
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable luminescent iridium(III) complexes with bis(N-heterocyclic carbene) ligands: photo-stability, excited state properties, visible-light-driven radical cyclization and CO2 reduction, and cellular imaging†
Chen
Yang
ab,
Faisal
Mehmood
a,
Tsz Lung
Lam
cd,
Sharon Lai-Fung
Chan
*cd,
Yuan
Wu
a,
Chi-Shun
Yeung
a,
Xiangguo
Guan
a,
Kai
Li
ab,
Clive Yik-Sham
Chung
a,
Cong-Ying
Zhou
ab,
Taotao
Zou
a and
Chi-Ming
Che
*ab
aState Key Laboratory of Synthetic Chemistry, Institute of Molecular Functional Materials, HKU-CAS Joint Laboratory on New Materials and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: cmche@hku.hk
bHKU Shenzhen Institute of Research and Innovation, Shenzhen, China
cThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen, PR China
dDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, China. E-mail: sharonlf.chan@polyu.edu.hk
First published on 20th January 2016
Abstract
A new class of cyclometalated Ir(III) complexes supported by various bidentate C-deprotonated (C^N) and cis-chelating bis(N-heterocyclic carbene) (bis-NHC) ligands has been synthesized. These complexes display strong emission in deaerated solutions at room temperature with photoluminescence quantum yields up to 89% and emission lifetimes up to 96 μs. A photo-stable complex containing C-deprotonated fluorenyl-substituted C^N shows no significant decomposition even upon irradiation for over 120 h by blue LEDs (12 W). These, together with the strong absorption in the visible region and rich photo-redox properties, allow the bis-NHC Ir(III) complexes to act as good photo-catalysts for reductive C–C bond formation from C(sp3/sp2)–Br bonds cleavage using visible-light irradiation (λ > 440 nm). A water-soluble complex with a glucose-functionalized bis-NHC ligand catalysed a visible-light-driven radical cyclization for the synthesis of pyrrolidine in aqueous media. Also, the bis-NHC Ir(III) complex in combination with a cobalt catalyst can catalyse the visible-light-driven CO2 reduction with excellent turnover numbers (>2400) and selectivity (CO over H2 in gas phase: >95%). Additionally, this series of bis-NHC Ir(III) complexes are found to localize in and stain endoplasmic reticulum (ER) of various cell lines with high selectivity, and exhibit high cytotoxicity towards cancer cells, revealing their potential uses as bioimaging and/or anti-cancer agents.
Introduction
Luminescent organometallic complexes of 3rd row transition metals, such as Ir,1–5 Pt,6–11 and Au12–19 are currently receiving burgeoning interest due to their profound applications in materials science,20–22 biology,23,24 and organic synthesis.25–27 In particular, the favourable emission properties of cyclometalated Ir(III) complexes have been harnessed for applications by many research groups with examples such as the development of high performance OLEDs by Thompson and co-workers3–5,28–31 and the design of bioimaging and cellular probes by Lo and co-workers.32–36 Over the past decade, as a result of extensive work most notably by MacMillan,37,38 Yoon,27,39–41 and Stephenson and co-workers,42–45 luminescent cyclometalated iridium(III) complexes are widely used in photo-redox catalysis, which has been shown to have useful applications in organic synthesis.In 2008, Yoon and co-workers40 reported [2 + 2] enone cycloadditions, and MacMillan and co-workers38 published the alkylation of aldehydes, both of which were catalysed by triplet metal-to-ligand-charge-transfer (3MLCT) excited state of [Ru(bpy)3]2+ generated upon visible-light irradiation. Subsequently, Stephenson and co-workers achieved the reductive dehalogenation of activated alkyl halides catalysed by [Ru(bpy)3]2+,44 and reductive dehalogenation of alkyl, alkenyl and aryl iodides by using fac-Ir(ppy)3 as photo-redox catalyst.45 Compared to [Ru(bpy)3]2+ and fac-Ir(ppy)3, the application of luminescent platinum(II) photo-catalysts is still nascent. Recently, our group and Wu's group demonstrated that pincer Pt(II) complexes are capable of catalysing light induced C–C bond formation.46,47
The important features that allow luminescent transition metal complexes to act as useful photo-redox catalysts or photo-sensitizers for light induced reactions include: (1) the long lifetime of their electronic excited states, thus allowing bimolecular reaction to proceed in solution; (2) their electronic excited states as both strong reducing and oxidizing reagents with reduction potentials systematically varied by the auxiliary ligands.41 For the photo-catalysis to have practical interest, the design of highly stable photo-redox catalysts with long-lived electronic excited states in solution is desirable.
Our endeavour to develop transition metal photo-catalysis is to use visible light for activation of small molecules such as CO2, described in this work, and for C–X bond functionalization. A major consideration is to utilize visible light, which falls within the solar spectrum and avoids deleterious high-energy UV-initialized photochemical side reactions.48,49 In the literature, platinum group metal complexes and semi-conductors are usually used as photo-redox catalysts for the photochemical reduction of CO2.50 Earlier examples of transition metal photo-catalysts used for the photochemical reduction of CO2 include cobalt porphyrins,51 Re(bpy)(CO)3Cl52,53 and Ir(terpy)(ppy)Cl.54 More recently, systems comprising fac-Ir(ppy)3 in conjunction with [Ni(Prbimiq1)]2+,55 Fe(porphyrin),56 [Co(TPA)Cl]Cl,57 (TPA = tris(2-pyridylmethyl)amine) and [Co(N5)]2+ (N5 = 2,13-dimethyl-3,6,9,12,18-pentaazabicyclo-[12.3.1]octadeca-1(18),2,12,14,16-pentaene)58 were reported for photochemical reduction of CO2.
The stability of photo-catalysts is an important issue for the practical application of transition metal photochemistry. Numerous studies revealed that the photochemically active excited states of [Ru(bpy)3]2+,59–61 [Ir(ppy)2(bpy)]+,60,62 and fac-Ir(ppy)355,57,63 are not stable under light irradiation for a long period of time as a result of dissociation of coordinated ligand(s) presumably via low lying d–d excited state(s). To address the photo-stability issue, we considered the use of N-heterocyclic carbene (NHC) ligands which have been receiving burgeoning attention in coordination chemistry due to their strong σ-donor strength to develop robust metal photo-sensitizers and photo-catalysts.64–70 Also, the N-substituent of NHC ligands can be used to tune both the physical and chemical properties of the resultant photo-active transition metal complexes such as their solubility in various solvents including water. NHC ligands functionalized with carboxylate, sulfonate, amine/ammonium, and alcohol motifs have been reported for the development of water-soluble transition metal catalysts for Suzuki coupling, hydrosilylation, hydrogenation, olefin metathesis and CO2 reduction.65,71
Compared with Ir(III) complexes supported by bidentate acetylacetonate28 and/or 2,2′-bypyridine1 ligands, the photophysical and application studies of the related bis-NHC Ir(III) complexes are relatively scarce. In 2010, cationic bis-NHC Ir(III) complexes were reported by De Cola and co-workers72 to have application in blue-light emitting electrochemical cells; subsequently, a number of bis-NHC Ir(III) complexes were reported for photophysical and biological studies.65,67,72–74 In this work, a series of strongly luminescent Ir(III) complexes (Chart 1) containing bis-NHC ligands and visible light absorbing C-deprotonated (C^N) ligands was synthesized and their photophysical and electrochemical properties were examined. These complexes display high photo-stability and are strongly emissive with long lifetimes of up to 96 μs in solution at room temperature. The water-soluble luminescent Ir(III) complexes, containing the glucose-functionalized NHC ligand, were found to be active photo-catalysts for radical cyclization leading to the formation of 5-membered pyrrole rings in aqueous media with high substrate conversions and yields. One of the photo-stable Ir(III) complexes was utilized as a photo-sensitizer and in conjunction with a recently reported catalyst [Co(TPA)Cl]Cl to convert CO2 into CO with a turnover number (TON) > 2400, selectivity in gas phase > 95% and yield of 5.6% (1 mL out of 18 mL of CO2 was converted into CO at 5 μM concentration of Co complex). Some of the complexes were also demonstrated as potential bioimaging and/or anti-cancer agents.
 | ||
| Chart 1 Iridium(III) complexes in this work. Complexes 1a,73,741b,731d28 and 2d31 have been reported in the literature. | ||
Results
Synthesis, characterization and photo-stability of [(C^N)2Ir(NHC)]X complexes
The structures of 18 bis-NHC Ir(III) complexes synthesized in this work (1c and 2a, 2b, 3–9), together with previously reported Ir(III) complexes 1a,73,741b,731d28 and 2d,31 are depicted in Chart 1. Complexes 1–9 were synthesized in good yields by refluxing [(C^N)2Ir(μ-Cl)]2 with bis(imidazolium) salts in the presence of silver(I) oxide in 2-methoxylethanol. Details of synthesis and characterization data of ligands and complexes are provided in the ESI.† Notably, 1H NMR spectra of 7b and 7c show poorly resolved peaks in the aromatic region (6.7–8.2 ppm) at ambient temperature, and hence 1H NMR spectra are recorded from 238 K to 300 K (Fig. S1, ESI†) in order to assure the purity of 7b and 7c.Complexes 1a–4Mea, 1b–4Meb, 4Hexb–9b with N-methyl or N-butyl substituent on bis-NHC ligands are soluble in most common aprotic solvents, but not in protic solvents e.g. methanol (MeOH) or water. Complexes 1c, 4Mec, 6Hc, 7c and 9c with glucose functionalized bis-NHC ligand are soluble in MeOH, ethanol (EtOH) and water.
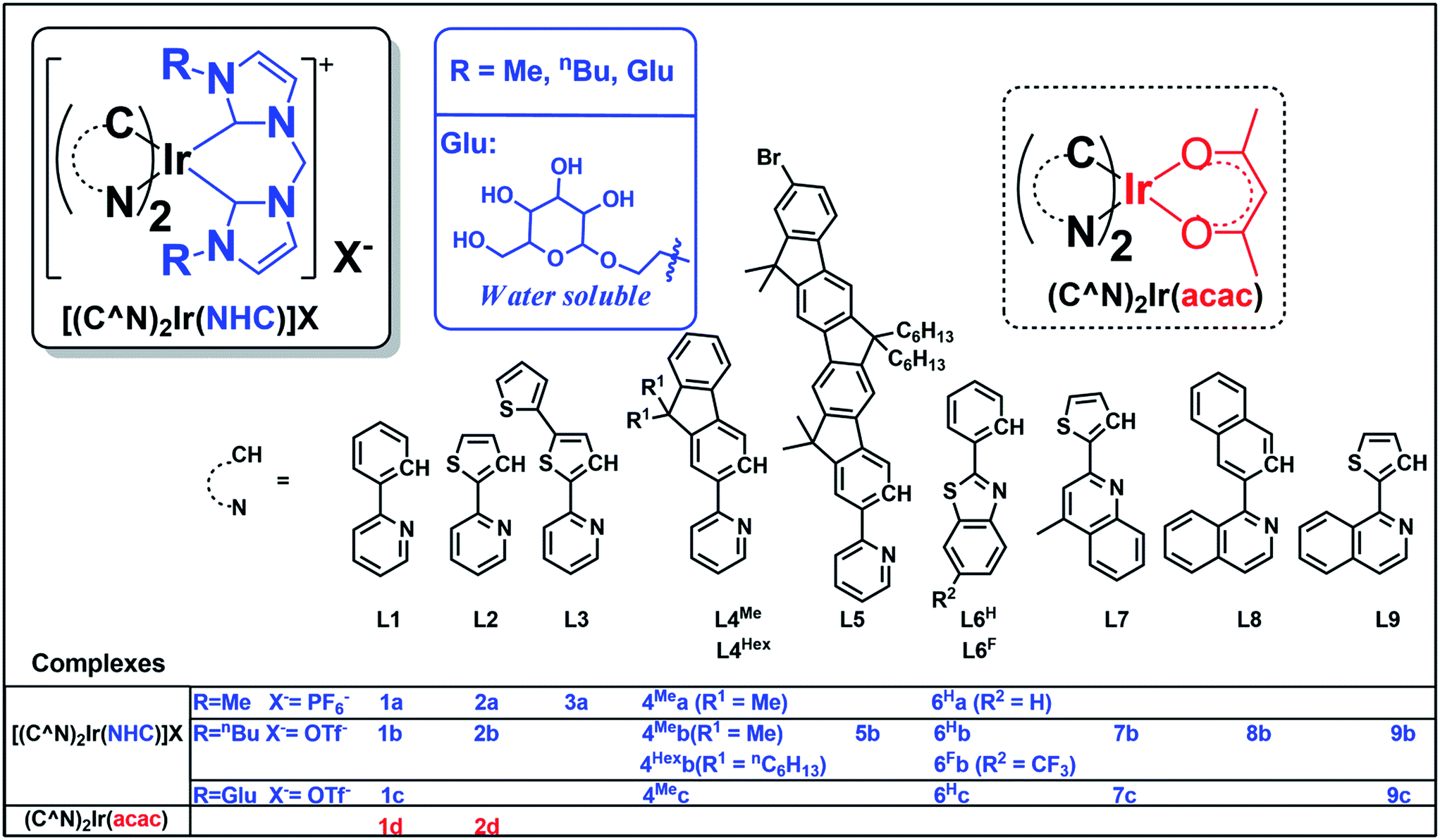
The photo-stability of these Ir(III) complexes with bis-NHC ligands was examined by using 4Meb and 6Hb as representative examples. 4Meb and 6Hb in degassed deuterated MeCN were irradiated using blue light (12 W blue LEDs) for 5 days. The photolysis was monitored by 1H NMR spectroscopy. As depicted in Fig. 1a, in the case of 4Meb, less than 5% of the complex was observed to undergo photochemical decomposition after irradiation of 120 h, revealing its outstanding photo-stability. Under the same conditions, Ru(bpy)3Cl2, fac-Ir(ppy)3 and [(dFCF3ppy)2Ir(dtbbpy)]PF6 (dFCF3ppy = 3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl]phenyl; dtbbpy = 4,4′-bis(1,1-dimethylethyl)-2,2′-bipyridine) were found to decompose after 10 h irradiation as revealed by the changes of their 1H NMR spectra (Fig. 1c).
 | ||
| Fig. 1 1H NMR spectra for (a) 4Meb; (b) 6Hb; (c) Ru(bpy)3Cl2, fac-Ir(ppy)3 and [(dFCF3ppy)2Ir(dtbbpy)]PF6 in deuterated MeCN solution for irradiating by blue light (12 W, λmax = 462 nm)46 (all solutions are degased by nitrogen gas for 10 min); (d) crystal structure diagrams showing the photoinduced transformation of coordination for Ir metals in 6Hb and cis-6Hb (butyl group on bis-NHC ligand and hydrogen atoms are omitted for clarity); (e) UV/Vis absorption (dotted line), excitation (dashed line) and emission (solid line) spectra of solutions of 6Hb and cis-6Hb (concentration of 2.0 × 10−5 M) in degassed DCM at 298 K. | ||
Interestingly, irradiation of 6Hb using blue LEDs for 10 h led to a clean and quantitative conversion to a new species which did not show further changes upon subsequent irradiation for another 90 h (Fig. 1b, this species is noted as cis-6Hb). The yield of scale synthesis of cis-6Hb from the irradiation of degassed MeCN solution of 6Hb (100 mg in 4 mL MeCN) was 94%. cis-6Hb was found to be stable upon standing in solution in the dark for another 40 h, or exposed to air for another 20 h (Fig. S2, ESI†). To verify that the transformation of 6Hb to cis-6Hb was caused by visible-light irradiation, a negative control experiment was conducted by keeping 6Hb in deuterated MeCN in the dark (Fig. S2, ESI†), and no structural changes were detected by 1H NMR spectroscopy.
The UV-Vis absorption, emission and excitation spectra of cis-6Hb measured in DCM solution at 298 K are different from those of 6Hb with hypsochromic shifts in peak maxima (Fig. 1e). The emission band for cis-6Hb displays vibronic spacings of 1245 cm−1 while that of 6Hb shows spacings of 908 cm−1. Taking into consideration the ESI-MS and spectroscopic data of cis-6Hb, cis-6Hb is likely a structural isomer for 6Hb. The exact structure of cis-6Hb has been determined by X-ray crystallography.
X-Ray crystallography
Crystals of 4Meb (with PF6− counter anion), 6Hb, cis-6Hb and 7b suitable for X-ray crystallographic analysis were obtained by slow diffusion of diethyl ether into DCM (6Hb), MeCN (4Meb and 7b) and chloroform (cis-6Hb) solution of these complexes, respectively. Perspective views of 6Hb and cis-6Hb are shown in Fig. 1d (those of 4Meb, 7b are shown Fig. S3 in ESI†). Selected bond lengths and angles are compiled in Table S1 (ESI†). Similar to the reported examples,726Hb adopts a distorted octahedral geometry (Fig. 1), the iridium atom is coordinated by two cyclometalated C^N ligands and one bis-NHC ligand. The two C^N ligands adopt a mutually eclipsed configuration, with the two N atoms (N1 and N2) trans to each other and with Ir–N bond length of 2.067(2) and 2.071(2) Å. The two substituted phenyl rings are oriented cis to each other with Ir–C bond length of 2.054(2) and 2.077(2) Å. The CNHC atoms of bis-NHC ligand are trans to two C atoms from C^N ligands. The Ir–CNHC distances of 2.105(2) Å and 2.128(2) Å are comparable to the literature values of Ir–CNHC bonds trans to phenyl groups.72 The CNHC–Ir–CNHC bite angles of 4Meb (84.7(3)o), 6Hb (85.10(9)o), 7b (85.58(6)o) and that of reported [(dfppy)2Ir(NHCMe)]PF6 (85.28(15)o) (dfppy = 4,6-difluoro-phenylpyridine) are consistent.72 The structures of 4Meb and 7b are in line with that of 6Hb. Interestingly, for cis-6Hb the N atoms from the two C^N ligand are cis to each other (Fig. 1d), indicating that 6Hb and cis-6Hb are structural isomers. The crystallographic refinement parameters for 4Meb, 6Hb, cis-6Hb and 7b are summarized in Table S2 (ESI†).Electronic absorption and emission spectroscopy
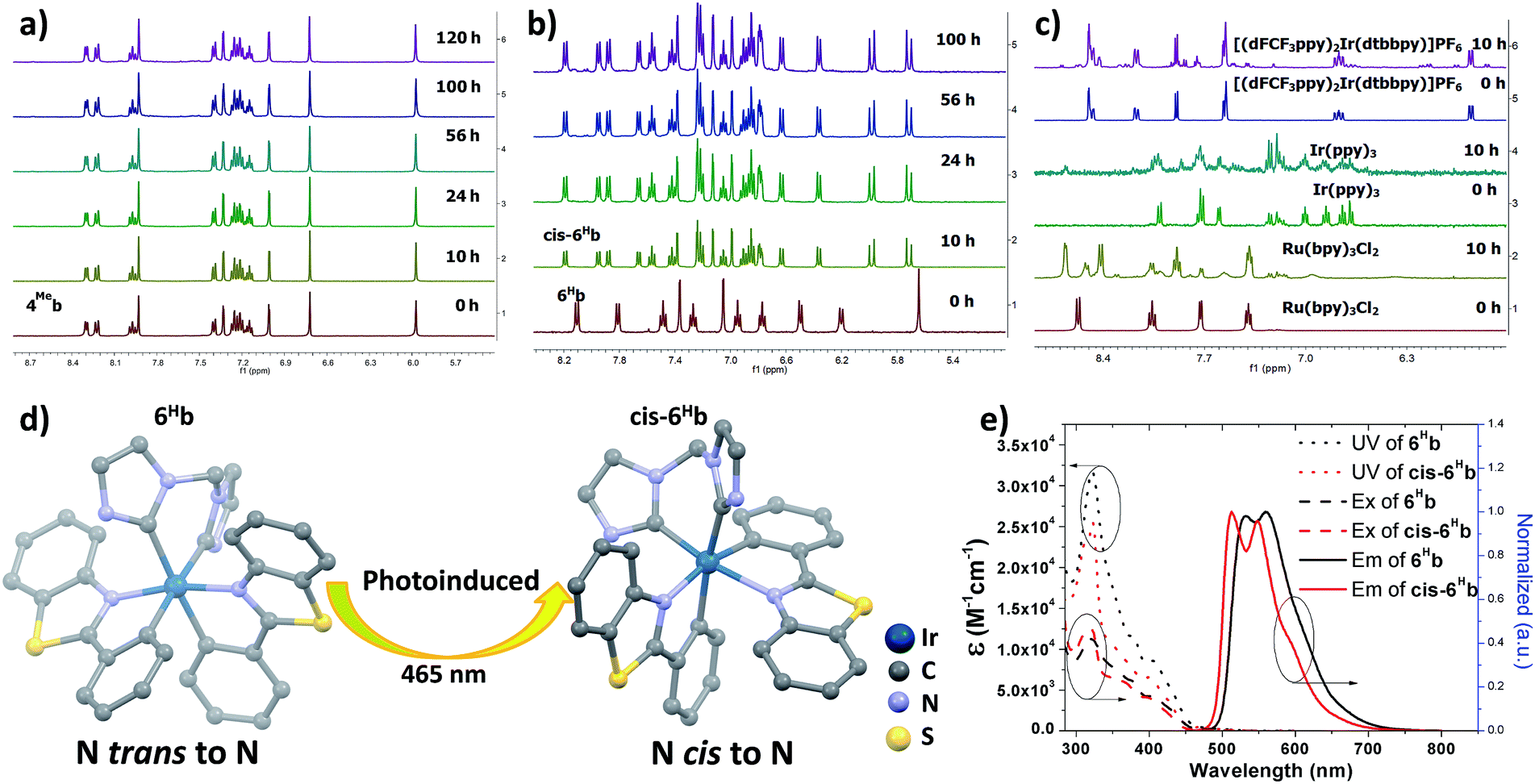
Photophysical data of 1–9 including their UV/Vis absorption and emission maxima, emission lifetimes and photo-luminescent quantum yields are tabulated in Table 1. All the complexes show intense high-energy absorptions ranging from 250 to 380 nm and lower-energy absorptions at 416–494 nm (Fig. 2, 3 and S4 in ESI†). The high-energy absorptions are attributed to the 1π → π* transition of the ligands, whereas the low energy absorption bands should be ascribed to the admixture of metal-to-ligand charge transfer (MLCT) transitions and ligand centered (LC) π–π* transitions.72 The charge transfer (CT) assignment is supported by the energy trend observed in the complexes with different C^N ligands. For example, the low-energy absorptions of 8b and 5b reveal significant red-shifts from those of 1b and 4Meb respectively. This can be rationalized by the extended π-conjugation of the C^N ligand in 8b and 5b, leading to the lowering of the π*(C^N) orbitals and hence decreases in CT energy. These red-shifts in the UV-Vis absorptions, together with the large molar absorptivities in the visible region (e.g., 5b: 7.2 × 104 M−1 cm−1), allow the complexes to show strong absorption in the visible region. This is believed to be crucial for harvesting visible light in the solar spectrum as well as avoiding UV-initiated side reactions due to the use of UV in the photo-catalysis by the Ir(III) complexes. As revealed by the UV absorption spectra of 6Fb and 6Hb (Fig. S4b, ESI†), a bathochromic shift about 500 cm−1 is observed for the lower-energy absorption band(s). These could be attributed to the electron-withdrawing group of CF3 on the cyclometalated ligand which lowers the C^N based LUMO level.| Complex | Mediuma | Absorption (λmax/nm) (10−3ε/M−1 cm−1) | Emission λem/nm (τ/μs) | Φ /% |
|---|---|---|---|---|
| a Measured in degassed CH2Cl2 and water (2.0 × 10−5 M) at 298 K. b 4Mea, 4Meb and 4Hexb were measured at the concentration of 5 × 10−6 M. c 5b was measured at the concentration of 1 × 10−6 M. d Phosphorescence quantum yields were measured by using [Ru(bpy)3](PF6)2 (Φ = 0.062 in MeCN) as standard. | ||||
| 1a | CH2Cl2 | 255 (26.8), 267 (24.9), 311 (10.6), 342 (6.1), 380 (3.7), 416 (1.2) | 470 (2.1), 499, 534 | 89 |
| 1b | CH2Cl2 | 254 (36.8), 266 (35.5), 311 (15.5), 342 (9.1), 381 (5.82), 416 (2.16) | 470 (2.1), 500, 534 | 89 |
| 1c | H2O | 254 (36.8), 266 (35.5), 311 (15.5), 342 (9.1), 381 (5.82), 416 (2.16) | 469 (2.0), 498, 534 | 89 |
| 2a | CH2Cl2 | 252 (13.5), 290 (21.7), 332 (12.6), 369 (6.07), 405 (4.92) | 530, 547, 570 (3.03) | 11.1 |
| 3a | CH2Cl2 | 252 (15.5), 282 (15.1), 313 (20.5), 335 (23.1), 375 (21.9), 389 (20.9), 440 (20.9) | 472, 674 (4.8), 824 | 0.2 |
| 4Mea | CH2Cl2b | 260 (31.4), 297 (30.0), 316 (33.5), 334 (32.3), 360 (22.4), 378 (16.8), 421 (10.5) | 524, 564 (28.6), 614 | 65 |
| 4Meb | CH2Cl2b | 267 (52.2), 297 (46.8), 316 (50.9), 335 (49.0), 358 (36.3), 376 (27.3), 421 (16.2) | 525, 566 (28.7), 614 | 75 |
| 4Hexb | CH2Cl2b | 268 (42.1), 299 (42.3), 320 (50.4), 338 (49.2), 364 (32.6), 379 (25.6), 421 (14.6) | 527, 568 (32.7), 616 | 82 |
| 5b | CH2Cl2c | 260 (54.1), 273 (42.5), 310 (32.9), 323 (34.8), 377 (67.2), 398 (97.0), 436 (71.6) | 576 (96.1), 625, 683 | 22.6 |
| 6Hb | CH2Cl2 | 271 (23.0), 321 (25.0), 377 (9.57), 407 (6.81), 436 (3.38) | 531, 560 (6.4) | 78 |
| 6Hc | H2O | 268 (21.4), 321 (22.1), 377 (8.06), 407 (5.49), 436 (2.42) | 536, 560 (5.0) | 66 |
| 6Fb | CH2Cl2 | 270 (23.0), 323 (32.1), 383 (10.7), 415 (7.88), 447 (4.08) | 530, 558 (3.2) | 68 |
| 7b | CH2Cl2 | 296 (26.8), 333 (18.5), 388 (9.31), 437 (7.76), 467 (4.60) | 582, 623 (6.2) | 36 |
| 7c | H2O | 293 (25.9), 330 (17.4), 386 (9.63), 432 (6.92), 464 (3.71) | 583, 622 (5.9) | 32 |
| 8b | CH2Cl2 | 260 (80.1), 290 (48.5), 315 (35.4), 376 (34.5), 429 (7.52), 485 (3.47), 494 (2.88) | 618, 659 (5.2) | 3 |
| 9b | CH2Cl2 | 313 (24.2), 334 (23.8), 384 (12.7), 428 (10.7), 457 (8.20), 485 (3.47) | 617, 667 (7.4) | 13 |
| 9c | H2O | 315 (18.4), 380 (9.69), 420 (7.6), 454 (4.95), 475 (2.15) | 620 (4.5), 668 | 9 |
 | ||
| Fig. 2 UV-Vis absorption (top) and emission (bottom) spectra of solutions of 1a–3a and 1d–2d in degassed DCM (concentration of 2.0 × 10−5 M) at 298 K. | ||
 | ||
| Fig. 3 UV/Vis absorption (black dashed line), excitation (red solid line) and emission (green solid line) spectra of solutions of (a) 4Meb (5.0 × 10−6 M); (b) 5b (1.0 × 10−6 M) in degassed DCM at 298 K. | ||
On the other hand, the N-alkyl substituents on the bis-NHC ligand are found to have only minor effects, if any, on the UV/Vis absorption spectra of the Ir(III) complexes, as revealed by the overlaid spectra of the group of 1a, 1b, 1c (Fig. S4a, ESI†). Complex 1a absorbs weakly at the wavelength from 430 nm to 500 nm with molar absorptivity less than 500 M−1 cm−1 in contrast to the high values of 2500 M−1 cm−1 for 1d with the same C^N luminophore. The calculated wavelength for ground state HOMO → LUMO (S0 → S1 transition) is 369 nm and 414 nm for 1a and 1d, respectively (Fig. S4e and f, ESI†). These calculated values are in reasonable agreement with the corresponding experimental absorption λmax values 410 and 460 nm respectively.
Complexes 1–9 display strong phosphorescence in deaerated solution at room temperature (Fig. 2, 3 and S4 (ESI†); Table 1). All the complexes show vibronic-structured emissions, and their emission lifetimes are found to be in the microsecond regime. For example, 4Meb exhibits structured emission bands with vibrational spacings of ∼1380 cm−1 and a long emission lifetime of 28.2 μs. Only small negative solvatochromic effects on the emission at 524 nm are found (±5 nm; Fig. S4d, ESI†). These findings, together with TD-DFT calculations, suggest that the photoluminescence of the complexes is derived from triplet metal-perturbed ligand-centred (3LC) π–π* excited states. On the other hand, the structureless emission of 1d (the acetylacetonate (acac) analogous of 1a) at 516 nm should be ascribed to the 3MLCT/LLCT emission.2 Interestingly, complex 3a (Φ = 0.2%; τ = 4.8 μs) displays dual emission (Fig. 2) in contrast to the single emission of the acac analogue 3d,75 suggesting the possibility of modulation of photophysical properties of Ir(III) complexes by the NHC ligands.
Changes in chemical structure of the cyclometalated ligands also result in a profound effect on the photophysical properties of the Ir(III) complexes. For example, 5b displays a significant red-shift in emission maximum (576 nm) when compared to 4Meb and 4Hexb (525 and 527 nm respectively). This can be rationalized by the extended π-conjugation of the C^N ligands, leading to the decrease in energy of metal-perturbed 3LC emission. Moreover, for 4Mea, 4Meb and 4Hexb, their photoluminescence quantum yields in solution are affected by the substituents on the fluorenyl moiety as well as the N-alkyl groups of the bis-NHC ligands, e.g. complex 4Hexb exhibits a higher photoluminescence quantum yield than 4Mea and 4Meb. This is probably attributed to the fact that the long hexyl and N-butyl chains disfavour intermolecular stacking interactions among the planar C^N ligands, leading to reduced triplet–triplet annihilation and a higher photoluminescence quantum yield.
In addition, 5b shows a significantly longer emission lifetime (96.1 μs) than 4Meb (28.7 μs). This might be due to the reduced metal character in the electronic excited state of 5b, and hence slower triplet radiative decay. Similarly, with a smaller parentage of metal character in the frontier molecular orbitals, 6Hb (6.4 μs) shows a longer emission lifetime than 6Fb (3.2 μs). These long-lived triplet excited states allow the Ir(III) NHC complexes to undergo a variety of photochemical reactions, notably for visible-light-driven photo-catalytic reductive C–Br bond cleavage and CO2 reduction which will be illustrated later.
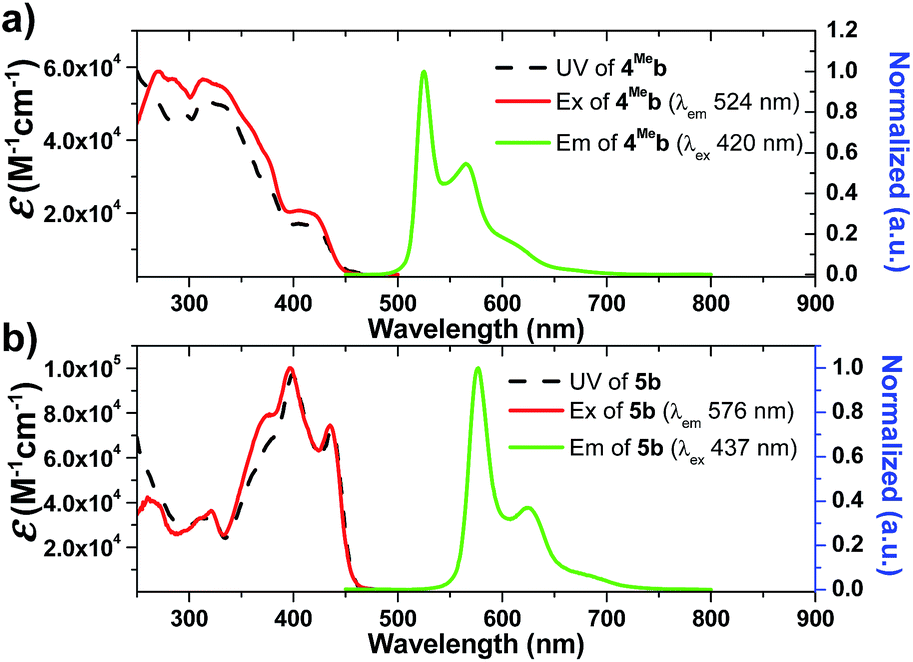
Electrochemistry
The electrochemical data of 1b–9b are summarized in Table 2 (all values versus Ag/AgNO3, scan rate of 100 mV s−1, 0.1 M nBu4NPF6 in MeCN as supporting electrolyte) and Table S3† (values vs. Cp2Fe+/0, ESI†). These complexes display one irreversible oxidative wave at Epa = 0.62–1.04 V and one irreversible reductive wave at Epc = −2.52 to −1.94 V (vs. Ag/AgNO3). The cyclic voltammograms of 1a have been reported elsewhere74 with the oxidation potential of +1.16 V (vs. Ag/AgNO3) in CH2Cl2. By comparing with the electrochemical data of 1d and 2d2, both of which have the ancillary acac ligand, the first oxidation wave of 1a should originate from Ir(III)-centred/C^N ligand-centred oxidation. This assignment is further supported by the observation of the more positive oxidation potential of 6Fb (+1.04 V) than 6Hb (+0.98 V). The electron-withdrawing –CF3 group of 6Fb will lower the energy levels of the π(C^N) orbitals, leading to the more positive oxidation potential of 6Fb. On the other hand, 4Meb and 5b have more extended π-conjugated C^N ligands than 1b. This will result in an increase in the energy level of π(C^N) orbitals, thus 4Meb and 5b can undergo oxidation more readily than 1b and hence less positive oxidation potentials are found.| Complex | E pa /V | E pc /V |
|---|---|---|
| a Supporting electrolyte: 0.1 M nBu4NPF6 in MeCN and values are recorded vs. Ag/AgNO3 (0.1 M) in MeCN; Cp2Fe+/0 occurs at the range of 0.05–0.08 (V) vs. Ag/AgNO3. b Values refer to oxidation peak potential (Epa) at 25 °C for irreversible couples at a scan rate of 100 mV s−1. c Values refer to reduction peak potential (Epc) for the irreversible reduction waves. | ||
| 1b | 0.87 | −2.52 |
| 2a | 0.64 | −2.24 |
| 4Mea | 0.74 | −2.41 |
| 4Meb | 0.79 | −2.40 |
| 4Hexb | 0.74 | −2.44 |
| 5b | 0.72 | −2.25 |
| 6Hb | 0.98 | −2.14 |
| 6Fb | 1.04 | −1.94 |
| 7b | 0.69 | −2.09 |
| 8b | 0.62 | −1.94 |
| 9b | 0.62 | −2.16 |
Compared with 1b, [(dfppy)2Ir(bis-NHCBu)]PF6 (ref. 72) displays a more anodic oxidation potential of Eox = 1.04 V and a similar reduction potential of Ere = −2.37 V (vs. Cp2Fe+/0), which is attributed to stabilization of HOMOs as a result of the presence of highly electron-withdrawing F substitution on the C^N ligand (dfppy). For the irreversible reduction wave, a less negative reduction potential is found for 6Hb (Epc = −2.14 V vs. Ag/AgNO3, Table 2, Fig. S4†), as compared to that of 1b (Epc = −2.52 V vs. Ag/AgNO3, Table 2, Fig. 4) which has a less extended π-conjugation of the C^N ligand. The reductive wave is further anodically shifted in the case of 6Fb (Epc = −1.94 V vs. Ag/AgNO3, Table 2, Fig. S5a, ESI†). Therefore, the reduction process should be localized on the C^N ligands.
 | ||
| Fig. 4 Cyclic voltammograms of 1b, 2a, 4Meb, 5b and 6Hb in MeCN with nBu4NPF6 (0.1 M) as supporting electrolyte. Conditions: glass-carbon, working electrode, scan rate: 100 mV s−1. | ||
Excited state properties
Nano-second transient absorption and emission spectroscopic (tr-abs and tr-em) measurements were undertaken for the highly emissive bis-NHC Ir(III) complexes with long-lived electronic excited states i.e.4Meb (Fig. 5) and 4Mec (Fig. 6a) in MeCN and water respectively. These two complexes have the same lumophore but different functionalized NHC groups, leading to a different solubility in solvents, such as one is soluble in organic solvents and the other in water. The tr-abs and tr-em spectra of the other bis-NHC Ir(III) complexes e.g.5b in MeCN (Fig. S6a, ESI†), and 6Hc (Fig. S6b, ESI†) in water were also recorded and the results can be found in the ESI.†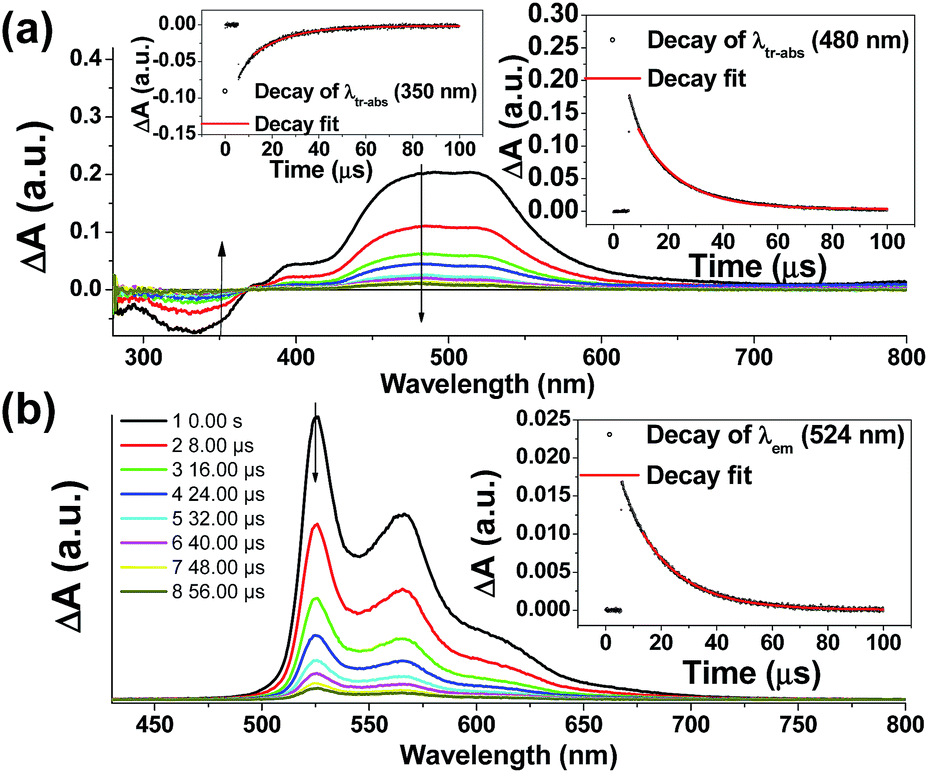 | ||
| Fig. 5 Time-resolved spectra of 4Meb (a) tr-abs (insets: decay of tr-abs at λ = 350 nm and 480 nm); (b) tr-em (decay of tr-em at λ = 524 nm) spectra recorded at specified times after laser pulse excitation (355 nm) in degassed MeCN at 298 K. | ||
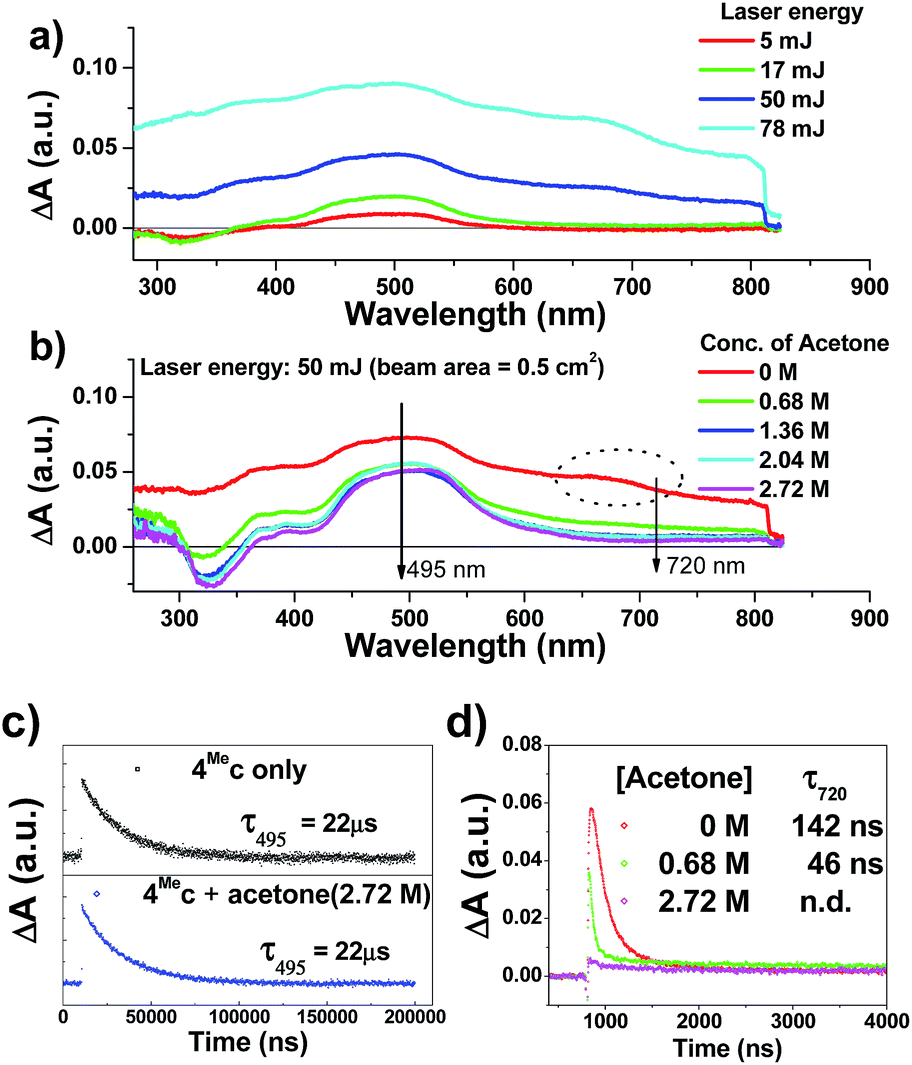 | ||
| Fig. 6 Transient absorption spectra recorded (a) under specified laser energy; (b) in presence of specified concentrations of acetone; of a degassed aqueous solution of 4Mec (about 1 × 10−5 M). Kinetic studies of (c) λtr-abs (495 nm) and (d) λtr-abs (720 nm) in the absence/presence of specified concentrations of acetone. | ||
The time-resolved absorption and emission spectra of 4Meb recorded at various time intervals after excitation at 355 nm are depicted in Fig. 5. The kinetic decay analysis of bleaching of ground-state of 4Meb (τ350 nm = 15.4 μs, Fig. 5a, left inset) matches well with the growth of the absorption of triplet excited state (τ480 nm = 15.2 μs, Fig. 5a, right inset) as well as the emission lifetime (τ524 nm = 16.5 μs, Fig. 5b, right inset) in MeCN at 298 K.
The transient absorption spectra of 4Mec in aqueous solution recorded at different energies of laser beams (355 nm) reveal different spectral changes. As depicted in Fig. 6a, in addition to the growth of the absorption of triplet excited state of 4Mec from 380 to 700 nm, the emergence of an absorption band from 650 to 730 nm was observed at laser pulse energy ≥ 50 mJ (beam area: 0.5 cm2). This absorption band could be quenched upon addition of acetone (Fig. 6b and S7a and b, ESI†), but no quenching of transient absorption at 495 nm and emission at 524 nm were observed in the presence of acetone (Fig. 6c and S7, ESI†). The decay rate constant monitored at 720 nm of 4Mec (Fig. 6d, absence of acetone) was much faster than that measured at 495 nm in Fig. 6c/S7c† and decay of emission at 524 nm (Fig. S7d, ESI†). In view of these different kinetic behaviours, the transient absorption from 650 to 730 nm depicted in Fig. 6b might originate from hydrated electrons eaq−,76,77 which were formed by the photo-induced ionization of 4Mec in aqueous solution upon excitation with high energy laser beams. This is in line with a reported photoionization of [Pt2(POP)4]4−,78 as well as the findings in the studies of solvated electron with acetone.79,80 Similarly, complex 6Hc in aqueous solution was also observed to undergo photo-ionization as revealed by the increase in transient absorption in the region of 600 to 700 nm (Fig. S8b, ESI†). For 4Meb, its transient absorption spectra monitored at high laser pulse energy in MeCN exhibit similar profiles as for lower energy (Fig. S8b and c, ESI†). However, newly generated long-lived species have been observed at high laser pulse energies after 100 μs (Fig. S8b–e, ESI†), revealing that photo-ionization of 4Meb with the generation of [4Meb]+ likely occurs. The accompanying solvated electrons are not observed in this case due to ready quenching by MeCN.81
Based on the electrochemical data and the determination of E0–0 from the spectroscopic measurements, the excited state redox properties of the bis-NHC Ir(III) complexes can be estimated (Table S3, ESI†). The triplet excited states of the complexes are found to be powerful oxidants and reductants, and some of them are even more reactive towards photoredox reactions than [Ru(bpy)3]2+ and fac-Ir(ppy)3. A representative example is 4Meb (E(IrIV/III*) = −1.51 V vs. SCE) (here IrIV is a simple notation to denote the oxidized IrIII species, the site of the oxidation can be the metal and/or C^N ligand), which is a stronger reductant than [Ru(bpy)3]2+(E(RuIII/II*) of −0.81 vs. SCE).37,82 As a result, it is anticipated that the bis-NHC Ir(III) complexes described herein, upon photoexcitation in the visible-light region, can catalyse a number of reactions which are not feasible by the widely used [Ru(bpy)3]2+.
Photo-catalysis
With good photo-stability, long excited state lifetime, favourable absorption in the spectral region of blue LED and tunable photo-redox properties, these bis-NHC Ir(III) complexes have been investigated for photo-redox reactions (Chart 2), examples of which are described in the following section.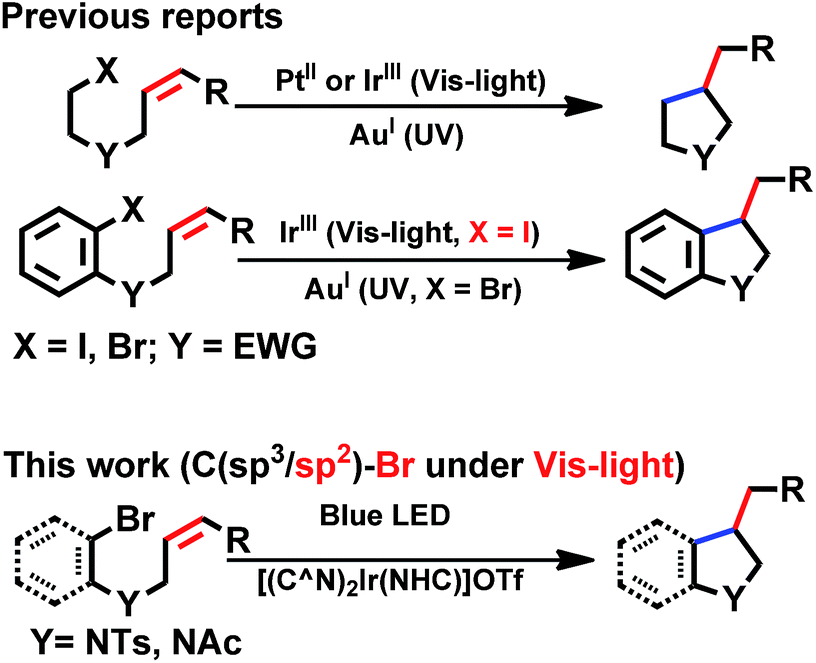 | ||
| Chart 2 General photo-catalytic reactions by cyclometalated complexes. EWG = electron-withdrawing group. | ||
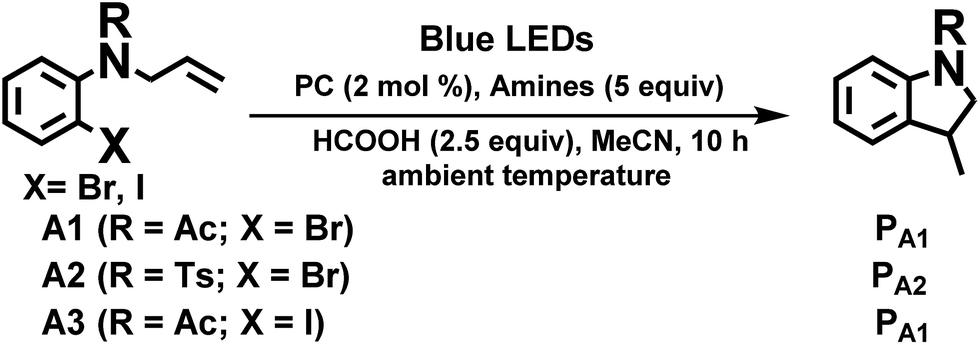
We considered a recent work by Lee and co-workers83 on visible-light-induced reductive cyclization of aromatic iodides and bromides to form indoline using [(ppy)2Ir(dtbbpy)]PF6 as photo-catalyst (PC). Yet, aryl bromides are found to be less reactive than iodides.84 Barriault and co-workers addressed this issue by using dinuclear gold(I) complexes ([Au2(dppm)2]OTf2) as photo-catalysts.85 However, this gold complex only shows absorption in the high-energy UV region (λ < 300 nm), which may result in destructive effects on the products and/or lead to undesired side reactions.
As the present bis-NHC Ir(III) complexes show strong absorption at λ > 400 nm, they were used to photo-catalyse the reductive cyclization of aryl bromides using blue LEDs. Among the bis-NHC Ir(III) complexes examined, 1b, 4Meb and 6Hb displayed good photo-catalytic activity for reductive cyclization of aryl bromide (A1) in terms of both substrate conversion and product yield (Table 3).
|
|
|||
|---|---|---|---|
| Entryb | PC | Conversionc/% | Yieldc/% |
| a Complex 3a was not tested because of the low quantum yield (0.2%, see Table 1). b Procedure: substrate 50 μmol, PC (1 mol%), DIPEA (5 equiv.), HCOOH (2.5 equiv.) in 4 mL MeCN solution was degassed by nitrogen, and irradiated by blue light (12 W, λmax = 462 nm) at ambient temperature for 4 h. c Determined by 1H NMR spectroscopy by adding internal standard of 5,5′-dimethyl-2,2′-bipyridine. | |||
| 1 | 1b | 93 | 59 |
| 2 | 2b | 1 | 0 |
| 3 | 4Meb | 90 | 59 |
| 4 | 5b | 90 | 52 |
| 5 | 6Hb | 93 | 59 |
| 6 | 6Fb | 80 | 44 |
| 7 | 7b | 49 | 24 |
| 8 | 8b | 11 | 7.3 |
| 9 | 9b | 0.1 | 0 |
In the case of another aryl bromide substrate A2, 4Meb showed both good substrate conversion and product yield (Table 4, entries 4–6). It was chosen as photo-sensitizer for optimization of the reaction conditions. A number of control experiments were performed, and no reaction was observed in the absence of amine or light (entries 13 and 14). Lowering the loading of PC (Table 3, entry 4), the absence of HCOOH or exposure to air (entries 11 and 12) were observed to decrease the substrate conversion of this reaction. Interestingly, using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, entry 10, Eonset+/0 = 0.60 V vs. Cp2Fe+/0), also led to excellent substrate conversion and good product yield comparable to what was obtained with tetramethylethylenediamine (TMEDA, entry 9, Eonset+/0 = 0.11 V vs. Cp2Fe+/0, Fig. S9, ESI†). In contrast, the widely used photo-catalysts [Ru(bpy)3]Cl2 and fac-Ir(ppy)3 (Table 4, entries 15 and 16) showed little or no conversion under similar conditions.
|
|
||||
|---|---|---|---|---|
| Entrya | PC (substrate) | Amines | Conversionb/% | Yieldb/% |
| a Procedure: substrate 50 μmol, PC (2 mol%), amine (5 equiv.), HCOOH (2.5 equiv.) in 4 mL aqueous solution was degassed by nitrogen, and irradiated by blue light (12 W, λmax = 462 nm) at ambient temperature for 10 h. b Determined by 1H NMR spectroscopy by adding internal standard of 5,5′-dimethyl-2,2′-bipyridine. c Irradiated for 4 h. d Absence of HCOOH. e Presence of air (no degassing). f Absence of amine. g Absence of light. | ||||
| 1 | 1b (A1) | DIPEA | 97 | 67 |
| 2 | 4Meb (A1) | DIPEA | 97 | 64 |
| 3 | 6Hb (A1) | DIPEA | 96 | 64 |
| 4 | 4Meb (A2) | DIPEA | 98 | 54 |
| 5 | 1b (A2) | DIPEA | 99 | <15 |
| 6 | 6Hb (A2) | DIPEA | 84 | 51 |
| 7c | 4Meb (A3) | TEA | 97 | 79 |
| 8 | 4Meb (A1) | TEA | 91 | 64 |
| 9 | 4Meb (A1) | TMEDA | 52 | 32 |
| 10 | 4Meb (A1) | DBU | 96 | 72 |
| 11d | 4Meb (A1) | DIPEA | 85 | 54 |
| 12e | 4Meb (A1) | DIPEA | 62 | 28 |
| 13f | 4Meb (A1) | — | 0 | 0 |
| 14g | 4Meb (A1) | DIPEA | 0 | 0 |
| 15 | Ru(bpy)3Cl2 (A1) | DIPEA | 0 | 0 |
| 16 | fac-Ir(ppy)3 (A1) | DIPEA | 27 | 16 |
The radical cyclization of the alkyl bromides, B1 and B2, catalysed by 1b, 4Meb or 6Hb proceeded smoothly, with reasonable to excellent substrate conversion and product yields. The yield of cyclization of B1 was improved to up to 90% (entry 5, Table 3) when formic acid was added and 6Hb was used as a photo-catalyst (PC). When 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was added, the reaction was totally inhibited, indicating the involvement of a radical intermediate in the reaction (entry 12).
The photo-catalytic reaction could be initiated from the oxidative quenching of 4Meb* with aryl/alkyl halides. This is because the excited state reduction potential of 4Meb* (E(IrIV/III*)pc = −1.51 V (vs. SCE), Table S3, Fig. S9†) can allow a direct one electron reduction of aryl/alkyl halides by 4Meb*, leading to carbon–halogen (σ*(C–X)) bond cleavage to give alkyl radical in the case of sp3 carbon or radical anion intermediate for sp2 carbon.86 Subsequent reactions of the alkyl radical or radical anion intermediate with C(sp2)–H bond lead to C–C bond formation. An aminium radical cation generated from the oxidation of amine by [4Meb]+83,85 could serve as an electron donor to complete the reductive process. In the case of [(ppy)2Ir(dtbbpy)]PF6,83 its triplet excited state ([(ppy)2Ir(dtbbpy)]+*) reacts with DIPEA via a reductive quenching mechanism to generate Ir(II) species (E(IrIII/II)pc = −1.51 V (vs. SCE)1, Fig. S9, ESI†), which initiates the subsequent reducing catalytic reaction (Table 5).
|
|
||||
|---|---|---|---|---|
| Entrya | PC | Amines | Conversionb/% | Yieldb/% |
| a Entry 1–12: R = H (substrate B1); procedure: substrate 50 μmol, PC (2 mol%), amine (5 equiv.) and HCOOH (2.5 equiv.) in 4 mL MeCN solution was degassed by nitrogen, and irradiated by blue light (12 W, λmax = 462 nm) at 25 °C. b Determined by 1H NMR spectroscopy by adding an internal standard of 5,5′-dimethyl-2,2′-bipyridine. c Absence of HCOOH (20 equiv.). d Absence of light. e Presence of TEMPO (radical trapping reagent, 2 equiv.). f Entry 13: R = Me (substrate B2). | ||||
| 1 | Ru(bpy)3Cl2 | DIPEA | 19 | 5 |
| 2 | fac-Ir(ppy)3 | DIPEA | 90 | 72 |
| 3 | 1b | DIPEA | 99 | 85 |
| 4 | 4Meb | DIPEA | 99 | 76 |
| 5 | 6Hb | DIPEA | 99 | 90 |
| 6 | 4Meb | TEA | 99 | 72 |
| 7 | 4Meb | TMEDA | 84 | 55 |
| 8 | 4Meb | DBU | 99 | 64 |
| 9 | 4Meb | — | 0 | 0 |
| 10c | 4Meb | DIPEA | 27 | 15 |
| 11d | 4Meb | DIPEA | 0 | 0 |
| 12e | 4Meb | DIPEA | 0 | 0 |
| 13f | 4Meb | DIPEA | 99 | 59 |

Interestingly, modifying the N-substituent of bis-NHC ligand from an alkyl group to a glucose moiety renders photo-catalyst 6Hc soluble in aqueous media. At the outset, we examined the 6Hc-catalysed reductive cyclization of B1 in a mixture of H2O/MeOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with ascorbic acid as reductant, but both the substrate conversion and product yield were low. The use of diisopropylethylamine (DIPEA) as reductant instead of ascorbic acid and addition of tetrabutylammonium chloride improved the conversion and yield to 98% and 49%, respectively. Increasing the vol% of methanol in aqueous solution to 75% leads to 99% conversion and 87% product yield. To the best of our knowledge, this is the first example of visible-light-driven radical cyclization for synthesis of pyrrolidine in aqueous media (Table 6).
:1) with ascorbic acid as reductant, but both the substrate conversion and product yield were low. The use of diisopropylethylamine (DIPEA) as reductant instead of ascorbic acid and addition of tetrabutylammonium chloride improved the conversion and yield to 98% and 49%, respectively. Increasing the vol% of methanol in aqueous solution to 75% leads to 99% conversion and 87% product yield. To the best of our knowledge, this is the first example of visible-light-driven radical cyclization for synthesis of pyrrolidine in aqueous media (Table 6).
|
|
||||
|---|---|---|---|---|
| Entrya | Solvents (H2O/MeOH)b | Reductant | Conversionc/% | Yieldc/% |
| a Procedure: substrate 50 μmol, 6Hc (2 mol%), reductant (5 equiv.), nBu4NCl (5 equiv.) in 4 mL aqueous solution was degassed by nitrogen, and irradiated by blue light (12 W, λmax = 462 nm) at 25 °C. b Solvent system used is water/methanol in volume ratio (v/v). c Determined by 1H NMR spectroscopy by adding internal standard of 5,5′-dimethyl-2,2′-bipyridine. d Absence of HCOOH. e Absence of nBu4NCl. | ||||
| 1 | 3/1 | Ascorbic acid | 23 | 10 |
| 2d | 3/1 | Ascorbic acid | 20 | 14 |
| 3 | 3/1 | DIPEA | 98 | 49 |
| 4d,e | 3/1 | DIPEA | 90 | 21 |
| 5d | 3/1 | DIPEA | 79 | 31 |
| 6 | 1/1 | DIPEA | 99 | 64 |
| 7 | 1/3 | DIPEA | 99 | 87 |
| 8 | 0/1 | DIPEA | 99 | 66 |

Visible-light-driven CO2 reduction
There is a surge of interest in developing photo-catalytic CO2 reduction using earth abundant metal complexes as catalysts and luminescent cyclometalated Ir(III) complexes particularly fac-Ir(ppy)3 as PC. Nevertheless, several recent reports drew attention to the instability of fac-Ir(ppy)3, which is a challenge for achieving efficient light-driven CO2 reduction in the long run.55,57,63 In view of the good photo-stability of 4Meb, we investigated the visible-light-driven CO2 reduction by utilizing 4Meb in combination with the recently reported [Co(TPA)Cl]Cl complex.57A CO2-saturated MeCN/triethylamine solution (4:1, v/v; 4 mL) containing catalytic amounts of 4Meb and [Co(TPA)Cl]Cl was irradiated by blue LEDs (12 W) for a specified time period, and the evolved gases were separated and identified by GC-TCD equipped with a molecular sieve column. The volume of H2 and CO gases were calculated by using CH4 as the internal standard.
As shown in Fig. 7a and b, the amount of CO and H2 generated from the reaction mixture is found to show strong dependence on the concentrations of 4Meb and Co(II) catalysts. Particularly, the highest TON value of over 5000 can be accomplished at 0.5 μM of Co(II), while no generation of gases is found at low concentration of Co(II) (50 nM; Fig. S11, ESI†). Similarly, only negligible amount of product gases can be detected after irradiation for 24 h when [Ir] (0.005 mM) is lower than [Co] (0.05 mM). With the representative system containing 4Meb (0.5 mM) and Co(II) (0.005 mM), (Fig. 7c) the visible-light-driven CO2 reduction in three parallel reaction runs gives TON (CO) > 2400 (conversion of about 18 mL of CO2 into 1 mL of CO) with excellent selectivity in generating CO over H2 in the gaseous phase (>95%) after reaction for 72 h. This result is better than for the system utilizing fac-Ir(ppy)3 as PC, which reveals only TON (CO) > 900 and selectivity (CO) of 85% under similar reaction conditions.57
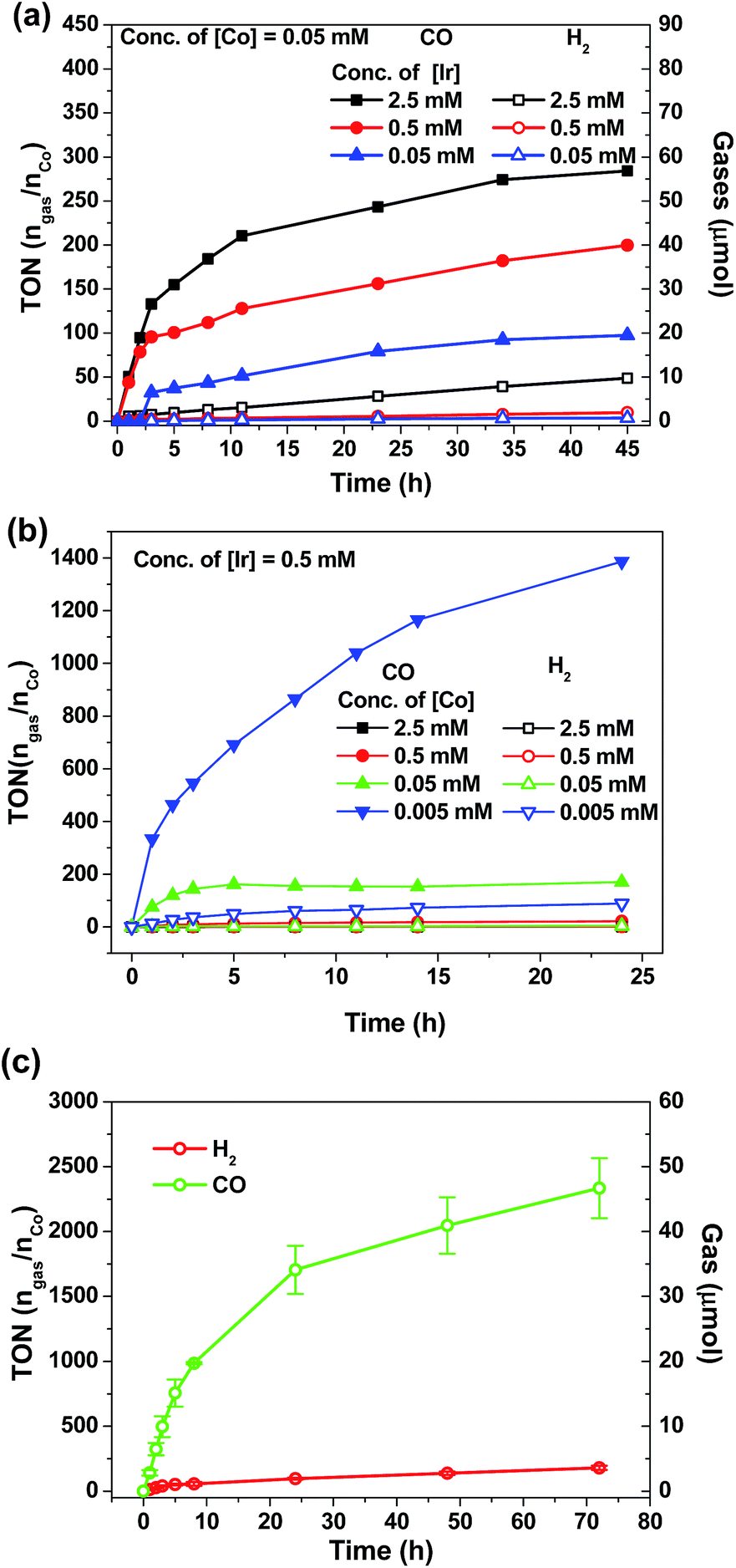 | ||
| Fig. 7 TON value and amounts of gases (CO and H2) generation from CO2 in a CO2-saturated MeCN/TEA (4/1, v/v, 4 mL in total) solution as a function of irradiation time: concentration dependence of (a) PC 4Meb and (b) catalyst [Co(TPA)Cl]Cl; (c) a solution containing 0.005 mM [Co(TPA)Cl]Cl, 0.5 mM 4Meb was irradiated using blue LEDs (12 W) based on the averaged results from three parallel reaction runs. | ||
In order to confirm the roles of catalysts in photo-driven CO2 reduction, several control experiments were performed (Table S5, ESI†). Firstly, in the absence of Co(II) complex, no CO gas was observed in a CO2-saturated MeCN/TEA (4:1, v/v; 4 mL) solution after irradiation for 24 h. On the other hand, in the absence of light, sacrificial amine or PC, the reaction mixture only gives negligible amounts of CO. To ascertain the catalytic role of the Co(II) complex in the reaction, mercury was added to the reaction mixture in order to exclude the possibility of CO generation from heterogeneous Co nanoparticles. To specify, 29.2 μmol of CO (0.714 mL, TON 146) could be generated from the solution with [Ir] (0.5 mM), [Co] (0.05 mM) and elementary Hg (1 mL) after irradiation for 18 h, and this result is comparable with that using the solution mixture without Hg (28.4 μmol of CO formed, TON 142).
Cellular imaging and cell viability assay
We have a long-standing interest in luminescent transition metal complexes, particularly those supported by NHC ligands that display anti-cancer activities.9,16,87–90 The luminescence would allow direct monitoring of cellular uptake and tracking of cellular location in cancer cells by fluorescence microscopy, and such properties of transition metal NHC complexes have been demonstrated to be useful in the elucidation of the anti-cancer mechanism of action. In this work, in view of their favourable photophysical properties, the in vitro cytotoxicity of the bis-NHC Ir(III) complexes against HeLa cells was investigated by MTT assay. As shown in Table S6 (ESI†), the Ir(III) complexes display potent cytotoxicity, with IC50 values ranging from 0.5 to 56.1 μM depending on the lipophilicity and structures of the Ir(III) complexes. Those complexes with butyl groups on the bis-NHC ligand are more cytotoxic than those with glucose units.Since complexes 1b, 4Meb, 6Ha, 6Hb and 6Hc demonstrate outstanding photophysical properties i.e. high quantum yield with long-lived electronic excited states, cellular imaging of these complexes in HeLa cells were performed. After treatment of human cervical cancer cells (HeLa) with the Ir(III) complexes for 15 min, strong green/yellow luminescence was observed in the cytoplasm (but not nucleus) of cancer cells, as revealed by fluorescence microscopy (Fig. 8). Co-localization analyses indicate that the emission of these complexes are mainly localized in the endoplasmic reticulum (ER), which is stained by red-emissive ER-specific ER-Tracker™; a high Pearson's correlation coefficient (R) between the emissions of complexes and ER-Tracker™ is found (for example, 6Hb shows a high R value of 0.80). Consistently, these complexes do not accumulate in other organelles such as lysosome or mitochondria, as shown by the poor co-localization of the emissions of the complexes with the emission of Lyso-Tracker® and Mito-Tracker® respectively (Fig. S13, ESI†). Noticeably, 1b+ (both the counter anions of triflate and chloride) was found in our laboratory to be specifically localized in the ER of cancer cells (Fig. S14†), but not, as reported elsewhere, in mitochondria.73 With the specific accumulation of the complexes in ER, the cytotoxic properties of the complexes may originate from the induction of ER stress88 and immunogenic cell death.91
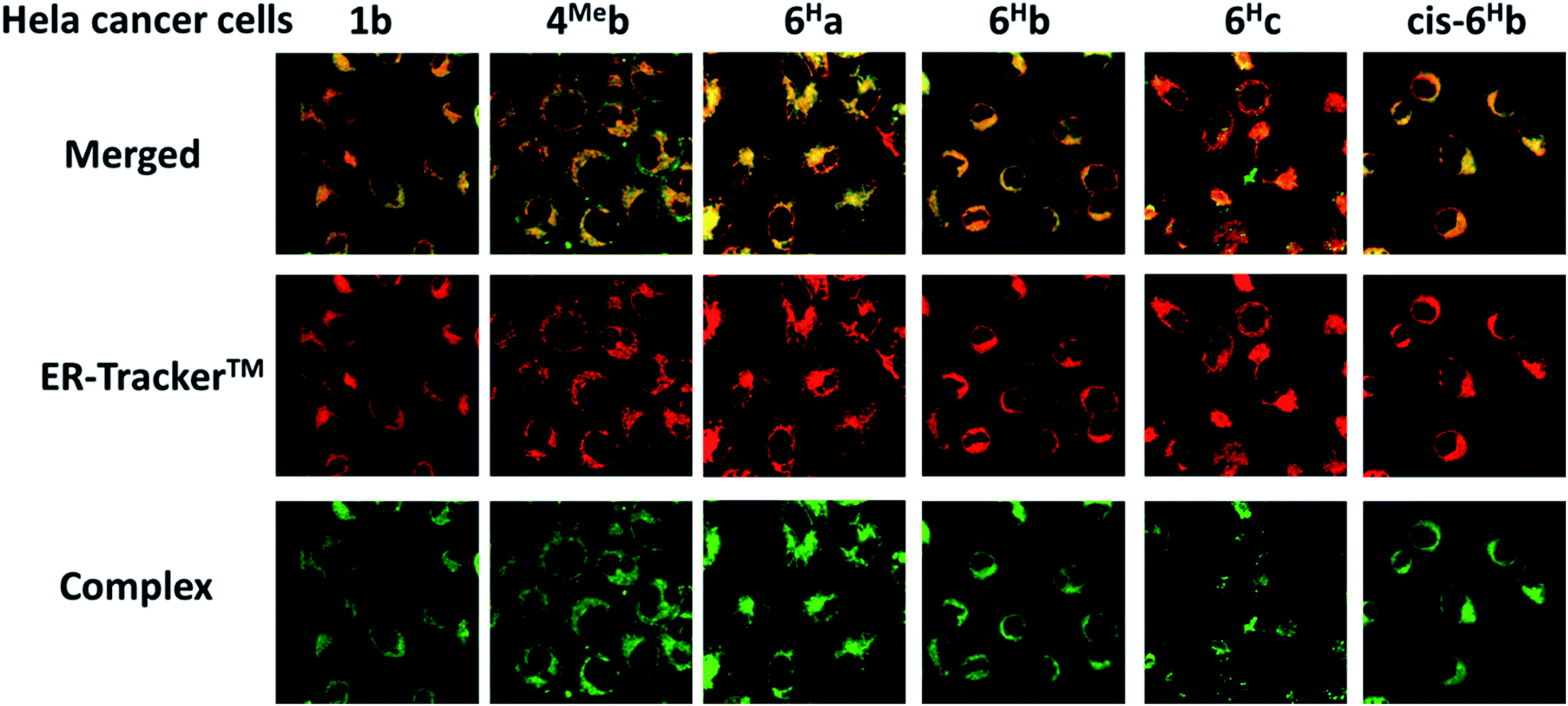 | ||
| Fig. 8 Fluorescence microscopy images of HeLa cancer cells incubated with the Ir(III) NHC complexes: merged (top), ER-tracker™ (middle) and complexes (bottom). Complexes were excited at 340 nm using an emission filter of 510 nm. ER-tracker™ was excited at 546 nm using an emission filter of >580 nm. | ||
Discussion
This work reveals the potential impact and usefulness of bis-NHC ligands for the development of robust metal photo-catalysts. Compared with the well-known complexes fac-Ir(ppy)3, this series of bis-NHC Ir(III) complexes exhibits: (i) outstanding photo-stability under visible light irradiation; (ii) good photo-catalytic performance for several photo-electrochemically reactions; and (iii) long-lived emissive excited states.Stability of NHC metal complexes
The most important feature of metal–ligand bonding between transition metals and NHCs can be rationalized as the σ-coordination (sp2-hybridized lone pair electron) from NHCs. The contribution of both π-back-bonding into carbene p-orbital and π-donation from the carbene p-orbital might be considered as less significant for NHC metal complexes. These behaviours are similar to the coordination characteristics of phosphines, but NHCs are in general better electron-donors than phosphines. Thus, the stronger metal–ligand interaction renders NHC–metal coordination less labile than metal–phosphine bonding and the NHC complexes are more thermally stable.92 The distinct electronic properties and coordination chemistry of NHCs can also lead to improved catalytic activity of the metal complexes, owing to the increased catalyst stability and consequently lower rates of catalyst decomposition.68 In the previous section, the studies on the photo-stability of 4Meb and fac-Ir(ppy)3 have revealed the outstanding stabilization contribution from bis-NHC carbene ligands. Thus this might be one reason that explains the better performance for photo-catalytic reactions using the present bis-NHC Ir(III) complexes as PC.Role of bis-NHC ligands in tuning emission energy and lifetimes
A comparison between the photophysical data of bis-NHC Ir(III) complexes with two notable Ir(III) complexes (fac-Ir(ppy)3 and (ppy)2Ir(acac) (1d)) will be helpful in understanding the role of NHC on the strong absorptivities, high emission efficiencies and long lifetimes of the present bis-NHC Ir(III) complexes. On the basis of DFT calculations on 1a as the representative example (Fig. S4, ESI†), the lowest-energy absorption bands of 1a, 1d and fac-Ir(ppy)3 are ascribed to the HOMO → LUMO transitions, the energy of which is sensitive to the charge of ancillary ligands. The transition energy of fac-Ir(ppy)3 and 1d with the negatively charge ancillary ligands (C^N and acac) are quite close with wavelengths of 416 and 414 nm respectively. However, for 1a with the neutral ancillary ligand (bis-NHC), the HOMO → LUMO transition is markedly hypsochromically shifted to 369 nm, which is consistent with the experimental observations. Fig. S10 (ESI†) shows the surfaces and the energies of HOMO and LUMO of 1a, fac-Ir(ppy)3 and 1d. On the whole, the HOMO contains comparable components of iridium and C^N ligand, and the LUMO is mainly localized on the C^N ligand. Thus, the transition can be assigned as an admixture of metal-to-ligand charge transfer (MLCT) and ligand centered (LC) π–π* transition, which is in accordance with the assignments reported in the literature.The different amounts of metal character in the frontier molecular orbitals of 1a, 1d and Ir(ppy)3, as deduced by TD-DFT calculations, can account for the photophysical properties and long emission lifetimes of the Ir(III) NHC complexes. As shown in Fig. S10 (ESI†), fac-Ir(ppy)3 and 1d have similar energy levels in HOMO and LUMO (around −4.5 eV and −2.0 eV). By simple substitution of the negatively charged ancillary ligands (C^N and acac) in Ir(ppy)3 and 1d by neutral NHC ligand, 1a is found to show lower energy levels of HOMO and LUMO (about −5.2 eV and −2.4 eV). This can be explained by the less electron-donating effect of the neutral NHC ligand compared to the negatively charged auxiliary ligands, as well as a certain contribution of the stabilization of dπ(Ir) by the π-acceptor orbitals of NHC ligand of 1a.68 As a result, the energy level of the HOMO of 1a is 700 mV lower than for 1d, which coincides with the experimental observation that the oxidation potential of 1a is 410 mV more positive than that of 1d. On the other hand, the transition energy of HOMO → LUMO in 1a is estimated to be 0.3 eV larger than in Ir(ppy)3 and 1d, which may account for the blue shift of low-energy absorption of 1a (369 nm vs. 410 nm), as compared to that of 1d and Ir(ppy)3, in UV-Vis absorption spectra. Notably, TD-DFT calculation reveals that frontier molecular orbitals of 1a show a smaller metal character than those of 1d (Ir character in the HOMO of 1a and 1d are found to be ∼30 and ∼40% respectively), and hence the triplet excited states of 1a are likely to have smaller metal character. This would probably slow down the spin–orbit coupling, resulting in slower radiative and non-radiative T1 → S0 decay for 1a. As a result, 1a shows a longer emission lifetime than 1d.
Triplet excited states for photo-catalytic reactivity
In addition to the photo-stability and strong absorptivity in the visible light region, the long-lived triplet excited states, especially their propensity to lose an electron, are to a large extent believed to be crucial for the photo-catalytic properties of the complexes. The long triplet excited state lifetime of 4Meb is beneficial for bi-molecular photochemical reactions allowing the electron-transfer pathways to have sufficient time to take place prior to the decay of the excited state to the ground state.Considering the fact that the excited state of [Ru(bpy)3]2+ (E(RuII*/I) = 0.77 V vs. SCE) has a more powerful oxidative potential than for 4Meb (E(IrIII*/II) = 0.51 V vs. SCE, Table S3†) and no radical cyclization products are obtained when using [Ru(bpy)3]2+, the photo-catalytic route via reductive quenching cycle is not feasible. The photo-catalytic reaction would possibly be initialized via an oxidative quenching cycle of excited states of the photo-catalyst. By carefully examining the transient-absorption spectra of triplet state of 4Meb in MeCN, newly generated long-lived species were observed by using higher energy laser beams (Fig. S8b–g†). These long-lived species were found to be increased in the presence of substrate A1, and could be quenched by DIPEA. This long-lived species might be Ir(IV), which is generated by single electron transfer from [4Meb]* to A1. The calculated excited-state reduction potential of 4Meb (E(IrIV/III*) = −1.51 V vs. SCE, Table S3†) reveals that 4Meb is not a stronger photo-reductant than fac-Ir(ppy)3 (E(IrIV/III*) = −1.73 V vs. SCE).37,93 However, the performance of 4Meb in visible-light-driven radical cyclization and CO2 reduction prove to be better than for fac-Ir(ppy)3. For example, the conversion of substrates A1/B1 to indoline/pyrrolidine by 4Meb (97%/99%) are higher than those by fac-Ir(ppy)3 (27%/90%). Therefore, there should be other reasons for the good performance of 4Meb in photo-catalysis. Plausible reasons could be: (i) the generated Ir(IV) [4Meb]+ (E(IrIV/III) = 0.96 V vs. SCE, Table S3†) are more easily reduced by amines than [fac-Ir(ppy)3]+ (E(IrIV/III) = 0.77 V vs. SCE); and (ii) the higher photo-stability of 4Meb compared with that of fac-Ir(ppy)3.
Consequently, the excellent photo-stability, strong absorptivity, long lifetimes and the photo-ionization behaviours of our bis-NHC Ir(III) complexes possibly enable the photolysis of long-lived excited states of Ir(III)* to Ir(IV) more easily and thus promote the radical cyclization in a catalytic cycle.
Diversities of biological activities of Ir(III) complexes with different N-substituents on NHC ligands
Although N-substituents of NHC ligands are found to show negligible effects on the luminescent properties of the bis-NHC Ir(III) complexes, they have a determinant role on the physical properties and hence the anti-cancer activities of the complexes. For example, complexes with N-butyl substituents on the bis-NHC ligands generally display lower IC50 values than those with N-methyl substituents (Table S6 in ESI†). This is probably attributed to the increase in lipophilicity of the complexes, resulting in better permeability through cellular membrane and higher accumulation of the complexes in cancer cells. The fast cellular uptake of the complexes is supported by the strong luminescence observed in HeLa cells after incubation of the cells with the complexes for 15 min (Fig. 8 and S12 in ESI†). On the other hand, the glucose-functionalized NHC ligands give the complexes good aqueous solubility and a more hydrophilic nature, leading to likely slower cellular uptake as well as reduced cytotoxicity toward cancer cells. It is noteworthy that the complexes accumulate in cellular ER as revealed by fluorescence microscopy images of the co-staining experiments. This can consequently induce ER stress88 and immunogenic cell death, which probably accounts for the high cytotoxicity of the complexes. Further manipulations of the functionalities on NHC ligands of the Ir(III) complexes may realize the development of a new class of diagnostic and/or therapeutic agents.Conclusions
A new series of cyclometalated Ir(III) complexes bearing bis-NHC ligands has been demonstrated as strongly luminescent materials and promising photo-catalysts for visible-light-driven radical cyclization and CO2 reduction and as biological theranostic agents. Owing to the high stability of the Ir–CNHC bond, these bis-NHC Ir(III) complexes show excellent photo-stability compared with widely-used PC, such as fac-Ir(ppy)3 and [Ru(bpy)3]2+. With long-lived triplet excited states and rich photoredox properties, 4Mec can undergo photo-ionization in aqueous media as supported by transient absorption experiments, while 4Meb is found to be a more effective catalyst for radical cyclization and CO2 reduction than fac-Ir(ppy)3 under visible-light irradiation. Interestingly, water-soluble 6Hc, which contains a glucose moiety on the bis-NHC ligands, has been demonstrated as the first PC for the synthesis of pyrrolidine in aqueous media. In addition, through modulations of the chemical structures of the cyclometalated ligands and/or N-substituents on the bis-NHC ligands, the Ir(III) complexes have been found to show different luminescent properties as well as anti-cancer activities, indicating the potential of the complexes as theranostic agents.Author contribution
Chen Yang, Faisal Mehmood, Tsz-Lung Lam, Yuan Wu, Chi-Shun Yeung, Xiangguo Guan, Kai Li, Clive Yik-Sham Chung, Cong-Ying Zhou and Taotao Zou carried out all the experiments and performed data analysis. Chen Yang, Chi-Ming Che and Sharon Lai-Fung Chan designed the experiments, analysed the data and wrote the manuscript. All authors reviewed the manuscript.Acknowledgements
This work was supported by the National Key Basic Research Program of China (No. 2013CB834802), the University Grants Committee of the HKSAR Area of Excellence Scheme (AoE/P-03/08), and the CAS-Croucher Foundation Funding Scheme for Joint Laboratories. C. Yang acknowledges the support of the postgraduate studentships from the University of Hong Kong and Miss Yingshuo Zhang for the synthesis of part of the substrates. S. L.-F. Chan thanks the Hong Kong Research Grants Council (PolyU 253038/15P) and the National Science Foundation of China (21401157) for financial support. Cellular imaging data were acquired using equipment maintained by the University of Hong Kong Li Ka Shing Faculty of Medicine Faculty Core Facility.Notes and references
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, J. Robert, A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- C. Yang, S.-L. Lai, S. L.-F. Chan, K.-H. Low, G. Cheng, K.-T. Yeung, C.-C. Kwok and C.-M. Che, Chem.–Asian J., 2014, 9, 3572–3585 CrossRef CAS PubMed.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed.
- S. C. F. Kui, P. K. Chow, G. Cheng, C.-C. Kwok, C. L. Kwong, K.-H. Low and C.-M. Che, Chem. Commun., 2013, 49, 1497–1499 RSC.
- K. Li, G. Cheng, C. Ma, X. Guan, W.-M. Kwok, Y. Chen, W. Lu and C.-M. Che, Chem. Sci., 2013, 4, 2630–2644 RSC.
- S. C. F. Kui, P. K. Chow, G. S. M. Tong, S.-L. Lai, G. Cheng, C.-C. Kwok, K.-H. Low, M. Y. Ko and C.-M. Che, Chem.–Eur. J., 2013, 19, 69–73 CrossRef CAS PubMed.
- K. Li, T. Zou, Y. Chen, X. Guan and C. M. Che, Chem.–Eur. J., 2015, 21, 7441–7453 CrossRef CAS PubMed.
- E. S.-H. Lam, D. P.-K. Tsang, W. H. Lam, A. Y.-Y. Tam, M.-Y. Chan, W.-T. Wong and V. W.-W. Yam, Chem.–Eur. J., 2013, 19, 6385–6397 CrossRef CAS PubMed.
- Z. He, W.-Y. Wong, X. Yu, H.-S. Kwok and Z. Lin, Inorg. Chem., 2006, 45, 10922–10937 CrossRef CAS PubMed.
- S.-L. Lai, L. Wang, C. Yang, M.-Y. Chan, X. Guan, C.-C. Kwok and C.-M. Che, Adv. Funct. Mater., 2014, 24, 4655–4665 CrossRef CAS.
- W. X. Ni, M. Li, J. Zheng, S. Z. Zhan, Y. M. Qiu, S. W. Ng and D. Li, Angew. Chem., Int. Ed., 2013, 52, 13472–13476 CrossRef CAS PubMed.
- M.-C. Tang, D. P.-K. Tsang, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446–449 CrossRef CAS PubMed.
- W.-P. To, K. T. Chan, G. S. M. Tong, C. Ma, W.-M. Kwok, X. Guan, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 6648–6652 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930–2933 CrossRef CAS PubMed.
- F.-F. Hung, W.-P. To, J.-J. Zhang, C. Ma, W.-Y. Wong and C.-M. Che, Chem.–Eur. J., 2014, 20, 8604–8614 CrossRef CAS PubMed.
- X.-S. Xiao, W.-L. Kwong, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem.–Eur. J., 2013, 19, 9457–9462 CrossRef CAS PubMed.
- R. Visbal, J. M. Lopez-de-Luzuriaga, A. Laguna and M. C. Gimeno, Dalton Trans., 2014, 43, 328–334 RSC.
- C.-M. Che, C.-C. Kwok, S. C. F. Kui, S.-L. Lai and K.-H. Low, in Compr. Inorg. Chem. II, ed. J. R. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 607–655 Search PubMed.
- H. Yersin and W. J. Finkenzeller, in Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 1–97 Search PubMed.
- Y. Ma, H. Zhang, J. Shen and C. Che, Synth. Met., 1998, 94, 245–248 CrossRef CAS.
- K. K.-W. Lo, A. W.-T. Choi and W. H.-T. Law, Dalton Trans., 2012, 41, 6021–6047 RSC.
- F. L. Thorp-Greenwood, R. G. Balasingham and M. P. Coogan, J. Organomet. Chem., 2012, 714, 12–21 CrossRef CAS.
- J. N. Demas and B. A. DeGraff, Anal. Chem., 1991, 63, 829A–837A CrossRef CAS.
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704–1711 CrossRef CAS PubMed.
- J. Li, P. I. Djurovich, B. D. Alleyne, M. Yousufuddin, N. N. Ho, C. J. Thomas, J. C. Peters, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 1713–1727 CrossRef CAS PubMed.
- A. F. Rausch, M. E. Thompson and H. Yersin, J. Phys. Chem. A, 2009, 113, 5927–5932 CrossRef CAS PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- K. K.-W. Lo, B. T.-N. Chan, H.-W. Liu, K. Y. Zhang, S. P.-Y. Li and T. S.-M. Tang, Chem. Commun., 2013, 49, 4271–4273 RSC.
- S. P.-Y. Li, T. S.-M. Tang, K. S.-M. Yiu and K. K.-W. Lo, Chem.–Eur. J., 2012, 18, 13342–13354 CrossRef CAS PubMed.
- W. H.-T. Law, L. C.-C. Lee, M.-W. Louie, H.-W. Liu, T. W.-H. Ang and K. K.-W. Lo, Inorg. Chem., 2013, 52, 13029–13041 CrossRef CAS PubMed.
- K. K.-W. Lo and K. K.-S. Tso, Inorg. Chem. Front., 2015, 2, 510–524 RSC.
- K. K.-W. Lo and K. Y. Zhang, RSC Adv., 2012, 2, 12069–12083 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- J. W. Tucker, J. D. Nguyen, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Chem. Commun., 2010, 46, 4985–4987 RSC.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084–2089 CrossRef CAS PubMed.
- J.-J. Zhong, Q.-Y. Meng, G.-X. Wang, Q. Liu, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Chem.–Eur. J., 2013, 19, 6443–6450 CrossRef CAS PubMed.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- C. D. Windle and R. N. Perutz, Coord. Chem. Rev., 2012, 256, 2562–2570 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- D. Behar, A. T. Dhanasekaran, P. Neta, C. M. Hosten, A. D. Ejeh, P. Hambright and E. Fujita, J. Phys. Chem. A, 1998, 102, 2870–2877 CrossRef CAS.
- R. Ziessel, J. Hawecker and J.-M. Lehn, Helv. Chim. Acta, 1986, 69, 1065–1084 CrossRef CAS.
- H. Hori, F. P. A. Johnson, K. Koike, O. Ishitani and T. Ibusuki, J. Photochem. Photobiol., A, 1996, 96, 171–174 CrossRef CAS.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 14413–14424 CrossRef CAS PubMed.
- J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed.
- S. L.-F. Chan, T. L. Lam, C. Yang, S.-C. Yan and N. M. Cheng, Chem. Commun., 2015, 51, 7799–7801 RSC.
- L. Chen, Z. Guo, X.-G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabéhère-Mallart, K.-C. Lau, T.-C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918–10921 CrossRef CAS PubMed.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano, Energy Environ. Sci., 2014, 7, 1477–1488 CAS.
- C.-F. Leung, S.-M. Ng, C.-C. Ko, W.-L. Man, J. Wu, L. Chen and T.-C. Lau, Energy Environ. Sci., 2012, 5, 7903–7907 CAS.
- A. B. Tossi and H. Görner, J. Photochem. Photobiol., B, 1993, 17, 115–125 CrossRef CAS.
- Y.-J. Yuan, Z.-T. Yu, X.-J. Liu, J.-G. Cai, Z.-J. Guan and Z.-G. Zou, Sci. Rep., 2014, 4, 4045 Search PubMed.
- S. Schmidbauer, A. Hohenleutner and B. König, Beilstein J. Org. Chem., 2013, 9, 2088–2096 CrossRef PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS.
- A. Azua, S. Sanz and E. Peris, Chem.–Eur. J., 2011, 17, 3963–3967 CrossRef CAS PubMed.
- Y. Unger, A. Zeller, M. A. Taige and T. Strassner, Dalton Trans., 2009, 4786–4794 RSC.
- A. G. Tennyson, E. L. Rosen, M. S. Collins, V. M. Lynch and C. W. Bielawski, Inorg. Chem., 2009, 48, 6924–6933 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304–5313 CrossRef CAS PubMed.
- B. Rao, H. Tang, X. Zeng, L. Liu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2015, 54, 14915–14919 CrossRef CAS PubMed.
- L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed.
- C.-H. Yang, J. Beltran, V. Lemaur, J. Cornil, D. Hartmann, W. Sarfert, R. Fröhlich, C. Bizzarri and L. De Cola, Inorg. Chem., 2010, 49, 9891–9901 CrossRef CAS PubMed.
- Y. Li, C.-P. Tan, W. Zhang, L. He, L.-N. Ji and Z.-W. Mao, Biomaterials, 2015, 39, 95–104 CrossRef CAS PubMed.
- J. Jin, H.-W. Shin, J. H. Park, J. H. Park, E. Kim, T. K. Ahn, D. H. Ryu and S. U. Son, Organometallics, 2013, 32, 3954–3959 CrossRef CAS.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. Gareth Williams, Inorg. Chem., 2011, 50, 3804–3815 CrossRef CAS PubMed.
- E. J. Hart and J. W. Boag, J. Am. Chem. Soc., 1962, 84, 4090–4095 CrossRef CAS.
- M. H. Elkins, H. L. Williams, A. T. Shreve and D. M. Neumark, Science, 2013, 342, 1496–1499 CrossRef CAS PubMed.
- K. C. Cho and C. M. Che, Chem. Phys. Lett., 1986, 124, 313–315 CrossRef CAS.
- R. Y. N. Gengler, D. S. Badali, D. Zhang, K. Dimos, K. Spyrou, D. Gournis and R. J. D. Miller, Nat. Commun., 2013, 4, 2560 Search PubMed.
- A. K. El Omar, U. Schmidhammer, P. Pernot, S. Murata and M. Mostafavi, J. Phys. Chem. A, 2012, 116, 11989–11996 CrossRef CAS PubMed.
- C. Xia, J. Peon and B. Kohler, J. Chem. Phys., 2002, 117, 8855–8866 CrossRef CAS.
- A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS.
- H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS PubMed.
- M. Majek, U. Faltermeier, B. Dick, R. Pérez-Ruiz and A. Jacobivon Wangelin, Chem.–Eur. J., 2015, 21, 15496–15501 CrossRef CAS PubMed.
- G. Revol, T. McCallum, M. Morin, F. Gagosz and L. Barriault, Angew. Chem., Int. Ed., 2013, 52, 13342–13345 CrossRef CAS PubMed.
- M. Arun Prasad and M. V. Sangaranarayanan, Chem. Phys. Lett., 2005, 414, 55–60 CrossRef.
- R. W.-Y. Sun, A. L.-F. Chow, X.-H. Li, J. J. Yan, S. S.-Y. Chui and C.-M. Che, Chem. Sci., 2011, 2, 728–736 RSC.
- T. Zou, C.-N. Lok, Y. M. E. Fung and C.-M. Che, Chem. Commun., 2013, 49, 5423–5425 RSC.
- T. Zou, C. T. Lum, C.-N. Lok, W.-P. To, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 5810–5814 CrossRef CAS PubMed.
- J. L.-L. Tsai, A. O.-Y. Chan and C.-M. Che, Chem. Commun., 2015, 51, 8547–8550 RSC.
- D. Y. Q. Wong, W. W. F. Ong and W. H. Ang, Angew. Chem., Int. Ed., 2015, 54, 6483–6487 CrossRef CAS PubMed.
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., ed. V. Balzani and S. Campagna, Springer, Berlin/Heidelberg, 2007, vol. 281, pp. 143–203 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, figures and tables. CCDC 1428476–1428479. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc04458h |
| This journal is © The Royal Society of Chemistry 2016 |