
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Luminescent metal complexes featuring photophysically innocent boron cluster ligands†
Kent O.
Kirlikovali
,
Jonathan C.
Axtell
,
Alejandra
Gonzalez
,
Alice C.
Phung
,
Saeed I.
Khan
and
Alexander M.
Spokoyny
*
Department of Chemistry and Biochemistry, University of California, 607 Charles E. Young Drive East, Los Angeles, California 90025-1569, USA. E-mail: spokoyny@chem.ucla.edu; Web: https://www.organomimetic.com
First published on 27th April 2016
Abstract
We report the synthesis and characterization of a series of d8 metal complexes featuring robust and photophysically innocent strong-field chelating 1,1′-bis(o-carborane) (bc) ligand frameworks. A combination of UV-Vis spectroscopy, single crystal X-ray structural analysis, and DFT calculations of these species suggest that the dianionic bc ligand does not contribute to any visible metal-to-ligand charge transfer (MLCT) transitions, yet it provides a strong ligand field in these complexes. Furthermore, a bc-based Pt(II) complex containing a 4,4′-di-tert-butyl-2,2′-bipyridine ligand (dtb-bpy) has been prepared and was found to display blue phosphorescent emission dominated by MLCT from the Pt(II) center to the dtb-bpy ligand. Importantly, the bulky three-dimensional nature of the bc ligand precludes intermolecular Pt(II)⋯Pt(II) interactions in the solid state where the resulting compounds retain their emission properties. This study opens a potentially new avenue for designing organic light-emitting diode (OLED) materials with tunable properties featuring photophysically innocent boron-rich cluster ligands.
Introduction
Many square planar platinum complexes containing chelating biaryl ligands have been established as efficient phosphorescent emitters, making them desirable dopants in organic light-emitting diodes (OLEDs).1,2 The emissive properties of these Pt(II) complexes can be tuned by varying the electronic nature of the ligands surrounding the metal center; however, complete control over desired photophysical properties is still limited. For example, these square planar complexes are susceptible to aggregation as a consequence of the lack of steric bulk above and below the square plane, leading to intermolecular Pt(II)⋯Pt(II) interactions that bring about luminescence quenching and red-shifting of the solid-state emission frequency.2,3 Furthermore, in prototypical mixed ligand complexes, (L)(L′)Pt(II), the HOMO is generally both metal- and ligand-based (Pt(II) and L) while the LUMO is located on the remaining ligand (L′).4 This ultimately complicates the predictability of ligand effects on emission properties. A class of compounds that could kinetically stabilize the Pt(II) square planar framework without participating in electronic transitions would prove very useful for creating improved phosphorescent emitters for the next generation of OLED devices.Icosahedral dicarba-closo-dodecaboranes (C2B10H12, carborane) are robust, charge-neutral boron clusters that are often viewed as 3D aromatic analogues of arenes. Unfunctionalized carborane species have an extremely large HOMO–LUMO gap (∼8 eV, see ESI†),5 making them potentially useful building blocks for probing their photophysical innocence in the context of metal-based phosphorescent emitters. This is especially appealing given the available functionalization routes through either carbon or boron vertices in these clusters, enabling the synthesis of tailored ligand frameworks for transition metal complexes.6 For example, Lee and co-workers have recently demonstrated that κ2-C,N-bound 1-(2-pyridyl)-o-carboranyl7a and κ2-C,P-bound 1-(iPr2PCH2)-o-carboranyl7b can be strong ancillary ligands that contribute to the electronic stabilization of bis(heteroleptic) Ir(III) species (see ESI† for molecular structures), leading to an arylpyridine-dominant phosphorescent emission. From DFT calculations and analysis of the emission spectra, these authors determined the C-bound o-carboranyl unit remains uninvolved in electronic transitions and that phosphorescent emission results from MLCT of the Ir(III)-based HOMO to the arylpyridine-based LUMO. Furthermore, several groups have functionalized biaryl ligands with C-connected carboranyl moieties (ortho, meta, para, and nido) to tune luminescent properties8 (see ESI† for molecular structures). To the best of our knowledge, attempts to design a tunable, exclusively carborane-based ligand scaffold for phosphorescent emitter molecules have not been explored thus far.9 Such a ligand framework would be an ideal system for a rational design of metal-based luminescent complexes (vide supra).
In 1964, Hawthorne reported the first synthesis of 1,1′-bis(o-carborane) (2),10a effectively a 3D analogue of biphenyl (Fig. 1), and in 1973, Zakharkin showed that the oxidative coupling of two o-carboranes (1) through carbon vertices yields 2.10d Later, Hawthorne demonstrated that the deprotonation of 2 results in a dianionic species bc, which was shown to bind several transition metals in bidentate or monodentate fashions.10b–f Ligand bc possesses similar electronic and physical properties as the parent o-carborane (see ESI†), and behaves as a robust transition metal ligand. More recently, several groups have improved the synthesis of 2 (ref. 10j) and further expanded the series of heteroleptic late-transition metal complexes containing bc.11 However, fundamental electronic characterization and potential applications for these compounds as electronic materials have yet to be disclosed.
 | ||
| Fig. 1 Oxidative coupling of o-carborane (1) yields 1,1′-bis(o-carborane) (2). Deprotonation of 2 yields the dianionic bc ligand, which can be conceptually thought of as a 3D aromatic analogue of a dianionic biphenyl ligand (bph). | ||
Two monoanionic bidentate ligands are commonly used in Pt(II) architectures employed for OLEDs, but there are few examples of Pt(II)-based emitters containing a dianionic bidentate ligand and a neutral bidentate ligand.12 One such example is Pt(bph)(bpy)12a–c (where bph = biphenyl, bpy = 2,2′-bipyridine, see ESI† for a molecular structure). We hypothesized that employing bc in place of bph could introduce sufficient steric bulk above and below the square plane to eliminate intermolecular Pt(II)⋯Pt(II) interactions, which are often responsible for non-radiative decay pathways that lower luminescent efficiency.2,3 Additionally, the lack of an exposed π-aromatic system in bc should help in reducing these undesirable intermolecular interactions and unwanted substitution and degradation pathways. Furthermore, the high-lying LUMO of bc should be inaccessible for orbital mixing and MLCT processes. Therefore, the bc ligand was hypothesized to provide kinetic stability while maintaining photophysical innocence in the context of designing OLEDs. Finally, emission originating from a single ligand will yield greater color purity, as mixing of emissions from multiple delocalized excited states will not be possible. Together, these properties should allow for the rational tuning of other ancillary ligands without electronic interference from bc.
Results and discussion
To test our hypothesis, we first proceeded to evaluate the photophysical behavior of bc as a ligand in a series of d8 model transition metal complexes of the type M(bc)(dppe) (M = Ni(II), Pd(II), Pt(II); dppe = 1,2-bis(diphenylphosphino)ethane; Fig. 2). The dilithio salt of bc (Li2[bc]) was generated in THF and transferred into a slurry of M(dppe)Cl2 in THF at −80 °C. The reaction was allowed to slowly warm to room temperature overnight, yielding a dark brown solution. Pure compounds were isolated in the yields reported in Fig. 2 as crystalline yellow-orange solids after purification using column chromatography on silica or alumina (see ESI†). All reactions can be easily monitored via31P NMR spectroscopy. For example, 3a and 3b exhibit a significant (>10 ppm) upfield shift in their observed 31P NMR singlet resonance compared to the starting metal-based precursors (see ESI†). These results are also consistent with the recent work by Welch and co-workers who independently synthesized 3a.11a Observed 1JPt–P coupling values for 3c change significantly compared to the starting material, exhibiting a decrease of 1133 Hz (1JPt–P value changes from 3624 Hz to 2491 Hz). The substantial reduction in the magnitude of the 1JPt–P coupling for 3c can be attributed to the strong ligand field of bcversus that of the chloride ligands.13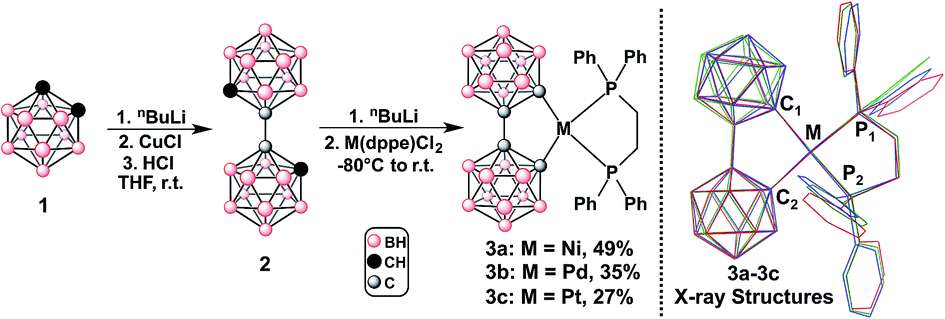 | ||
| Fig. 2 Left: Synthesis of M(bc)(dppe), where M = Ni (3a), Pd (3b), Pt (3c). The synthesis of 2 was adapted from ref. 10j. Right: Stacked X-ray crystal structures of compounds 3a–3c (CCDC 1446940–1446942), illustrating structural similarity down the group. Red = 3a, green = 3b, blue = 3c (see ESI† for thermal ellipsoid plots of 3a–3c). | ||
To demonstrate that the bc framework is structurally similar to the biphenyl (bph) framework, we compared bond distances, angles, and molecular geometries of 3a–3c to those of a series of cyclometalated M(bph)(L^L) in reported X-ray crystal structures, and determined that bc does not influence the intramolecular geometry much differently than the bph ligand. Furthermore, the M–P bond lengths in 3a–3c are also consistent with the strong-field ligand nature of bc in these complexes (see ESI† for specific discussion).
UV-Vis spectroscopic measurements were performed on 3a–3c and revealed strong transitions in the UV region below 360 nm corresponding to π–π* transitions on the ligand. UV-Vis spectra of 3a–3c also feature weaker intensity transitions in the visible region ranging from 400–500 nm that result from M(II)-(dppe) MLCT. DFT calculations were performed on the geometry optimized X-ray crystal structure inputs of 3a–3c in order to confirm the nature of the observed electronic transitions (see ESI†). Our computational studies reveal an almost entirely metal-based HOMO and dppe ligand-based LUMO with negligible contribution from the bc ligand in all frontier orbitals. As we hypothesized previously, bc chelated to the d8 transition metals in our model complexes remains uninvolved in all MLCT-based visible transitions, suggesting its photophysical innocence in the UV-Vis region.
Next, we sought to synthesize a Pt(II)-bpy (bpy = 2,2′-bipyridine) complex chelated by bc, as Pt(II) complexes containing this class of ligands are known to exhibit phosphorescent emission.12a–c Starting with the addition of Li2[bc] to a slurry of Pt(bpy)Cl2, a large amount of emissive, insoluble product was obtained. The extremely low solubility of this product in common organic solvents hampered its characterization. In order to potentially circumvent this issue, we then chose the 4,4′-di-tert-butyl-2,2′-bipyridine (dtb-bpy) ligand as an alternative, anticipating more favorable solubility properties. Using the same synthetic route yielded, again, a largely insoluble, emissive solid (Fig. 3A). After dissolving the crude product in hot 1,2-difluorobenzene and passing the solution through a Celite plug, a non-emissive solid was left on the Celite; a yellow solid that emitted blue-green under UV excitation (365 nm) remained after all volatiles were removed in vacuo. Surprisingly, 1H NMR spectroscopic data suggests that the isolated product consists of a mixture of two species (4a/4b) with a bc ligand chelated to the Pt(II) center in both κ2-C,C-bound (4a) and κ2-B,C-bound modes (4b) (Fig. 3). The κ2-C,C-bound species 4a, derived from the symmetric binding of the bc ligand, is consistent with the presence of three resonances of equal integration in the aromatic region (dtb-bpy ligand) of the 1H NMR spectrum (Fig. 3B, label A). The six remaining resonances in the aromatic region (Fig. 3B, label B) of the 1H NMR spectrum are consistent with an asymmetric κ2-B,C-bound bc species 4b (vide infra). From the relative integration of these two sets of resonances, we estimate that the produced mixture contained a ratio of 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 of 4a to 4b. Repeated attempts to optimize this reaction produced the same mixture in varying ratios of 4a and 4b (see ESI†). Attempts to drive the formation of one isomer from the mixture of isomers while heating under forcing conditions produced no observable change in both 1H and 11B NMR spectra. Notably, during the preparation of this manuscript, Welch and co-workers reported the synthesis of a series of Ru(II) complexes chelated by the κ2-B,C-bound bc ligand.11c The authors explained this B–Ru bond formation results from a competitive B–H activation process, which we think is reminiscent of the observed formation of 4b in this work.
:1.0 of 4a to 4b. Repeated attempts to optimize this reaction produced the same mixture in varying ratios of 4a and 4b (see ESI†). Attempts to drive the formation of one isomer from the mixture of isomers while heating under forcing conditions produced no observable change in both 1H and 11B NMR spectra. Notably, during the preparation of this manuscript, Welch and co-workers reported the synthesis of a series of Ru(II) complexes chelated by the κ2-B,C-bound bc ligand.11c The authors explained this B–Ru bond formation results from a competitive B–H activation process, which we think is reminiscent of the observed formation of 4b in this work.
 | ||
| Fig. 3 (A) Synthesis of the Pt(bc)(dtb-bpy) complex leads to a mixture containing two product isomers featuring a κ2-C,C-bound bc (4a) and κ2-B,C-bound bc (4b). (B) 1H NMR spectrum of the aryl region for the isolated mixture of 4a (label A) and 4b (label B). (C) Single crystal X-ray structure of 4a drawn with 50% thermal ellipsoid probability (CCDC 1446943). H atoms are omitted for clarity. | ||
A mixture of 4a and 4b was dissolved in hot 2-MeTHF and allowed to cool to room temperature, yielding single crystals of 4a (Fig. 3C). The molecule adopts a minimally distorted square planar geometry with the C–Pt–Ntrans angles at 174.2(7)° and 173.0(8)°. The bulky bc ligand forces the molecule to pack “head-to-tail” with Pt(II)⋯Pt(II) distances ranging between 5.87(0) Å and 5.52(0) Å (see ESI†), which far exceed the reported 3.15–3.76 Å expected for intermolecular Pt(II)⋯Pt(II) interactions2,3 (see ESI† for further discussion about the single crystal X-ray structure of 4a).
Given that the bc framework is amenable to substitution, we hypothesized that functionalizing this scaffold with alkyl groups would increase the solubility of the resulting Pt(II) complexes, ultimately allowing us to better characterize these emissive species. We therefore installed ethyl groups at the B(9) and B(12) positions of the parent 9,12-B-diiodo-o-carborane (5) using Kumada cross-coupling conditions producing bis(alkylated) species 6 (Fig. 4A and ESI†). Compound 6 was then subjected to Cu-mediated oxidative coupling conditions, ultimately producing the tetralkylated-bc (7) in 50% isolated yield (Fig. 4A and ESI†).10j Compound 7 was dilithiated and added to Pt(dtb-bpy)Cl2 in a similar manner as with 2 (Fig. 4A). Surprisingly, after the reaction mixture was stirred for a day at 60 °C, predominantly a κ2-B,C-bound isomer 8 was observed by 1H and 2D 13C–1H HSQC NMR spectroscopy (>80%). Purification of the resulting mixture further afforded pure κ2-B,C-bound species as a pale orange solid which exhibits blue-green emission in the solid state and, as hypothesized, is extremely soluble in the majority of common organic solvents. To our knowledge, this is the first reported example of a functionalized bc bound to a metal.10i
 | ||
| Fig. 4 (A) Synthetic route to 9,9′,12,12′-tetraethyl-1,1′-bis(o-carborane) (7), syntheses of 5 and 6 from ref. 6k. (B) X-ray crystal structure of 8 (CCDC 1446944) with thermal ellipsoids drawn at 50% probability, H atoms omitted for clarity. (C) Stacking of 8 with Pt(II)⋯Pt(II) distances of 5.981 Å and 7.979 Å. | ||
Crystals of 8 suitable for X-ray analysis were grown by slow evaporation of diethyl ether over the course of one week. The diffraction study confirmed the presence of the asymmetric isomer with one Pt–C(1) bond and one Pt–B(4) bond (Fig. 4B). At 2.07(3) Å, the Pt–B(4) bond is slightly longer than the 2.03(6) Å Pt–C(1) bond. Furthermore, the greater trans influence of the carborane-based boryl moiety14 can be seen in the elongation of the Pt–N bond lengths: the Pt–N(1) bond is 2.17(5) Å, whereas the Pt–N(2) bond is only 2.05(3) Å. As a result of the asymmetric binding of the bc-based ligand in 8, one carborane cage rotates and forces the ethyl group about 30° out of the plane created by C(1)–Pt–B(4), whereas the other 3 ethyl groups sit in the square plane (Fig. 4C). This protruding ethyl group likely forces the dtb-bpy out of the square plane, causing the molecule to adopt a slightly distorted square planar structure; however, bond angles of 176.5(5)° for C(1)–Pt–N(2) and 168.8(2)° for B(4)–Pt–N(1) are well within the range of corresponding angles in previously reported 4-coordinate Pt(bph)(N^N) compounds (see ESI†).
Importantly, the intermolecular Pt(II)⋯Pt(II) distances were augmented even more in the solid-state than in 4a through the introduction of ethyl groups, yielding Pt(II)⋯Pt(II) distances of 5.891 Å (when ethyl groups face away from each other) and 7.979 Å (when ethyl groups point towards each other), effectively preventing any potential intermolecular Pt(II)⋯Pt(II) interactions (Fig. 4C). Furthermore, the solid-state packing adopts a “head-to-tail” arrangement such that dtb-bpy lies above and below the bc-based ligand in the crystal lattice, eliminating the potential for any π–π stacking interactions, which have also been reported to result in deleterious non-radiative emission quenching.2c,3d
Cyclic voltammetry (CV) of 8 reveals a reversible, one-electron reduction (ERed1/2 = −1.92 V) and an irreversible one-electron oxidation (EOx1/2 = 0.85 V), as shown in Table 1 and Fig. 5A. This electrochemical behavior is consistent with other square planar Pt(II) species undergoing a reversible ligand-centered reduction and irreversible metal-centered oxidation.2–4,12a,c,15a Further, DFT calculations support these data (vide infra).
| Compound | E Red1/2 (V) | E Ox1/2 (V) | Solvent | Reference |
|---|---|---|---|---|
| a Values reported relative to the ferrocene/ferrocenium couple (Fc/Fc+). b Values were corrected according to ref. 15b. c Reversible. d Irreversible. | ||||
| 8 | −1.92c | 0.85d | MeCN | This work |
| Pt(bph)(bpy)b | −1.87c | −0.33d | MeCN | 12a |
| Pt(bph)(en)b | −2.13c | 0.25d | CH2Cl2 | 12c |
 | ||
| Fig. 5 (A) Cyclic voltammogram of 8versus Fc/Fc+ in MeCN with a glassy carbon working electrode, scan rate = 0.1 V s−1. (B) UV-Vis absorption spectrum in CH2Cl2 (dotted line) and emission spectra of 8 in 2-MeTHF at 77 K (blue), 2 wt% PMMA film (red), and neat solid (black), λexc = 380 nm; inset: picture of 8 in 2-MeTHF at 77 K (λexc = 365 nm). | ||
Though electrochemical characterization for heteroleptic Pt(II) complexes bound by a dianionic bidentate ligand and a neutral bidentate ligand are scarce, Table 1 presents redox potentials for two such examples, Pt(bph)(bpy) and Pt(bph)(en) (where en = 1,2-ethylenediamine).15b The reduction potential for 8 is similar to the other two compounds (Table 1, column 2); however, the oxidation potential of 8 is significantly greater (Table 1, column 3). This is consistent with the strong field ligand character of the bc-based framework, which should make it more difficult to remove an electron from the Pt(II) HOMO level in 8.
Similar to 4a/4b, we observed that 8 emits an intense blue-green color upon irradiation with a table-top UV lamp at 365 nm at room temperature. Given the improved solubility properties of 8, we were able to carry out a detailed series of photophysical measurements in order to ascertain the efficiency and nature of this luminescent behaviour. The UV-Vis and phosphorescent emission spectra for 8 are presented in Fig. 5B with corresponding data in Table 2. The absorption spectrum reveals strong transitions in the UV region (≤330 nm) that arise from π–π* transitions on the dtb-bpy ligand. The broad, lower intensity band from 340–420 nm can be assigned to both singlet and triplet metal-to-ligand charge transfers (1MLCT and 3MLCT). Compound 8 is non-emissive in solution at room temperature, suggesting emission might be thermally quenched through interaction with solvent molecules. However, at 77 K in 2-MeTHF, bright blue phosphorescence is observed (λmax = 485 nm, τ = 11.4 μs). The well-defined vibrational features suggest ligand-centered emission resulting from an MLCT.1a
| Medium | Em. λmax (nm) | ϕ | τ (μs) | k r (104 s−1) | k nr (104 s−1) |
|---|---|---|---|---|---|
| a Solutions at room temperature were non-emissive, and 77 K spectra were measured in 2-MeTHF. b PMMA film was prepared as 2 wt%, neat solid was 8 in powder form. c Quantum yields were measured using an integrating sphere under N2. d 77 K lifetime was measured in 2-MeTHF, PMMA film and neat solid lifetimes were measured in the absence of air. e Values obtained from the weighted average of a multi-exponential decay. f Calculated according to the equations kr = ϕ/τ and knr = (1 − ϕ)/τ, where kr is the radiative rate constant, knr is the non-radiative rate constant, ϕ is the quantum yield, and τ is the luminescence lifetime. | |||||
| 77 K | 456, 486, 514 | — | 11.4 | — | — |
| PMMA film | 497 | 0.07 | 4.24e | 1.67 | 22.1 |
| Neat solid | 476, 505, 540 | 0.03 | 0.94e | 3.20 | 103.1 |
Similarly, the neat solid 8 also exhibits an emission profile with a resolved vibronic fine structure, further suggesting the ligand-centered emission. Compared to the emission profile from the neat solid, emission from the solution at 77 K is hypsochromically shifted by roughly 20 nm. This shift is expected as vibrational relaxations to a lower energy excited state will not be favorable at lower temperatures, resulting in a higher energy, blue-shifted emission observed for 8 at 77 K in 2-MeTHF. When 8 is doped in a PMMA matrix (2 wt%), the emission profile is broadened and the peak is blue-shifted by about 8 nm versus the emission of the neat solid. The excited-state lifetime (τ) for 8 increases as the environment becomes more rigid. This increase is significant, going from 0.94 μs as a neat solid, to 4.24 μs doped in PMMA, further to 11.4 μs at 77 K. This evidence suggests that decreasing vibrational motion through a more rigid and ordered surrounding environment can preserve the excited state, possibly by minimizing the energy loss via non-radiative relaxation pathways.1a
From the measured excited state lifetimes and quantum yields for 8 doped in the PMMA matrix and neat solid, the radiative rate constant (kr) and non-radiative rate constant (knr) could be calculated (Table 2). Though kr for the doped PMMA film is half that of the neat solid, knr for the doped PMMA film decreased by about a factor of 5, which supports the trend seen for measured τ values (vide supra). Additionally, the doped PMMA film exhibits a quantum yield (ϕ) more than twice that of the neat solid, as well as a lifetime that is about 4.5 times greater. Based on these data, it is likely that the PMMA film decreases access to a non-radiative decay pathway through its behavior as a rigid matrix.
To further our understanding of the photophysical properties of 8, we performed a DFT computational study at the BP86-D3 level using the TZP basis set (Fig. 6). The optimized geometry of the singlet state displays a slightly distorted square planar structure, which is in agreement with the obtained single crystal X-ray structure. The frontier orbital diagram indicates a HOMO and HOMO−1 almost completely localized on the Pt(II) with negligible contribution from the bc fragment. Both the LUMO and LUMO+1 are isolated on dtb-bpy, which corroborates the observed ligand-centered phosphorescence of 8 without observed contribution from the bc fragment. The optimized geometry of the triplet state, however, reveals an almost tetrahedral structure that is extremely distorted from the favorable square planar geometry seen in the ground state (Fig. 6). In the excited state, the complex twists via a non-radiative decay pathway, resulting in a large value for knr. This hypothesis supports the observed decrease in knr from the pure solid to the PMMA matrix: as the rigidity of the environment increases, the geometry of the molecule will be more difficult to distort. These calculations suggest that future molecular designs should incorporate a large degree of steric bulk to potentially minimize this excited state distortion, thereby improving phosphorescence efficiency in these compounds.
 | ||
| Fig. 6 Frontier orbitals of 8 based on optimized geometries of S0 and T1 states (BP86-D3, TZP). | ||
In general, phosphorescent blue OLEDs suffer from short lifetimes (∼600 hours) relative to their red and green counterparts (106 hours).16a Several groups have previously investigated the degradation of blue OLEDs and found that the decomposition of the phosphorescent dopant molecules occurs during regular use, greatly inhibiting the overall lifetime and efficiency of the device.16b–e Thermogravimetric analysis of 8 suggests the bc ligand framework remains intact upon heating to 500 °C (see ESI†). This observation suggests that chelating boron cluster scaffolds may be potentially appropriate ligands that can ameliorate previously described stability issues in OLED devices.
Conclusions
In conclusion, for the first time, we have presented a detailed study on the behavior of 1,1′-bis(o-carborane) as a dianionic ligand (bc) in group 10 metal complexes. From the synthesis and characterization of model complexes, we have discovered that unlike the structurally reminiscent biphenyl ligand, bc displays a unique photophysical innocence and remains uninvolved in relevant photophysical transitions when bound to the group 10 transition metals. Furthermore, since bc introduces sufficient steric bulk above and below the square plane of the metal center, it effectively shuts down undesired intermolecular interactions in the solid-state. For Pt(II) complexes featuring phosphorescent emission properties, this unique ligand design aspect allowed us to remove any possible Pt(II)⋯Pt(II) interactions, which commonly lead to luminescence quenching. The organomimetic6e,17 properties of carboranes in general, and bc in particular, enabled us to rationally tune the processability of a blue phosphorescent emitting Pt(II) species. We are currently investigating the substitution of carborane-based ligands with bulkier functional groups that will minimize the excited state distortion. This work opens a new avenue in designing luminescent materials with improved properties incorporating robust and photophysically innocent multidentate ligand platforms.Acknowledgements
We would like to thank the Department of Chemistry and Biochemistry at UCLA for start up funds. Portions of this material are based upon work supported by the National Science Foundation (CHE-1048804). We are indebted to Prof. Peter I. Djurovich and Prof. Mark E. Thompson for assistance with photophysical measurements and helpful discussions. A. G. thanks the UCLA CARE Fellows & Scholars Program for funding. We thank Dr Liban M. A. Saleh for helpful comments and Rafal M. Dziedzic for assistance with the syntheses of 5 and 6. The authors declare no competing financial interest. A patent application covering the content of this manuscript was filed by UCLA on February 18, 2016.References
- (a) Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) P.-T. Chou and Y. Chi, Chem.–Eur. J., 2007, 13, 380–395 CrossRef CAS PubMed; (c) Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- For selected examples of phosphorescent cyclometallated Pt(II) complexes, see: (a) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed; (b) J. Kavitha, S.-Y. Chang, Y. Chi, J.-K. Yu, Y.-H. Hu, P.-T. Chou, S.-M. Peng, G.-H. Lee, Y.-T. Tao, C.-H. Chien and A. J. Carty, Adv. Funct. Mater., 2005, 15, 223–229 CrossRef CAS; (c) S.-Y. Chang, J. Kavitha, S.-W. Li, C.-S. Hsu, Y. Chi, Y.-S. Yeh, P.-T. Chou, G.-H. Lee, A. J. Carty, Y.-T. Tao and C.-H. Chien, Inorg. Chem., 2006, 45, 137–146 CrossRef CAS PubMed; (d) S.-Y. Chang, Y.-M. Cheng, Y. Chi, Y.-C. Lin, C.-M. Jiang, G.-H. Lee and P.-T. Chou, Dalton Trans., 2008, 6901–6911 RSC; (e) Z. M. Hudson, C. Sun, M. G. Helander, H. Amarne, Z.-H. Lu and S. Wang, Adv. Funct. Mater., 2010, 20, 3426–3439 CrossRef CAS; (f) X.-C. Hang, T. Fleetham, E. Turner, J. Brooks and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6753–6756 CrossRef CAS PubMed; (g) Y. Unger, D. Meyer, O. Molt, C. Schildknecht, I. Münster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2010, 49, 10214–10216 CrossRef CAS PubMed; (h) M. Tenne, S. Metz, G. Wagenblast, I. Münster and T. Strassner, Organometallics, 2015, 34, 4433–4440 CrossRef CAS.
- (a) D. Bandyopadhyay, P. Bandyopadhyay, A. Chakravorty, F. A. Cotton and L. R. Falvello, Inorg. Chem., 1983, 22, 1315–1321 CrossRef CAS; (b) L. Chassot, E. Müller and A. von Zelewsky, Inorg. Chem., 1984, 23, 4249–4253 CrossRef CAS; (c) M. Ghedini, D. Pucci, A. Crispini and G. Barberio, Organometallics, 1999, 18, 2116–2124 CrossRef CAS; (d) K. E. Dungey, B. E. Thompson, N. A. P. Kane-Maguire and L. L. Wright, Inorg. Chem., 2000, 39, 5192–5196 CrossRef CAS PubMed.
- (a) D. P. Rillema, A. J. Cruz, C. Moore, K. Siam, A. Jehan, D. Base, T. Nguyen and W. Huang, Inorg. Chem., 2013, 52, 596–607 CrossRef CAS PubMed; (b) D. P. Rillema, S. R. Stoyanov, A. J. Cruz, H. Nguyen, C. Moore, W. Huang, K. Siam, A. Jehan and V. KomReddy, Dalton Trans., 2015, 44, 17075–17090 RSC.
- (a) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (b) J. M. Oliva, P. v. R. Schleyer, G. Aullón, J. I. Burgos, A. Fernández-Barbero and I. Alkorta, Phys. Chem. Chem. Phys., 2010, 12, 5101–5108 RSC.
- For select examples, see: (a) Y. Quan and Z. Xie, Angew. Chem., 2016, 128, 1317–1320 CrossRef; (b) R. Cheng, J. Zhang, J. Zhang, Z. Qiu and Z. Xie, Angew. Chem., 2016, 128, 1783–1786 CrossRef; (c) C. Tang and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 7662–7665 CrossRef CAS PubMed; (d) H. Lyu, Y. Quan and Z. Xie, Angew. Chem., 2015, 127, 10769–10772 CrossRef; (e) A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903–919 CrossRef CAS PubMed; (f) A. M. Spokoyny, C. D. Lewis, G. Teverovskiy and S. L. Buchwald, Organometallics, 2012, 31, 8478–8481 CrossRef CAS PubMed; (g) C. A. Lugo, C. E. Moore, A. L. Rheingold and V. Lavallo, Inorg. Chem., 2015, 54, 2094–2096 CrossRef CAS PubMed; (h) A. R. Popescu, F. Teixidor and C. Viñas, Coord. Chem. Rev., 2014, 269, 54–84 CrossRef CAS; (i) B. Xu, Y.-P. Wang, Z.-J. Yao and G.-X. Jin, Dalton Trans., 2015, 44, 1530–1533 RSC; (j) J. Kahlert, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, L. M. Rendina, P. J. Low, L. Weber and M. A. Fox, Dalton Trans., 2015, 44, 9766–9781 RSC; (k) Z. Zheng, W. Jiang, A. A. Zinn, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1995, 34, 2095–2100 CrossRef CAS; (l) P. A. Jelliss, J. Mason, J. M. Nazzoli, J. H. Orlando, A. Vinson, N. P. Rath and M. J. Shaw, Inorg. Chem., 2006, 45, 370–385 CrossRef CAS PubMed; (m) S. W. Buckner, M. J. Fischer, P. A. Jelliss, R. Luo, S. D. Minteer, N. P. Rath and A. Siemiarczuk, Inorg. Chem., 2006, 45, 7339–7347 CrossRef CAS PubMed.
- (a) H. J. Bae, H. Kim, K. M. Lee, T. Kim, M. Eo, Y. S. Lee, Y. Do and M. H. Lee, Dalton Trans., 2013, 42, 8549–8552 RSC; (b) T. Kim, J. Lee, S. U. Lee and M. H. Lee, Organometallics, 2015, 34, 3455–3458 CrossRef CAS; (c) A. M. Prokhorov, P. A. Slepukhin, V. L. Rusinov, V. N. Kalinin and D. N. Kozhevnikov, Chem. Commun., 2011, 47, 7713–7715 RSC; (d) A. M. Prokhorov, T. Hofbeck, R. Czerwieniec, A. F. Suleymanova, D. N. Kozhevnikov and H. Yersin, J. Am. Chem. Soc., 2014, 136, 9637–9642 CrossRef CAS PubMed.
- (a) C. Shi, H. Sun, Q. Jiang, J. Wang, W. Huang and H. Yan, Chem. Commun., 2013, 49, 4746–4748 RSC; (b) C. Shi, H. Sun, X. Tang, W. Lv, H. Yan, Q. Zhao, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2013, 52, 13434–13438 CrossRef CAS PubMed; (c) T. Kim, H. Kim, K. M. Lee, Y. S. Lee and M. H. Lee, Inorg. Chem., 2013, 52, 160–168 CrossRef CAS PubMed; (d) H. J. Bae, J. Chung, H. Kim, J. Park, K. M. Lee, T.-W. Koh, Y. S. Lee, S. Yoo, Y. Do and M. H. Lee, Inorg. Chem., 2014, 53, 128–138 CrossRef CAS PubMed.
- For recent, comprehensive reviews on boron cluster-based luminescent materials, see: (a) X. Li, H. Yan and Q. Zhao, Chem.–Eur. J., 2016, 22, 1888–1898 CrossRef CAS PubMed; (b) S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC.
- (a) R. A. Wiesboeck and M. F. Hawthorne, J. Am. Chem. Soc., 1964, 86, 1642–1643 CrossRef CAS; (b) D. A. Owen and M. F. Hawthorne, J. Am. Chem. Soc., 1970, 92, 3194–3196 CrossRef CAS; (c) D. A. Owen and M. F. Hawthorne, J. Am. Chem. Soc., 1971, 93, 873–880 CrossRef; (d) First example of oxidative coupling of two o-carboranes: L. I. Zakharkin and A. I. Kovredov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1973, 22, 1396 CrossRef; (e) X. Yang, W. Jiang, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1992, 114, 9719–9721 CrossRef CAS; (f) Bis-carboranes that feature both C–C and B–C bonds: X. Yang, W. Jiang, C. B. Knobler, M. D. Mortimer and M. F. Hawthorne, Inorg. Chim. Acta, 1995, 240, 371–378 CrossRef CAS; (g) D. E. Harwell, M. D. Mortimer, C. B. Knobler, F. A. L. Anet and M. F. Hawthorne, J. Am. Chem. Soc., 1996, 118, 2679–2685 CrossRef CAS; (h) D. E. Harwell, J. McMillan, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1997, 36, 5951–5955 CrossRef CAS PubMed; (i) Only report of functionalized 2: A. Herzog, A. Maderna, G. N. Harakas, C. B. Knobler and M. F. Hawthorne, Chem.–Eur. J., 1999, 5, 1212–1217 CrossRef CAS; (j) Optimized synthesis of 2: S. Ren and Z. Xie, Organometallics, 2008, 27, 5167–5168 CrossRef CAS.
- (a) M. J. Martin, W. Y. Man, G. M. Rosair and A. J. Welch, J. Organomet. Chem., 2015, 798, 36–40 CrossRef CAS; (b) Z.-J. Yao, Y.-Y. Zhang and G.-X. Jin, J. Organomet. Chem., 2015, 798, 274–277 CrossRef CAS; (c) L. E. Riley, A. P. Y. Chan, J. Taylor, W. Y. Man, D. Ellis, G. M. Rosair, A. J. Welch and I. B. Sivaev, Dalton Trans., 2016, 45, 1127–1137 RSC.
- (a) C. Cornioley-Deuschel and A. von Zelewsky, Inorg. Chem., 1987, 26, 3354–3358 CrossRef CAS; (b) M. Maestri, D. Sandrini, V. Balzani, A. von Zelewsky, C. Deuschel-Cornioley and P. Jolliet, Helv. Chim. Acta, 1988, 71, 1053–1059 CrossRef CAS; (c) C. B. Blanton, Z. Murtaza, R. J. Shaver and D. P. Rillema, Inorg. Chem., 1992, 31, 3230–3235 CrossRef CAS; (d) J. A. Zuleta, C. A. Chesta and R. Eisenberg, J. Am. Chem. Soc., 1989, 111, 8916–8917 CrossRef CAS; (e) J. M. Bevilacqua and R. Eisenberg, Inorg. Chem., 1994, 33, 2913–2923 CrossRef CAS; (f) S. D. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949–1960 CrossRef CAS; (g) W. Paw, S. D. Cummings, M. A. Mansour, W. B. Connick, D. K. Geiger and R. Eisenberg, Coord. Chem. Rev., 1998, 171, 125–150 CrossRef CAS; (h) M. Hissler, J. E. McGarrah, W. B. Connick, D. K. Geiger, S. D. Cummings and R. Eisenberg, Coord. Chem. Rev., 2000, 208, 115–137 CrossRef CAS.
- These results are consistent with the decrease in the 1JPt–P value for other Pt(P^P)Cl2 to Pt(P^P)L2 complexes, where P^P = chelating diphosphine and L = strong-field ligand. (a) P. E. Garrou, Chem. Rev., 1981, 81, 229–266 CrossRef CAS; (b) B. L. Edelbach, D. A. Vicic, R. J. Lachicotte and W. D. Jones, Organometallics, 1998, 17, 4784–4794 CrossRef CAS; (c) P. Sgarbossa, A. Scarso, R. A. Michelin and G. Strukul, Organometallics, 2007, 26, 2714–2719 CrossRef CAS.
- For selected examples of molecules featuring carborane-based B–M bonds, see: (a) A. M. Spokoyny, M. G. Reuter, C. L. Stern, M. A. Ratner, T. Seideman and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 9482–9483 CrossRef CAS PubMed; (b) Z.-J. Yao, W.-Y. Yu, Y.-J. Lin, S.-L. Huang, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2014, 136, 2825–2832 CrossRef CAS PubMed; (c) N. Fey, M. F. Haddow, R. Mistry, N. C. Norman, A. G. Orpen, T. J. Reynolds and P. G. Pringle, Organometallics, 2012, 31, 2907–2913 CrossRef CAS; (d) M. E. El-Zaria, H. Arii and H. Nakamura, Inorg. Chem., 2011, 50, 4149–4161 CrossRef CAS PubMed; (e) B. J. Eleazer, M. D. Smith and D. V. Peryshkov, Organometallics, 2016, 35, 106–112 CrossRef CAS.
- (a) P.-I. Kvam, M. V. Puzyk, K. P. Balashev and J. Songstad, Acta Chem. Scand., 1995, 49, 335–343 CrossRef CAS; (b) N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- (a) Y. Zhang, S. Lee and S. R. Forrest, Nat. Commun., 2014, 5, 1–7 CAS; (b) S. Scholz, R. Meerheim, K. Walzer and K. Leo, Proc. SPIE, 2008, 69991B CrossRef; (c) V. Sivasubramaniam, F. Brodkorb, S. Hanning, H. P. Loebl, V. van Elsbergen, H. Boerner, U. Scerf and M. Kreyenschmidt, J. Fluorine Chem., 2009, 130, 640–649 CrossRef CAS; (d) I. R. de Moraes, S. Scholz, B. Lüssem and K. Leo, Org. Electron., 2011, 12, 341–347 CrossRef; (e) R. Seifert, I. R. de Moraes, S. Scholz, M. C. Gather, B. Lüssem and K. Leo, Org. Electron., 2013, 14, 115–123 CrossRef CAS.
- M. F. Hawthorne, J. Chem. Educ., 2009, 86, 1131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See ESI for full experimental details. CCDC 1446940–1446945. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01146b |
| This journal is © The Royal Society of Chemistry 2016 |