
Measurement issues associated with quantitative molecular biology analysis of complex food matrices for the detection of food fraud
Malcolm
Burns
*a,
Gordon
Wiseman
b,
Angus
Knight
c,
Peter
Bramley
d,
Lucy
Foster
e,
Sophie
Rollinson
e,
Andrew
Damant
f and
Sandy
Primrose
g
aLGC, Queens Road, Teddington, Middlesex, TW11 0LY, UK. E-mail: Malcolm.Burns@lgcgroup.com; Tel: +44 (0)208 943 7000
bPremier Analytical Services, Premier Foods Group Ltd., The Lord Rank Centre, Lincoln Road, High Wycombe, Bucks HP12 3QS, UK
cLeatherhead Food Research, Randalls Road, Leatherhead, Surrey KT22 7RY, UK
dSchool of Biological Sciences, Royal Holloway, University of London, Egham, Surrey TW20 0EX, UK
eDepartment for the Environment, Food and Rural Affairs, 17 Smith Square, London SW1P 3JR, UK
fFood Standards Agency, Aviation House, 125 Kingsway, London, WC2B 6NH, UK
gBusiness & Technology Management, 21 Amersham Road, High Wycombe, Bucks HP13 6QS, UK
First published on 23rd November 2015
Abstract
Following a report on a significant amount of horse DNA being detected in a beef burger product on sale to the public at a UK supermarket in early 2013, the Elliott report was published in 2014 and contained a list of recommendations for helping ensure food integrity. One of the recommendations included improving laboratory testing capacity and capability to ensure a harmonised approach for testing for food authenticity. Molecular biologists have developed exquisitely sensitive methods based on the polymerase chain reaction (PCR) or mass spectrometry for detecting the presence of particular nucleic acid or peptide/protein sequences. These methods have been shown to be specific and sensitive in terms of lower limits of applicability, but they are largely qualitative in nature. Historically, the conversion of these qualitative techniques into reliable quantitative methods has been beset with problems even when used on relatively simple sample matrices. When the methods are applied to complex sample matrices, as found in many foods, the problems are magnified resulting in a high measurement uncertainty associated with the result which may mean that the assay is not fit for purpose. However, recent advances in the technology and the understanding of molecular biology approaches have further given rise to the re-assessment of these methods for their quantitative potential. This review focuses on important issues for consideration when validating a molecular biology assay and the various factors that can impact on the measurement uncertainty of a result associated with molecular biology approaches used in detection of food fraud, with a particular focus on quantitative PCR-based and proteomics assays.
 Malcolm Burns | Malcolm Burns is the Principal Scientist and Special Adviser to the Government Chemist, based at LGC in Teddington (London, UK). He specialises in using molecular biology approaches for food authenticity testing, inclusive of quantitation of genetically modified ingredients in food and feed. Malcolm has published over 30 peer reviewed papers and EU guidance notes on detecting food adulteration. He is a member of a number of international working groups and advisory committees, providing regular consultancy and training on a range of food authenticity testing issues, inclusive of development and validation of molecular biology methods. |
 Sandy Primrose | Sandy Primrose has held senior management positions in academia and various industry sectors including pharmaceuticals, specialty chemicals, instrumentation and diagnostics. He now manages a technology consultancy specialising in product development and manufacturing and business strategy. For the past 13 years he has been the Independent Advisor to the UK government's food authenticity programme (currently managed by Defra). |
1. Introduction
On the 15th January 2013, the Food Safety Authority of Ireland (FSAI) published a report which stated that a significant amount of horse DNA had been found in some beef burger products, which were on sale at a supermarket.1 In response to this, the UK Government commissioned an independent review into the integrity and assurance of the food supply network. HM Government Elliott Review into the Integrity and Assurance of Food Supply Networks was published on Thursday 4th September 2014 and included recommendations with respect to improving systems to deter, identify and prosecute food adulteration.2 This report included advice on improving laboratory testing capability to ensure a standardised approach for testing for food authenticity. It was apparent there was a greater need to develop sensitive, specific and harmonised detection methods for meat ingredients, inclusive of those techniques that had quantitative potential. In response to this review, the Department for the Environment, Food and Rural Affairs’ (Defra's) independent Authenticity Methods Working Group (AMWG) published a report addressing aspects of harmonisation in food authenticity testing.3 The report provided pragmatic and practical guidance for stakeholders regarding ensuring that testing for food authenticity was reliable and consistent between testing laboratories.The fraudulent misdescription of foods for economic gain can mislead the consumer and impact on businesses, and can occur by the substitution of high-added value products that command a premium price with cheaper products which claim to be authentic. To prove conclusively that fraud has occurred it is necessary first to identify the authenticity of its composition as claimed on the label and then to quantify the analytes of interest, or provide evidence that they are present above a legislative or agreed threshold. Often the substituents are very similar biochemically to the materials that they replace and this makes their identification and quantification problematic. The fact that food matrices are extremely complex, variable, and can be subject to varying degrees of processing and treatment, further adds to the issue. Recently, methods based on the polymerase chain reaction (PCR) and proteomics have been shown to have the required discriminatory capability for the purposes of identification.4 These methods can also be used quantitatively.
A key issue with the misdescription of foods is distinguishing between adventitious contamination and deliberate substitution. The former can occur as a result of inadequate cleaning of equipment between processing different batches but often is not expected to exceed more than 5% on a weight or volume basis.5 On the other hand, if deliberate adulteration has occurred, the undeclared ingredient is likely to be present at more than 5%, in order to gain an economic advantage in make the deliberate substitution. Below the 5–10% level the economic gain probably is insufficient to make substitution worthwhile.
A reporting level of 1% (w/w) of meat species was adopted in the UK and the European Union (EU) following the findings of a significant amount of horse DNA found in beef burgers.1 This level for enforcement action was a pragmatic approach based on the experience of regulators, enforcement and industry of an appropriate level at which to distinguish trace contamination from deliberate adulteration.
In the EU, all materials originating from genetically modified (GM) sources must be labelled accordingly, subject to a threshold of 0.9% for adventitious presence of material from EU approved GM varieties.6 Basmati rice is a different case for in Europe a number of varieties can be imported tariff-free but adventitious contamination with unapproved varieties must be below 7% w/w according to the Basmati Code of Practice.7 If international trade is not to be disrupted it is essential that competent authorities have access to validated analytical methods.
Lack of harmonised best practice often leads to high measurement uncertainty associated with a result. However, the implications of poor practice frequently go beyond this, and have the potential to cause confusion in the minds of those who commission analytical and molecular biological services in food authenticity. This review makes a number of recommendations with respect to best practice guidance for the detection of food fraud, with a particular emphasis on quantitative approaches.
2. Method validation and interpretation of results
A significant challenge for industry, analytical laboratories, and regulatory authorities exists when evidence for fraudulent activity is uncovered using a method that has not undergone validation or a new uncharacterised adulterant is identified for the first time. Orthogonal confirmation is desirable. However accurate identification and quantification is only possible using validated methods and agreed standards. The use of validated test methods allows the precision and trueness of measurement to be obtained in relation to a defined standard.In order to demonstrate the methods a laboratory implements are fit for the purpose for which they were originally intended, method validation must be undertaken. This comprises both the process of obtaining data for the fitness for purpose of a method as well as documenting this evidence. Method validation is an essential component of the actions that a laboratory should implement to allow it to produce reliable analytical data. Methods of analysis of food are governed by EU legislation8 which describes the required validation. “Full” validation for an analytical method is usually taken to comprise an examination of the characteristics of the method in an inter-laboratory method performance study (also known as a collaborative study or collaborative trial). Internationally accepted protocols have been established for the “full” validation of a method of analysis by such a collaborative trial.9,10 These protocols/standards require a minimum number of laboratories and test materials to be included in the collaborative trial to validate fully the analytical method.
Most published literature on analytical method development, validation and quality control is focussed on classic analytical chemistry methodology rather than molecular biology or proteomics/metabolomics. However, many of the guiding principles can be applied to molecular biology methods, which form a key part of the food authenticity detection “tool kit”. For example, The Codex Committee for Methods of Analysis and Sampling (CCMAS) have developed guidelines on criteria for methods for the detection and identification of foods derived from biotechnology.11 These guidelines provide information for the validation of methods for the detection, identification, and quantification of specific DNA sequences and specific proteins in foods derived from modern biotechnology. They may also provide information on the validation of methods for other specific DNA sequences and proteins of interest in other foods. Information relating to general considerations for the validation of methods for the analysis of specific DNA sequences and specific protein in foods is given in the first part of the CCMAS guidelines. Specific annexes are provided that contain information on definitions, validation of qualitative and quantitative PCR methods, validation of protein-based methods, and proficiency testing. A similar set of method-acceptance criteria and method-performance requirements has been compiled by the European Network of GMO Laboratories (ENGL). Method-acceptance criteria are criteria that have to be fulfilled prior to the initiation of any method validation by the EU Reference Laboratory for GMOs in feed and food (EU-RL-GMFF).12 The method performance requirements define the minimum performance characteristics of the method that have to be demonstrated upon completion of a validation study carried out according to internationally accepted technical provisions. This latter requirement is needed in order to certify that the method validated is fit for the purpose of enforcement of Regulation (EC) No 1829/2003.6
In the field of genetically-modified organisms (GMOs), the modular approach to method validation has been discussed in great depth by molecular biologists.13 According to this approach, the analytical procedure can be described as a series of successive steps: sampling, sample processing, analyte extraction, and ending in interpretation of an analytical result produced with, for example, the real-time polymerase chain reaction. Precision estimates for each stage can be combined into a total precision estimate. In theory, this approach allows the analyst to tailor individual analytical steps to the analyte/matrix combination being analysed. Holst-Jensen and Berdal13 comment that the final analytical result is dependent on proper method selection and execution and is valid only if valid methods (modules) are used throughout the analytical procedure.
3. Procedures for the estimation of measurement uncertainty
All analytical results take the form “a ± ku” or “a ± U” where “a” is the best estimate of the true value of the concentration of the measurand (the analytical result), “u” is the standard uncertainty, “k” is a coverage factor based on the number of independent estimates from which “u” is derived, and “U” (equal to ku) is the expanded uncertainty. The standard uncertainty is identical to the estimated standard deviation. Whilst the coverage factor “k” can take a number of values, it is often stated as 2 in order to equate to an approximate 95% confidence interval. The range within which the true value is estimated to fall is usually given by “4u”. The value of “U” or “2u” is the value which is normally used and reported by analysts, and may be estimated and expressed in a number of different ways.Within the molecular biology area, the major work on measurement uncertainty estimation has again been undertaken within the GMO sector. Trapmann et al.14 presented two approaches for the estimation of measurement uncertainty associated with a result. The first approach uses collaborative trial data in combination with in-house quality control data for the estimation of measurement uncertainty of a result. An alternative approach using data obtained from within-laboratory sample analysis is also presented. The approaches laid down by Trapmann et al.14 are being widely implemented by European laboratories undertaking GMO analyses and the principles proposed are widely applicable to other molecular biology analyses. Despite these measures, a recent report published by the EU-RL-GMFF regarding an international comparative test for detection and quantification of GM events in rice noodles in 2014, revealed that only 58% of participants to the study provided measurement uncertainty estimates associated with a result in a complete and consistent manner.15 This highlighted the need for improvements and harmonisation in the way that analytical testing laboratories report their measurement uncertainty estimates.
There is concern that some laboratories underestimate the size of their measurement uncertainty associated with a result. For chemical analyses, using the results from collaborative trials (i.e. the top-down approach), it would not be unreasonable to anticipate that the (expanded) uncertainties reported by laboratories would be of the orders shown in Table 1.9 Within the molecular biology sector the analyte concentration being determined is often less than 100 μg kg−1. Consequently, it is not uncommon to expect expanded relative measurement uncertainties of at least 44% for analytical results obtained using PCR-based approaches.
| Concentration | Expanded uncertainty | Range of acceptable concentrations |
|---|---|---|
| 100 g/100 g | 4% | 96 to 104 g/100 g |
| 10 g/100 g | 5% | 9.5 to 10.5 g/100 g |
| 1 g/100 g | 8% | 0.92 to 1.08 g/100 g |
| 1 g kg−1 | 11% | 0.89 to 1.11 g kg−1 |
| 100 mg kg−1 | 16% | 84 to 116 mg kg−1 |
| 10 mg kg−1 | 22% | 7.8 to 12.2 mg kg−1 |
| 1 mg kg−1 | 32% | 0.68 to 1.32 mg kg−1 |
| <100 μg kg−1 | 44% | 56 to 144 μg kg−1 |
Measurement uncertainty is probably the most important single parameter that describes the quality of measurement associated with a result. However, many laboratories reporting results only report the measurement uncertainty associated with the final analysis and do not normally include the measurement uncertainty associated with sampling itself. It is widely recognised that major portion of the total measurement uncertainty budget can arise from the upstream sampling stage. The EURACHEM-CITAC Guide16 on the estimation of measurement uncertainty arising from sampling provides a set of useful tools with which the analyst can determine sampling uncertainty and thereby the total measurement uncertainty associated with a result.
4. Uncertainty in compliance assessment
In order to assess whether or not an analytical value exceeds a threshold, the measurement uncertainty of that result needs to be determined and reported. The procedure adopted by most control analysts is to report samples as containing not less than “a − 2u” in situations where the statutory limit is a maximum permissible concentration. Here any enforcement action is only taken when the analyst is sure that the specification has been exceeded. This is consistent with the requirement to prove beyond reasonable doubt that a limit has been exceeded if the case should come to Court. This means that the effective enforcement level is not identical to the numerical value given in the EU legislation. Thus the enforcement level is the tool and equates to the maximum level plus the expanded uncertainty.It is essential that the measurement uncertainty of the test result be known before deciding if the test result shows compliance or non-compliance with a specification. The reason for this is shown in Fig. 1 where four different results for the concentration of an analyte are assessed for their compliance with an agreed limit. For each result, the vertical lines show the expanded uncertainty ± U associated with a result. Based on the assumption of a normal distribution, there is a higher probability that the concentration of the analyte will lie nearer the centre of the expanded uncertainty interval than nearer the ends. For results (a) and (d) the analyte concentrations respectively are well above and well below the limit. However, for result (b) there is a high probability that the value of the analyte is above the limit but the limit is within the uncertainty interval. Similarly, for result (c) the probability that the analyte is below the limit is high but not absolute.
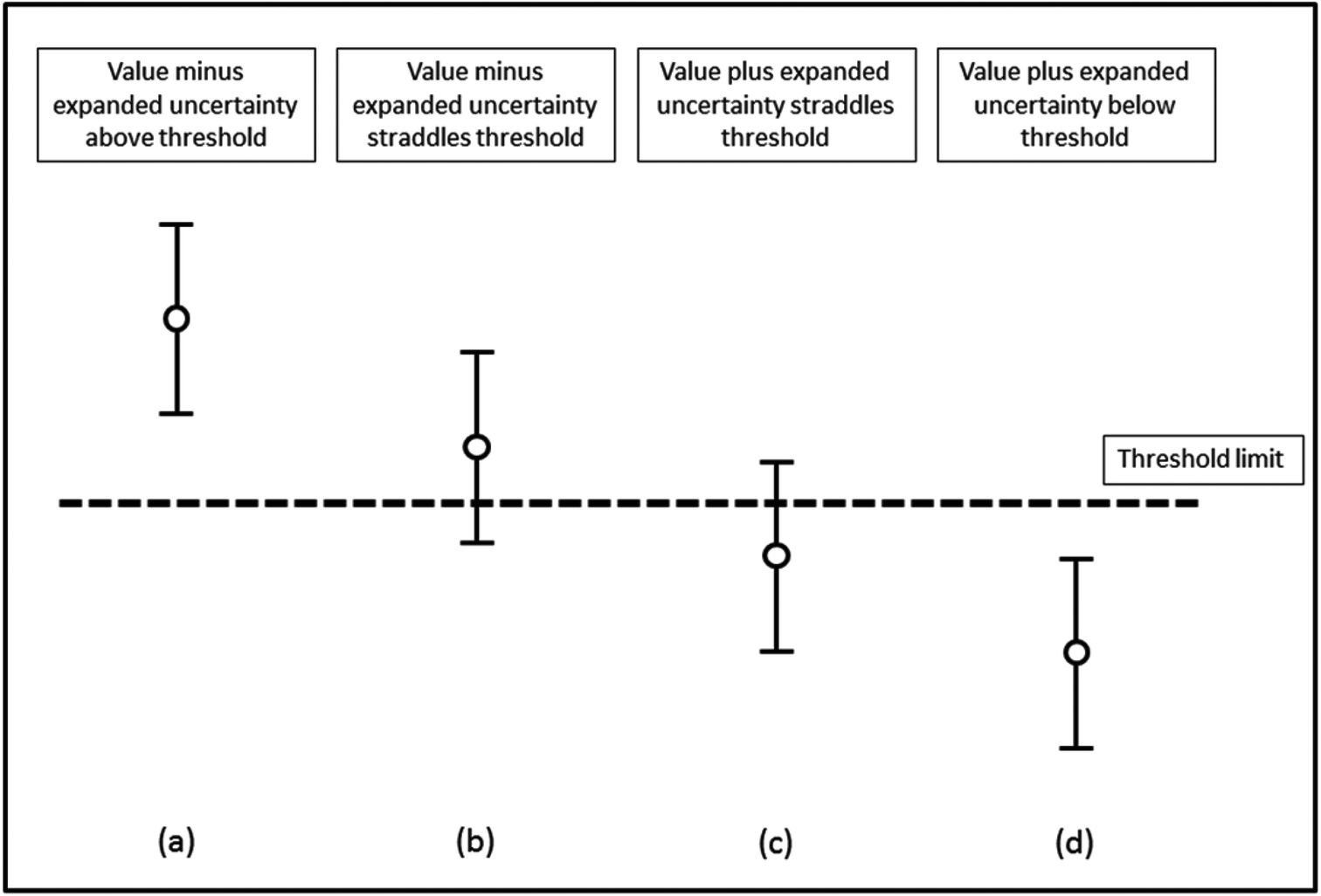 | ||
| Fig. 1 Assessment of compliance with a specification limit. Mean values and associated 95% confidence intervals are shown. | ||
It is a relatively simple matter to determine the factors contributing to uncertainty associated with the reported result for an assay where highly purified reagents are used. However, when real samples are to be analysed it is necessary to consider the total analytical procedure (Fig. 2). For example, when implementing a bottom-up approach to determine the measurement uncertainty of results obtained using a PCR-based method this will include sample preparation, DNA extraction and DNA purification steps. If the material to be analysed is blood (e.g. in a clinical assay) there will be relatively little variation in different samples and this reduces uncertainty. In the case of foodstuffs the matrices are very complex and variable and any processing that occurs only increases the variability. Consequently one expects the measurement uncertainty associated with the reported result to be high. Contributions to the overall measurement uncertainty can also occur during the PCR setup, equipment operation, software analysis, manual analysis and user interpretation stages.17 Such aspects of plasticware consumables, use of reference materials and quality of primer/probes must be carefully considered in order to minimise the uncertainty associated with the analytical result. In particular, care must be taken to ensure all analytical instruments (e.g. balances, thermal cyclers, centrifuges, etc.) are serviced and calibrated correctly.
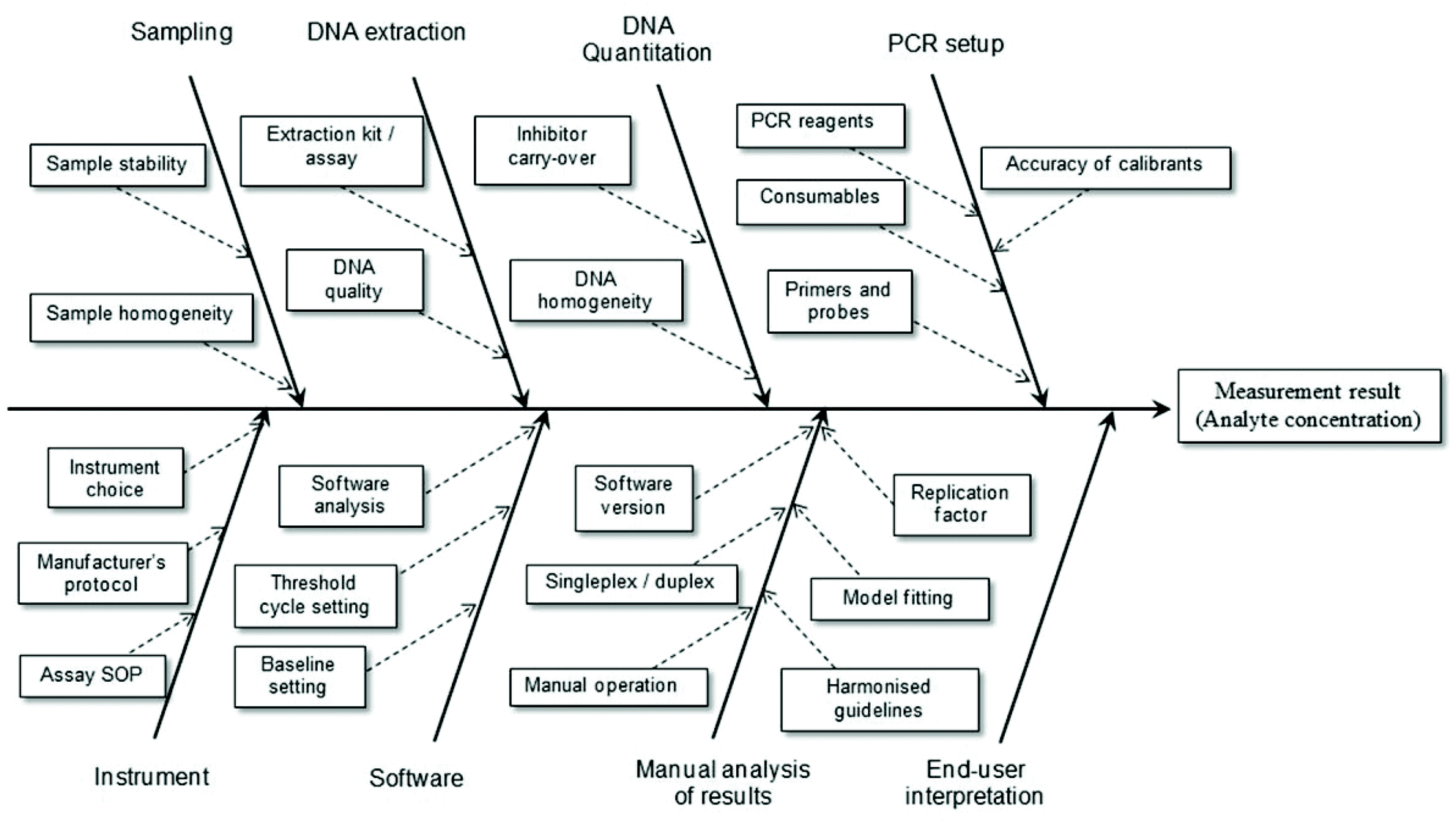 | ||
| Fig. 2 Example factors contributing to measurement uncertainty of a test result involving the use of real-time PCR. Adapted from Burns and Valdivia.17 | ||
Special attention should be paid to pipettes as their accuracy and precision need to be determined more frequently than for other instruments. Using gravimetric analysis, the performance of individual pipettes should be compared with manufacturer's specifications according to a routine schedule: for example, accuracy checks involving individual measurements may have to be conducted weekly, and precision tests involving multiple measurements may have to be done bi-annually. In addition, leak tests may have to be performed on a more regular and frequent basis.
5. Standard operating procedures (SOPs)
An essential first step in reducing analytical uncertainty is to have one or more SOPs covering all of the steps from sample selection to data evaluation. A properly written SOP is unambiguous and should ensure that different individuals in different laboratories use the same reagents and glassware and perform all the manipulative steps in exactly the same way. The UK Government's Food Authenticity Programme has prepared an SOP for writing SOPs and this is available on request from foodauthenticity@defra.gsi.gov.uk.6. Sampling
The samples chosen for analysis must be appropriate for the nature and complexity of the product. The more complex the product and/or the larger the product components, the more thought needs to be given to sampling. In this context it should be noted that there can be sampling issues even with an apparently homogeneous material such as bulk grain. A bulk load of, say, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes will be a combination of material from many different truckloads. If one of these truckloads is contaminated with GM grain at the 10% w/w level, or even is 100% GM, will this GM material be present in any of the samples that are taken? Within the GMO sector significant work has been undertaken on investigating and developing sampling strategies for the analysis of GMOs in bulk consignments.18–20 Within the Kernel lot distribution assessment (KeLDA) project20 the GMO content of 15 soybean lots imported into the EU was estimated by analysing 100 increment samples systematically sampled from each lot at predetermined time intervals during the whole off-loading process. The distribution of GMO material was inferred by the temporal distribution of contaminated increments. All the lots analysed displayed significant spatial structuring therefore indicating that randomness cannot be assumed. Evidence from the KeLDA highlights the need to develop sampling protocols for GMO analytes based upon statistical models free of distribution requirements.
000 tonnes will be a combination of material from many different truckloads. If one of these truckloads is contaminated with GM grain at the 10% w/w level, or even is 100% GM, will this GM material be present in any of the samples that are taken? Within the GMO sector significant work has been undertaken on investigating and developing sampling strategies for the analysis of GMOs in bulk consignments.18–20 Within the Kernel lot distribution assessment (KeLDA) project20 the GMO content of 15 soybean lots imported into the EU was estimated by analysing 100 increment samples systematically sampled from each lot at predetermined time intervals during the whole off-loading process. The distribution of GMO material was inferred by the temporal distribution of contaminated increments. All the lots analysed displayed significant spatial structuring therefore indicating that randomness cannot be assumed. Evidence from the KeLDA highlights the need to develop sampling protocols for GMO analytes based upon statistical models free of distribution requirements.
7. Sample preparation
Sample preparation is an essential first step in the analysis of food and can be a major source of uncertainty. Raw materials such as cereal grains and vegetable oils are reasonably homogenous and there should be little difference in extraction behaviour between a GM and a non-GM cereal grain. However, if one is looking for offal or different meat species in a meat pie then consideration needs to be given to the mechanical properties of the key components. For example, chicken is a much softer meat than pork and the two may not homogenise in the same way and thus the key analytes (DNA or protein) may not be extracted with the same efficiency. Similarly, heart, liver and kidney will not behave the same as muscle tissue. There are reports that the quantification of GM material in grain is influenced by the particle size of milled samples.21 Accurate quantification only was possible in mixtures of conventional and transgenic material in the form of analogous milling fractions. Where processing such as cooking has taken place, the degree of degradation of the analytes also may differ between meat species or tissues. This could be particularly significant with test procedures involving the PCR. Even with unprocessed materials there could be differences in extraction behaviour that reflect different growing conditions or seasonal variation. This variation cannot be eliminated or controlled. Rather, it is essential that during method validation due consideration is given to this variation when designing method validation protocols.8. Nucleic acid extraction and purification
If an analytical method is going to be validated then the repeatability of the extraction procedure needs to be determined. However, there is no definitive answer as to an acceptable value. Whereas a twofold range in the amount of analyte purified might be acceptable a tenfold range almost certainly would not. A small number of certified reference materials are available for determining the GM content of cereals. One would expect the uncertainty in the amount extracted from these reference materials to be much less than for a more complex food. The key question is what one measures when determining the repeatability of extraction. The PCR is influenced by many different factors and so it is not sufficient to measure the quantity of DNA extracted. The integrity of the DNA and its purity are of equal importance.The quality and quantity of DNA extracted from food products tend to decrease with the extent to which the food is processed because physical, chemical and enzymatic treatment of food can result in a marked decrease in DNA fragment size.22–24 With highly sheared DNA there may not be enough template DNA available for the PCR.25 An added complication is that the amount of DNA extracted is governed by the particle size of the food: as particle size diminishes the amount of DNA extracted increases.21,26 However, homogenisation of the food sample to reduce particle size might result in shearing of the DNA. The preferred method for determining if DNA has been extensively degraded is to determine its size using gel or capillary electrophoresis to ensure that there is a high mean fragment size, and minimal smear or a “tail” present which is indicative of fragmented DNA.
A number of methods have been used for quantifying either the amount of DNA that has been extracted or the amount being added to a PCR reaction. These methods are: spectrophotometry, fluorimetry and chemiluminescence. For a solution of purified double-stranded DNA that is not degraded, an absorbance value of one at 260 nm (A260) wavelength corresponds to a concentration of 50 μg mL−1.27 However, as the DNA becomes degraded the absorbance increases and this probably is due to the presence of single-stranded DNA. Note that single-stranded DNA can occur even in the absence of size degradation.28 If fluorimetry is used to determine DNA concentration then the samples first need to be incubated with a fluorescent dye such as PicoGreen®. There are three advantages of fluorimetry for determining DNA concentration. First, it is ∼100 times more sensitive than UV spectrophotometry. Second, the linear concentration range extends over four orders of magnitude. Third, it is relatively insensitive to the presence of contaminants with the notable exception of CTAB which is used in many DNA extraction protocols.28 Chemiluminescence can be used to quantify DNA. It has a sensitivity similar to that of fluorimetry but the DNA must be smaller than 6000 base pairs in length. If the DNA is larger than this then it must be reduced in size by treatment with an appropriate restriction enzyme. Also, the degree of sensitivity to quenching by other constituents of the solution is not known.
There also are issues associated with determining sample purity and this particularly is critical if the PCR is going to be used. A standard method of assessing DNA purity is to determine the A260:A280 ratio, which refers to the ratio of the absorbances at 260 and 280 nm wavelengths. The value obtained indicates if the DNA is contaminated with RNA, protein or aromatic compounds. However, many different substances can inhibit the PCR, even when present in trace amounts, and most of them will not be detected by simple spectrophotometry.29 These inhibitors can come from the test sample or the quality of reagents and plasticware used. The uncertainty associated with the quality of the reagents and plasticware can be minimised by specifying the grade and source in SOPs. Residual amounts of reagents such as CTAB, EDTA, ethanol, isopropanol and phenol also can be inhibitory to the PCR. Food ingredients such as acidic plant polysaccharides, polyphenolics, fat and protein also are inhibitory. Thus SOPs for nucleic acid purification need to ensure that these inhibitory materials are removed and the efficiency of removal needs to be demonstrated. This is best done by performing an inhibition test using either internal controls or evaluating the linearity of calibration curves.30,31 It should be noted that amplification of an endogenous positive control, if taken on its own, does not necessarily indicate the absence of PCR inhibitors.26 Equally well, examination of the A260:A230 ratio can be used as a quality metric to determine the likely presence of organic compounds or chaotropic salts (e.g. phenolate ions, EDTA and polysaccharides) that may have been co-extracted with the DNA and can inhibit the downstream PCR on that sample. If the A260:A280 or A260:A230 ratios are much lower than a value of around 2.0, then this is indicative of the presence of inhibitors. In such cases corrective action must be undertaken to remove these (e.g. by cleaning, re-precipitating and re-suspending the DNA pellet) or the DNA extraction procedure should be repeated.
Many different methods have been used for extracting and purifying DNA prior to amplification in the PCR and these have been reviewed.29 These methods fall into two main categories: variations on “home made” protocols, usually involving the use of cetyltrimethylammonium bromide (CTAB) or sodium dodecyl sulphate (SDS), and commercial kits. Within these two main categories, numerous variations on the exact type of DNA extraction exist, including solution based approaches (e.g. phenol/chloroform), solid based approaches (e.g. magnetic beads) or any combination of the two (e.g. CTAB followed by a column based clean up). The ideal method is the one that yields the greatest amount of DNA of the highest molecular weight and the lowest concentration of PCR inhibitors. Given the wide range of food matrices that are likely to be encountered this means that there is no generic method. For every new matrix examined it is essential to optimise the extraction and purification procedure and validate it.
The uncertainty associated with the DNA extraction phase has been minimised in some real-time PCR approaches for food authenticity testing. For example, for the quantitation of GMO ingredients, real-time PCR is used to quantify the amount of GM target analyte (e.g. DNA from GM soya) relative to the total amount of species specific DNA present (e.g. DNA from the total soya content). In this manner a relative expression is derived and reported for GMO content, and the impact of reduced DNA extraction efficiency may often be minimised as the sources of measurement uncertainty tend to effect all DNA targets in a consistent manner.
Recognising the importance of the DNA extraction phase and the impact this can have upon downstream molecular biology analyses, the Department for the Environment, Food and Rural Affairs (Defra) commissioned a one day workshop in 2014 to discuss harmonised approaches to this area between UK enforcement laboratories.32
9. The polymerase chain reaction (PCR)
The Royal Society of Chemistry Analytical Methods Committee has published a technical brief explaining the basic theory of PCR.33 This document highlights the large number of acronyms for PCR variations which can cause some confusion to the analyst. When DNA analysis is used to discriminate between species or varieties the effort is directed at one or a small number of polymorphisms. These represent a miniscule part of the total genome and so before analysis can proceed it is necessary to selectively amplify them. This amplification is conducted using the PCR, which can be a major source of uncertainty. The process occurs in three phases as shown in Fig. 3. In the first phase, products accumulate exponentially. In theory, the product should double in concentration with every cycle but in practice many factors can affect the efficiency of the process (see below). In the second phase the reaction begins to slow down and the product accumulates linearly. This happens because the reagents are being consumed and there is end-product inhibition and other complex kinetic effects. In the third phase the reaction has stopped and no more products are made. It is normal practice to quantify DNA during the exponential amplification phase of PCR (using real-time PCR) as opposed to the plateau phase (end-point PCR), as samples containing exactly the same starting amount of DNA can exhibit different reaction kinetics at the plateau phase. However, there are other times when end-point PCR can be used (see later section).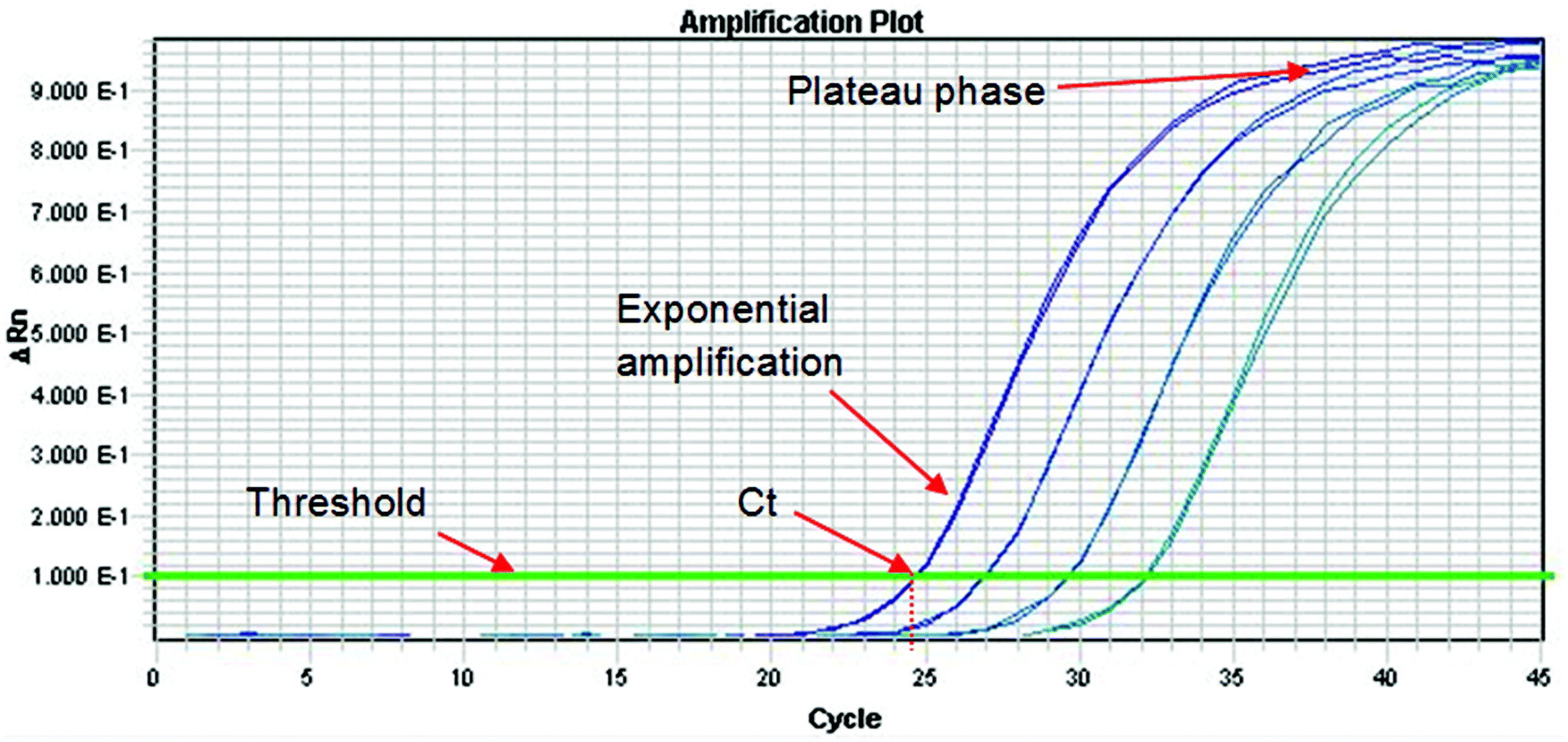 | ||
| Fig. 3 A typical real-time PCR amplification curve using a fluorescently labelled probe. The PCR cycle number is shown on the x-axis, and the logarithm of the change in intensity of the fluorescence response from the probe (equal to the amount of target DNA present) is shown on the y-axis. The threshold is marked on the graph and is the point above which any measurable signal is assumed to originate from amplification of the target sequence, as opposed to any background interference. The Cycle threshold value (Ct) represents the fraction of a PCR cycle at which point the fluorescence of a sample passes the fixed common threshold. The exponential and plateau phases of the PCR cycle are labelled. In this example, a four point 1 in 6 serial dilution series of a sample is run, where each dilution is represented by two PCR replicates. | ||
As with all aspects involved in producing an analytical result, it is good practice to put in place quality criteria associated with each phase of an analytical approach to ensure measurement uncertainty is minimised and results are produced that are fit for purpose. Such quality criteria for the PCR phase can involve use of an internal positive control (IPC) in the PCR, and testing that the correlation coefficient (r2) and PCR efficiency of any dilution series of calibrants or test samples to ensure that these are close to the ideal expected values of 1 and 100% respectively, using real-time PCR.
9.1. Real-time PCR
In real-time PCR one determines the cycle at which the fluorescence signal of the sample reaches an intensity above a background (or threshold). This is the cycle threshold (Ct) value, which is also increasingly being referred to as the quantification cycle (Cq) in recent texts. In a well-controlled PCR experiment, replicates should not differ by more than 0.3 cycles34 and the efficiency should be 100 ± 10%. The efficiency is calculated by determining the Ct values for dilutions of the test sample. If the efficiency is 100% then the Ct values of a tenfold dilution will be 3.3 cycles apart and the amplification curves will be parallel to each other. If the Ct values are more than 3.6 cycles apart then the PCR has poor efficiency. Factors that affect the efficiency include the presence of inhibitors in samples, sub-optimal PCR primer and probe design and inaccurate sample and reagent pipetting. Primer and probe design can be optimized during method development but the other factors are contributors to assay uncertainty. Ideally, the extraction and purification method selected will always remove PCR inhibitors but with complex and highly processed foods this might not be possible. Inaccurate pipetting can be minimised with proper training but never can be eliminated.9.2. Quantifying DNA using real-time PCR
Because PCR involves amplification of DNA, quantifying a particular sequence can usually only be done by reference to another material that is subjected to the same procedure – the exception being digital PCR (see next section). There are two basic methods: determination of comparative Ct values and a calibration curve approach. In the comparative method one compares the Ct value of one target gene to another, e.g. an internal control or reference gene, in a single sample. If TaqMan® chemistry is used then this comparison can be done in a single tube. Because a standard curve is not used dilution errors are minimised. However, it is essential that the efficiencies of amplification of the target and endogenous control genes are approximately equal. The greater the difference in efficiencies the more uncertainty there will be in the measurement and the reported test result. Key factors affecting relative efficiencies include amplicon size and primer design. It also is essential to identify limiting primer concentrations and to ensure that these do not affect Ct values, especially if multiplex PCR is being used.The more usual approach for quantification is to express the measurement response of a test sample relative to a calibration curve. Methods using a calibration curve are ideal if one wishes to quantify a single substance in a sample relative to a reference material. However, in food authenticity work it usually is necessary to determine the relative proportions of one analyte versus another. In this case it is necessary to have standard curves for both analytes. The selection and development of suitable standards is made difficult by natural variation and any effects of processing. Ideally one uses a certified reference material (CRM) as the source of DNA for the standard curve but only a few such materials are available and only for GMOs.35
Some of the more recent certified reference materials commercially available are available only as 100% GMO. With these, quantification only can be achieved using a “relative copy number” method. This involves making logarithmic dilutions of the reference material with the PCR being carried out on each dilution to specifically amplify the event specific and endogenous gene sequences. The Ct values obtained for the dilution series are plotted against arbitrary copy numbers for each dilution to generate a linear calibration curve. Test samples are assessed within the same series of PCR and the calibration curves used to determine the “relative copy number” of each of the event specific sequence and endogenous gene sequences present in the test sample. It is important to note that, if the original CRM used to construct the calibration curve had its GM content certified on a mass per mass (m/m) basis, then the result from the test sample will also be expressed in terms of a m/m basis.
Plasmids have been investigated as an alternative calibration source to CRMs for use in detecting GMOs. These plasmids contain specific GM sequences and endogenous (reference) gene sequences. A comparison of genomic and plasmid-based calibrants concluded that plasmid calibration gave a closer mean estimate of the expected % GM content of samples and exhibited less variation.36 Plasmid calibrants also gave more accurate results in terms of trueness and precision when assessed using an inter-laboratory study. However, plasmids generated by gene manipulation can be unstable and it is necessary to be sure that there are no changes over time in the cloned genes. This could be a significant issue if the amounts of two species (e.g. chicken and beef) are being determined by exploiting nucleotide differences in the same gene. If both genes are on the same plasmid then deletions could occur through homologous recombination. Finally, quantification is only possible if the amplification efficiencies of DNA from test samples are the same as DNA used in construction of the standard curve. To be sure of this it is necessary to run a dilution series of the test sample.
A potential source of error when quantifying DNA is the concentration of magnesium ions in the buffer used in the amplification step. An assumption often is made that hybridization of the primers is highly specific but this may not be the case. If, as is usual, the magnesium is present at 5 mM then this permits non-specific PCR and the amount of amplicon may be over-estimated. This problem can be detected by measuring the melting temperature of the end product or analysing it by gel electrophoresis. If a probe is present (as in real-time PCR) then this gives added selectivity to help ensure that only DNA from the correct amplicon is quantified.
The only well documented example of the use of real-time PCR to quantify food adulteration, other than with GMOs, is the measurement of bread wheat (T. aestivum) in durum wheat (T. durum) used to make pasta.37 Durum wheat is tetraploid (AABB) whereas bread wheat is hexaploid (AABBDD). All three genomes carry the psr128 sequence and this shows little or no polymorphism except for the presence of a 53 basepair insertion in an intron sequence in the D-genome. Primers were selected that permit amplification of a 117 base-pair D-genome specific amplicon and a 121 base-pair amplicon in the coding region of psr128. The latter is used to normalise for the amount of total amplifiable wheat DNA present in the sample.
To facilitate an understanding of the analytical variation involved in quantification, two pasta standards were prepared from flour mixtures containing 0.2% and 5.89% bread wheat in durum wheat. In an “in house” study the lower performance standard gave a value of 0.19% ± 0.04% bread wheat based on 36 replicates. The coefficient of variation was 21% corresponding to an uncertainty at an approximate 95% confidence limit of 0.11 to 0.26%. Hence, for a single analytical determination of a material known to contain 0.19% contamination, the result could be expected to be in the range 0.11–0.26%, 19 times in every 20 analyses. The higher performance standard (value 5.89% ± 1.9% based on 12 replicates) had a coefficient of variation of 33% corresponding to an uncertainty at an approximate 95% confidence limit of 2.02% to 9.75%. Given that these results were generated in a laboratory that fully understands all the factors that affect the PCR, they highlight the breadth of responses where the true value may actually lie when using real-time PCR for quantification in food authenticity investigations.
The 2013 horse meat incident provided evidence for the need to develop molecular biology approaches for the quantitative determination of important food ingredients. During the same year, Defra commissioned work at LGC to develop a real-time PCR approach for the quantitation of horse DNA.38 This approach used best measurement practice guidance in the area of real-time PCR to develop a method that would quantitate the amount of horse DNA relative to the total amount of mammalian DNA present in a sample. Sets of primers and probes were chosen that were equine specific and also targeted a universal growth differentiation factor gene. A range of gravimetrically prepared horse in beef meat mixtures, as well as horse and beef DNA mixtures, were prepared and used to demonstrate the trueness and precision associated with the quantitative estimation using the real-time PCR assay across a range of concentrations.
Given the importance and prevalence of real-time PCR as an analytical and diagnostic aid, inclusive and outside of food authenticity testing, it is of paramount importance to ensure results are reported to the highest level of quality and are repeatable and reproducible. The publication of the MIQE guidelines (minimum information for publication of quantitative real-time PCR experiments)39 have helped to address harmonisation in this area, and provide a set of criteria to address and abide by when reporting results from real-time PCR.
The choice of DNA target for species detection and quantitation is equally important. The weight of current scientific evidence suggests that mitochondrial DNA, being in very high abundance within a cell, are suitable targets to facilitate sensitive detection of a species.40 However, due to the high variability in the number of mitochondria per cell (between species, within species and even between tissues within an organism), they may not be the most suitable targets for species quantitation. Nuclear DNA targets, being less abundant but generally of a stable copy number between cells, may provide a better target for species quantitation.41
9.3. Digital PCR
As noted above, real-time PCR is not without problems and these include: initial amplification cycles may not be exponential; low initial concentrations of nucleic acid molecules from adulterants may not amplify to detectable levels; and quantitation is relative to a calibration curve and PCR amplification efficiency in a sample of interest may be different from that of reference samples. Some of the above issues can be minimised or even negated through the use of digital PCR.42Digital PCR helps facilitate absolute single molecule detection without reference to a calibration curve. It achieves this through the process of limiting dilutions: the real-time PCR reaction is split into thousands of individual reactions, and by counting the number of positive reactions relative to negative ones, an accurate estimate of the starting number of molecules can be made. As a calibration curve is no longer a necessity in digital PCR, this therefore mitigates any matrix differences between calibrant and test sample that may cause differential PCR amplification. As digital PCR allows absolute single molecule detection, it also has the advantage of producing results which are more traceable to the SI unit, instead of providing a result that is relative to a calibrant or expressed as a relative percentage. Additionally, because of the very high level of sample replication afforded, digital PCR can produce results with very tight precision. There are a number of commercially available digital PCR instruments currently on the market (including chamber and droplet based digital PCR), providing evidence of the importance of this new technology in quantitative molecular biology approaches. Burns and colleagues42 pioneered some early work of applying digital PCR for food authenticity testing and demonstrated the applicability of the technique to estimate absolute limits of detection and quantifying plasmid copy number associated with GMO analysis. In 2011, Sanders et al., examined some of the underlying factors that influenced accurate measurements in a digital PCR instrument, and provided guidance on important issues to consider when designing digital PCR experiments.43 Corbisier et al.,44 examined the suitability of this methodology for the absolute quantification of genetically modified maize and found the results to be identical to those obtained by real-time PCR. The major advantage of the digital PCR method was that it permitted accurate measurement without the need for a reference calibrator.
The growth in interest of digital PCR, both as an aid in metrological traceability and as a real-life application across a range of sectors inclusive of food testing, has meant that a plethora of data is being produced. This has led to the establishment of a set of guidelines for the production and publication of digital PCR data, as an aid to helping harmonise the approach and provide meaningful results which can be readily interpreted.45
9.4. Isothermal technologies
PCR approaches could be criticised for their reliance upon the need for complex thermal cycling instruments and profiles, and the impact that inhibitors can have upon the subsequent PCR amplification efficiency which assumes a doubling of target template each cycle. These limitations, in part, have driven the need for the development of isothermal technologies for nucleic acid amplification, which are not dependent upon complex thermal cycling parameters. Isothermal technologies typically employ just the one single temperature for amplification of target molecules, facilitating an increased choice of enzymes to use to help catalyse the reaction and also the choice of nucleic acid template. As well as negating the requirement for complex thermal cycling instrumentation, isothermal approaches have demonstrated rapid analytical turnaround times coupled with a reduced susceptibility to inhibitors, lending themselves well to development of point of test devices. The miniaturisation and portability of some of the isothermal technologies and integration into compact microfluidic-type devices has shown application in the areas of food safety, environmental, and GMO testing. The importance of isothermal technologies is evidenced by the fact that the number of publications regarding this technology increased over four fold between 2004 and 2011 to well over 400 publications a year.There are a number of isothermal instruments currently available based on differing technologies, such as nucleic acid sequences-based amplification, single primer isothermal amplification, strand displacement amplification, rolling circle amplification, loop-mediated isothermal amplification (LAMP) and even whole genome amplification. Whilst there has been an increased interest in the development and application of isothermal technologies in recent years, the process itself is not without its own limitations. Background noise can often interfere with an isothermal amplification, and nonspecific priming has also been an issue. Agreement on a harmonised approach for regulating and inferring the starting point of an isothermal reaction would also be beneficial. Production of a set of harmonised guidelines for production of data from isothermal technologies could help towards standardisation and expression of results in this interesting area, as well as fuelling debate about possible quantitative applications in the future.46
Reports in the published literature provide evidence for the application of isothermal technologies for speciation and food analysis. The application of Loop-Mediated Isothermal Amplification (LAMP) for meat species detection with potential quantitative capabilities has previously been described,47 as well as its application to detection of horse meat in raw and processed meat products.48 In 2010 a LAMP based approach for detection of pork, chicken and beef was published,49 and isothermal approaches have also been described for identification of mushroom species.50
There are a number of publications describing the application of isothermal technologies for the detection of Genetically Modified Organisms.48,51,52
Whilst still considered a new and emerging technology, the current state of the art associated with isothermal approaches means that results produced from such technologies are still largely qualitative in nature, and their quantitative potential has yet to be fully realised.
9.5. Quantitative end-point PCR
As noted earlier, quantification based on end-point PCR has a much higher uncertainty compared with real-time PCR. Nevertheless, if the analytical protocols are carefully designed it is possible to obtain results that meet the needs of enforcement authorities. However, to date, the only validated protocol for determining food adulteration based on end-point PCR is one developed by Colyer et al.53 for determining non-Basmati rice varieties in admixture with Basmati rice. This method has been shown to be fit for purpose based on a ring trial involving 11 laboratories.54 When the laboratories were presented with standard rice mixes and three unknown mixtures the absolute expanded measurement uncertainty was estimated as being ∼6% across the concentration range 8–35% non-Basmati rice in Basmati rice. For each of the three mixtures, the average value of the non-Basmati rice was within 5% of the true value indicating that there was insignificant bias.Analytical chemistry is a well-established discipline but analytical molecular biology is still in an early stage of development. Although the situation is rapidly improving, only a limited range of laboratories have the requisite skills to undertake quantification using real-time PCR and most of these have applied the technique only to the determination of GM material in relatively simple matrices. An alternative and much simpler analytical platform is laboratory-on-a-chip capillary electrophoresis (LOC) and this has been used successfully by analytical chemists to identify a range of food materials.4,55 LOC analysis is based on end-point PCR and as noted above will have a higher uncertainty than methods that use real-time PCR if used for quantitative purposes. However, the LOC approach has been successfully applied for the detection of adulteration across a range of matrices when used as a qualitative tool, inclusive of fish speciation, GMO identification, durum wheat determination, basmati rice identification, and fruit juice adulteration. A number of protocols for food authenticity testing using the LOC approach have been published by the Food Standards Agency.56 However, there is another consideration and this relates to heteroduplex formation.57
The objective in many investigations of food authenticity is to determine the amount of an undeclared ingredient that is present in a sample versus a declared ingredient. If the two ingredients are similar then the PCR may amplify DNA targets that have a high degree of homology. The consequence of this is that when the PCR plateau phase is reached the predominant product will be a heteroduplex. The amount of heteroduplex can be calculated from the ratio p2:2pq:q2 where p and q represent the concentration of authentic and adulterant homoduplexes and pq represents that of each heteroduplex. It should be noted that this ratio only is valid if: the amplification efficiencies are equal for the two targets; the two sources of DNA are haploid such as mitochondrial or chloroplast DNA markers that are frequently used in PCR based tests for authenticity; and the intercalator dye used for quantification binds to heteroduplex and homoduplex molecules with the same efficiency.
An alternative method for quantifying adulterants using end-point PCR is the use of Pyrosequencing™. This is a sequencing-by-synthesis method and the results are presented as a series of peaks where peak height corresponds to the number of nucleotides incorporated. The close correlation between nucleotide incorporation and peak height can be used to determine how many of the template molecules have incorporated the added nucleotide, thereby allowing for allele (SNP) frequency determination in a mixed sample.58,59 Ortola-Vidal et al.,60 used this method to detect and quantify “undeclared” fruit in fruit yogurts. The limit of detection of the assay was 2% w/w rhubarb yoghurt in raspberry yoghurt and the limit of quantification was 5% w/w. As with all PCR-based methods it is important to have equal amplification efficiency for the different alleles.
This method of quantifying alleles using pyrosequencing has not been fully validated but it is very attractive for a number of reasons. First, reactions are internally controlled using the authentic species as control and allow the simultaneous detection of multiple adulterants. Second, the method is definitive since it depends on sequence determination rather than indirect characterisation using probes. Finally, the method is quick and simple with minimal operator intervention.
9.6. Additional DNA technologies
DNA arrays represent a well-established technology for the qualitative detection of specific targets, particularly with respect to clinical applications. Arrays typically consist of a highly ordered pattern of spots containing DNA, immobilised in a regular high-density pattern on a solid support and fabricated by high-speed robotics. However, their use in the food authenticity testing area is poorly documented. There is a general belief that the multiplexing capability of arrays coupled with their relatively low costs could provide a suitable platform for quantitative ingredient determination should the technology continue to develop.Advances in modern technologies now mean that whole genome sequencing is a reality, and this may help facilitate species identification in food samples based on Next Generation Sequencing (NGS). However, at the current time, there are only a limited number of papers describing the use of NGS for food authenticity testing, and the current high costs and complex workflow associated with NGS precludes its use for quantitative ingredient determination as part of routine food authenticity testing.
10. ELISA
ELISA (Enzyme-Linked ImmunoSorbent Assay) is a type of immunoassay, which is often used for food and feed analysis. ELISA technologies are reliant upon the use of enzymes to detect target antibodies or antigens in an assay. Applications in the food authenticity testing area include detection of allergens (e.g. soya) skeletal meat proteins, proteins associated with Genetic Modification, fish speciation, dairy products and feedstuff origin determination. Performance characteristics associated with ELISAs include good sensitivity, cost effectiveness and easy application, as indicated by the plethora of commercially available ELISA tests which are currently available. ELISA has successfully been applied for the identification of fish species in processed foods and feeds.61However, generating antibodies with the ability to discriminate target analytes from closely-related species can be extremely difficult and this is the major limitation in the use of ELISA in food authenticity applications. ELISA approaches also can suffer from interference from other ingredients. Since ELISA is considered as an immunological technique rather than a molecular biology approach it is not discussed further in this review.
11. Quantitative proteomics
Guidance in the field of best practice for the development of mass spectrometry analysis for the determination of allergens in foods has previously been reviewed.62 The review describes an overview of some of the experimental design and methodological challenges encountered when using mass spectrometry, including multiplexing target analytes, bioinformatics and choice of peptide, markers for quantitation, optimisation of protein digestion, and the importance of harmonised methods and results. The review concludes with a list of recommendations on how to address these aspects and what the likely impact of these would be.The invention of SDS-polyacrylamide gel electrophoresis (SDS-PAGE) in the 1970s and later, the development of 2-dimensional PAGE (2-DE), were major breakthroughs in the analysis of proteins, allowing many individual proteins to be separated and analysed in a single experiment. Utilising mass spectrometry (MS), following the invention of electrospray ionisation (ESI) and matrix-associated laser desorption ionisation (MALDI) in the 1980s, allowed tryptic peptides and small proteins to be studied, as reviewed by Domon and Aebersold.63 However, it became apparent that 2-DE had limitations with respect to the range of relative abundance and solubility of the proteins under investigation. These problems can be overcome by coupling liquid chromatography (LC) with tandem mass spectrometry (MS/MS), using so-called multidimensional protein identification technology (MudPIT). The use of cation exchange and reverse phase LC, linked to MS/MS, has greatly extended the coverage of the proteome, including quantitative measurements.64
There are several reviews on the principles and applications of quantitative proteomics using 2-DE or LC-MS/MS,65–68 whilst a comprehensive text on all aspects of proteomics in foods has recently been published.69
11.1. Quantitation and labelling methods
The basic methodology that is used for quantification using LC-MS/MS is simple conceptually. It involves purification of the target protein, cleavage with a proteolytic enzyme and separation of the resultant peptides by LC. The mass and identity of each peptide then is determined by MS/MS and the amount of one or more peptides calculated from the intensity of the ion signals. However, there is a fundamental problem: mass spectrometry inherently is not quantitative. The intensity of a peptide ion signal does not accurately reflect the amount of peptide in a sample because different peptides vary in size, charge, hydrophobicity, etc. and this leads to large differences in mass spectrometric response. This problem can be overcome by introducing a calibrant in the form of an identical peptide that has been labelled with one or more heavy isotopes. The light and heavy variants of the peptide will have identical chemical properties but can be distinguished by their mass differences. The ratio of the light and heavy peptide ions gives the relative abundance of the peptide of interest. This approach eliminates run-to-run variations in performance of LC and MS, amounts of injected sample and ion-suppressing effects.A number of methods have been developed for labelling proteins or peptides with stable isotopes. In the context of analysis of complex matrices these include a number of chemical methods, e.g. isotope-coded affinity tag (ICAT), isotope-coded protein labelling (ICPL), and isobaric tag for relative and absolute quantification (iTRAQ). For ICAT and ICPL the tagging reaction occurs before proteolytic digestion, whereas with iTRAQ it is the peptides that are labelled. When the identity of the protein to be quantified is known, as often is the case with issues of food authenticity, the ideal method is to use isotopically labelled synthetic reference peptides. In this absolute quantification (AQUA) method the reference peptide is synthesised with one of its amino acids labelled with 13C or 15N. Additionally, there are “label-free” approaches to quantitation. Two protocols have been reported, one based on the frequency of identification, known as spectral counting70 and the other uses peak intensity in which the peak areas of peptides correlate to the amount of the parent protein from which they were derived.71 A recent application of this has been to the assessment of GM tomato fruit,72 whilst Gong and Wang73 have reviewed the use of proteomics to identify unintended effects in GM crops.
11.2. Sources of variability
If LC-MS/MS is to be used quantitatively then a number of key issues need to be considered. These include the extraction protocol for the target protein, the selection of the peptide to be quantified, the digestion step and the design of the MS analysis. Of these, the extraction protocol is the greatest source of uncertainty. Ocaña et al.74 undertook an evaluation of the iTRAQ and AQUA methods for the quantification of enolpyruvylshikimate-3-phosphate (EPSPS) in genetically modified (GM) soya. This involved protein extraction, precipitation and fractionation by anion exchange chromatography. When the anion exchange fractions containing EPSPS were combined they retained between 11 and 33% of the total protein in the precipitated fractions indicating that this one step alone can be the source of considerable variability.Ocaña et al.74 found another source of variability associated with sample handling. They extracted EPSPS from soya containing 0.5, 0.9, 2 and 5% GM material and determined the signal ratios for the target and labelled peptides using the AQUA method. Although the area ratios showed a good linear relationship with the amount of transgenic material present, the correlation coefficient indicated some divergence from a perfect linear correlation. Furthermore, the coefficients of variation for three replicate analyses of the different samples varied from 16–29%. When the ESPS was extracted from the 5% GM material and then diluted to 0.5, 0.9 and 2% before analysis there was a strong correlation (R2 = 0.9999) between the signal area ratios and the percentage of transgenic material. In this case, the coefficients of variation for four replicate analyses were 3% (0.9, 2 and 5% GM) and 14% (0.5% GM). These improved results are attributable to the elimination of potential variability from sample handling during extraction, precipitation and fractionation. Other groups have reported similar levels of variation from this source.74
The peptide that is used as the analyte must be unique to the protein of interest. If it is not, then over-estimation will occur. The selected peptide also must be efficiently liberated by digestion of the protein and must be stable in solution during the whole process. It also must chromatograph well and be easily detectable by MS. Finally, the selected peptide must withstand modification by any industrial processes used in the manufacture of the test sample.
The efficiency of digestion of the target protein by the selected protease is critically important as incomplete digestion will lead to underestimation of the analyte. Usually, the target protein will have multiple cleavage sites for the protease and some will be more readily cleaved than others. In an ideal situation the peptide selected as the analyte will be flanked by readily-cleavable sites and this should be tested using purified protein of known provenance. In addition, when test samples are subjected to MS analysis a search should be made for larger peptides that incorporate the target sequence as these will indicate missed cleavages and make accurate quantification very difficult. In the case of the AQUA method this is not a problem. With the iTRAQ method all the peptides are labelled and one or more that always are produced need to be selected, even before ensuring that complete cleavage has occurred. In the case of the EPSPS study of Ocaña et al.,74 only one peptide (and its isotopomer) was consistently found.
A key factor affecting accuracy and dynamic range of quantification is the choice of mass spectrometer. With some instruments the definition of very low and very strong signals can be problematic. Low intensity spectra result in higher uncertainty of measurement because of poor ion statistics. Saturation is more of a problem with quadrupole TOF instruments than ion traps but if it occurs will lead to erroneous quantification. The recent introduction of high resolution/high mass accuracy instruments should facilitate accurate quantification. This is because the increased instrument performance permits the exact discrimination of peptide isotope clusters from interfering signals caused by near isobaric peptides. Interference also can be reduced by improving the purification of the target protein prior to digestion and LC-MS/MS analysis but this can lead to increased losses and hence underestimation.
From their work on EPSPS, Ocaña et al.74 concluded that both the iTRAQ and AQUA methods had the potential to determine whether the presence of GM material is above the 0.9% limit set by the European Union. However, iTRAQ requires much more experimental and data analysis than AQUA and hence AQUA is the preferred approach when only a single protein is being quantified. Even so, the data obtained (Table 2) indicates the limitations of the method. Some of the discrepancies observed will be due to differential sample handling and processing, particularly as the reference standard is added at a late stage in the workflow.
| GM ratio | Theoretical ratio | Observed ratio | % Inaccuracy |
|---|---|---|---|
| 5/0.9 | 5.56 | 4.73 | −15 |
| 2/0.9 | 2.22 | 2.41 | 9 |
| 0.5/0.9 | 0.56 | 0.40 | −28 |
As noted earlier, the development of quantitative proteomics is at a much earlier stage compared with quantitative PCR and many issues affecting measurement uncertainty of a reported result remain to be addressed. Whilst the results shown in Table 2 are encouraging it needs to be borne in mind that they were obtained with a single food component (soya). If the methods are transferred to complex and processed foods then the problems to be overcome will be considerably greater. Highly processed foods provide a challenging complex matrix in which to extract the analyte from, and further work will highlight if the issues associated with analysis of nucleic acids from such matrices may be resolved in the future using proteomics approaches.
12. Conclusions
This review has examined a number of important measurement issues associated with the use and development of molecular biology approaches for food authenticity analysis, with particular emphasis on quantitative approaches. Table 3 summarises some of the measurement issues and recommendations associated with addressing these issues, which have been discussed in this paper.| Topic | Issue | Recommendation |
|---|---|---|
| Ensuring food integrity in the supply chain | Improving laboratory testing capacity and capability to ensure a harmonised approach for testing for food authenticity | General recommendations outlined in the: |
| • HM Government Elliott Review into the Integrity and Assurance of Food Supply Networks2 | ||
| • Defra's AMWG: Response to Elliott review on “integrity and assurance of food supply networks” – recommendation 43 | ||
| Method validation and interpretation of results | When evidence for fraudulent activity is uncovered using a method that has not undergone validation | Development of validated methods and agreed standards |
| Agreement on values and criteria for minimum performance characteristics of a method | ||
| Procedures for the estimation of measurement uncertainty | Measurement uncertainty estimates may not be consistently reported and may be significant underestimates | Need for harmonised guidance in estimating and reporting measurement uncertainty |
| Use of SOPs | ||
| Servicing and calibration of analytical instruments | ||
| Choice of specific consumables and reference materials | ||
| Sampling | Uncertainty from sampling and sample preparation | Requirement to develop sampling protocols tailored to specific analytical areas (e.g. GMO analysis) |
| Samples chosen must be appropriate for the nature and complexity of the product | ||
| Nucleic acid extraction and purification | Ensuring integrity and purity of the DNA and efficiency of DNA extraction | Use of SOPs |
| Determine DNA purity using absorbances at 230, 260 and 280 nm wavelengths | ||
| Check degradation by gel/capillary electrophoresis | ||
| Relative quantitation of a sample (relative to both a target specific and a normalising reference gene) can reduce impact of poor DNA extraction efficiency | ||
| The polymerase chain reaction (PCR) and real-time PCR | Confidence in results and accurate quantitation | Use of SOPs |
| Use of suitable reference materials as controls and calibrants | ||
| Harmonisation regarding reporting of results (e.g. MIQE guidelines39) | ||
| Choice of DNA target (e.g. mitochondrial vs. chromosomal DNA) | ||
| Correlation coefficient (r2) and PCR efficiency associated with calibrant and test sample | ||
| Optimisation of primer and probe design | ||
| Use of an internal positive control (IPC) | ||
| New and emerging technologies (e.g. digital PCR, NGS, Isothermal approaches) | Technologies yet to firmly establish themselves for quantitative analysis of foods | Establishment of a set of harmonised guidelines for the production and publication of results (e.g. dMIQE guidelines45) |
| Quantitative proteomics | Developing the quantitative potential of mass spectrometry for food analysis | Use of an identical peptide labelled isotopically to be used as a calibrant |
| Production of harmonised guidance for: extraction protocol; target peptide selection; digestion stage; design of the mass spectrometry analysis; choice of mass spectrometer |
Methods based on quantitative PCR that have the necessary precision and trueness for use in detection of food fraud have been developed but only for use in relatively unprocessed foods, e.g. GMOs in flour, bread wheat in pasta, non-Basmati varieties in Basmati rice and raw meat samples. Attempts to extend quantitative PCR to more processed food have met with additional challenges. Pyrosequencing might be a viable alternative to quantitative PCR for the evaluation of complex and highly processed foods but much more work on this method is required. Quantitative proteomics is at an early stage of development and its full potential remains unknown but it could provide an alternative to PCR for the examination of unprocessed ingredients.
There is an increased requirement to develop approaches for the quantitative determination of food ingredients, to help detect food fraud and ensure the traceability of materials in the food chain. A number of molecular biology approaches, for example digital PCR, show good potential for sensitive, specific and traceable detection of target molecules. With the rapid pace at which these methods are being developed, it is equally important to ensure these methods are fully validated and the measurement uncertainty associated with a result is correctly characterised, so that objective data is generated to provide evidence of the fitness for purpose of these methods and help towards harmonisation of molecular biology results and the interpretation of data.
Acknowledgements
Part of the work associated with this review was supported through the UK Department for Business, Innovation & Skills (BIS), Government Chemist Programme 2014–2017.References
- Food Safety Authority of Ireland. FSAI Survey Finds Horse DNA in Some Beef Burger Products. https://www.fsai.ie/news_centre/press_releases/horseDNA15012013.html.
- “Elliott Review into the Integrity and Assurance of Food Supply Networks – Final report. A National Food Crime Prevention Framework” July 2014, HM Government. https://www.gov.uk/government/publications/elliott-review-into-the-integrity-and-assurance-of-food-supply-networks-final-report.
- Defra”s independent Authenticity Methods Working Group (AMWG). Response to Elliott review on “integrity and assurance of food supply networks” – recommendation 4 (March 2015) https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/409253/amwg-elliott-response.pdf.
- M. Woolfe and S. Primrose, Trends Biotechnol., 2004, 22, 222–226 CrossRef CAS PubMed.
- Defra cross contamination project: A project to establish whether carry-over of meat species occurs in UK meat processing plants during the GMP production of mince meat, Defra project FA0137, 2014.
- COMMISSION REGULATION (EC) No 1829/2003 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 22 September 2003 on genetically modified food and feed.
- The rice association, BRMA, British Retail Consortium “Code of Practice on Basmati Rice”, http://www.brc.org.uk/Downloads/Basmati_Code.pdf.
- Commission Regulation (EC) No. 882/2004 of the European Parliament and Council of 29 April 2004 on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules.
- W. Horwitz, Pure Appl. Chem., 1995, 67, 13 CrossRef.
- ISO 5725-1:1994. Accuracy (trueness and precision) of measurement methods and results – Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method. ISO 5725-2, 1-42.
- Codex Alimentarius. CAC/GL 74–2010 GUIDELINES ON PERFORMANCE CRITERIA AND VALIDATION OF METHODS FOR DETECTION, IDENTIFICATION AND QUANTIFICATION OF SPECIFIC DNA SEQUENCES AND SPECIFIC PROTEINS IN FOODS. http://www.codexalimentarius.org/standards/list-of-standards/.
- European Union Reference Laboratory for Genetically Modified Food and Feed, http://gmo-crl.jrc.ec.europa.eu/.
- A. Holst-Jensen and K. G. Berdal, J. AOAC Int., 2004, 87, 927–936 CAS.
- S. Trapmann, M. Burns, H. Broll, R. Macarthur, R. Wood and J. Zel, JRC Scientific and Technical Reports - Guidance document on measurement uncertainty for GMO testing laboratories, European Commission, Joint Research Centre, Instutute for Reference Materials and Measurements, DOI:10.2787/18988. https://ec.europa.eu/jrc/sites/default/files/eur22756en.pdf.
- European Commission: Comparative Testing Report on the Detection and Quantification of GM Events in Rice Noodles, 2014. http://publications.jrc.ec.europa.eu/repository/handle/JRC91953.
- EURACHEM/CITAC Guide - Measurement uncertainty arising from sampling - A guide to methods and approaches (First Edition), 2007 Search PubMed.
- M. Burns and H. Valdivia, Eur. Food Res. Technol., 2007, 226, 7–18 CrossRef CAS.
- Commission Recommendation (EC) No 787/2004 of 4 October 2004 on technical guidance for sampling and detection of genetically modified organisms and material produced from genetically modified organisms as or in products in the context of Regulation (EC) No 1830/2003.
- R. Macarthur, A. W. Murray, T. R. Allnutt, C. Deppe, H. J. Hird, G. M. Kerins, J. Blackburn, J. Brown, R. Stones and S. Hugo, Nat. Biotechnol., 2007, 25, 169–170 CrossRef CAS PubMed.
- C. Paoletti, A. Heissenberger, M. Mazzara, S. Larcher, E. Grazioli, P. Corbisier, N. Hess, G. Berben, P. Lübeck, M. De Loose, G. Moran, C. Henry, C. Brera, I. Folch, J. Ovesna and G. Van den Eede, Eur. Food Res. Technol., 2006, 224, 129–139 CrossRef CAS.
- F. Moreano, U. Busch and K. H. Engel, J. Agric. Food Chem., 2005, 53, 9971–9979 CrossRef CAS PubMed.
- C. Peano, M. C. Samson, L. Palmieri, M. Gulli and N. Marmiroli, J. Agric. Food Chem., 2004, 52, 6962–6968 CrossRef CAS PubMed.
- D. S. Smith, P. W. Maxwell and S. H. De Boer, J. Agric. Food Chem., 2005, 53, 9848–9859 CrossRef CAS PubMed.
- C. F. Terry, N. Harris and H. C. Parkes, J. AOAC Int., 2002, 85, 768–774 CAS.
- T. Yoshimura, H. Kuribara, T. Matsuoka, T. Kodama, M. Iida, T. Watanabe, H. Akiyama, T. Maitani, S. Furui and A. Hino, J. Agric. Food Chem., 2005, 53, 2052–2059 CrossRef CAS PubMed.
- M. J. Holden, J. R. Blasic Jr., L. Bussjaeger, C. Kao, L. A. Shokere, D. C. Kendall, L. Freese and G. R. Jenkins, J. Agric. Food Chem., 2003, 51, 2468–2474 CrossRef CAS PubMed.
- S. Priyanka and S. Namita, Molecular Biology: Principles and Practices, Laxmi Publications, 2010 Search PubMed.
- M. J. Holden, R. J. Haynes, S. A. Rabb, N. Satija, K. Yang and J. R. Blasic Jr., J. Agric. Food Chem., 2009, 57, 7221–7226 CrossRef CAS PubMed.
- T. Demeke and G. R. Jenkins, Anal. Bioanal. Chem., 2010, 396, 1977–1990 CrossRef CAS PubMed.
- P. Corbisier, W. Broothaerts, S. Gioria, H. Schimmel, M. Burns, A. Baoutina, K. R. Emslie, S. Furui, Y. Kurosawa, M. J. Holden, H. H. Kim, Y. M. Lee, M. Kawaharasaki, D. Sin and J. Wang, J. Agric. Food Chem., 2007, 55, 3249–3257 CrossRef CAS PubMed.
- M. Lipp, R. Shilito, R. Giroux, F. Spiegelhalter, S. Charlton, D. Pinero and P. Song, J. AOAC Int., 2005, 88, 20 Search PubMed.
- Defra: Knowledge Transfer event for DNA extraction approaches to support food labelling enforcement - FA0144, 2014, http://randd.defra.gov.uk/Default.aspx?Menu=Menu&Module=More&Location=None&Completed=0&ProjectID=19082.
- Royal Society of Chemistry, Analytical Methods - AMC Technical Briefs. PCR- the polymerase chain reaction, 2014, DOI: 10.1039/c3ay90101g. http://www.rsc.org/images/TB%2059_tcm18–241515.pdf.
- PCR Troubleshooting and Optimization: The Essential Guide, Caister Academic Press, 2011 Search PubMed.
- N. Marmiroli, E. Maestri, M. Gulli, A. Malcevschi, C. Peano, R. Bordoni and G. De Bellis, Anal. Bioanal. Chem., 2008, 392, 369–384 CrossRef CAS PubMed.
- M. Burns, P. Corbisier, G. Wiseman, H. Valdivia, P. McDonald, P. Bowler, K. Ohara, H. Schimmel, D. Charels, A. Damant and N. Harris, Eur. Food Res. Technol., 2006, 224, 249–258 CrossRef CAS.
- G. Wiseman, in Real-Time PCR: Current Technology and Applications, ed. J. Logan, K. Edwards and N. Saunders, Caister Academic Press, 2009, pp. 253–267 Search PubMed.
- Defra: Method development for the quantitation of equine DNA and feasibility of establishing objective comparisons between measurement expression units (DNA/DNA compared to w/w tissue) - FA0135, 2013, http://randd.defra.gov.uk/Default.aspx?Menu=Menu&Module=More&Location=None&Completed=0&ProjectID=18741.
- S. A. Bustin, V. Benes, J. A. Garson, J. Hellemans, J. Huggett, M. Kubista, R. Mueller, T. Nolan, M. W. Pfaffl, G. L. Shipley, J. Vandesompele and C. T. Wittwer, Clin. Chem., 2009, 55, 611–622 CAS.
- N. Z. Ballin, F. K. Vogensen and A. H. Karlsson, Meat Sci., 2009, 83, 165–174 CrossRef CAS PubMed.
- C. Floren, I. Wiedemann, B. Brenig, E. Schutz and J. Beck, Food Chem., 2015, 173, 1054–1058 CrossRef CAS PubMed.
- M. J. Burns, A. M. Burrell and C. A. Foy, Eur. Food Res. Technol., 2010, 231, 353–362 CrossRef CAS.
- R. Sanders, J. F. Huggett, C. A. Bushell, S. Cowen, D. J. Scott and C. A. Foy, Anal. Chem., 2011, 83, 6474–6484 CrossRef CAS PubMed.
- P. Corbisier, S. Bhat, L. Partis, V. R. Xie and K. R. Emslie, Anal. Bioanal. Chem., 2010, 396, 2143–2150 CrossRef CAS PubMed.
- J. Huggett, C. Foy, V. Benes, K. Emslie, J. Garson, R. Haynes, J. Hellemens, M. Kubista, R. Mueller, T. Nolan, M. Pfaffl, G. Shipley, J. Vandesomple, C. Wittwer and S. Bustin, Clin. Chem., 2013, 59 Search PubMed.
- G. Nixon and C. Bushell, in PCR Technology - Current Innovations, ed. T. Nolan and S. Bustin, CRC Press - Taylor & Francis Group, 3rd edn, 2013, ch. 26, pp. 363–391 Search PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- C. Zahradnik, R. Martzy, R. L. Mach, R. Krska, A. H. Farnleitner and K. Brunner, Food Anal. Methods, 2014, 8, 1576–1581 CrossRef.
- M. U. Ahmed, Q. Hasan, M. Mosharraf Hossain, M. Saito and E. Tamiya, Food Control, 2010, 21 Search PubMed.
- F. Vaagt, I. Haase and M. Fischer, J. Agric. Food Chem., 2013, 61, 1833–1840 CrossRef CAS PubMed.
- D. Morisset, D. Dobnik and K. Gruden, NASBA-based detection: a new tool for high-throughput GMO diagnostics in food and feedstuffs, Conference paper from Rapid Methods Europe, 2008 Search PubMed.
- D. Lee, M. La Mura, T. R. Allnutt and W. Powell, BMC Biotechnol., 2009, 9(7) DOI:10.1186/1472-6750-9-7.
- A. Colyer, R. Macarthur, J. Lloyd and H. Hird, Food Addit. Contam., Part A, 2008, 25, 1189–1194 CrossRef CAS PubMed.
- Food Standards Agency Information Bulletin on Methods of Analysis and Sampling of Foodstuffs No. 75, Report on the InterLaboratory Trial of the Microsatellite Method for the Identification of Certain Basmati Rice Varieties, 2007, http://tna.europarchive.org/20111030113958/http://www.food.gov.uk/multimedia/pdfs/075a.pdf.
- J. J. Dooley, H. D. Sage, M. A. Clarke, H. M. Brown and S. D. Garrett, J. Agric. Food Chem., 2005, 53, 3348–3357 CrossRef CAS PubMed.
- Food Standards Agency - Programme of Work. http://tna.europarchive.org/20141103165934/http://www.foodbase.org.uk/category.php?action=programme&f_category_id=2&f_community_id=26.
- M. Scott and A. Knight, J. Agric. Food Chem., 2009, 57, 4545–4551 CrossRef CAS PubMed.
- S. Shifman, A. Pisante-Shalom, B. Yakir and A. Darvasi, Mol. Cell. Probes, 2002, 16, 429–434 CrossRef CAS PubMed.
- J. Wasson, G. Skolnick, L. Love-Gregory and M. A. Permutt, BioTechniques, 2002, 32(5), 1144–1152 CAS.
- A. Ortola-Vidal, H. Schnerr, A. Knight, M. Rojmyr and F. Lysholm, Food Control, 2007, 18, 6 CrossRef.
- C. G. Sotelo, C. Piñeiro, J. M. Gallardo and R. I. Pérez-Martin, Trends Food Sci. Technol., 1993, 4, 395–401 CrossRef.
- P. E. Johnson, S. Baumgartner, T. Aldick, C. Bessant, V. Giosafatto, J. Heick, G. Mamone, G. O'Connor, R. Poms, B. Popping, A. Reuter, F. Ulberth, A. Watson, L. Monaci and E. N. Mills, J. AOAC Int., 2011, 94, 1026–1033 CAS.
- B. Domon and R. Aebersold, Science, 2006, 312, 5 CrossRef PubMed.
- Q. Wu, H. Yuan, L. Zhang and Y. Zhang, Anal. Chim. Acta, 2012, 731, 1–10 CrossRef CAS PubMed.
- M. Bantscheff, S. Lemeer, M. M. Savitski and B. Kuster, Anal. Bioanal. Chem., 2012, 404, 939–965 CrossRef CAS PubMed.
- F. Beck, J. M. Burkhart, J. Geiger, R. P. Zahedi and A. Sickmann, Methods Mol. Biol., 2012, 893, 101–113 CAS.
- J. Cox and M. Mann, Annu. Rev. Biochem., 2011, 80, 273–299 CrossRef CAS PubMed.
- K. Kito and T. Ito, Curr. Genomics, 2008, 9, 263–274 CrossRef CAS PubMed.
- F. Toldrá and L. Nollet, Proteomics in Foods - Principles and Applications, Springer, New York, 2013 Search PubMed.
- S. P. Rodrigues, J. A. Ventura, C. Aguilar, E. S. Nakayasu, H. Choi, T. J. Sobreira, L. L. Nohara, L. S. Wermelinger, I. C. Almeida, R. B. Zingali and P. M. Fernandes, J. Proteomics, 2012, 75, 3191–3198 CrossRef CAS PubMed.
- W. Zhu, J. W. Smith and C. M. Huang, J. Biomed. Biotechnol., 2010, 2010, 840518 Search PubMed.
- L. Mora, P. M. Bramley and P. D. Fraser, Proteomics, 2013, 13, 2016–2030 CrossRef CAS PubMed.
- C. Y. Gong and T. Wang, Front. Plant Sci., 2013, 4, 41 Search PubMed.
- M. F. Ocaña, P. D. Fraser, R. K. Patel, J. M. Halket and P. M. Bramley, Anal. Chim. Acta, 2009, 634, 75–82 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |