cytosolic lipid droplets during the process of macrophage foam cell formation,56 to study carbon flow from an amino acid source to predatory nematodes via E. coli,57 and to, for the first time, observe the exchange of amino acids between a host cell and an infecting parasite, T. gondii, in real time.58
As well as improvements in collection times for conventional Raman spectroscopy two further Raman approaches have been shown to significantly reduce acquisition times: coherent anti-Stokes Raman spectroscopy (CARS) and surface enhanced Raman spectroscopy (SERS).
CARS is a nonlinear technique that measures the anti-Stokes shift rather than the Stokes shift. CARS achieves this by using a pump beam at frequency ωp and a Stokes beam at frequency ωs to tightly focus onto a cell, generating an anti-Stokes signal at a frequency of 2ωp − ωs. If this frequency is equal to the frequency of the molecular vibration of a particular chemical bond, then a strong signal is generated as all these molecules are converted into a vibrational state at the same time.59 As only a single vibrational frequency is examined at any one time, CARS is a highly selective imaging technique with a signal that is 5–6 orders of magnitude greater than that observed in the spontaneous Raman scattering process, meaning that CARS is particularly useful for the study of specific molecules within a cell. As traditional CARS is limited to study only a single Raman peak, it has most commonly been used to examine lipid distribution in cells, due to the abundance of C–H bonds within lipids. It has been used to map lipid droplet distribution by measuring the vibrational frequency at 2845 cm−1, and to follow cellular differentiation of live fibroblast cells into fat cells over a period of several days.60 It has also been used to visualise the nuclear membrane during interphase and chromosomes during metaphase in living cells, as well as to observe the process of apoptosis, again in fibroblast cells.61
SERS is a technique that aims to increase the weak Raman intensity associated with spontaneous scattering, and in some cases has increased the intensity to such an extent that single molecule detection has been achievable.27 In SERS, a metallic nanostructure is used as a substrate (typically silver or gold); when this surface interacts with incident photons, an enhanced electric field is generated, increasing Raman scattering from molecules near to or adsorbed onto the substrate (reviewed in detail in ref. 62). For cell-based studies, cells can be incubated with a metal colloid suspension in order to give high resolution intracellular images with a short acquisition time.63 These nanoparticles can be targeted towards organelles of interest in order to map organelles within living cells, and this technique has been used in a number of different studies. Gold nanoparticles have been used in order to visualise the nucleus of human oral squamous carcinoma cells as they passed through the cell cycle over a period of 24 hours, showing clearly the changes in nuclear structure as cells progressed from G1 through to M phase.64 Another recent study used dye-coded gold nanoparticles modified with peptides to target the cytoplasm, mitochondria and nucleus of human oral cancer cells. Cells were incubated with these particles for either three, six or twelve hours prior to Raman spectral acquisition. The use of gold nanoparticles increased the Raman intensity by such a factor that acquisition times of 10 ms per pixel could be used to map mitochondria and the nucleus, resulting in a total spectral acquisition time of just 27.5 seconds.65
It is worth noting that a number of other instrumental or sampling approaches can be used in order to increase imaging speed without resorting to CARS and SERS in order to obtain a full spectrum across the whole cell without the introduction of nanoparticles. Selective sampling can be used in order to reduce acquisition times, and involves taking the Raman spectra at selected points rather than raster scanning the whole sample, and is particularly useful in studies were only a particular part or structure of the cell is of interest. This methodology has been used in order to measure Raman spectra of a parasite, Neospora caninum, invading host cells, reducing acquisition times by up to ten times in comparison to raster scanning.66 Multi-focal scanning is another technique that can increase the speed of spectral acquisition, and has been able to acquire Raman maps of bacterial spores in ∼1 minute.67
Together, these studies demonstrate the ability of Raman spectroscopy to provide valuable biochemical information from living cells in a relatively short space of time, allowing for the real-time observation and monitoring of cellular processes in vitro in a way that other current imaging techniques cannot.
Current and potential applications of live-cell imaging
Different cell types express different molecular markers, and these can be exploited in order to characterise and sort specific cell types from a heterogeneous population of cells. Pascut et al. utilised molecular markers specific for hESC-derived cardiomyocytes, glycogen (band at 860 cm−1) and myofibril proteins (band at 938 cm−1), in order to specifically image cardiomyocytes in a population of other hESC-derived cells with a high level of accuracy for phenotypic identification.70 This study demonstrated the potential of Raman spectroscopy in characterising hESC phenotype in a non-invasive manner, which is vital for the cell sorting and purification required for the clinical application of stem cell based therapies, but acquisition times were long and therefore inappropriate for translation into the clinic. However, the same group managed to decrease acquisition times to just 5 seconds per cell in a more recent study,71 making strides towards Raman spectroscopy mediated cell identification and sorting to be used in the clinic. The same technique has been applied to neural stem cells, using RNA content as a molecular marker (peak at 813 cm−1) in order to map them and to distinguish these cells from glial cells72 (see Fig. 4), demonstrating the versatility of this technique to detect different stem cell phenotypes.
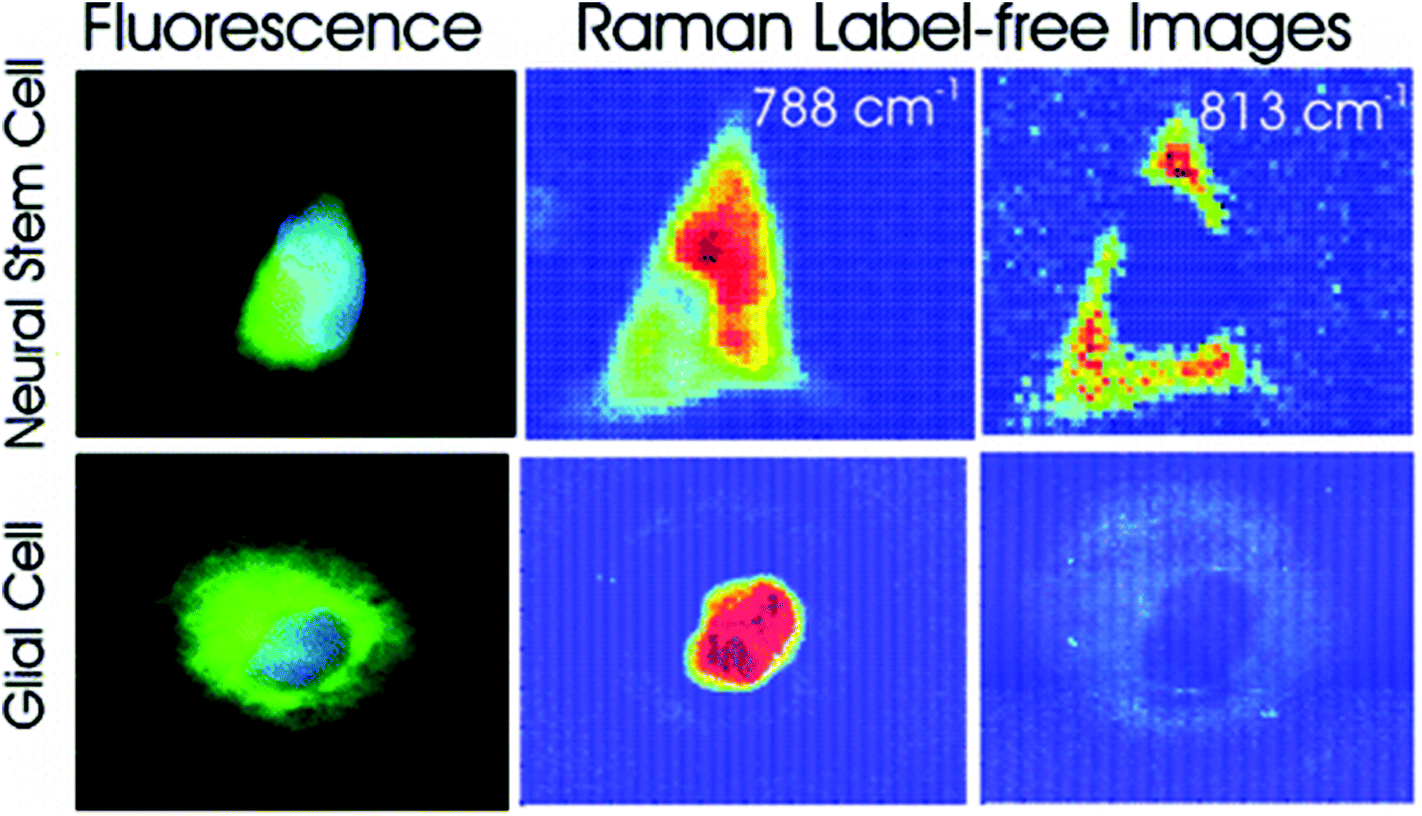 | ||
| Fig. 4 Fluorescent and Raman images demonstrating the differences between glial and neural stem cells. The Raman band at 788 cm−1 is associated with the nucleic acids of DNA and RNA, whilst the band at 813 cm−1 is associated with RNA only. High levels of RNA are observed in the cytoplasm neural stem cells, but RNA is absent in the cytoplasm of glial cells, demonstrating that Raman spectroscopy can be used to non-invasively distinguish cell phenotype. Reproduced with permission from the American Chemical Society, Copyright 2012, ref. 72. | ||
More recently, Raman spectroscopy has been used to characterise the differentiation of adipose derived stem cells (ADSCs) over a period of 14 days73 and dental pulp stromal cells (DPSCs) over a period of 28 days74 under aseptic conditions. The authors used customised, sterilised cell culture flasks with quartz windows in order to achieve this, and showed that it is possible to follow the differentiation of these cells by monitoring key biochemical changes that occur during the process. The fact that the sterility of the culture was maintained in these experiments is significant, as the same cells can be expanded and used in further experiments, saving time and money over traditional characterisation techniques such as flow cytometry and immunocytochemistry.75 While these methods are not yet applicable to the clinic with a reduction in differentiation seen in one study73 and long acquisition times in the other,74 they further demonstrate the potential of Raman spectroscopy in the identification and characterisation of stem cell phenotype and, with further developments, may be able to be translated into the clinic.
Pharmaceutical applications
Perhaps one of the most exciting potential applications of Raman spectroscopy is in the study of pharmaceutical compounds. In vitro studies of drug distribution and cellular response can be monitored in real-time, potentially reducing both the financial burden of the early stages of drug development and the need for animal studies, especially in regards to toxicity testing. Apoptosis, the cellular response to toxic agents, can be detected by changes in the Raman spectra of cells, and visualised by Raman mapping. This technique has been used to show a build-up of lipids in the cytoplasm of etoposide treated breast cancer cells,76 and the loss of cell volume and cell shrinkage associated with docetaxel treatment in a human adenocarcinoma cell line.77 If the drug of interest has a peak in its Raman spectra that differs from or is outside the range of the typical cellular components, then the drug itself can be visualised intracellularly in a label-free manner. This technique has been used to show the distribution of the anti-cancer drug, paclitaxel, in a breast cancer cell line by measuring the Raman peak at 1740 cm−1, representative of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond vibration, to show the movement of the drug through the cell membrane and into the cytoplasm.78
O bond vibration, to show the movement of the drug through the cell membrane and into the cytoplasm.78
SERS is a particularly useful technique in this field, as nanoparticles can be used not only to enhance the Raman signal, but to aid cellular uptake of the drug of interest and allow for targeted drug delivery. Gold-coated silver nanoparticles have been used to track the movement and cellular metabolism of an anti-tumour drug, 6MP, in a human lung adenocarcinoma cell line over a period of 24 hours,79 suggesting that SERS is an efficient method for studying drug efficacy (demonstrated in Fig. 5). In addition, SERS nanoparticles can be designed not only to be conjugated to the drug of interest, but also to have an outer layer that is pH sensitive, allowing for pH-dependent drug release. This technique has been used to deliver doxorubin to living cervical cancer cells in vitro. The nanoparticles were conjugated with transferrin, which is overexpressed in these cells, for specific targeting, and SERS maps were constructed to show drug internalisation.80 As most cancer cells exist within an acidic environment due to the nature of their metabolism,81 pH-sensitive nanoparticles conjugated to toxic agents may represent a novel way to develop new anti-cancer treatments.
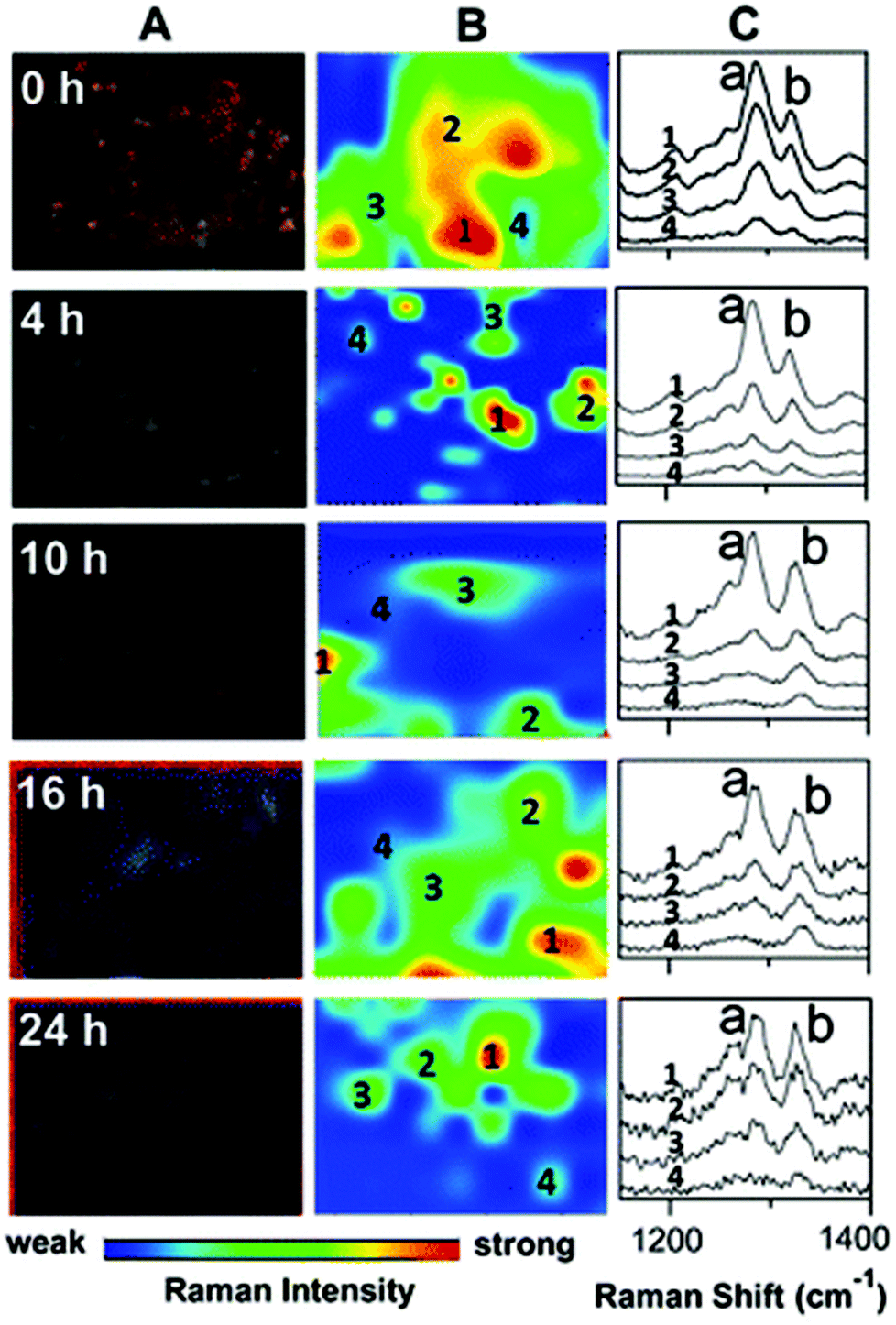 | ||
| Fig. 5 Darkfield microscopy images (A), Raman maps (B) and Raman spectra (C) to show the metabolism of 6MP in A549 cells over a period of 24 hours. At 0 hour Raman intensity was mapped at 1286 cm−1 (peak a) to show the distribution of 6MP. Subsequent maps use the Raman intensity of 1330 cm−1 (peak b), representative of 6MPR, one of the active anti-cancer agents formed from the breakdown of 6MP. 6MPR began to form at 4 hours, and the concentration increased with increasing time as the concentration of 6MP decreased. This demonstrates the successful development of a label-free Raman based methodology to monitor and visualise the metabolism of 6MP in living cells. Reproduced with permission from the American Chemical Society, Copyright 2014, ref. 79. | ||
Together, all of the above studies show the potential of Raman microscopy in the development of pharmaceutical compounds, not only for toxicity studies but to assess intracellular distribution and the effect different drugs have on different cell lines in addition to allowing for targeted delivery, and suggest that Raman microscopy may be a feasible method of predicting the in vivo response of a drug in vitro.
Diagnostics
Another major potential application of Raman spectroscopy is in the diagnosis of disease. Many diseases originate from underlying biochemical changes within cells, and as Raman spectroscopy is able to detect these subtle changes, sometimes with a high level of sensitivity, its potential in diagnostics cannot be ignored. Raman spectroscopy could potentially be used in the diagnosis of a number of different diseases from atherosclerosis82,83 to osteoarthritis,84 but it is particularly relevant in cancer research. As with fixed cells, Raman spectroscopy has been used to examine the differences between normal and neoplastic living cells, mainly by their Raman spectra,85,86 and SERS can be used to probe living cells for specific markers. Many cancer cells express proteins that are not usually found in normal cells of the same phenotype, and detection of these proteins may aid in the diagnosis of disease. The feasibility of SERS in this field has previously been demonstrated in several studies. Both silver87 and gold88 nanoparticles conjugated to an antibody specific for HER2, a receptor overexpressed on the surface of many breast cancer cells, have been used in order to map these cells live (see Fig. 6). There is the potential for this technique to be developed further and to be used in in vivo studies. Already, several studies have been conducted on murine models, primarily collecting Raman spectra rather than mapping individual cells themselves,89,90 and there is the real possibility that these techniques may be able to be developed further until they are clinically relevant. The combination of Raman spectroscopy and endoscopy has already been employed in the diagnosis of oesophageal cancer with an accuracy of 96%,91 suggesting that, with further development in order to distinguish between healthy and cancerous tissue of other cells and tissues within the body, Raman spectroscopy could become an important clinical diagnostic tool in the future.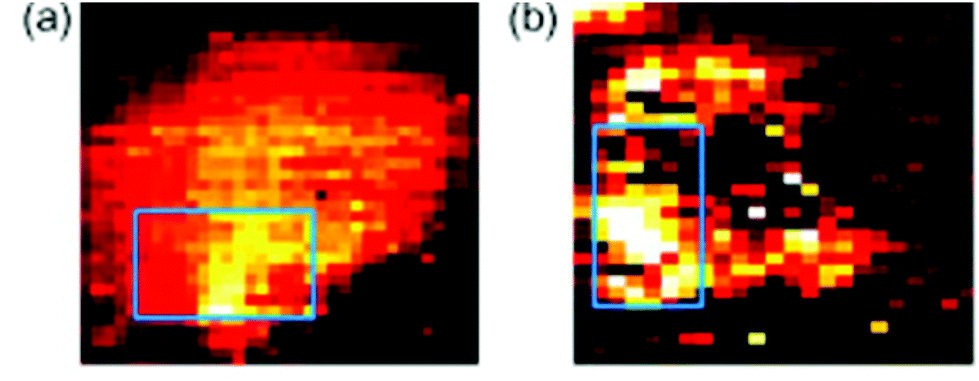 | ||
| Fig. 6 SERS mapping images of a MCF7 cell expressing HER-2 using hollow gold nanoparticles (a) and silver nanoparticles (b) specific for HER-2 at 1620 cm−1, demonstrating the feasibility of using SERS probes for cancer detection. Reproduced, with permission from Elsevier, Copyright 2009, ref. 87. | ||
The use of SERS probes may also be particularly useful in the detection of cancerous cells within solution, such as blood or urine for non-invasive diagnosis of disease,92 with relevance to leukaemia,93 and bladder cancer.94 The development of optofluidic cell sorting systems has meant that single cells in solution can be identified and sorted according to their Raman spectra95 with a high specificity of 99% for the discrimination of leukocytes, leukemic cells, and two different breast cancer cell lines,96 giving further evidence towards the use of Raman spectroscopy in this type of diagnosis.
Overall, these studies demonstrate the potential of Raman spectroscopy in cancer diagnosis. Current diagnostic techniques do not give accurate biochemical information about a sample in the way that Raman spectroscopy is able to, and often rely on examining visual differences that may be misinterpreted. There is therefore an interest in developing more accurate techniques, and there is evidence that Raman spectroscopy may be a viable, label-free and non-invasive alternative.
3D imaging
3D cell cultures are becoming an increasingly used and important in vitro model in a number of different biological applications, from cellular differentiation to drug development, due to their ability to more closely replicate the in vivo environment than 2D models are currently able.97 While 2D cultures exist in a single plane and can be captured by a single image, 3D cultures are distributed over a number of planes in z-direction; as such, analysis of 3D cultures has proved difficult as most techniques currently in use were developed for 2D cultures. To image 3D cultures, a number of images need to be acquired at different focal positions (z-stack), and as confocal Raman spectroscopy is able to do this, it is an attractive option for the 3D imaging of cell cultures.98 A good example of this approach is demonstrated by Majzner et al. (Fig. 7) who successfully used confocal Raman spectroscopy for the 3D mapping of individual endothelial cells in vitro and ex vivo within the vascular wall.99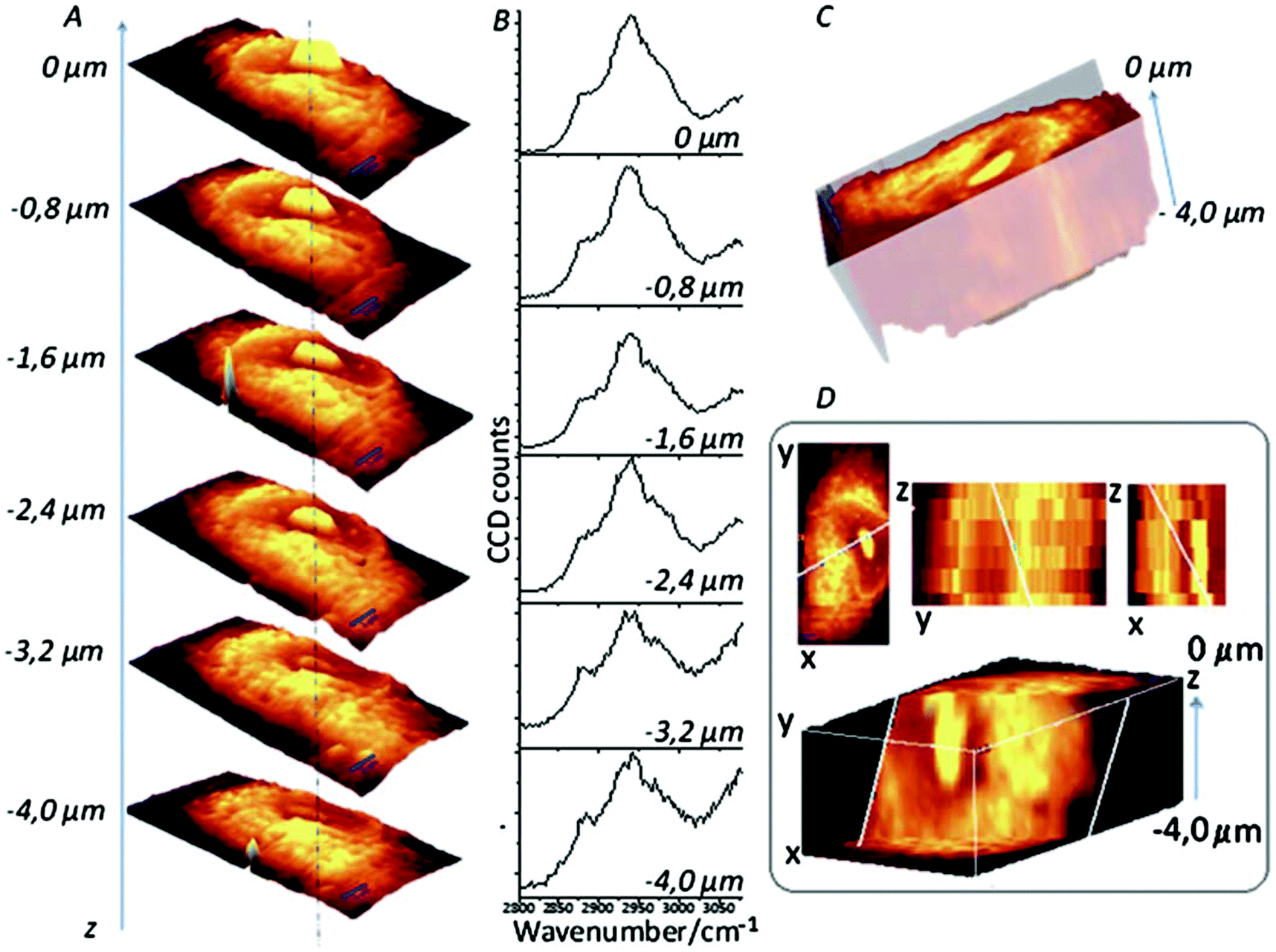 | ||
| Fig. 7 Confocal Raman imaging-stack of an EA.hy 926 cell covering 16.7 × 34.1 × 4 μm3. Integration map (A) over the νC–H band (2800–3020 cm−1 range), single spectra (B) extracted from the same point in the maps (dotted line), a reconstructed 3D image seen from the top (C) and a cross-section of the reconstructed 3D image (D). Reproduced with permission from the Royal Society of Chemistry, Copyright 2013, ref. 99. | ||
SERS nanotags have also been utilised to construct a 3D image of Chinese Hamster Ovarian cells, demonstrating the potential of SERS tags in detecting and mapping intracellular structures.100 While in both of these studies, cells were fixed, they show that Raman spectroscopy is a viable technique for 3D imaging in a non-disruptive manner that, if applied to live cells, could lead to an increased understanding of cellular dysfunction in disease by detecting changes in the underlying architecture. In the case of endothelial cells, it could lead to the development of suitable models for vascular diseases, and outlines the potential of 3D Raman mapping in diagnostics. SERS has been used for the 3D imaging of cellular pathways within living cells. A gold nanoparticle was endocytosed by macrophages and its movement tracked over time to show interactions with intracellular structures associated with cellular transport e.g. microtubule-associated proteins dynein and kinesin.101 The ability to track intracellular movement and measure the interaction between nanoparticles and intracellular molecules in 3D is an exciting prospect in cell-based studies, but particularly in drug development as it may allow for the visualisation of drugs of interest in a 3D environment close to in vivo conditions.
Conclusion
To summarise, Raman spectroscopy is a spectroscopic technique that detects molecular vibrations in order to characterise the underlying chemical structure of a sample. It is particularly relevant in biology as it allows for the non-invasive, label-free observation and imaging of cells under normal physiological conditions in a way that current imaging techniques cannot. Raman imaging, especially live cell imaging, is a developing technique, and as we have demonstrated already shows potential application in a number of different biomedical fields. These include (but are not limited to): cell characterisation and sorting; the development and testing of new pharmaceutical compounds; the diagnosis and detection of disease; and in 3D imaging. While there are still some limitations of Raman spectroscopy, such as the time it takes to acquire maps and a lack of robustness and consistency in the way Raman maps are reported,34 there is hope that new developments and advancements can overcome this and lead to Raman spectroscopy becoming a more accessible and widely-used imaging technique in the future.References
- H. Lodish, C. Kaiser, A. Bretscher, A. Amon, A. Berk, M. Krieger, H. Ploegh and M. Scott, Molecular Cell Biology, W. H. Freeman, 7th edn, 2012 Search PubMed.
- C. V. Raman and K. S. Krishnan, Nature, 1928, 121, 501–502 CrossRef CAS.
- E. Smith and G. Dent, Modern Raman Spectroscopy - A Practical Approach, John Wiley & Sons, Chichester, 2005 Search PubMed.
- A. F. Palonpon, M. Sodeoka and K. Fujita, Curr. Opin. Chem. Biol., 2013, 17, 708–715 CrossRef CAS PubMed.
- A. J. Koster and J. Klumperman, Nat. Rev. Mol. Cell Biol., 2003, Ss6–Ss10, DOI:10.1038/nrm1194.
- D. J. Stephens and V. J. Allan, Science, 2003, 300, 82–86 CrossRef CAS PubMed.
- S. M. Wilson and A. Bacic, Nat. Protoc., 2012, 7, 1716–1727 CrossRef CAS PubMed.
- R. Henriques, C. Griffiths, E. H. Rego and M. M. Mhlanga, Biopolymers, 2011, 95, 322–331 CrossRef CAS PubMed.
- S. T. Hess, T. J. Gould, M. V. Gudheti, S. A. Maas, K. D. Mills and J. Zimmerberg, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17370–17375 CrossRef CAS PubMed.
- R. Wombacher, M. Heidbreder, S. van de Linde, M. P. Sheetz, M. Heilemann, V. W. Cornish and M. Sauer, Nat. Methods, 2010, 7, 717–719 CrossRef CAS PubMed.
- S. A. Jones, S. H. Shim, J. He and X. W. Zhuang, Nat. Methods, 2011, 8, 499–U496 CrossRef CAS PubMed.
- R. B. Sekar and A. Periasamy, J. Cell Biol., 2003, 160, 629–633 CrossRef CAS PubMed.
- K. Xu, H. P. Babcock and X. W. Zhuang, Nat. Methods, 2012, 9, 185–188 CrossRef CAS PubMed.
- E. M. Denholm and G. P. Stankus, Cytometry, 1995, 19, 366–369 CrossRef CAS PubMed.
- K. B. Abbitt, G. E. Rainger and G. B. Nash, J. Immunol. Methods, 2000, 239, 109–119 CrossRef CAS PubMed.
- S. M. Ali, F. Bonnier, H. Lambkin, K. Flynn, V. McDonagh, C. Healy, T. C. Lee, F. M. Lyng and H. J. Byrne, Anal. Methods, 2013, 5, 2281–2291 RSC.
- D. H. Rank and R. V. Wiegand, J. Opt. Soc. Am., 1946, 36, 325–334 CrossRef PubMed.
- R. F. Stamm and C. F. Salzman, J. Opt. Soc. Am., 1953, 43, 126–137 CrossRef CAS.
- M. Minsky, Microscopy Apparatus, US3013467, 1961.
- A. L. Schawlow and C. H. Townes, US2929922, 1960.
- P. D. Maker and R. W. Terhune, Phys. Rev. Online Archive, 1965, 137, A801–A818 Search PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- M. Delhaye and P. Dhamelincourt, J. Raman Spectrosc., 1975, 3, 33–43 CrossRef CAS.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- S. B. Dierker, C. A. Murray, J. D. Legrange and N. E. Schlotter, Chem. Phys. Lett., 1987, 137, 453–457 CrossRef CAS.
- G. J. Puppels, F. F. de Mul, C. Otto, J. Greve, M. Robert-Nicoud, D. J. Arndt-Jovin and T. M. Jovin, Nature, 1990, 347, 301–303 CrossRef CAS PubMed.
- S. Nie, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- A. Zumbusch, G. R. Holtom and X. S. Xie, Phys. Rev. Lett., 1999, 82, 4142–4145 CrossRef CAS.
- C. L. Evans, E. O. Potma, M. Puoris'haag, D. Cote, C. P. Lin and X. S. Xie, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16807–16812 CrossRef CAS PubMed.
- G. J. Puppels, J. H. F. Olminkhof, G. M. J. Segers-Nolten, C. Otto, F. F. M. De Mul and J. Greve, Exp. Cell Res., 1991, 195, 361–367 CrossRef CAS PubMed.
- A. Zoladek, F. Pascut, P. Patel and I. Notingher, Spectroscopy, 2010, 24, 131–136 CrossRef CAS.
- S. Schlucker, M. D. Schaeberle, S. W. Huffman and I. W. Levin, Anal. Chem., 2003, 75, 4312–4318 CrossRef PubMed.
- L. Ashton, K. A. Hollywood and R. Goodacre, Analyst, 2015, 140, 1852–1858 RSC.
- W. J. Howat and B. A. Wilson, Methods, 2014, 70, 12–19 CrossRef CAS PubMed.
- N. Uzunbajakava, A. Lenferink, Y. Kraan, E. Volokhina, G. Vrensen, J. Greve and C. Otto, Biophys. J., 2003, 84, 3968–3981 CrossRef CAS PubMed.
- N. Uzunbajakava, A. Lenferink, Y. Kraan, B. Willekens, G. Vrensen, J. Greve and C. Otto, Biopolymers, 2003, 72, 1–9 CrossRef CAS PubMed.
- H. Manen, Y. Kraan, D. Roos and C. Otto, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10159–10164 CrossRef PubMed.
- M. Okada, N. I. Smith, A. F. Palonpon, H. Endo, S. Kawata, M. Sodeoka and K. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 28–32 CrossRef CAS PubMed.
- C. Matthäus, S. Boydston-White, M. Miljković, M. Romeo and M. Diem, Appl. Spectrosc., 2006, 60, 1–8 CrossRef PubMed.
- C. Krafft, T. Knetschke, R. H. W. Funk and R. Salzer, Anal. Chem., 2006, 78, 4424–4429 CrossRef CAS PubMed.
- A. S. Haka, K. E. Shafer-Peltier, M. Fitzmaurice, J. Crowe, R. R. Dasari and M. S. Feld, Cancer Res., 2002, 62, 5375–5380 CAS.
- J. Chan, D. Taylor and D. Thompson, Biopolymers, 2009, 91, 132–139 CrossRef CAS PubMed.
- A. N. Kuzmin, A. Pliss and P. N. Prasad, Anal. Chem., 2014, 10909–10916 CrossRef CAS PubMed.
- F. Draux, C. Gobinet, J. Sulé-Suso, A. Trussardi, M. Manfait, P. Jeannesson and G. D. Sockalingum, Anal. Bioanal. Chem., 2010, 397, 2727–2737 CrossRef CAS PubMed.
- A. D. Meade, C. Clarke, F. Draux, G. D. Sockalingum, M. Manfait, F. M. Lyng and H. J. Byrne, Anal. Bioanal. Chem., 2010, 396, 1781–1791 CrossRef CAS PubMed.
- B. R. Wood, L. Hammer, L. Davis and D. McNaughton, J. Biomed. Opt., 2005, 10, 14005–14005 CrossRef PubMed.
- I. Notingher, S. Verrier, H. Romanska, A. E. Bishop, J. M. Polak and L. L. Hench, Spectroscopy, 2002, 16, 43–51 CrossRef CAS.
- F. Draux, P. Jeannesson, A. Beljebbar, A. Tfayli, N. Fourre, M. Manfait, J. Sulé-Suso and G. D. Sockalingum, Analyst, 2009, 134, 542–548 RSC.
- K. Klein, A. M. Gigler, T. Aschenbrenner, R. Monetti, W. Bunk, F. Jamitzky, G. Morfill, R. W. Stark and J. Schlegel, Biophys. J., 2012, 102, 360–368 CrossRef CAS PubMed.
- A. J. Hobro, N. Pavillon, K. Fujita, M. Ozkan, C. Coban and N. I. Smith, Analyst, 2015, 140, 2350–2359 RSC.
- H. J. van Manen, N. Uzunbajakava, R. van Bruggen, D. Roos and C. Otto, J. Am. Chem. Soc., 2003, 125, 12112–12113 CrossRef CAS PubMed.
- H. J. van Manen, Y. M. Kraan, D. Roos and C. Otto, J. Phys. Chem. B, 2004, 108, 18762–18771 CrossRef CAS.
- S. E. Ong, B. Blagoev, I. Kratchmarova, D. B. Kristensen, H. Steen, A. Pandey and M. Mann, Mol. Cell Proteomics, 2002, 1, 376–386 CAS.
- H. N. N. Venkata and S. Shigeto, Chem. Biol., 2012, 19, 1373–1380 CrossRef PubMed.
- C. Matthaus, C. Krafft, B. Dietzek, B. R. Brehm, S. Lorkowski and J. Popp, Anal. Chem., 2012, 84, 8549–8556 CrossRef PubMed.
- M. Q. Li, W. E. Huang, C. M. Gibson, P. W. Fowler and A. Jousset, Anal. Chem., 2013, 85, 1642–1649 CrossRef CAS PubMed.
- A. Naemat, H. M. Elsheikha, R. A. Boitor and I. Notingher, Sci. Rep., 2016, 6 DOI:10.1038/srep20811.
- F. El-Diasty, Vib. Spectrosc., 2011, 55, 1–37 CrossRef CAS.
- X. Nan, J.-X. Cheng and X. S. Xie, J. Lipid Res., 2003, 44, 2202–2208 CrossRef CAS PubMed.
- J.-X. Cheng, Y. K. Jia, G. Zheng and X. S. Xie, Biophys. J., 2002, 83, 502–509 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- A. F. Palonpon, J. Ando, H. Yamakoshi, K. Dodo, M. Sodeoka, S. Kawata and K. Fujita, Nat. Protoc., 2013, 8, 677–692 CrossRef CAS PubMed.
- B. Kang, L. A. Austin and M. A. El-Sayed, Nano Lett., 2012, 12, 5369–5375 CrossRef CAS PubMed.
- J. W. Kang, P. T. C. So, R. R. Dasari and D.-K. Lim, Nano Lett., 2015, 15(3), 1766–1772 CrossRef CAS PubMed.
- K. Kong, C. J. Rowlands, H. Elsheikha and I. Notingher, Analyst, 2012, 137, 4119–4122 RSC.
- L. B. Kong, P. F. Zhang, J. Yu, P. Setlow and Y. Q. Li, Appl. Phys. Lett., 2011, 98, 213703 CrossRef.
- I. J. Fox, G. Q. Daley, S. A. Goldman, J. Huard, T. J. Kamp and M. Trucco, Science, 2014, 345 CrossRef CAS PubMed , 1247391.
- B. E. Reubinoff, M. F. Pera, C. Y. Fong, A. Trounson and A. Bongso, Nat. Biotechnol., 2000, 18, 559–559 CrossRef.
- F. C. Pascut, H. T. Goh, N. Welch, L. D. Buttery, C. Denning and I. Notingher, Biophys. J., 2011, 100, 251–259 CrossRef CAS PubMed.
- F. C. Pascut, H. T. Goh, V. George, C. Denning and I. Notingher, J. Biomed. Opt., 2011, 16 CrossRef PubMed , 045002.
- A. Ghita, F. C. Pascut, M. Mather, V. Sottile and I. Notingher, Anal. Chem., 2012, 84, 3155–3162 CrossRef CAS PubMed.
- A. Mitchell, L. Ashton, X. B. B. Yang, R. Goodacre, A. Smith and J. Kirkham, Cytometry, Part A, 2015, 87, 1012–1019 CrossRef CAS PubMed.
- A. Mitchell, L. Ashton, X. B. B. Yang, R. Goodacre, M. J. Tomlinson, A. Smith and J. Kirkham, Analyst, 2015, 140, 7347–7354 RSC.
- M. Marti, L. Mulero, C. Pardo, C. Morera, M. Carrio, L. Laricchia-Robbio, C. R. Esteban and J. C. I. Belmonte, Nat. Protoc., 2013, 8, 223–253 CrossRef CAS PubMed.
- A. Zoladek, F. C. Pascut, P. Patel and I. Notingher, J. Raman Spectrosc., 2011, 42, 251–258 CrossRef CAS.
- K. Bräutigam, T. Bocklitz, M. Schmitt, P. Rösch and J. Popp, ChemPhysChem, 2013, 14, 550–553 CrossRef PubMed.
- H. Salehi, E. Middendorp, I. Panayotov, P.-Y. C. Dutilleul, A.-G. Vegh, S. Ramakrishnan, C. Gergely and F. Cuisinier, J. Biomed. Opt., 2013, 18, 56010–56010 CrossRef PubMed.
- G. Han, R. Liu, M. Y. Han, C. Jiang, J. Wang, S. Du, B. Liu and Z. Zhang, Anal. Chem., 2014, 86, 11503–11507 CrossRef CAS PubMed.
- W. Fang, Z. Wang, S. Zong, H. Chen, D. Zhu, Y. Zhong and Y. Cui, Biosens. Bioelectron., 2014, 57, 10–15 CrossRef CAS PubMed.
- O. Warburg, F. Wind and E. Negelein, J. Gen. Physiol., 1927, 8, 519–530 CrossRef CAS PubMed.
- A. Lattermann, C. Matthäus, N. Bergner, C. Beleites, B. F. Romeike, C. Krafft, B. R. Brehm and J. Popp, J. Biophotonics, 2013, 6, 110–121 CrossRef CAS PubMed.
- K. Marzec, T. Wrobel, A. Rygula, E. Maslak, A. Jasztal, A. Fedorowicz, S. Chlopicki and M. Baranska, Biophotonics, 2014, 7, 744–756 CrossRef CAS PubMed.
- R. Kumar, G. Singh, K. Grønhaug, N. Afseth, C. de Lange Davies, J. Drogset and M. Lilledahl, Int. J. Mol. Sci., 2015, 16, 9341–9353 CrossRef CAS PubMed.
- K. M. Omberg, J. C. Osborn, S. L. L. Zhang, J. P. Freyer, J. R. Mourant and J. R. Schoonover, Appl. Spectrosc., 2002, 56, 813–819 CrossRef CAS.
- J. W. Chan, D. S. Taylor, T. Zwerdling, S. M. Lane, K. Ihara and T. Huser, Biophys. J., 2006, 90, 648–656 CrossRef CAS PubMed.
- S. Lee, H. Chon, M. Lee, J. Choo, S. Y. Shin, Y. H. Lee, I. J. Rhyu, S. W. Son and C. H. Oh, Biosens. Bioelectron., 2009, 24, 2260–2263 CrossRef CAS PubMed.
- J. Yang, Z. Wang, S. Zong, C. Song, R. Zhang and Y. Cui, Anal. Bioanal. Chem., 2012, 402, 1093–1100 CrossRef CAS PubMed.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- U. S. Dinish, G. Balasundaram, Y.-T. Chang and M. Olivo, Sci. Rep., 2014, 4, 4075–4075 CAS.
- M. S. Bergholt, W. Zheng, K. Lin, K. Y. Ho, M. Teh, K. G. Yeoh, J. B. So and Z. Huang, Technol. Cancer Res. Treat., 2011, 10, 103–112 CAS.
- D. M. Good, V. Thongboonkerd, J. Novak, J.-L. Bascands, J. P. Schanstra, J. J. Coon, A. Dominiczak and H. Mischak, J. Proteome Res., 2007, 6, 4549–4555 CrossRef CAS PubMed.
- C. M. MacLaughlin, N. Mullaithilaga, G. Yang, S. Y. Ip, C. Wang and G. C. Walker, Langmuir, 2013, 29, 1908–1919 CrossRef CAS PubMed.
- S. Li, L. Li, Q. Zeng, Y. Zhang, Z. Guo, Z. Liu, M. Jin, C. Su, L. Lin, J. Xu and S. Liu, Sci. Rep., 2015, 5, 9582–9582 CrossRef CAS PubMed.
- A. Y. Lau, L. P. Lee and J. W. Chan, Lab Chip, 2008, 8, 1116–1120 RSC.
- S. Dochow, C. Beleites, T. Henkel, G. Mayer, J. Albert, J. Clement, C. Krafft and J. Popp, Anal. Bioanal. Chem., 2013, 405, 2743–2746 CrossRef CAS PubMed.
- M. Ravi, V. Paramesh, S. R. Kaviya, E. Anuradha and F. D. P. Solomon, J. Cell. Physiol., 2015, 230, 16–26 CrossRef CAS PubMed.
- V. Charwat, K. Schütze, W. Holnthoner, A. Lavrentieva, R. Gangnus, P. Hofbauer, C. Hoffmann, B. Angres and C. Kasper, J. Biotechnol., 2015, 205, 70–81 CrossRef CAS PubMed.
- K. Majzner, A. Kaczor, N. Kachamakova-Trojanowska, A. Fedorowicz, S. Chlopicki and M. Baranska, Analyst, 2013, 138, 603–610 RSC.
- S. McAughtrie, K. Lau, K. Faulds and D. Graham, Chem. Sci., 2013, 4, 3566–3566 RSC.
- K. C. Huang, K. Bando, J. Ando, N. I. Smith, K. Fujita and S. Kawata, Methods, 2014, 68, 348–353 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |