
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric 18F-fluorination for applications in positron emission tomography
Faye
Buckingham
and
Véronique
Gouverneur
*
University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, OX1 3UQ, Oxford, UK. E-mail: veronique.gouverneur@chem.ox.ac.uk
First published on 17th December 2015
Abstract
Positron emission tomography (PET) is becoming more frequently used by medicinal chemists to facilitate the selection of the most promising lead compounds for further evaluation. For PET, this entails the preparation of 11C- or 18F-labeled drugs or radioligands. With the importance of chirality and fluorine substitution in drug development, chemists can be faced with the challenge of preparing enantiopure molecules featuring the 18F-tag on a stereogenic carbon. Asymmetric 18F-fluorination is an emerging field of research that provides an alternative to resolution or conventional SN2-based radiochemistry. To date, both transition metal complexes and organomediators have been successfully employed for 18F-incorporation at a stereogenic carbon.
 Faye Buckingham | Faye Buckingham completed her MChem in 2011 at the University of Oxford before joining the group of Prof. V. Gouverneur (University of Oxford, U.K.) to work towards a DPhil in Organic Chemistry. Her research is focused on the development of metal-free approaches for 18F-radiosynthesis. |
 Véronique Gouverneur | Véronique Gouverneur received her PhD in chemistry at the UCL (Belgium) with Prof. L. Ghosez. She moved to a postdoctoral position at the Scripps Research Institute (CA, USA) with Prof. R. A. Lerner. She then returned to Europe with a position at the University Louis Pasteur (France). She started her research career at the University of Oxford (UK) in 1998, and became Professor in 2008. Her research on fluorine (radio)chemistry disclosed in more than 160 peer-reviewed publications was recently recognised with the ACS Award for Creative Work in Fluorine Chemistry 2015. |
1. Introduction
It is universally recognized that molecular chirality has a direct impact on function. In nature, many essential biological molecules exist only in one of two possible mirror-image structures, either because they possess a chiral unit or through their overall structure.1 In the context of drug development, chirality dominates and it has been accepted since the early 1980s that most of the biological activity observed for a racemate often resides within a single enantiomer.2 As a result, it was anticipated that the proportion of racemic new molecular entities (NMEs) would decrease over time and possibly vanish. A recent survey indicates that the number of enantiopure NMEs approved since the mid-1990s has indeed increased, but the development and approval of racemic compounds remains a viable approach.3 The deeply rooted importance of chirality in the pharmaceutical industry4 and other areas, such as material science, has encouraged much research in asymmetric synthesis and catalysis, two active fields of modern chemistry.5The scientific complexity of drug discovery and the commercial challenges currently facing the pharmaceutical industry have led medicinal chemists to consider Positron Emission Tomography (PET)6 more frequently as a technology for the identification of the most promising lead compounds much earlier in the drug discovery pipeline.7 PET is a non-invasive quantitative imaging modality that can be employed to study drug pharmacokinetics and pharmacodynamics, and the relationship of these pharmacological characteristics to the behavioral, therapeutic and toxic properties of drugs. With the current trend in the pharmaceutical industry to develop optically pure products, PET can also assess the behavior of individual enantiomers in living systems.8 For chiral radio-pharmaceuticals used in the clinic, the administration of a single enantiomer is beneficial in terms of minimizing amount of radioactivity for the patient and, in some cases, by reducing background uptake due to non-specific retention of the inactive enantiomer. The advantageous characteristics of the positron emitting isotope 18F,9 and the prominent position of fluorine substitution in drug discovery10 have fuelled an upsurge of interest in 18F-radiochemistry, with the appearance of novel methods for 18F-labeling inspired by modern 19F chemistry. Despite these advances, the production of chiral non-racemic 18F-labeled drugs remains challenging, especially when the 18F-tag is located on a stereogenic carbon. Fig. 1 presents the various approaches one may consider for this latter scenario.
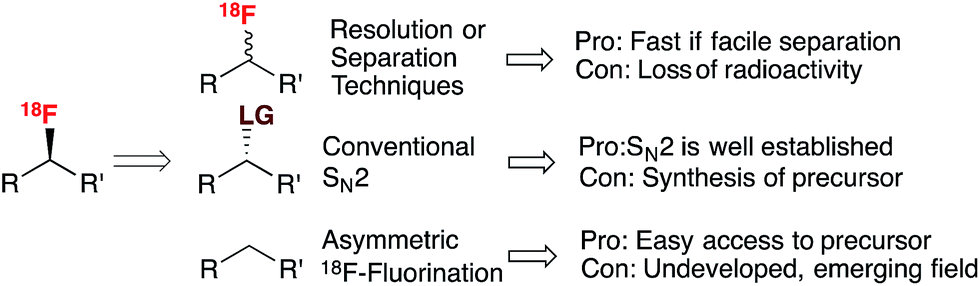 | ||
| Fig. 1 18F–C stereogenicity: resolution or separation techniques, conventional SN2, and asymmetric 18F-fluorination. | ||
Radiochemists would typically consider the separation of 18F-labeled stereoisomers using High Performance Liquid Chromatography (HPLC) in preference to overcoming the obstacles associated with stereoselective or asymmetric 18F-fluorination; this is despite the fact that such separation leads to a substantial loss of radioactivity (50% loss for the separation of two enantiomers). In this essay, we discuss the challenges associated with 18F-incorporation onto a stereogenic carbon and the current state of play of this field of research; in the conclusive remarks, we question how important such developments are for drug developers and PET radiochemists.
2. 18F–C bond formation and stereogenicity
The slow progress of 18F-radiochemistry in comparison with 19F-chemistry is commensurate with the various hurdles associated with 18F-labeling. Most academic and clinical research laboratories are not equipped to handle the cyclotron produced radioisotope 18F, a non-trivial limitation preventing fast development in 18F-radiochemistry. The half-life of the positron emitter 18F (109 min) imposes time constraints that are not compatible with the lengthy reaction time required for many late stage 19F-fluorinations, and the stoichiometry of 18F-radiochemical processes may lead to significant differences in terms of reaction kinetics, in addition to complications for purification.9 The 18F source is indeed employed in nano- or picomolar quantity and is therefore in large sub-stoichiometry with respect to the precursor. Furthermore, for radiopharmaceuticals, additional complications may arise during isolation and formulation due to radiolytic decomposition.11 Another significant difference between 18F and 19F chemistries stems from the preference for reactions using [18F]fluoride instead of [18F]F2. [18F]Fluoride is easier to produce and to handle than [18F]F2 and is therefore widely available. Importantly, use of a nucleophilic 18F source leads to 18F-labeled molecules in higher specific activity, an advantageous property that widens considerably the range of PET studies possible to support drug discovery programs as well as clinical studies. Despite the high strength of the C–F bond (for CH3F, BDE = 109.9 kcal mol−1),12 metabolic paths leading to radiodefluorination with release of [18F]fluoride are problematic for tracers formulated with high specific activity and concentration, because [18F]fluoride binds strongly to the skeletal system. This effect is minimized with 18F-labeled aryl fluorides, which are more stable towards defluorination than alkyl fluoride. The intrinsic sp3 hybridization of stereogenic carbons makes chiral 18F-labeled tracers susceptible to rapid metabolic degradation through oxidation and/or elimination pathways. Some reports suggest that the use of 18F-labeled cycloalkyl fluoride, and more generally 18F-incorporation onto secondary instead of primary carbon atoms, typically enhances metabolic resistance;13 these structurally refined compounds may feature 18F–C stereogenicity and pose additional radiosynthetic challenges.2.1 The conventional SN2 approach
The most commonly employed radiotracer in the clinic is 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG), a non-racemic chiral molecule which presents the 18F-substituent itself on a stereogenic carbon.14 Its preparation, and more generally 18F-fluorination at a stereogenic carbon, involves SN2 displacement of a leaving group positioned on an enantiomerically pure precursor (Scheme 1A). The leaving groups typically selected for displacement are triflate, tosylate or halide functionalities. Alternatively, one could consider the regio- and stereoselective ring opening of a cyclic sulfate, a reaction that was successfully applied for the radiosynthesis of 16-α-[18F]fluoroestradiol ([18F]FES) (Scheme 1B).15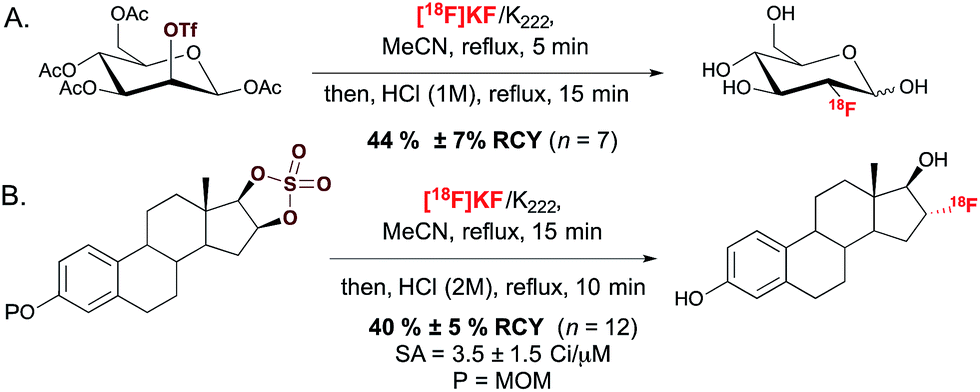 | ||
| Scheme 1 (A.) Radiosynthesis of 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG). (B.) Radiosynthesis of 16-α-[18F]fluoroestradiol ([18F]FES). SA = specific activity. MOM = methoxymethyl. | ||
This SN2-based approach typically employs high temperatures and is limited to substrates that are not prone to decomposition under the reaction conditions. Also, since fluoride is a potent base as well as a nucleophile, both the substrates and newly formed 18F-labeled product should be resistant to elimination and racemization (or epimerization) under the 18F-fluorination conditions. The possibility of incomplete inversion and the time-consuming synthesis of enantiopure precursors are further complications associated with this conventional SN2 strategy. Such challenges are possibly best illustrated with the synthesis of 18F-labeled 4-fluoro-L-glutamine (4F-GLN) and 4-fluoro-L-glutamic acid (4F-GLU) (Scheme 2).16
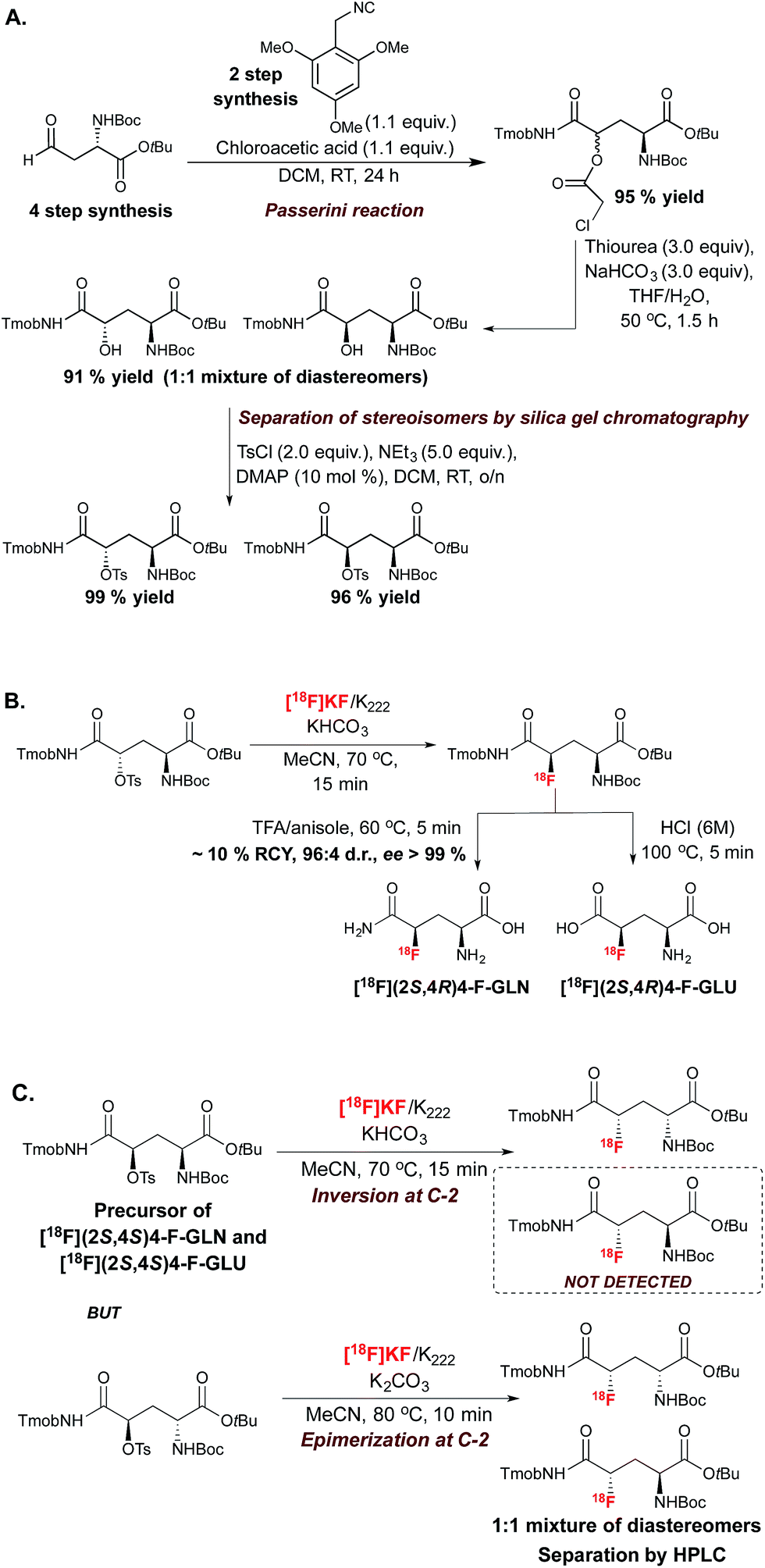 | ||
| Scheme 2 (A.) Synthesis of the substrates for the radiosynthesis of [18F]4 F-GLU and [18F]4-F-GLN. (B.) Radiosynthesis of [18F](2S,4R)4-F-GLU and [18F](2S,4R)4-F-GLN. (C.) Radiosynthesis of [18F](2S,4S)4-F-GLU and [18F](2S,4S)4-F-GLN precursors. Tmob = 2,4,6-trimethoxybenzyl. | ||
Protected precursors for the two possible diastereomers for these radiotracers were prepared by nucleophilic displacement with [18F]fluoride of the tosyl leaving group installed onto the requisite protected substrates. The challenges imposed by this strategy include the multi-step synthesis and fragile stability of the substrates, the occurrence of stereochemical erosion at C-2, and competitive cyclization upon 18F-fluorination. In vitro and in vivo studies were conducted with 18F-(2S,4R)4F-GLN and 18F-(2S,4R)4F-GLU, which are easier to access and to purify than diastereomers 18F-(2S,4S)4F-GLN and 18F-(2S,4S)4F-GLU. The development of these demanding 18F-labeling experiments was however worthwhile, as evaluation studies showed that tumor cell uptake of 18F-(2S,4R)4F-GLN is higher than that of 18F-(2S,4R)4F-GLU, likely due to increased amino acid transport activity, protein incorporation, and non-protein metabolic pathways such as glutaminolysis. In contrast, 18F-(2S,4R)4F-GLU is not incorporated into protein, with the uptake believed to be controlled by the transporter. In vivo studies showed that although 18F-(2S,4R)4F-GLN exhibited a higher uptake and longer retention in rats bearing 9L tumor xenographs, 18F-(2S,4R)4F-GLU showed a slightly higher tumor-to-background ratio due to a faster background clearance. Both 18F-(2S,4R)4F-GLN and 18F-(2S,4R)4F-GLU are useful as tumor metabolic imaging agents.17
2.2. Asymmetric 18F-fluorination with metals
More recent reports have sidestepped the challenges associated with stereoselective SN2 substitution, by favouring an alternative approach in which the product stereochemistry is set by an enantioselective 18F-fluorination. In 2011, the demonstration that a transition metal allowed for regioselective allylic 18F-incorporation opened numerous opportunities towards stereoselective or asymmetric 18F-fluorination directly inspired by research carried out with the non-radioactive isotope 19F.18 Allyl carbonates were found to react with the [18F]fluoride source [18F]TBAF in the presence of Pd(dba)219 or [Ir(COD)Cl]2,20 leading to branched, linear E or linear Z18F-labeled allyl fluorides with clean control over product selectivity. The demonstration that metal mediated 18F–Csp3 bond formation is possible, boded well for the use of these and other transition metals for stereocontrolled 18F-fluorination.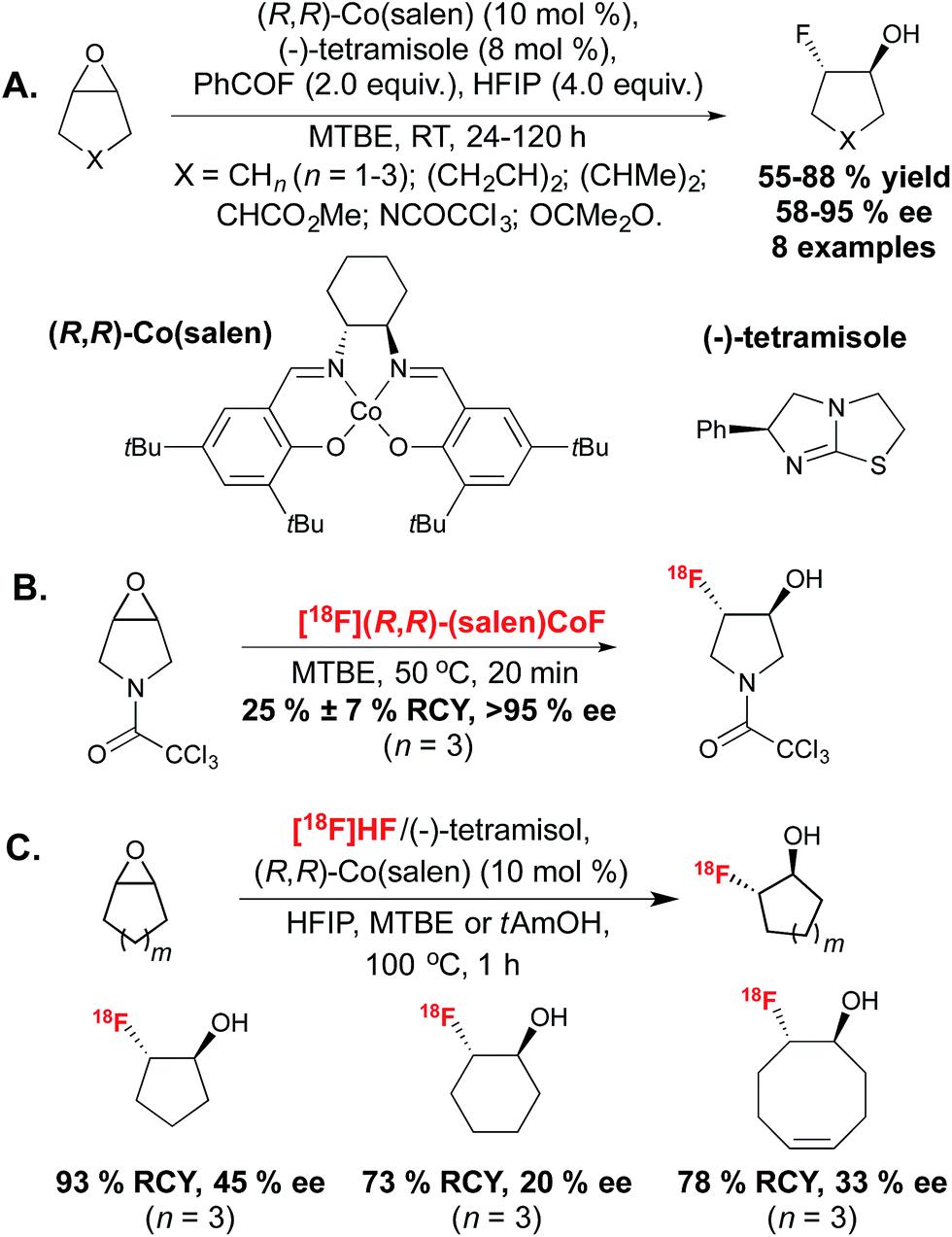 | ||
| Scheme 3 (A.) Enantioselective opening of meso epoxides by fluoride catalysed by (R,R)-Co(salen) and the chiral amine (−)-tetramisole. (B.) Radiosynthesis of an enantiopure fluorohydrin with [18F](R,R)-Co(salen). (C.) Enantioselective desymmetrization of meso epoxides with [18F]HF and (R,R)-Co(salen). | ||
Whilst the previous work had identified that chiral Lewis acid mediated epoxide ring-opening reactions with HF·pyridine complex suffered from low enantioselectivity due to racemic background reaction and catalyst degradation, Doyle discovered that a combination of benzoyl fluoride and hexafluoroisopropanol (HFIP) provided mild release of fluoride, thereby enabling the formation of the fluorohydrin product with excellent enantiocontrol. The reaction likely progressed via amine catalyzed in situ slow formation of HF, which in turn could generate the active (salen)Co(III) fluorine complex. The desymmetrization of a range of meso epoxides occurred with enantiomeric excesses (ee) reaching up to 95%. In a later report, more detailed mechanistic studies on this reaction led to an improved protocol with low catalyst loading.23 In 2014, Doyle in collaboration with Kung described extension of this methodology to 18F-radiofluorination.24 The [18F](salen)CoF complex was synthesized by reaction of (R,R)-(salen)CoOTs with [18F]fluoride eluted from an ion-exchange cartridge, without the requirement for azeotropic drying, and was employed for reaction with epoxide precursors in methyl-tert-butylether (MTBE) to achieve hydrofluorination with high RCY within 20 minutes. An excellent level of enantiocontrol was demonstrated, despite an increase of reaction temperature to 50 °C. The synthesis of a range of radiotracers was well tolerated. In this report, the majority of substrates were terminal epoxides and only one example in which desymmetrization of a meso epoxide led to a product with a 18F-substituted carbon stereocenter was disclosed (Scheme 3B). In 2013, Revunov and Zhuravlev took inspiration from the cobalt mediated enantioselective epoxide opening reaction.25 This work employed [18F]fluoride treated with H2SO4, proposed by the authors to form [18F]HF, which could be trapped by addition of (−)-tetramisole. The reaction took place by addition of (R,R)-Co(salen), HFIP and an epoxide precursor in either MTBE or 2-methyl-2-butanol (tAmOH) as solvent (Scheme 3C). The use of HFIP was found to be crucial to product formation. The reaction was performed at 100 °C for 1 hour and afforded 3 examples of [18F]fluorohydrin products from cyclic epoxides with good RCY but low ee values, which the authors hypothesized was due to the high reaction temperature.
This elegant transformation installs fluorine substitution onto a stereogenic carbon. In an extension of the 19F-fluorination reactions reported by the same group,27 this procedure facilitated the 18F-fluorination of a wide range of precursors containing benzylic C–H bonds. Analogously to the cobalt mediated reaction of Doyle, the reaction was proposed to proceed via formation of a [18F](salen)Mn fluorine complex, generated by reaction of Mn(salen)OTs with [18F]fluoride from an ion-exchange cartridge. In this case, this complex was proposed to undergo oxidation in the presence of iodosobenzene. Reaction at 50 °C in acetone for 10 minutes afforded 18F-labeled products with RCY reaching up to 72%, with an array of functional groups tolerated under these conditions. Starting with 3.5 mCi of [18F]fluoride, 18F-labeled celestolide was obtained in 10% non-decay corrected RCY with a specific activity of 2.68 Ci μmol−1 (end of bombardment). In a singular example of the potential for synthesis of enantioenriched products employing this protocol, the Mn(salen)OTs mediated 18F-fluorination of celestolide afforded the radiolabeled product with an ee of 25% (Scheme 4). Currently the optimization of this reaction in terms of ee has yet to be reported. Nevertheless, this preliminary example highlights the potential opportunity for asymmetric 18F-fluorination with this methodology, which advantageously utilizes nucleophilic [18F]fluoride and employs precursors which do not require pre-functionalization with a leaving group.
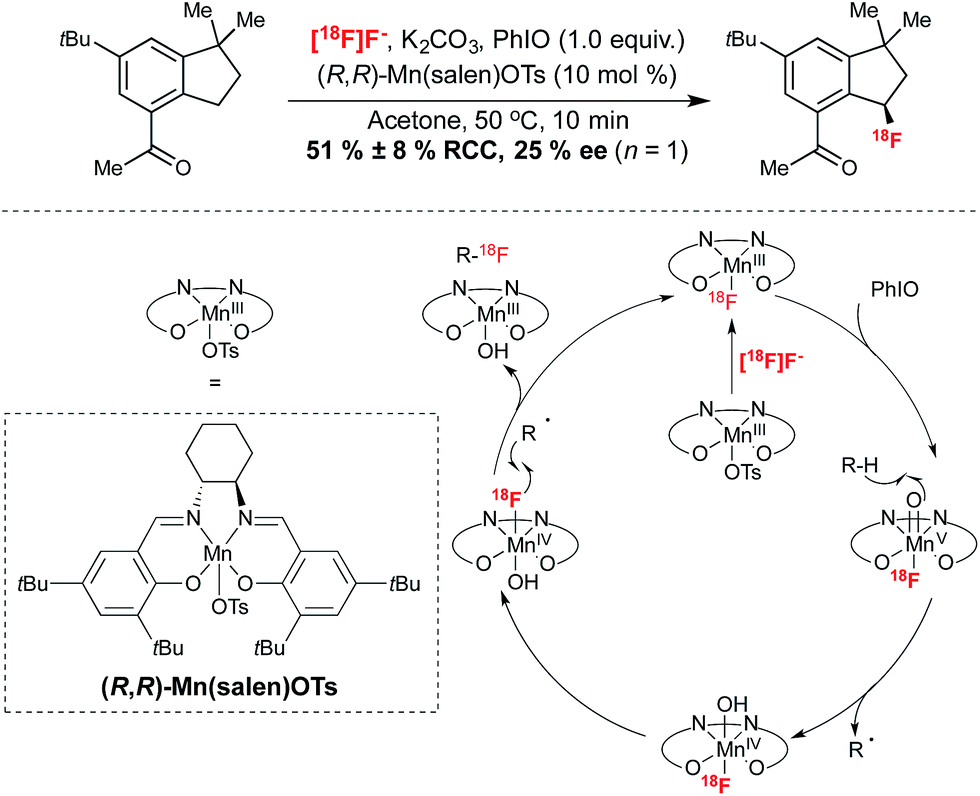 | ||
| Scheme 4 Enantioselective benzylic 18F-fluorination with [18F]fluoride through C–H functionalization. | ||
2.3. Organomediated asymmetric 18F-fluorination
Encouraged by the benefits associated with organocatalysis,28 our research group recently reported a metal-free approach29 to asymmetric 18F-fluorination by taking inspiration from the wealth of literature surrounding organocatalyzed asymmetric fluorination.30 Today, this field of research is largely limited to processes relying on electrophilic fluorination. A notable exception is the asymmetric nucleophilic oxidative fluorination of ketoesters and aminofluorination of alkenes reported by Shibata and co-workers.31 An additional remarkable case of metal free catalytic nucleophilic fluorination involves the natural fluorinase discovered by O'Hagan and co-workers, an enzyme capable of inducing SN2 substitution with fluoride in water, a property exploited for the 18F-labeling of small molecules and peptides under mild conditions; this enzyme has not been used in the context of asymmetric fluorination.32 Electrophilic 18F-fluorination remains a challenging process for radiochemists due to the narrow range of 18F+ sources available to date and the difficulties associated with their preparation.33 Nevertheless, 18F+ radiochemistry offers great opportunities in asymmetric 18F-fluorination. An early example of organocatalyzed asymmetric fluorination is the chiral amine mediated electrophilic α-fluorination of aldehydes, a reaction independently reported by four research groups in 2005.34 Translation of this reaction to radiofluorination posed multiple challenges, especially in terms of avoiding the low temperature conditions and long reaction times associated with organocatalysis. Since α-fluoroaldehydes are prone to decomposition and racemization, they are generally further derivatized prior to analysis. For application to radiosynthesis this two-step procedure should ideally occur in one-pot, without time-consuming purification of the intermediate. Our laboratory recently reported that prochiral aldehyde substrates are amenable to asymmetric 18F-fluorination upon treatment in MTBE with stoichiometric chiral imidazolidinone (S)-I and [18F]NFSI,35 an 18F+ reagent synthesized from post-target produced [18F]F2.36 Notably, [18F]Selectfluor bis(triflate)37 was not a suitable 18F+ source for this transformation. After stirring for 20 minutes at room temperature, reagents for derivatization of the enantioenriched 18F-labeled aldehydes intermediates were added directly. The use of benzhydrazide in methanol afforded the corresponding hydrazone products with good radiochemical conversion (RCC) from [18F]NFSI and ee of up to 92% in a one-pot procedure (Scheme 5).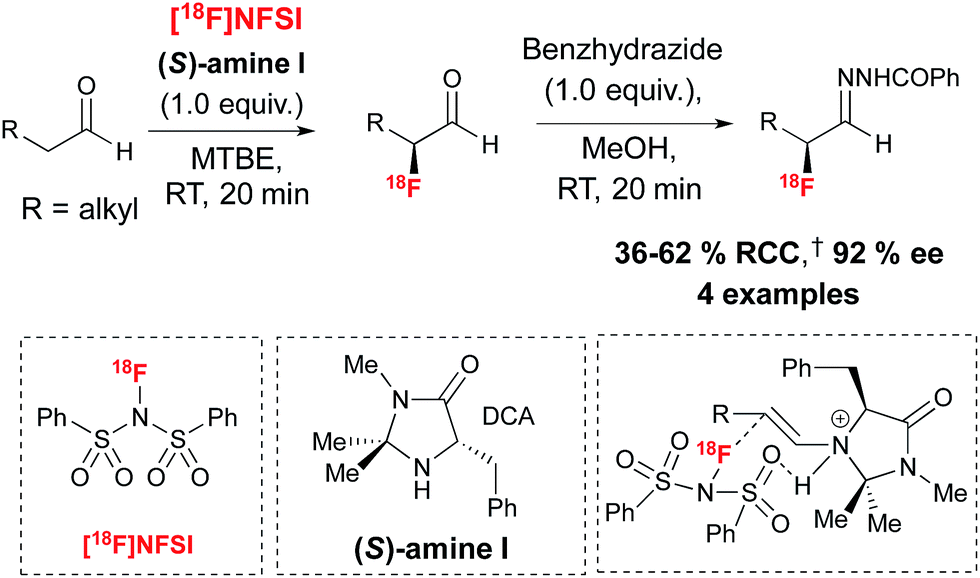 | ||
| Scheme 5 Chiral imidazolidinone (S)-I for the enantioselective 18F-fluorination of aldehydes with [18F]NFSI. †RCC determined by radio-HPLC relative to [18F]NFSI. DCA = dichloroacetic acid. For [18F]NFSI, SA = 0.05 Ci μmol−1, (n = 3). | ||
The utility of α-[18F]fluoroaldehyde synthons was further demonstrated with the preparation of an enantioenriched α-[18F]fluoro carboxylic acid, primary and secondary amides and a secondary amine product (Scheme 6).
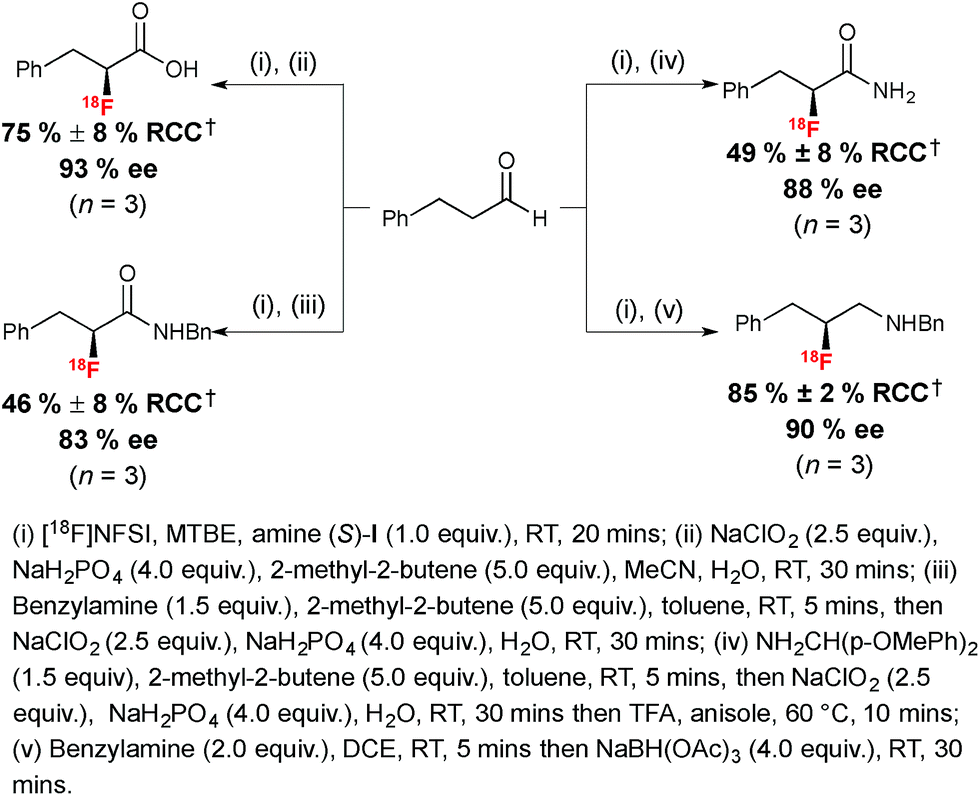 | ||
| Scheme 6 Radiosynthesis of enantioenriched 18F-labeled carboxylic acid, amides and amine. †RCC determined by radio-HPLC relative to [18F]NFSI. | ||
The Pinnick–Lindgren oxidation performed with sodium hypochlorite in acetonitrile in the presence of sodium dihydrogen phosphate and 2-methyl-2-butene led to the desired 18F-labeled carboxylic acid with no erosion of ee. Oxidative amidation was performed in one pot; this process involved formation of an imine in the first instance, followed by a Pinnick–Lindgren oxidation affording the desired 18F-labeled amide with a slightly eroded ee of 83%. A representative enantioenriched 18F-labeled primary α-fluoroamide was also within reach in 49% RCC and 88% ee. Finally, a chiral β-fluoroamine was prepared from the enantioenriched aldehyde via imine formation in dichloroethane (DCE) followed by reduction with sodium triacetoxyborohydride.
The method was subsequently applied to access (2S,4S)-4-[18F]fluoroglutamic acid (Scheme 7). The 18F-labeling of an enantiopure aldehyde precursor derived from L-glutamic acid under the optimized conditions, followed by a Pinnick–Lindgren type oxidation and TFA deprotection, afforded 4-(2S,4S)[18F]fluoroglutamic acid with very good RCC and d.r.; unlike the SN2 approach described in Scheme 2, this method did not lead to unwanted epimerization at C-2.38 Further experiments demonstrated a match/mismatch effect between the chiral imidazolidinone of opposite absolute configuration and the aldehyde substrate; therefore with (R)-amine I, significant erosion of d.r. was observed.
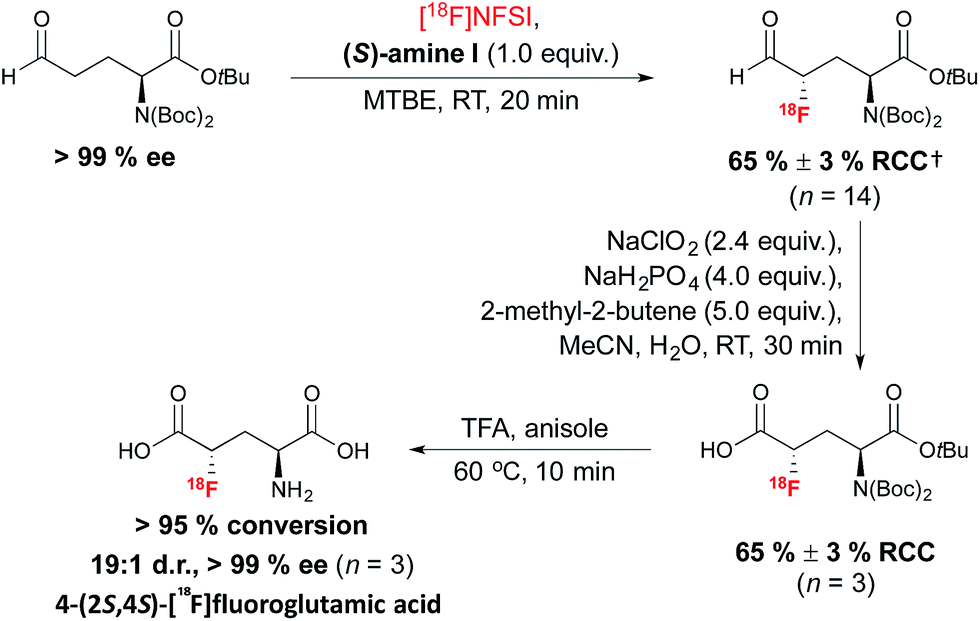 | ||
| Scheme 7 Organomediated radiosynthesis of [18F](2S,4S)-4-F-GLU. †RCC determined by radio-HPLC relative to [18F]NFSI. | ||
3. Conclusion
There is ample evidence in the literature that PET imaging can facilitate the process of drug discovery and development. However, from a pragmatic viewpoint, the routine use of this imaging technology imposes non-trivial radiosynthetic challenges for medicinal chemists, especially when the drug candidate under study is a chiral non-racemic entity. Classical laboratory scale synthesis must be entirely revisited to allow for the nanoscale nature and challenges characteristic of 18F-radiochemistry necessary to evaluate potential drug candidates in a living system. Following these imaging studies and after passing all the hurdles of preclinical and clinical evaluation, large-scale production must then be implemented for the chiral non-racemic drugs selected for manufacturing operations (Fig. 2). | ||
| Fig. 2 Synthetic scales in drug development. | ||
Such difficulties would be particularly stringent for chiral molecules with a stereogenic fluorinated carbon. These considerations pose a fundamental question on the real value of asymmetric 18F-fluorination for radiotracer production since one could argue that separation techniques of stereoisomers may be more rapid or cost effective; this is assuming that the identification of suitable separation conditions is fast and facile. A related debate arose with the realization that although asymmetric catalysis is an attractive method to control stereochemistry into pharmaceutically active molecules, in practice, other techniques are often used for pilot plant or production operations.39 This dichotomy results from industrial constraints for the application of asymmetric catalysis to the large-scale synthesis of drug candidates and commercial drugs. Such constraints include economic considerations, the speed to implement a particular process, the freedom to operate and process robustness. Today, most medicinal chemists would accept that the enormous progress made in asymmetric catalysis will be used by the pharmaceutical industry on a large scale when the catalysts employed are inexpensive, readily available and can be implemented rapidly. Similarly, it is likely that the development of a range of versatile and effective asymmetric 18F-fluorination reactions will be considered by radiochemists in the future as an alternative to separation techniques or conventional radiochemistry requiring multistep synthesis to access the chiral enantiopure precursors necessary for 18F-fluorination. To reach such a situation, it is important that our community continues to develop methods for effective asymmetric 18F-fluorination, employing readily available starting materials and mild reaction conditions, with a protocol that is easy to implement and ideally amenable to automation. The field of stereoselective and asymmetric labeling has been amply developed for a range of isotopes such as 11C, 2H or 3H.40 These advances have furthered our understanding of important biochemical processes and have highlighted the differential behavior of enantiomers in living systems. The radioisotope 18F is now receiving similar attention; this is an encouraging trend that, at a more fundamental level, should increase our understanding of the effect of chirality and F–C stereogenicity on living systems. New asymmetric catalytic fluorinations using transition metals, organocatalysts or other catalytic manifolds are being continuously developed in laboratories around the world; many of these methods offer attractive scope and selectivities. These will be used by radiochemists when the field of asymmetric 18F-fluorination has matured to a point where it is demonstrated that such strategy presents clear advantages over more conventional radiochemistry or separation techniques for rapid in vivo evaluation.
Acknowledgements
Financial support was provided by the BBSRC (F. B.) and GE Healthcare (F. B.). V. G. holds a Royal Society Wolfson Research Merit Award (2013–2018).Notes and references
- (a) G. H. Wagnière, On Chirality and the Universal Asymmetry: Reflections on Image and Mirror Image, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (b) P. L. Luisi, The Emergence of Life: From Chemical Origins to Synthetic Biology, Cambridge Univ. Press, Cambridge, UK, 2006 Search PubMed; (c) S. F. Mason, Nature, 1984, 311, 19–23 CrossRef CAS PubMed.
- (a) W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760–770 CrossRef CAS PubMed; (b) Chiral Drugs: Chemistry and Biological Action, ed. G.-Q. Lin, Q.-D. You and J.-F. Cheng, Wiley, Hoboken, NJ, 2011 Search PubMed; (c) L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2006, 2, 85–100 CAS.
- I. Agranat, S. R. Wainschtein and E. Z. Zusman, Nat. Rev. Drug Discovery, 2012, 11, 972–973 CrossRef CAS PubMed.
- (a) I. Agranat, H. Caner and J. Caldwell, Nat. Rev. Drug Discovery, 2002, 1, 753–768 CrossRef CAS PubMed; (b) H. Caner, E. Groner, L. Levy and I. Agranat, Drug Discovery Today, 2004, 9, 105–110 CrossRef CAS PubMed; (c) J. Gal, in Chirality in Drug Research, ed. E. Francotte and W. Lindner, Wiley-VCH, Weinheim, Germany, 2006, pp. 3–26 Search PubMed.
- (a) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, Hoboken, NJ, 3rd edn, 2010 Search PubMed; (b) Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques, and Applications, ed. M. Gruttadauria and F. Giacalone, Wiley, Hoboken, NJ, 2011 Search PubMed; (c) V. Caprio and J. M. J. Williams, Catalysis in Asymmetric Synthesis, Wiley, Chichester, UK, 2nd edn, 2009 Search PubMed.
- (a) F. R. Wrenn Jr., M. L. Good and P. Handler, Science, 1951, 113, 525–527 CAS; (b) M. E. Phelps, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9226–9233 CrossRef CAS PubMed; (c) B. Pichler, H. F. Wehrl, A. Kolb and M. S. Judenhofer, Semin. Nucl. Med., 2008, 38, 199–208 CrossRef PubMed.
- (a) P. M. Matthews, E. A. Rabiner, J. Passchier and R. N. Gunn, Br. J. Clin. Pharmacol., 2012, 73, 175–186 CrossRef CAS PubMed; (b) J. S. Fowler, N. D. Volkow, G.-J. Wang, Y.-S. Ding and S. L. Dewey, J. Nucl. Med., 1999, 40, 1154–1163 CAS; (c) R. E. Gibson, H. D. Burns, T. G. Hamill, W.-S. Eng, B. E. Francis and C. Ryan, Curr. Pharm. Des., 2000, 6, 973–989 CrossRef CAS PubMed; (d) A. M. J. Paans and W. Vaalburg, Curr. Pharm. Des., 2000, 6, 1583–1591 CrossRef CAS PubMed; (e) L. Hammond, L. Denis, U. Salman, P. Jerabek, C. Thomas Jr. and J. Kuhn, Invest. New Drugs, 2003, 21, 309–340 CrossRef CAS PubMed; (f) L. Cunha, K. Szigeti, D. Mathé and L. F. Metello, Drug Discovery Today, 2014, 19, 936–948 CrossRef CAS PubMed; (g) M. Bergström, A. Grahnén and B. Långström, Eur. J. Clin. Pharmacol., 2003, 59, 356–366 CrossRef PubMed.
- Y.-S. Ding and J. S. Fowler, Drug Dev. Res., 2003, 59, 227–239 CrossRef CAS.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., 2008, 120, 9136–9172 ( Angew. Chem. Int. Ed. , 2008 , 47 , 8998–9033 ) CrossRef.
- (a) H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, ed. V. Gouverneur and K. Müller, Imperial College Press, London, 2012 Search PubMed.
- (a) M. Kuchar and C. Mamat, Molecules, 2015, 20, 16186–16220 CrossRef CAS PubMed; (b) P. J. H. Scott, B. G. Hockley, H. F. Kung, R. Manchanda, W. Zhang and M. R. Kilbourn, Appl. Radiat. Isot., 2009, 67, 88–94 CrossRef CAS PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- (a) B. K. Park and N. R. Kitteringham, Drug Metab. Rev., 1994, 26, 605–643 CrossRef CAS PubMed; (b) M. Kuchar and C. Mamat, Molecules, 2015, 20, 16186–16220 CrossRef CAS PubMed; (c) M. Wrobleski, G. Reichard, S. Paliwal, S. Shah, H. Tsui, R. Duffy, J. Lachowicz, C. Morgan, G. Varty and N. Shih, Bioorg. Med. Chem. Lett., 2006, 16, 3859–3863 CrossRef CAS PubMed; (d) P. M. Manoury, J. Binet, J. Rousseau, F. Lefevre-Borg and I. Cavero, J. Med. Chem., 1987, 30, 1003–1011 CrossRef CAS PubMed; (e) B. Sorensen, J. Rohde, J. Wang, S. Fung, K. Monzon, W. Chiou, L. Pan, X. Deng, D. A. Stolarik and E. U. Frevert, Bioorg. Med. Chem. Lett., 2006, 16, 5958–5962 CrossRef CAS PubMed.
- (a) T. Ido, C.-N. Wan, V. Casella, J. S. Fowler, A. P. Wolf, M. Reivich and D. E. Kuhl, J. Labelled Compd. Radiopharm., 1978, 14, 175–183 CrossRef CAS; (b) K. Hamacher, H. H. Coenen and G. Stöcklin, J. Nucl. Med., 1986, 27, 235–238 CAS; (c) J. S. Fowler and T. Ido, Semin. Nucl. Med., 2002, 32, 6–12 CrossRef PubMed; (d) J. W. Fletcher, B. Djulbegovic, H. P. Soares, B. A. Siegel, V. J. Lowe, G. H. Lyman, R. E. Coleman, R. Wahl, J. C. Paschold, N. Avril, L. H. Einhorn, W. W. Suh, D. Samson, D. Delbeke, M. Gorman and A. F. Shields, J. Nucl. Med., 2008, 49, 480–508 CrossRef PubMed; (e) A. Almuhaideb, N. Papathanasiou and J. Bomanji, Annals of Saudi Medicine, 2011, 31, 3–13 CrossRef PubMed.
- (a) M. Dixit, J. Shi, L. Wei, G. Afari and S. Bhattacharyya, Int. J. Mol. Imaging, 2013, 278607 Search PubMed; (b) D. O. Kiesewetter, M. R. Kilbourn, S. W. Landvatter, D. F. Heiman, J. A. Katzenellenbogen and M. J. Welch, J. Nucl. Med., 1984, 25, 1212–1221 CAS; (c) J. L. Lim, L. Zheng, M. S. Berridge and T. J. Tewson, Nucl. Med. Biol., 1996, 23, 911–915 CrossRef CAS PubMed; (d) L. Sundararajan, H. M. Linden, J. M. Link, K. A. Krohn and D. A. Mankoff, Semin. Nucl. Med., 2007, 37, 470–476 CrossRef PubMed.
- (a) W. Qu, Z. Zha, K. Ploessl, B. P. Lieberman, L. Zhu, D. R. Wise, C. B. Thompson and H. F. Kung, J. Am. Chem. Soc., 2010, 133, 1122–1133 CrossRef PubMed; (b) B. P. Lieberman, K. Ploessl, L. Wang, W. Qu, Z. Zha, D. R. Wise, L. A. Chodosh, G. Belka, C. B. Thompson and H. F. Kung, J. Nucl. Med., 2011, 52, 1947–1955 CrossRef CAS PubMed; (c) K. Ploessl, L. Wang, B. P. Lieberman, W. Qu and H. F. Kung, J. Nucl. Med., 2012, 53, 1616–1624 CrossRef CAS PubMed; (d) S. Venneti, M. P. Dunphy, H. Zhang, K. L. Pitter, P. Zanzonico, C. Campos, S. D. Carlin, G. La Rocca, S. Lyashchenko, K. Ploessl, D. Rohle, A. M. Omuro, J. R. Cross, C. W. Brennan, W. A. Weber, E. C. Holland, I. K. Mellinghoff, H. F. Kung, J. S. Lewis and C. B. Thompson, Sci. Transl. Med., 2015, 7, 274 Search PubMed.
- K. Smolarz, B. J. Krause, F. P. Graner, F. M. Wagner, H.-J. Wester, T. Sell, C. Bacher-Stier, L. Fels, L. Dinkelborg and M. Schwaiger, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, 1861–1868 CrossRef CAS PubMed.
- C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC.
- C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., 2011, 123, 2661–2665 ( Angew. Chem. Int. Ed. , 2011 , 50 , 2613–2617 ) CrossRef.
- (a) E. Benedetto, M. Tredwell, C. Hollingworth, T. Khotavivattana, J. M. Brown and V. Gouverneur, Chem. Sci., 2013, 4, 89–96 RSC; (b) J. J. Topczewski, T. J. Tewson and H. M. Nguyen, J. Am. Chem. Soc., 2011, 133, 19318–19321 CrossRef CAS PubMed.
- S. Bruns and G. Haufe, J. Fluorine Chem., 2000, 104, 247–254 CrossRef CAS.
- J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2010, 132, 3268–3269 CrossRef CAS PubMed.
- J. A. Kalow and A. G. Doyle, J. Am. Chem. Soc., 2011, 133, 16001–16012 CrossRef CAS PubMed.
- T. J. A. Graham, R. F. Lambert, K. Ploessl, H. F. Kung and A. G. Doyle, J. Am. Chem. Soc., 2014, 136, 5291–5294 CrossRef CAS PubMed.
- E. Revunov and F. Zhuravlev, J. Fluorine Chem., 2013, 156, 130–135 CrossRef CAS.
- X. Huang, W. Liu, H. Ren, R. Neelamegam, J. M. Hooker and J. T. Groves, J. Am. Chem. Soc., 2014, 136, 6842–6845 CrossRef CAS PubMed.
- (a) W. Liu, X. Huang and J. T. Groves, Nat. Protoc., 2013, 8, 2348–2354 CrossRef CAS PubMed; (b) W. Liu and J. T. Groves, Angew. Chem., 2013, 125, 6140 ( Angew. Chem. Int. Ed. , 2013 , 52 , 6024–6027 ) CrossRef; (c) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed.
- (a) D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed; (b) M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Drug Discovery Today, 2007, 12, 8–26 CrossRef CAS PubMed.
- F. Buckingham, A. K. Kirjavainen, S. Forsback, A. Krzyczmonik, T. Keller, I. M. Newington, M. Glaser, S. K. Luthra, O. Solin and V. Gouverneur, Angew. Chem., 2015, 127, 13564–13567 ( Angew. Chem. Int. Ed. , 2015 , 54 , 13366–13369 ) CrossRef.
- (a) C. Bobbio and V. Gouverneur, Org. Biomol. Chem., 2006, 4, 2065–2075 RSC; (b) G. Valero, X. Companyó and R. Rios, Chem.–Eur. J., 2011, 17, 2018–2037 CrossRef CAS PubMed; (c) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1–PR43 CrossRef CAS PubMed.
- S. Suzuki, T. Kamo, K. Fukushi, T. Hiramatsu, E. Tokunaga, T. Dohi, Y. Kita and N. Shibata, Chem. Sci., 2014, 5, 2754–2760 RSC.
- (a) D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed; (b) C. Dong, F. Huang, H. Deng and C. Schaffrath, Nature, 2004, 427, 561–565 CrossRef CAS PubMed; (c) H. Deng and D. O'Hagan, Curr. Opin. Chem. Biol., 2008, 12, 582–592 CrossRef CAS PubMed; (d) D. O'Hagan, J. Fluorine Chem., 2006, 127, 1479–1483 CrossRef; (e) D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- M. Tredwell and V. Gouverneur, Angew. Chem., 2012, 124, 11590–11602 ( Angew. Chem. Int. Ed. , 2012 , 51 , 11426–11437 ) CrossRef.
- (a) D. Enders and M. R. M. Hüttl, Synlett, 2005, 991–993 CrossRef CAS; (b) M. Marigo, D. Fielenbach, A. Braunton, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., 2005, 117, 3769–3772 ( Angew. Chem. Int. Ed. , 2005 , 44 , 3703–3706 ) CrossRef; (c) D. D. Steiner, N. Mase and C. F. Barbas III, Angew. Chem., 2005, 117, 3772–3776 ( Angew. Chem. Int. Ed. , 2005 , 44 , 3706–3710 ) CrossRef; (d) T. D. Beeson and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 8826–8828 CrossRef CAS PubMed.
- H. Teare, E. G. Robins, E. Årstad, S. K. Luthra and V. Gouverneur, Chem. Commun., 2007, 2330–2332 RSC.
- J. Bergman and O. Solin, Nucl. Med. Biol., 1997, 24, 677–683 CrossRef CAS PubMed.
- H. Teare, E. G. Robins, A. Kirjavainen, S. Forsback, G. Sandford, O. Solin, S. K. Luthra and V. Gouverneur, Angew. Chem., 2010, 122, 6973–6976 ( Angew. Chem. Int. Ed. , 2010 , 49 , 6821–6824 ) CrossRef CAS; I. S. R. Stenhagen, A. K. Kirjavainen, S. J. Forsback, C. G. Jorgensen, E. G. Robins, S. K. Luthra, O. Solin and V. Gouverneur, Chem. Commun., 2013, 49, 1386–1388 RSC.
- The specific activity (SA) of 18F-NFSI is 1.9 GBq μmol−1 (n = 3) or 0.05 Ci μmol−1. Tracers prepared with this reagent are produced with SA in the same range. Radioligands employed to measure ligand binding to receptors should have high specific activity to detect low receptor densities; however, low specific activity is not a critical issue in PET studies targeting metabolic processes in vivo. 18F-Labeled glutamic acid is a metabolic imaging tracer.
- J. M. Hawkins and T. J. N. Watson, Angew. Chem., 2004, 116, 3286–3290 ( Angew. Chem. Int. Ed. , 2004 , 43 , 3224–3228 ) CrossRef.
- (a) D. Arigoni and E. L. Eliel, in Topics in Stereochemistry, Vol. 4, ed. E. L. Eliel and N. L. Allinger, Wiley, Hoboken, NJ, 1969, pp. 127–244 Search PubMed; (b) J. R. Hanson, The Organic Chemistry of Isotopic Labelling, RSC, Cambridge, UK, 2011 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |