
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
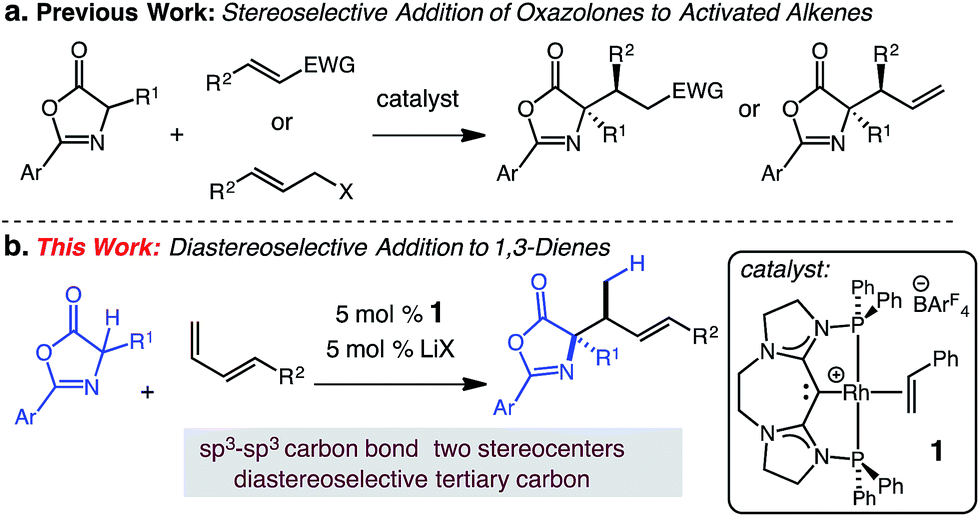
Diastereoselective synthesis of vicinal tertiary and N-substituted quaternary stereogenic centers by catalytic hydroalkylation of dienes†
Matthew J.
Goldfogel
and
Simon J.
Meek
*
Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290, USA. E-mail: sjmeek@unc.edu; Web: http://www.chem.unc.edu/people/faculty/meek/
First published on 11th March 2016
Abstract
An efficient and diastereoselective (CDC)–Rh-catalyzed hydroalkylation of dienes with 1,3-oxazol-5(4H)-ones is reported. Aryl and alkyl substituted dienes are converted to α,α-substituted oxazolones (24 examples) by the formation of N-substituted quaternary carbon stereogenic centers in good yields (up to 96%) and with high diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). The reaction is tolerant of a range of dienes and oxazolones bearing various functional groups. Utility of the oxazolone products is illustrated through hydrolysis to form α,β-substituted α-amino acid analogues and stereoselective epoxidation of the resultant alkene to create four contiguous stereocenters.
:1 dr). The reaction is tolerant of a range of dienes and oxazolones bearing various functional groups. Utility of the oxazolone products is illustrated through hydrolysis to form α,β-substituted α-amino acid analogues and stereoselective epoxidation of the resultant alkene to create four contiguous stereocenters.
Introduction
The catalytic hydroalkylation of alkenes is a valuable, atom-economical approach for the synthesis of C–C bonds from readily available starting materials.1 Pioneering studies have led to the development of intermolecular processes that employ styrenes,2 unactivated alkenes,3 allenes,4 and alkynes5 as effective substrates that can react with appropriate C-based nucleophiles. Despite recent advances in alkene hydroalkylation, intramolecular6 examples predominate and most transformations utilize nucleophiles that are malonate derived or trade atom-economy for reactivity.7 While this reaction is often applied to the transformation of alkenes, the intermolecular hydroalkylation of dienes remains relatively unexplored. The hydroalkylation of diene substrates is synthetically useful; such reactions convert readily available unsaturated hydrocarbons into versatile allyl-containing building blocks. Only a limited number of catalytic intermolecular hydroalkylations of dienes have been reported, with none able to effectively promote the diastereoselective addition of C-based nucleophiles to terminal dienes such as 3.Catalytic intermolecular diene hydroalkylation was first accomplished with a Pd catalyst by Takahashi.8 Subsequent Pd catalyzed hydroalkylations have introduced a variety of enolizable nucleophiles.9 These reactions selectively generate linear products via 1,4-addition with modest to excellent site-selectivity. The reactions work well with 2,3-substituted dienes but for 1,4-substituted dienes reactions are limited to methyl-substituted or cyclic substrates (e.g., cyclohexadiene).8
We postulated that the catalyst controlled γ-selective addition of a prochiral enol nucleophile to 1-substituted diene would enable C(sp3)–C(sp3) formation and generate vicinal strereogenic centers. Enantioselective Michael additions of 1,3-oxazol-5(4H)-ones to activated C–C π bonds10 has been demonstrated (aldehydes,11 ketones,12 amides,13 allenoates14), however, despite these notable advances, additions to unactivated alkenes or dienes substrates have not been reported. In this report, we describe a catalytic addition of substituted 1,3-oxazol-5(4H)-ones to 1-substituted dienes that is site- and diastereoselective (Scheme 1).15
 | ||
| Scheme 1 Diastereoselective (CDC)–Rh(I) catalyzed hydroalkylation of dienes with 1,3-oxazol-5(4H)-ones. | ||
We have previously developed carbodicarbene (CDC) Rh complexes for the catalytic hydroamination16 and hydroarylation17 of dienes under mild conditions in the presence of various polar functional groups.18 The reported carbodicarbene (CDC)-supported Rh catalyst 1 exhibits secondary activation through the reversible interaction of Lewis acids and the CDC ligand,19 which modulates electron donation to the Rh center (i.e., increasing the electrophilicity of the catalyst).17 This reversible activation lessens inhibition by the nucleophile. We sought to extend the use of this catalyst to the formation of C(sp3)–C(sp3) carbon bonds and the diastereoselective generation of two contiguous stereogenic centers.
Herein, we present the first diastereoselective catalytic addition of oxazolones to 1-substituted dienes. The reactions generate α,α-disubstituted allylic amino acid products in up to 96% yield and >20:1 dr. Products contain two contiguous stereocenters, including an N-substituted quaternary carbon. The transformation is catalyzed by 5 mol% of the (CDC)–Rh catalyst in the presence of 5 mol% lithium salt at 50 °C and affords the anti-addition products.
Results and discussion
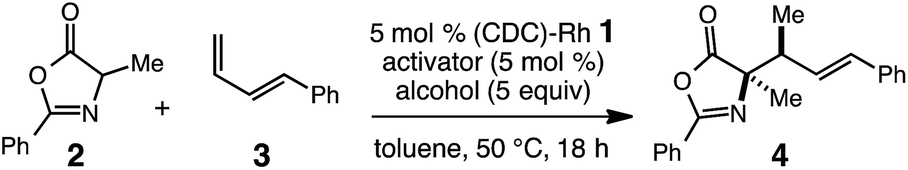
We began our studies for an efficient catalytic hydroalkylation of dienes by the reaction of oxazolone 2 with phenylbutadiene 3 in the presence of 5 mol% (CDC)–Rh complex 1 and 5 mol% LiPF6 activator in toluene at 50 °C (Table 1, entry 1). We were encouraged to observe the formation of 4 in 17% yield and 10:1 dr (anti/syn). A brief survey of solvents provided slightly higher yields (up to 21%), but reduced diastereoselectivities (<6:1 dr) in all cases (see ESI†). As such, toluene was used for further optimization. A variety of Lewis acid activators were screened (entries 2–4, Table 1) and demonstrated that lithium salts were most effective; 5 mol% of AgCl and LiBF4 resulted <10% conversion to 4 most likely the due to poor solubility of the metal salt in toluene. Weaker coordinating counter anions lead to dramatically improved reactivity as shown by LiBArF4, which furnishes 4 in 51% yield with 18:1 dr. Further increase in reaction efficiency can be achieved through the use of alcohol co-solvents (entries 5–8). While addition of i-PrOH led to no improvement in the LiBArF4 promoted reaction (50% yield, 11:1 dr, entry 5), treatment of 3 and 4 with 5 mol% (CDC)–Rh and 5 mol% LiPF6 in toluene/i-PrOH 40:1 at 50 °C proved optimal, delivering 4 in 85% yield and 19:1 dr (entry 6). Screening various alcohol co-solvents results in both decreased conversion and selectivity (entries 7 and 8); MeOH and t-BuOH afforded 4 in 26% yield (3:1 dr), and 29% yield (5:1 dr), respectively. It should be noted that MeOH as a co-solvent leads to competitive oxazolone decomposition via ring-opening. The conditions reported in entry 6 were identified as optimal and employed in further reaction development, although LiBArF4 was found to be optimal for certain substrates (vide infra). Additional control reactions run without LiPF6 (entry 9), or with 2.5 mol% [Rh(cod)Cl]2 and 5 mol NaBArF4 in place of (CDC)–Rh 1 (entry 10) result in no reaction, highlighting the importance of cationic (CDC)–Rh complex 1, in combination with a Lewis acid co-catalyst, for reactivity.
|
|
|||
|---|---|---|---|
| Entry | Activator; mol% | Alcoholb | Yieldc (%); drd |
|
a See ESI for experimental details; all reactions performed under N2 atm.
b A solvent ratio of 40:1 toluene/alcohol used.
c Yields of purified products are an average of two runs.
d Values determined by analysis of 400 or 600 MHz 1H NMR spectra of unpurified mixtures with hexamethyldisiloxane as an internal standard.
e Reaction run with 2.5 mol% [Rh(cod)Cl]2 and 5 mol% NaBArF4 as catalyst.
|
|||
| 1 | LiPF6; 5 | — | 17; 10:1 |
| 2 | AgCl; 5 | — | 0; — |
| 3 | LiBF4; 5 | — | 8; 4:1 |
| 4 | LiBArF4; 5 | — | 51; 18:1 |
| 5 | LiBArF4; 5 | i-PrOH | 50; 11:1 |
| 6 | LiPF 6 ; 5 | i-PrOH |
85; 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 7 | LiPF6; 5 | MeOH | 26; 3:1 |
| 8 | LiPF6; 5 | t-BuOH | 29; 5:1 |
| 9 | — | i-PrOH | 0; — |
| 10e | LiPF6; 5 | i-PrOH | 0; — |
Determining the role of the alcohol and its influence on reaction efficiency and diastereoselectivity cannot necessarily be decoupled, however, a number of observations can be made: (1) the lithium salt is required for the reaction to occur. (2) Considering the difference in reaction efficiency between LiPF6 and LiBArF4 without i-PrOH, the alcohol is likely assisting in solubilizing the lithium salt. (3) Addition of i-PrOH changes the product diastereoselectivity, either suggesting hydrogen bonding of the alcohol with the nucleophile or alcohol solvation of the lithium salt. In the case of LiPF6 dr increases where as for LiBArF4 the selectivity decreases. (4) Additional roles, such as formation of Brønsted acid, are disfavored by reactions run in the presence of 2,6-ditert-butyl pyridine, which show no deleterious effects (see ESI† for details). (5) Use of chiral alcohols and diols do not result in an enantioselective reaction (see ESI† for details).
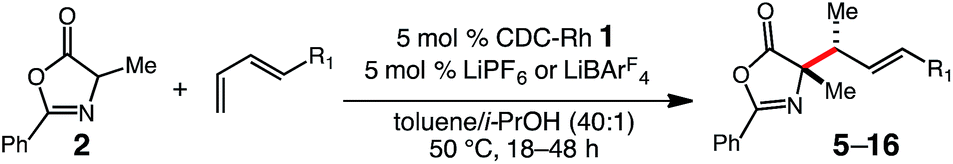
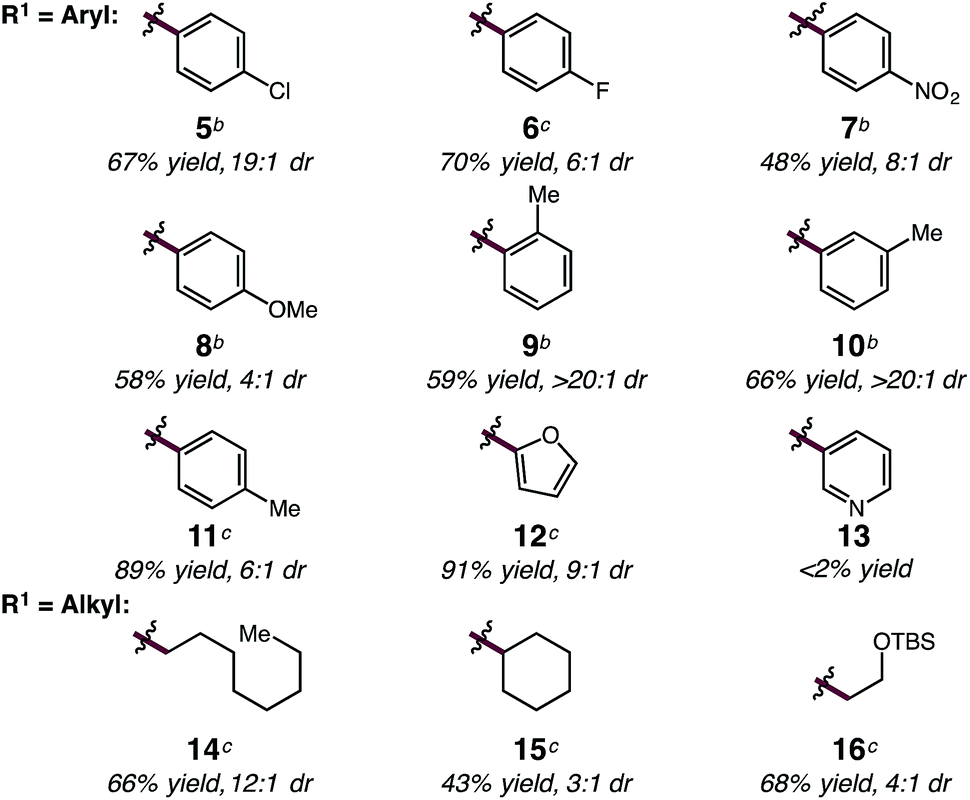
With optimized conditions in hand, we sought to explore the diene scope of the hydroalkylation with oxazolone 2. For certain diene substrates LiBArF4 proved to be the more effective lithium salt in order to obtain good yields and selectivities. As shown in Table 2, formation of the N-substituted quaternary carbon occurs readily with modest to excellent levels of selectivity with aryl (5–13) and alkyl dienes (14–16). Electronic modifications to the aryl ring were well tolerated by the reaction. Aryl rings bearing halogens or electron withdrawing groups react with only slight decreases to yield and diastereoselectivity; p-Cl-, p-F- and p-NO2-phenylbutadienes react to give 5 in 67% yield (19:1 dr), 6 in 70% yield (6:1 dr), and 7 in 48% yield (8:1 dr), respectively. Electron-rich arenes are also compatible, but result in reduced anti/syn diastereoselectivity; p-MeO-phenylbutadiene reacts to form 8 in 58% yield and 4:1 dr. Phenylbutadiene containing alkyl substitution at the ortho-, meta- and para-positions of the aryl ring are excellent substrates providing 9 in 59% yield (>20:1 dr), 10 in 66% yield (>20:1 dr), and 11 in 89% yield (6:1 dr). The high selectivity in the formation of 9 and 10 demonstrates the influence of sterics and its translation to increased diastereoselectivity in C–C bond formation with only slight decreases in yield. Dienes bearing oxygen heterocycles participate in the hydroalkylation reaction with 12 formed in 91% and in 9:1 dr; however, pyridyl groups appear to inhibit catalyst 1 as 13 does not form under the same reaction conditions.20 Alkyl-substituted dienes are effective substrates and react with oxazolone 2 to produce alkenyl products 14–16 in good yields and selectivities (Table 2). We anticipated that the decreased size of the alkyl chain, compared to an aryl ring, would result in diminished diastereoselectivity, however, the opposite was observed; 14 was formed in high diastereoselectivity (12:1 dr) in 66% yield. The increased α-branching in cyclohexylbutadiene results in lower reactivity and diastereoselectivity, providing 15 in 43% yield but in 3:1 dr. Additionally, the mild reaction conditions are tolerant of silyl ether functionality; for example homoallyl TBS ether 16 is delivered in 68% yield and 4:1 dr without silyl ether deprotection or elimination to form the conjugated diene. For some substrates lower conversions can be observed due to competitive ring-opening of the oxzalone by i-PrOH, however, yields can often be improved by increasing the equivalents of the nucleophile.
We next examined the scope of the oxazolone nucleophiles tolerated by (CDC)–Rh catalyst 1. To explore the interplay between the identity of the oxazolone substituent and diene, oxazolones were reacted with representative dienes bearing heterocyclic, alkyl and aryl motifs to afford 17–28 (Table 3). Extension from methyl- to n-propyl-substituted oxazolones (17–19) resulted in high conversions, similar to those obtained with 2, but with lower selectivity compared to 9, 12 and 14; 17 was produced in 89% yield with 6:1 dr, 18 was synthesized in 55% yield with 7:1 dr, and 19 was formed in 57% yield with 10:1 dr. The lower selectivity may be a consequence of increased sterics on the α-substituent influencing the orientation of the nucleophile as it approaches the activated diene. Reactions with sec-butyl-substituted oxazolone demonstrate that β-branched alkyl substituents work effectively as 20–22 are formed in good to high yields with varying selectivity; 20 is formed in 96% yield with 19:1 dr, while 21 is synthesized in 51% yield with 8:1 dr and 22 in 89% yield with 10:1 dr. To further demonstrate that increased substitution on the oxazolone is viable, phenethyl-oxazolone was reacted to give 23–25; 23 was formed in 57% yield with 5:1 dr, 24 in 21% yield with 7:1 dr and 25 in 50% yield with 10:1 dr. The (CDC)–Rh catalyst is compatible with alkenes as evidenced by the successful formation of 26 in 28% yield with 5:1 dr. The reduction in yield is from competitive isomerization of the allyl group to the internal alkene, which is not a competent nucleophile. Furthermore, formation of 27 (2:1 dr) demonstrates the subtle effect that the sterics of the diene play in obtaining a selective reaction (cf., 22, generated in 20:1 dr). We were able to modify the aryl substituent of the oxazolone, as demonstrated by the formation of 28; p-Cl-phenyl-oxazolone reacted to provide 28 in 71% yield and 3:1 dr, however, the site-selectivity of the reaction decreased to give a 11:1 mixture of γ,δ- and α,δ-regioisomers.


The substituted oxazolone products generated through the catalytic stereoselective hydroalkylation protocol can be readily transformed to other useful molecules (Scheme 2). Firstly, ring-opening of allyl-substituted oxazolone 4 with MeOH and K2CO3 at 22 °C delivers methyl esters 29 in 87% yield; synthesis of 29 confirmed the anti diastereoisomer as the major product by comparison to previously reported data.15d Two additional α,α-disubstituted products, 20 and 21, were converted to their corresponding methyl esters 30 and 31 in 99% and 84% yield, respectively. Second, conversion of the oxazolone moiety to benzoyl-protected amino acids can be accomplished by acid hydrolysis; the conversion of 24 to 32 with dilute HCl in 87% yield is representative. Finally, the vicinal tertiary allylic, and N-substituted quaternary stereocenters can be used to impart stereocontrol in further alkene functionalizations. In this regard, 18 (9:1 dr) was successively hydrolyzed to the methyl ester and subjected to m-CPBA epoxidation to form epoxide 33 in 58% yield over two steps with complete stereocontrol.21 The resulting product contains four contiguous stereocenters and a versatile epoxide ring, which could be opened stereoselectively to introduce a variety of nucleophiles.22
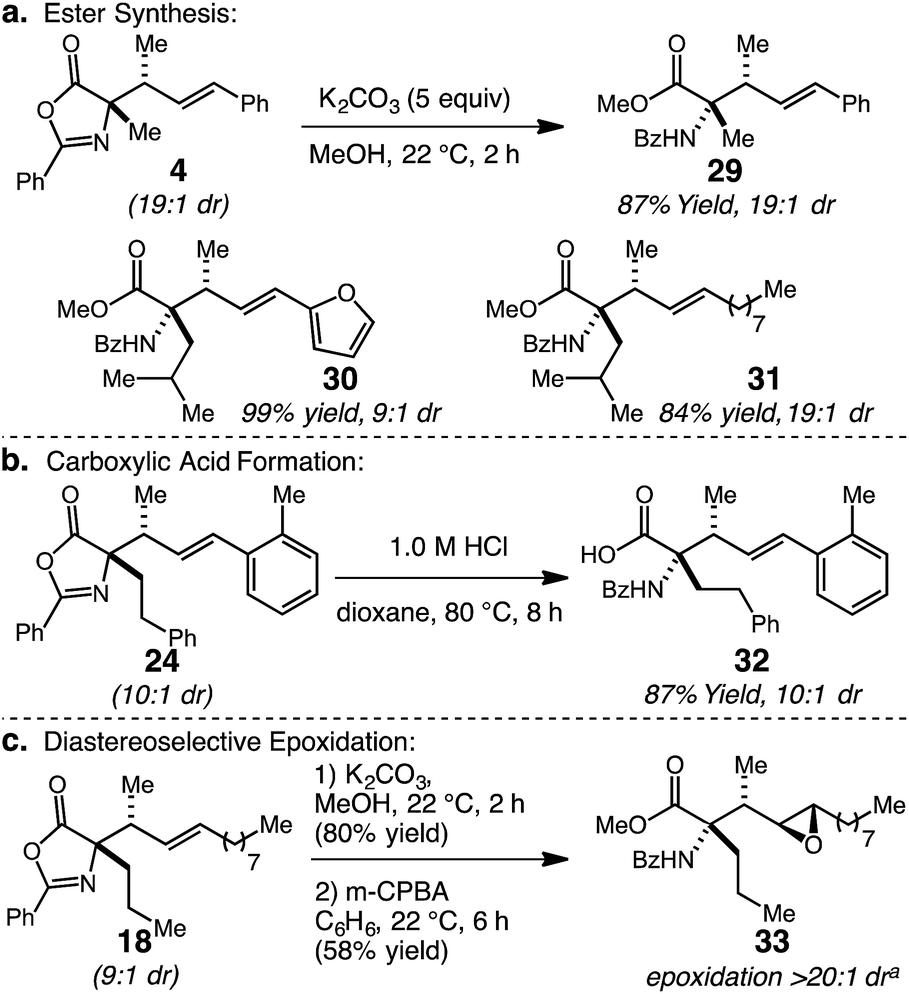 | ||
| Scheme 2 α-Allyl oxazol-5(4H)-one functionalizations. aDetermined by analysis of 600 MHz 1H NMR spectra of unpurified mixture. | ||
According to our data, a possible catalytic cycle for the reported hydroalkylation is depicted in Scheme 3. While the specific role of the lithium salt is not yet determined, previous studies indicate that secondary binding to the CDC carbon (e.g., II) decreases electron density at the Rh center, resulting in decreased π-back donation,17 and thus facilitating nucleophile addition (II → III).23 Product formation and regeneration of I could occur by two possible pathways: (a) direct Rh–alkyl protonation; (b) proton transfer to Rh and subsequent reductive elimination.24
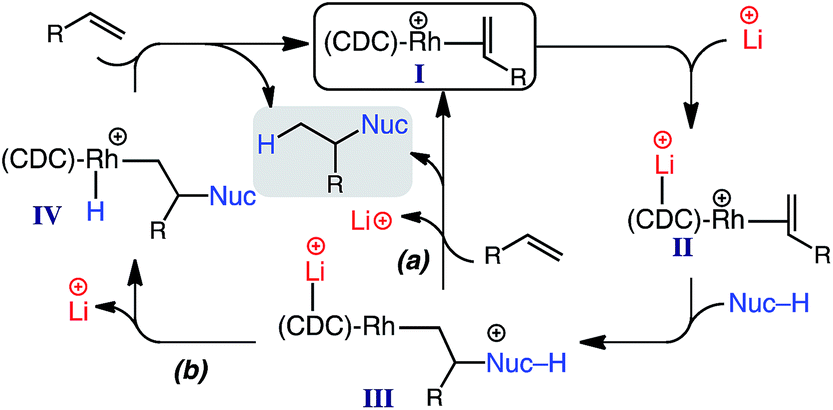 | ||
| Scheme 3 Proposed catalytic cycle. | ||
Conclusions
In summary, we have demonstrated the first diastereo- and siteselective hydroalkylation of 1-substituted 1,3-dienes with oxazolone nucleophiles promoted by a cationic (CDC)-Rh catalyst. The use of a catalytic lithium salt activator, and alcohol serve to provide optimal reactivity and good diastereoselectivity under mild conditions for a range of dienes with oxazolone nucleophiles. The resulting products contain two contiguous stereocenters and an N-substituted quaternary center, which can be deprotected to generate useful amino acid analogues, or exploited to impart acyclic stereocontrol in alkene epoxidation. Related studies are in progress to expand the scope of carbon-based nucleophiles and alkene electrophiles in hydroalkylation processes as well as development of enantioselective variants.Acknowledgements
Financial support was provided by the University of North Carolina at Chapel Hill, the Petroleum Research Fund of the American Chemical Society (52447-DNI1). We are grateful to C. Roberts (UNC) for her contributions to the synthesis of (CDC)–Rh complex 1.Notes and references
- J. F. Hartwig, Science, 2002, 297(5587), 1653 CrossRef CAS PubMed
.
-
(a) X. Yao and C.-J. Li, J. Org. Chem., 2005, 70, 5752 CrossRef CAS PubMed
-
(a) X. Wang and R. A. Widenhoefer, Chem. Commun., 2004, 660 RSC
-
(a) B. M. Trost, C. Jäkel and B. Plietker, J. Am. Chem. Soc., 2003, 125, 4438–4439 CrossRef CAS PubMed
-
(a) W.-B. Liu, H.-F. Jiang and C.-L. Qiao, Tetrahedron, 2009, 65, 2110 CrossRef CAS
-
(a) W. Fang, M. Presset, A. Guérinot, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Org. Chem. Front., 2014, 1, 608 RSC
-
(a) T. Iwasaki, R. Shimizu, R. Imanishi, H. Kuniyasu and N. Kambe, Angew. Chem., Int. Ed., 2015, 54, 9347 CrossRef CAS PubMed
- K. Takahashi, A. Miyake and G. Hata, Bull. Chem. Soc. Jpn., 1972, 45, 1183 CrossRef CAS
-
(a) P. W. Jolly and N. Kokel, Synthesis, 1990, 1990, 771 CrossRef
- R. A. Mosey, J. S. Fisk, T. L. Friebe and J. J. Tepe, Org. Lett., 2008, 10, 825 CrossRef CAS PubMed
- Y. Hayashi, K. Obi, Y. Ohta, D. Okamura and H. Ishikawa, Chem.–Asian J., 2009, 4(2), 246 CrossRef CAS PubMed
-
(a) E. P. Ávila, A. C. de Mello, R. Diniz and G. W. Amarante, Eur. J. Org. Chem., 2013, 2013, 1881 CrossRef
- For recent examples of related 1,6-Michael addition, see:
(a) D. Uraguchi, K. Yoshioka, Y. Ueki and T. Ooi, J. Am. Chem. Soc., 2012, 134, 19370 CrossRef CAS PubMed
- M. Kalek and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 9438 CrossRef CAS PubMed
- Allyl substituted oxazolones can also be accessed via metal-catalyzed allylic alkylations, however, products bear different substitution patterns to those obtain by the reported method. For examples, see:
(a) B. M. Trost, C. Heinemann, X. Ariza and S. Weigand, J. Am. Chem. Soc., 1999, 121, 8667–8668 CrossRef CAS
- M. J. Goldfogel, C. C. Roberts and S. J. Meek, J. Am. Chem. Soc., 2014, 136, 6227 CrossRef CAS PubMed
- C. C. Roberts, D. M. Matías, M. J. Goldfogel and S. J. Meek, J. Am. Chem. Soc., 2015, 137, 6488 CrossRef CAS PubMed
- For additional examples of CDC ligands used in catalysis, see:
(a) Y.-C. Hsu, J.-S. Shen, B.-C. Lin, W.-C. Chen, Y.-T. Chan, W.-M. Ching, G. P. A. Yap, C.-P. Hsu and T.-G. Ong, Angew. Chem., Int. Ed., 2015, 54, 2420–2424 CrossRef CAS PubMed
-
(a) C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206 CrossRef CAS PubMed
- Catalytic hydroalkylation of 3 in the presence of 10 mol% 2,6-di-tertbutylpyridine afford 4 in 86% conv., and >20:1 dr. This indicates that Lewis basic N-heteroarenes do not inhibit hydroalkylation due to their Bronsted basicity, but rather by binding to the (CDC)–Rh catalyst.
- For a review of substrate-directed epoxidations, see:
(a) A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307 CrossRef CAS
- For recent examples of stereospecific ring-opening of internal epoxides with carbon nucleophiles, see:
(a) E. M. Valentín, M. Mulero and J. A. Prieto, Tetrahedron Lett., 2012, 53, 2199–2201 CrossRef PubMed
-
(a) O. Eisenstein and R. Hoffmann, J. Am. Chem. Soc., 1980, 102, 6148–6149 CrossRef CAS
- For a DFT study regarding cleavage of Rh–alkyl bond in hydroamination, see: H. M. Senn, P. E. Blöchl and A. Togni, J. Am. Chem. Soc., 2000, 122, 4098–4107 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04908c |
| This journal is © The Royal Society of Chemistry 2016 |