
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A highly diastereoselective “super silyl” governed aldol reaction: synthesis of α,β-dioxyaldehydes and 1,2,3-triols†
Wafa
Gati
 * and
Hisashi
Yamamoto
*
* and
Hisashi
Yamamoto
*
Molecular Catalyst Research Center, Chubu University, 1200 Matsumoto-cho, Kasugai, Aichi 487-8501, Japan. E-mail: wafagati@isc.chubu.ac.jp; hyamamoto@isc.chubu.ac.jp
First published on 6th October 2015
Abstract
A highly diastereoselective approach for the synthesis of protected α,β-dioxyaldehydes derived from (Z)-tris(trimethylsilyl)silyl “super silyl” enol ethers is described. A general and highly syn-stereoselective aldol reaction directed by the “super silyl” group catalyzed by triflimide (HNTf2) is developed providing α,β-dioxyaldehydes and 1,2,3-triol fragments which can be a useful platform for the elaboration of natural and unnatural sugar derivatives.
Introduction
Polyols are among the most interesting motifs present in various natural and synthetic products. Over the past several decades, organic chemists have made great efforts to invent simpler and more efficient strategies to access stereodefined polyol motifs toward the synthesis of complex sugar frameworks. Because the majority of molecules containing this motif require a multistep protocol for access, chemists have been engaged with creating multiple stereocenters in a one-pot procedure. Although there are numerous routes to C–C bond formation, the aldol reaction remains the most promising and straightforward method for creating two new adjacent stereogenic centers toward the construction of the required polyol subunits.1,2Recently our group has actively investigated the Mukaiyama aldol reaction of tris(trimethylsilyl)silyl “super silyl” enol ethers for the highly diastereoselective synthesis of β-super siloxy aldehydes and α-halo-β-super siloxy aldehydes employing Lewis acid catalysis.3 This efficient methodology allows for a rapid and stereoselective construction of mono-, bis- and tris-hydroxyaldehydes through mono, double and triple cross aldol processes, respectively, affording polyketide-like scaffolds which are particularly useful for the oriented construction of complex natural polyketides. In our continuous studies on Mukaiyama aldol reactions of super silyl enol ethers, we questioned whether a similar strategy might provide access to α,β-dioxygenated aldehydes which could be a useful building block for construction of complex sugar moieties.4
Results and discussion
Herein we describe the first highly diastereoselective aldol reaction with dioxy enol ethers to give protected α,β-dioxygenated aldehydes in moderate to good yields and with exclusively high syn selectivities.The super silyl enol ether derived from silyloxy acetaldehyde5 was prepared according to the general procedure recently developed in our laboratory.2a We began our studies by establishing optimal conditions for the Mukaiyama aldol reaction of bissuper silyloxy enol ether 1a with 1-octanal using 1 mol% of HNTf2 as catalyst in dichloromethane at −40 °C (Scheme 1).
 | ||
| Scheme 1 Influence of the additive on the aldol reaction. Yields of isolated aldehydes are shown. The dr values are determined from crude 1H NMR. | ||
We were pleased to find that the aldol adduct was obtained in high diastereoselectivity (dr = 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) but with moderate yield (40%). Thus, in an attempt to optimize the conditions we performed the reaction in the presence of 10 mol% of iodobenzene, which has previously been found to be very useful for increasing the reactivity and the rate of the aldol reaction.3d Gratifyingly, we found that the reaction works more efficiently and the adduct 2a was obtained with much better yield (73%) and a slightly improved diastereoselectivity (dr = 95:5). Although we are not sure about the exact role of iodobenzene, we believe that it acts as a co-catalyst that stabilizes the silylenium cation formed in situ. Because the additive seemed to be playing a critical role in affecting the rate of the reaction, we conducted a 29Si NMR study with the hypothesis that [PhI-Si(TMS)3]+ is the real active catalytic species. We first recorded a reference 29Si NMR spectrum using a simple test substrate (allyltris(trimethylsilyl)silane) in the presence of triflimide (Scheme 2, (1)). We detected a first singlet corresponding to three trimethylsilyl groups that appears at −15.35 ppm and a second singlet corresponding to central silicon that appears at 4.61 ppm. Iodobenzene was then added to the NMR tube and a second 29Si NMR spectrum was recorded after 45 min at room temperature.
:9) but with moderate yield (40%). Thus, in an attempt to optimize the conditions we performed the reaction in the presence of 10 mol% of iodobenzene, which has previously been found to be very useful for increasing the reactivity and the rate of the aldol reaction.3d Gratifyingly, we found that the reaction works more efficiently and the adduct 2a was obtained with much better yield (73%) and a slightly improved diastereoselectivity (dr = 95:5). Although we are not sure about the exact role of iodobenzene, we believe that it acts as a co-catalyst that stabilizes the silylenium cation formed in situ. Because the additive seemed to be playing a critical role in affecting the rate of the reaction, we conducted a 29Si NMR study with the hypothesis that [PhI-Si(TMS)3]+ is the real active catalytic species. We first recorded a reference 29Si NMR spectrum using a simple test substrate (allyltris(trimethylsilyl)silane) in the presence of triflimide (Scheme 2, (1)). We detected a first singlet corresponding to three trimethylsilyl groups that appears at −15.35 ppm and a second singlet corresponding to central silicon that appears at 4.61 ppm. Iodobenzene was then added to the NMR tube and a second 29Si NMR spectrum was recorded after 45 min at room temperature.
 | ||
| Scheme 2 29Si NMR study on the influence of iodobenzene on the aldol reaction. 1 equiv. of iodobenzene was used. Experiments conducted in NMR tube in CD2Cl2 under nitrogen atmosphere and at room temperature. | ||
Surprisingly, we found that the second singlet was shifted up to 6.07 ppm (Scheme 2, (2)). Surprised by the large effect that iodobenzene had on the outcome of the NMR experiment, we decided to perform other experiments varying the stoichiometry and the reaction time. Interestingly, we found that with higher amounts of iodobenzene, the silicon shift is more pronounced, and the singlets also shift more with longer reaction times (see ESI†). To the best of our knowledge, this is the first NMR proof of the role of organoiodide compounds in the Mukaiyama aldol reaction and the 29Si NMR study was proof of our principle considering [PhI-Si(TMS)3]+ as a more active catalytic species than Tf2N-Si(TMS)3.
Satisfied with these results, we applied our general conditions to the reaction of various super silyl enol ethers with a broad array of aldehydes to afford protected α,β-dioxygenated aldehydes (Scheme 3). Most linear aliphatic aldehydes reacted very smoothly and selectively with super silyl enol ether 1a–e providing the desired α,β-dioxyaldehydes (2–8) in moderate to high yields (up to 83% for compound 5b) and with excellent and exclusive syn-selectivities (up to 98:2). Fortunately, the major diastereomer of compound 3a was crystalline, and the syn stereochemistry was directly determined from X-ray analysis.6 An aldehyde bearing an unsaturation (alkynyl group) in alpha to the carbonyl group was also tested and found to react rather sluggishly with super silyl enol ethers 1a and 1b to afford the corresponding adducts 9a and 9b respectively with low yields and poor selectivity. We next investigated aliphatic aldehydes bearing an additional substitution in alpha to the carbonyl which were also tolerated but with moderate yields (up to 51%) and selectivities (up to 71:29 dr) (10a, 12–13a) due to the presence of the extraordinarily bulky silyloxy group. Nevertheless when we tested these branched aldehydes with a less bulky silyl enol ether by substitution of one of the super silyloxy groups with a benzyloxy (1b) or a triethylsilyloxy group (1c), we found that the previously obtained yields and diastereoselectivities were incredibly improved (10avs.10b and 10c, 12avs.12b, 13avs.13b and 13c). (Z)-1-Supersilyloxy-2-benzyloxy enol ether 1b reacted as expected with remarkably high selectivities (up to >99:1) and better yields (up to 72%) obtained in almost all products (2–15). Notably, pivalaldehyde, which was unreactive with other super silyl enol ethers, was found to react smoothly with (Z)-1-supersilyloxy-2-benzyloxy enol ether 1b to afford the corresponding aldol adduct 15b with excellent yield and diastereoselectivity (81%, dr = 98:2). On the other hand, (Z)-1-supersilyloxy-2-triethylsilyloxy enol ether 1c was found to react less effectively affording the corresponding aldol adducts with diminished yields, probably due to the competitive reaction of the triethylsilyloxy group with our catalyst, although we did not observe the formation of the corresponding regioisomer, and with no remarkable changes in the diastereoselectivity ratios obtained with 1a or 1b. Subsequently, additional super silyl enol ethers bearing allyloxy (1d) or methoxy (1e) groups were also briefly investigated. We subjected (Z)-1-allyloxy-2-supersilyloxy enol ether 1d and (Z)-1-methoxy-2-supersilyloxy enol ether 1e to our optimized reaction conditions, affording the corresponding desired aldol adducts with comparable yields and diastereoselectivities.
 | ||
| Scheme 3 Synthesis of protected α,β-dioxyaldehydes: substrate scope of aliphatic aldehydes. Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale. Yields of isolated aldehydes are shown. The dr values are based on the integration of the 1H NMR signals of crude material. The attribution of syn and anti-ratios was based on the coupling constants of characteristic protons. | ||
After the exploration of the scope of aliphatic aldehydes, we next turned our attention to the scope of aromatic aldehydes which were found to be more challenging. When we first investigated the reactivity of benzaldehyde with 1a using 1 mol% of triflimide catalyst without any additive, we found that the reaction did not proceed and only trace amounts (<5%) of the desired adduct were detected. However, when the reaction was performed with 10 mol% of iodobenzene, the results were remarkably improved and the reaction provided the desired aldol adduct 16a in high yield (78%) but with moderate diastereoselectivity (Scheme 4). Despite the encouraging results regarding the increased yield using iodobenzene, all other attempts to improve the diastereoselectivity ratios for aromatic aldehydes failed, probably due to the presence of two very bulky super silyl groups. Even so, we were interested in examining the scope of aromatic substrates with our super silyl enol ethers. The reaction with 1a was found to have poor selectivity (16a, 18–23a) due to steric hindrance with the two silyloxy groups. Nevertheless, we were delighted to find that the diastereoselectivity could be improved up to 98% (for compound 20b) starting from 1b and up to 93% (for compound 18d) starting from 1d. Then we considered the use of heteroaromatic aldehydes and experiments have shown that an electron-withdrawing group on the heteroaromatic ring is necessary for the reaction to proceed. Our scope was then extended and compounds 22a-b and 23a were obtained in acceptable yields and diastereoselectivities. It is worth noting that all the protected syn-α,β-dioxyaldehydes obtained are stable in almost all cases and can be kept for weeks in the freezer, since these compounds are known to be rather sensitive to both elimination and epimerisation.
 | ||
| Scheme 4 Synthesis of protected α,β-dioxyaldehydes: substrate scope of aromatic aldehydes. Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale. Yields of isolated aldehydes are shown. The dr values are based on the integration of the 1H NMR signals of crude material. The attribution of syn and anti-ratios was based on the coupling constants of characteristic protons. | ||
The scope of the reaction was further examined by reacting an optically pure aldehyde with different super silyl enol ethers. In this case, it is known that the stereochemical outcome of the reaction can be controlled by the chirality of the substrate (1,2-asymmetric induction).7,8
Indeed, the use of (R)-2-phenylpropanal exhibited, as expected, a high Felkin control in conjunction with syn selectivity to afford 24a,b,d with three adjacent stereocenters in excellent diastereoselectivity ratios (up to >99:1 syn–syn for compound 24b) (Scheme 5).9
 | ||
| Scheme 5 1,2-Stereodirected aldol reaction. Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale. Yields of isolated aldehydes are shown. The dr values are based on the integration of the 1H NMR signals of crude material. The attribution of syn and anti-ratios was based on the coupling constants of characteristic protons. | ||
Next, we investigated the possibility of subsequent one-pot sequential transformation of the obtained protected α,β-dioxygenated aldehydes (Table 1). The addition of alkyl, vinyl, alkynyl, thiophen-2-yl or aryl Grignard reagents to the crude material proceeded smoothly to afford trishydroxy products 25–34 with good to excellent yields (50–84%) and exceptionally high syn–syn diastereoselectivities (>99:1:0) which was confirmed by single crystal X-ray analysis of triol 27.10 In the same fashion as in Scheme 5, we considered the use of an aldehyde with a defined α-stereocenter for a 1,2-asymmetric induction investigation. After reaction of (R)-2-phenylpropanal with super silyl enol ether 1b and addition of phenylmagnesium chloride we obtained the desired triol 33 in moderate yield (50%) and diastereoselectivity (dr = 83:17:0).
| Entry | R | R′ | Nucleophileb | Major product | %Yieldc (dr)d |
|---|---|---|---|---|---|
| a Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale. b 1.5 equiv. of nucleophile was used. c Yields of isolated products are shown. d The dr are based on the integration of the 1H NMR signals of crude material. e The reaction was slowly warmed to −20 °C after addition of 2.0 equiv. of nucleophile. | |||||
| 1 | Bn | CH2CH2Ph |

|

|
84% (dr > 99:1:0) |
| 2 | Bn | CH2CH2Ph |

|

|
81% (dr > 99:1:0) |
| 3 | Bn | CH2Ph |

|

|
78% (dr > 99:1:0) |
| 4 | Bn | CH2Ph |

|

|
59% (dr > 99:1:0) |
| 5 | Bn | CH2Ph |

|

|
68% (dr > 99:1:0) |
| 6 | Bn | CH2CH(CH3)2 |

|

|
75% (dr > 99:1:0) |
| 7 | Allyl | CH2CH2Ph |

|

|
82% (dr > 99:1:0) |
| 8 | Allyl | CH2CH(CH3)2 |

|

|
52% (dr = 88:12:0) |
| 9 | Bn |

|

|

|
50% (dr = 83:17:0) |
| 10 | Allyl |

|

|

|
63% (dr > 99:1:0) |
| 11e | Bn | CH2(CH3)3 |

|
|
33% (dr > 99:1:0) |
| 12e | Allyl | CH2(CH3)3 |

|

|
36% (dr > 99:1:0) |
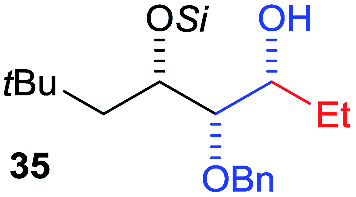
Then we decided to test the more reactive (Z)-supersilyloxy-2-allyloxy enol ether 1d and introduce an additional vinyl group, as it is a rather valuable handle for further transformations. By the addition of a vinyl Grignard reagent we were delighted to easily isolate the synthetically useful vinylic triol 34 generating three new adjacent stereocenters in a one-pot sequential manner in 63% yield and excellent all-syn diastereoselectivity (>99:1:0). Moreover, the reaction was also successful using the lithiated nucleophile EtLi affording the desired alkyl triols 35 and 36 in high diastereoselectivity but with dramatically decreased yields (33% and 36% respectively).
Inspired by the important skeleton of the vinylic all-syn triol 34 and in an attempt to further probe the utility of our highly diastereoselective one-pot sequential aldol reaction, we targeted pentose and hexose-like scaffolds which are usually difficult to access without employing natural sugar as starting material.11 We first applied our strategy to establish the desired α-allyloxy-β-supersilyloxyaldehyde 24d which was obtained at a slightly decreased yield (58%) on a 1 mmol scale but with no loss of selectivity (98:2 syn–syn). Olefination through Wittig reaction and ring closing metathesis using Grubbs second generation catalyst yielded the five member ring compound 37 in 61% yield (over 2 steps). The last step of the asymmetric dihydroxylation was performed under optimal conditions using catalytic AD-mix-β in biphasic solution at 0 °C for four days12 which afforded a single diastereomer of 38 in 73% yield containing five adjacent stereocenters in excellent all-syn selectivity (Scheme 6). The stereochemistry of compound 38 was determined based on 1H, NOE and NOESY experiments (see the ESI†) in comparison with the literature.
 | ||
| Scheme 6 Synthesis of pentose and hexose-like scaffolds. (a) (1) CH3PPh3, n-BuLi, THF (2) Grubbs 2nd generation (2 mol%), CH2Cl2, 40 °C, 2 h (b) AD-mix-β, MeSO2NH2, t-BuOH/H2O (1:1), 0 °C, 4 d (c). Grubbs 2nd generation (2 mol%), CH2Cl2, 40 °C, 2 h (d). OsO4, NMO, t-BuOH, acetone/H2O (1.7:1), r.t, 12 h. | ||
Finally, we considered the possibility of hexose-like scaffold construction, which can be a useful building block to access complex natural and unnatural sugar targets. First we employed our highly diastereoselective Lewis acid catalyzed one-pot sequential aldol scaled-up reaction (1 mmol scale) starting from (R)-2-phenylpropanal and silyl enol ether 1d followed by nucleophilic addition of vinyl magnesium bromide to obtain the desired vinylic triol 34 with no loss of reactivity or diastereoselectivity (62%, 99% dr). Next, a very low loading of the Grubbs second generation catalyst (2 mol%) gave access to six membered ring 39 in excellent yield (97%). A quick optimization of the asymmetric dihydroxylation step (see ESI†) showed that cis-osmilation using osmium tetroxide in presence of excess of N-methylmorpholine N-oxide provided the desired hexose-like structure.13 Thus, compound 40, containing six adjacent stereocenters, was obtained in 68% yield and with an exclusive 4,5-anti stereochemistry. The determination of the stereochemistry of the latter compound was based on the optimization reactions where we obtained the same single isomer using both chiral AD-mix-α or β with comparable selectivity but with a slower reaction rate (50% conversion after 4 days), which can be explained by the preferred attack of osmium from the opposite side of the free hydroxy group present in 39. In addition, a very high coupling constant value (J4-5 > 10.6 Hz) was detected which emphasizes an anti-like relationship between C4–H and C5–H.14
Conclusions
In summary, a very useful strategy to generate synthetically important protected syn-α,β-dioxyaldehydes using Lewis acid catalysis has been described. To the best of our knowledge, this is the first synthesis of α-hydroxyaldehydes using Mukaiyama aldol reaction. Furthermore, a 29Si NMR study was performed providing the first proof of the role of iodobenzene as additive in increasing the reactivity of the active silylenium cation formed in situ. Since the ability of using different protecting groups in the same molecule is an attractive tool to discriminate among chemically similar hydroxyl groups, super silyloxy, benzyloxy, triethylsilyloxy, allyloxy and methoxy have proved to be suitable for the construction of α,β-dioxyaldehydes and 1,2,3-triols. Various nucleophiles were found to react smoothly in a sequential manner allowing for the highly stereoselective construction of all-syn 1,2,3-triols. We have finally demonstrated the utility of our methodology as a key step for the elegant construction of pentose and hexose-like scaffolds. Further applications using super silyl governed aldol reactions targeting complex sugar construction are currently underway in our laboratory and will be reported in due course.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research (no. 23225002), Nippon Pharmaceutical Chemicals Co., Ltd, and Advance Electric Co., Inc.Notes and references
- For reviews on the aldol reaction in the synthesis of carbohydrates and polyketides, see: (a) D. A. Evans, J. V. Nelson and T. Taber, in Topics in Stereochemistry, Wiley, New York, 1982, vol. 13, p. 1 Search PubMed; (b) T. D. Machajewski and C.-H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352 CrossRef CAS; (c) C. Palomo, M. Oiarbide and J. M. García, Chem.–Eur. J., 2002, 8, 36 CrossRef CAS PubMed.
- For selected references on the Mukaiyama aldol reaction, see: (a) T. Mukaiyama, K. Narasaka and K. Banno, Chem. Lett., 1973, 1011 CrossRef CAS; (b) H. Gröger, E. M. Vogl and M. Shibasaki, Chem.–Eur. J., 1998, 47, 1137 CrossRef; (c) R. Mahrwald, Chem. Rev., 1999, 99, 1095 CrossRef CAS PubMed; (d) K. Ishihara and H. Yamamoto, in Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, 2004, vol. 2, p. 25 Search PubMed; (e) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (f) J.-I. Matsuo and M. Murukami, Angew. Chem., Int. Ed., 2013, 52, 9109 CrossRef CAS PubMed.
- (a) M. Boxer and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 48 CrossRef CAS PubMed; (b) M. Boxer and H. Yamamoto, J. Am. Chem. Soc., 2007, 129, 2762 CrossRef CAS PubMed; (c) M. Boxer and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 1580 CrossRef CAS PubMed; (d) B. J. Albert and H. Yamamoto, Angew. Chem., Int. Ed., 2010, 49, 2747 CrossRef CAS PubMed; (e) B. J. Albert, Y. Yamaoka and H. Yamamoto, Angew. Chem., Int. Ed., 2011, 50, 2610 CrossRef CAS PubMed; (f) J. Saadi, M. Akakura and H. Yamamoto, J. Am. Chem. Soc., 2011, 133, 14248 CrossRef CAS PubMed; (g) P. B. Brady and H. Yamamoto, Angew. Chem., Int. Ed., 2012, 51, 1942 CrossRef CAS PubMed; (h) P. B. Brady, B. J. Albert, M. Akakura and H. Yamamoto, Chem. Sci., 2013, 4, 3223 RSC; (i) A. Izumiseki and H. Yamamoto, J. Am. Chem. Soc., 2014, 136, 1308 CrossRef CAS PubMed; (j) P. B. Brady, S. Oda and H. Yamamoto, Org. Lett., 2014, 16, 3864 CrossRef CAS PubMed.
- For selected examples on the synthesis of sugar derivatives using the aldol reaction, see: (a) N. S. Chowdari, D. B. Ramachary, A. Córdova and F. Barbas, Tetrahedron Lett., 2002, 43, 9591 CrossRef CAS; (b) A. B. Northrup and D. W. C. MacMillan, Science, 2004, 305, 1752 CrossRef CAS PubMed; (c) J. Casas, M. Engqvist, I. Ibrahem, B. Kaynak and A. Córdova, Angew. Chem., Int. Ed., 2005, 44, 1343 CrossRef CAS PubMed; (d) J. Carpenter, A. B. Northrup, D. Chung, J. J. M. Wiener, S. Kim and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2008, 47, 3568 CrossRef CAS PubMed; (e) M. Peifer, R. Berger, V. W. Shurtleff, J. C. Conrad and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5900 CrossRef CAS PubMed.
- Supersilyloxy-and allyloxyacetaldehydes were prepared from ethyl glycolate following a described procedure: S. R. Angle, I. Choi and F. S. Tham, J. Org. Chem., 2008, 73, 6268 CrossRef CAS PubMed.
- The syn selectivity was confirmed by single crystal X-ray analysis of compound 3a.†.
- For references on stereodirected aldol reactions see: (a) R. Mahrwald, Chem. Rev., 1999, 99, 1095 CrossRef CAS PubMed; (b) L. C. Dias, E. C. Polo, E. C. de Lucca and M. A. B. Ferreira, in Modern Methods in Stereoselective Aldol Reactions, ed. R. Mahrwald, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, pp. 293–375 Search PubMed.
- IUPAC definition of asymmetric induction: IUPAC, Compendium of Chemical Terminology, (the “Gold Book”), ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997, http://goldbook.iupac.org, 2006, created by M. Nic, J. Jirat and B. Kosata, updates compiled by A. Jenkins, ISBN 0-9678550-9-8, DOI:10.1351/goldbook.
- The determination of the Felkin selectivity was achieved by comparison of coupling constant values with previous reports of similar work in our laboratory, see ref. 3a and 3h and references therein.
- The syn–syn selectivity was confirmed by single crystal X-ray analysis of compound 27.†.
- J. L. Clark, L. Hollecker, J. C. Mason, L. J. Stuyver, P. M. Tharnish, S. Lostia, T. R. McBrayer, R. F. Schinazi, K. A. Watanabe, M. J. Otto, P. A. Furman, W. J. Stec, S. E. Patterson and K. W. Pankiewicz, J. Med. Chem., 2005, 48, 5504 CrossRef CAS PubMed.
- For recent asymmetric dihydroxylation procedures using AD-mix-β in a five member ring see: (a) K. B. Lindsay and S. G. Pyne, J. Org. Chem., 2002, 67, 7774 CrossRef CAS PubMed; (b) N. S. Karanjule, S. D. Markad and D. D. Dhavale, J. Org. Chem., 2006, 71, 6273 CrossRef CAS PubMed.
- For recent asymmetric dihydroxylation procedures in six member rings see: (a) K. Matsumoto, T. Ebata, K. Koseki, H. Kawakami and H. Matsushita, Bull. Chem. Soc. Jpn., 1991, 64, 2309 CrossRef CAS; (b) F. Konno, T. Ishikawa, M. Kawahata and K. Yamaguchi, J. Org. Chem., 2006, 71, 9818 CrossRef CAS PubMed; (c) G. Errasti, C. Koundé, O. Mirguet, T. Lecourt and L. Micouin, Org. Lett., 2009, 11, 2912 CrossRef CAS PubMed; (d) M. T. Mwangi, M. D. Schultz and N. B. Bowden, Org. Lett., 2009, 11, 33 CrossRef CAS PubMed; (e) T. V. Robinson, D. S. Pederson, D. K. Taylor and E. R. T. Tiekink, J. Org. Chem., 2009, 74, 5093 CrossRef CAS PubMed; (f) C. Taffin, G. Kreutler, D. Bourgeois, E. Clot and C. Périgaud, New J. Chem., 2010, 34, 517 RSC; (g) W. Lim, J. Kim and Y. H. Rhee, J. Am. Chem. Soc., 2014, 136, 13618 CrossRef CAS PubMed.
- Please see the ESI† for more details.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental procedures and full compound characterization, including NMR spectra. CCDC 1409678 (27) and 1409680 (3a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03307a |
| This journal is © The Royal Society of Chemistry 2016 |