
Catalytic applications of [IPr·GaX2][SbF6] and related species†
Bastien
Michelet
a,
Shun
Tang
a,
Guillaume
Thiery
a,
Julien
Monot
a,
Huijing
Li
a,
Régis
Guillot
a,
Christophe
Bour
*a and
Vincent
Gandon
 *ab
*ab
aInstitut de Chimie Moléculaire et des Matériaux d'Orsay, CNRS UMR 8182, Univ. Paris-Sud, Université Paris-Saclay, bâtiment 420, 91405 Orsay cedex, France. E-mail: vincent.gandon@u-psud.fr; christophe.bour@u-psud.fr
bInstitut de Chimie des Substances Naturelles, CNRS UPR 2301, Univ. Paris-Sud, Université Paris-Saclay, 1, av. de la Terrasse, 91198 Gif-sur-Yvette, France
First published on 22nd September 2016
Abstract
The title cationic gallium(III) complexes are shown to be useful in homogeneous molecular catalysis. They have been used in catalytic reactions involving the activation of alcohols, alkynes and alkenes towards the addition of various nucleophiles (H2O, ROH, carbonyls, arenes, hydrides, etc.). The mechanism of the transformations of alkynes and alkenes are believed to involve carbocationic σ-vinyl and σ-alkyl gallium intermediates, which can be prone to unusual rearrangements and condensations. These species are likely to be formed reversibly, which confers a dynamic behavior to this chemistry. DFT computations shed light on these issues.
1. Introduction
The use of gallium complexes as catalysts for organic transformations is mostly limited to simple salts of type GaX3. Among them, GaCl3 and, to a lesser extent, GaBr3, have proven to be exquisite species for mediating a large array of reactions1,2 pertaining to σ- or π-Lewis acid catalysis.3 However, they are hard to handle moisture sensitive salts, which are easily destroyed by adventitious water. Loadings up to 100 mol% may be required, even for catalytic applications, which makes them little appreciated within the synthetic chemists’ community in spite of their capabilities. A way to make the chemistry of gallium more appealing would be to develop stable species that display the same catalytic activity than that of GaX3 salts or even surpass it. In the transition metals series, the replacement of simple neutral salts (MXn) by well-defined cationic organometallic complexes ([LnM]+[Y]−, L = organoligand, [Y]− = weakly coordinating anion) is a well-known strategy to increase the activity and the stability of a catalyst. This approach has not as yet imposed itself for main group metals. Cationic organometallic complexes of the main group elements are recognized as exquisite polymerization precatalysts4 but they are often considered to be too acidic to be useful in molecular catalysis. Over the past 4 years, we have developed cationic complexes of gallium and indium and studied in detail their structural properties and their stability.5,6 For instance, the NHC adducts IPr·GaX31 (Scheme 1; X = Cl,7 Br, I; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) easily react with AgSbF6 to give [IPr·GaX2][SbF6] 2.5a,d The IPr·GaCl2+ ion can be stabilized as the nitrile adduct 3 using 2,4,6-trifluorobenzonitrile (TFBN).5a | ||
| Scheme 1 Synthesis and stabilization of IPr·GaX2+ ions (ref. 5a and d). | ||
A few catalytic reactions were carried out with 2 (generated in situ with a slight excess of AgSbF6) and 3, which revealed the potential of the cationic gallium complexes in organic synthesis.8 The IPr·GaX2+ ions proved to be remarkable π-acids able to imitate noble metals-based catalysts by promoting intra- and/or intermolecular hydroarylations of alkynes and alkenes (Scheme 2, eqn (1)), skeletal reorganization of enynes (eqn (2)),5a and transfer hydrogenation of alkenes (eqn (3)).9
 | ||
| Scheme 2 Previously reported hydroarylations, cycloisomerizations and transfer hydrogenations promoted by cationic gallium complexes (ref. 5a and 9). | ||
They turn over at lower temperatures than GaCl3 and can be used in lower loadings. Moreover, the experimental procedure is much more practical since, unlike GaCl3, the gallium complexes are easy to handle, especially IPr·GaX3, which is bench-stable. Of note, while other carbon-based ligands can be used, the IPr carbene has provided the best results.5a The choice of the counterion is also of critical importance. The best results have been obtained with SbF6− because it can be easily exchanged by the substrate in the coordination sphere of the metal and is robust enough towards fluorine abstraction by Ga+.5d
In this full account, we report the fruits of our exploration of more reactions that can be promoted by cationic gallium complexes of type [IPr·GaX2][SbF6] in order to show their versatility as σ- or π-Lewis acids for homogeneous molecular catalysis. The reversibility of some of the transformations studied pertaining to Friedel–Crafts alkenylation and alkylation could be exploited in a rare type of dynamic covalent chemistry.
2. Results and discussion
2.1 Hydration of alkynes and alkenes
Examples of C–O bond formation from alkynes and alkenes are summarized in Scheme 3. The addition of water to alkynes to give ketones or aldehydes is a fundamental process in organic chemistry. It was first reported by Kutscheroff in 188110 and it established itself in spite of the use of Hg(II) salts and strong Brønsted acids. The past years have witnessed the development of catalytic versions based on the use of transition metal complexes,11 among which the IPr·AuCl/AgSbF6 catalytic system proved particularly efficient.12 With the IPr·GaCl3/AgSbF6 catalytic system, the hydration of phenylacetylene into acetophenone took place in wet 1,2-dichloroethane (DCE)‡ at 80 °C, yet in a low 25% yield. Interestingly, the yield could be raised up to 63% by using IPr·GaBr3. On the other hand, with IPr·GaI3, acetophenone was isolated in 41% yield only. The difference in the yields cannot be explained by the rate of halide abstraction. In each case, the cationic complexes were pre-formed by mixing the silver salt and the gallium complex for 5 min prior to adding the substrate. This period of time proved sufficient to reach full conversion of IPr·GaX3 into [IPr·GaX2][SbF6] with X = Cl, Br, or I. We attribute the halide effect to the stability of [IPr·GaX2][SbF6] in the presence of water. The hydrolysis of [IPr·GaCl2][SbF6] into the catalytically inactive complex [IPr·Ga(OH)2(H2O)2][SbF6] has already been described.5a This deactivation process might be slower with larger halides such as Br or I. The lower yield with X = I compared to X = Br could be due to the steric hindrance brought about by the iodine ligands. A strong dependence on the nature of the halide ligands was also found in the reaction of the diyne 12 under the same experimental conditions. Enone 14 was isolated in 50% yield with X = Cl, 79% yield with X = Br, and decomposition occurred with X = I. We suppose that this transformation involves the hydration of the two alkyne units followed by aldol condensation through 13.13,14 | ||
| Scheme 3 Activation of alkynes and alkenes towards C–O bond formation (adecomposition). | ||
In our initial work on GaCl3-catalyzed hydroarylation of arenynes,2b we noted that diol 15 does not lead to the expected product after C–C bond formation but to the cyclic enol ether 16 after addition of one OH group to the alkyne functionality. This compound proved too unstable to be isolated but it could be characterized by NMR. Its trapping with anisole allowed the isolation of 17 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers. The selectivity conferred by the gallium salt is different from that reported with gold complexes, which normally trigger the addition of the two OH groups onto the alkyne to give a bicyclic ketal.15 The use of the IPr·GaCl3/AgSbF6 catalytic mixture gave the same result as GaCl3 with a fairly improved yield of 90% (vs. 82%). The use of 3 as precatalyst also led to the same product, in 77% yield.
:1 mixture of diastereomers. The selectivity conferred by the gallium salt is different from that reported with gold complexes, which normally trigger the addition of the two OH groups onto the alkyne to give a bicyclic ketal.15 The use of the IPr·GaCl3/AgSbF6 catalytic mixture gave the same result as GaCl3 with a fairly improved yield of 90% (vs. 82%). The use of 3 as precatalyst also led to the same product, in 77% yield.
The gem-diester tethered arene-ene 18 did not react under dry conditions. In wet DCE, lactone 20 was obtained in 64% yield when submitted to the IPr·GaCl3/AgSbF6 catalytic mixture, likely through ketal 19. This kind of dealkylative lactonization has been reported as side reaction of Lewis acid-catalyzed cycloisomerizations.16 Excess of TfOH has also been used to promote such transformations.17 With precatalyst 3, lactone 20 was obtained in a low 14% yield.
2.2 Activation of alcohols
 | ||
| Scheme 4 Activation of alcohols towards C–C bond formation. | ||
The gallium ions behaved just like FeCl3 in the cyclization of 23 into allene 24 at rt.21 Raising up the temperature to 80 °C allowed to monitor the decumulation of the allene into the 1,3-diene 25. After 16 h, the latter was fully converted into the spiro compound 26 arising from an intramolecular Friedel–Crafts reaction. The isolated yield of 99% is not markedly higher than the one reported with 5 mol% FeCl3·6H2O at 80 °C in MeNO2 (90%), yet the fact that the Ga(III) complex is not an oxidizing agent contrary to Fe(III) makes it an appealing alternative.22
26 and B(C6F5)3,27 which show a good activity for the reduction of alcohols with Et3SiH or Ph2SiHCl. Table 1 shows that IPr·GaCl2+ ions are also very efficient, even with a low 0.5 mol% loading, which is much lower than the previously reported ones. We have used either the two-component approach with gallium and silver (method A) or simply the [IPr·GaCl2(TFBN)][SbF6] complex (method B). The two silanes Et3SiH and Ph2SiHCl were employed. While the gallium complexes showed similar catalytic activities, a strong dependence on the nature of the silane was found in the case of benzhydrol 34 (entries 8–11), Ph2SiHCl being far more efficient than Et3SiH. In other cases, both silanes could be used.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yielda (%) |
| a Isolated yield unless otherwise noted. b Et3SiH. c Ph2SiHCl. d Reaction performed in CD2Cl2, conversion determined by 1H NMR using mesitylene as internal standard (0.34 equiv.). e 20 h. f With 5 mol% [Ga] and 7 mol% [Ag] at 80 °C. g Conversion determined by 1H NMR using anisaldehyde as internal standard (1 equiv.). | |||
| 1 |

|

|
64 (A)b |
| 2d | 45 (A)c | ||
| 3d | 50 (B)b | ||
| 4d | 50 (B)c | ||
| 5 | 28, Ar = p-(MeO)Ph | 29 | 90 (B)b |
| 6 | 30, Ar = p-ClPh | 31 | 53 (B)b |
| 7 |

|
33 | 90 (B)b |
| 8 |

|

|
14 (A)b |
| 9d | 95 (A)c | ||
| 10d | 15 (B)b | ||
| 11d | 95 (B)c | ||
| 12 |
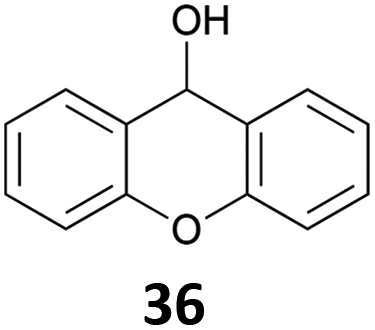
|

|
85 (B)b |
| 13 |

|

|
95 (B)c |
| 14 |

|

|
91 (B)c |
| 15e |

|

|
87 (A)b |
| 62 (B)b | |||
| 16f,g |

|

|
99 (A)b |

The reduction of propargyl, benzyl, and allyl alcohols was achieved in high yields at rt in 1 h, except for podophyllotoxin 42 into deoxypodophyllotoxin 43, a promising therapeutic agent,28 for which the reaction took 20 h. The reduction of 1-adamantanol required heating to 80 °C and a higher gallium loading of 5 mol% to reach completion.
2.3 Hydroarylation of alkynes and alkenes
:1 ratio of the regioisomers was measured by GC analysis. After 24 h at 40 °C, the ratio became 15:1, which shows that 50 can rearrange into the less sterically crowded para isomer 49. Very similar results were obtained at 80 °C. In the presence of anisole, the regioisomeric adducts 51 and 52 were obtained in a 1:1 ratio after 2 h at 40 °C. This suggests that 50 is more reactive than 49, which is corroborated by the 49:50 ratio of 71:1 instead of 6:1 without anisole. After 24 h, 51 is the major adduct (51:52 = 2.7:1). The fact that 50 is now preponderant over 49 (49:50 = 1:2) indicates that 52 can easily loose anisole. At 80 °C, no trace of 50 could be detected after 2 h, and the 51:52 ratio was 5.2:1. When full conversion was reached after 24 h, the 51:52 ratio changed to 28.3:1.
 | ||
| Scheme 5 Reversibility of the hydroarylation of compound 48 (aratio and conversion determined by GC analysis, b49 isolated in 92% yield, c51 isolated in 62% yield). | ||
With a deactivating chlorine atom instead of a methoxy group (53), the para isomer 54 was found to be even more preponderant than in the previous case (20:1 instead of 6:1), and the ortho anisole adduct 57 could not be detected (Scheme 6).
 | ||
| Scheme 6 Reversibility of the hydroarylation of compound 53 (aratio and conversion determined by GC analysis, b54 isolated in 84% yield, c56 isolated in 61% yield). | ||
The reversibility of the adducts could be demonstrated experimentally using compound 7 (Scheme 7). This product was obtained from arenyne 4 and anisole in 99% yield. To reach this yield, a three-fold excess of anisole had to be used. When 7 was isolated and subjected to the catalytic conditions, partial loss of anisole to give 6 could be ascertained. We decided to check whether the anisole moiety in 7 could be replaced by another nucleophile in excess. The Friedel–Crafts alkylation has been rarely exploited in dynamic covalent chemistry.30,31 However, with our catalytic system, such an exchange is actually possible. The indole adduct 59 could be formed either from arenyne 4 and the deactivated indole derivative 58 used in excess, or directly from 7 after substitution of anisole by 58, again used in excess.
 | ||
| Scheme 7 Reversibility of the hydroarylation of compound 6. | ||
Another potential application of dynamic Friedel–Crafts chemistry is shown in Scheme 8. As mentioned in the Introduction, the reaction of enyne 8 with a three-fold excess of anisole yields the adduct 9 (eqn (1), 3:1 regioisomeric mixture as far as the double bond is concerned). The putative intermediate for this transformation is the diene 61, which would arise from the cycloisomerization of 8. Attempt to isolate it or to trap it with 1,4-cyclohexadiene to afford alkene 60 after transfer hydrogenation failed (eqn (2)). However, the reduction of 9 worked well (eqn (3)). It should involve the elimination of anisole to give 61 and then transfer hydrogenation.
 | ||
| Scheme 8 Two-step formal transfer hydrogenative cyclization of enyne 8. | ||
 | ||
| Scheme 9 Hydroarylation of methyl-substituted arenynes (afull conversion). | ||
To the best of our knowledge, only one example of Friedel–Crafts alkylation involving an ipso attack and a 1,2-alkyl shift has already been reported.32 We decided to get a mechanistic insight into this intriguing process by means of DFT computations. Calculations were carried out using the Gaussian 09 software package at the M06-2X/6-311+G(d,p) level.33 Solvent correction was obtained by the CPCM method at the same level of theory. The values discussed are ΔG298 in kcal mol−1. To save computer time, GaCl3 was used as model complex instead of [IPr·GaCl2]+ which is too large to be treated at the high level of theory used. The relevance of this simplification has been shown elsewhere.2a Nevertheless, the key step of this study, calculated with GaCl3, has been recomputed with the model carbene 1,3-dimethylimidazol-2-ylidene, named NHC below. Scheme 10 shows the working hypothesis for the formation of products 66 and 67 either from 65 or 68. In this simple view, the gallium complex would react with 65 to give the alkyne complex A. This would trigger the nucleophilic attack of the arene moiety to give the Wheland intermediate B. A 1,3-proton shift would lead to C in which the aromaticity is restored. Dissociation of [Ga]+ from this σ complex would furnish 66 and then 67 after isomerization of the double bond. As for 68, its reaction with [Ga]+ would give the alkyne complex D, which would transform into the Wheland-type complex E after ipso attack. A 1,2-methyl shift would then take place to give B, which belongs to the previously described cycle.
 | ||
| Scheme 10 Mechanistic proposal accounting for the formation of 66 and 67 from 65 and 68. | ||
We anticipated that the calculations would actually point to a more complex scenario, notably because the 1,3 H-shift from B to C is symmetry forbidden. Our previous calculations on the gallium-catalyzed hydroarylation of arenyne 4 revealed that this shift can be modelled as a collapsing sequence of two 1,2-H shifts through a single transition state.2a In any case, an even more energetically favourable pathway involving the esters as proton shuttle was finally found (Scheme 11). After the formation of the Wheland intermediate G from F, a low-lying transition state corresponding to the abstraction of the arene proton by an ester group was located. It leads to H, which is able to transfer that proton to the carbon atom bearing the gallium atom to give I.
 | ||
| Scheme 11 Summary of our previous M06-2X/6-311+G(d,p) calculations of the cyclization of arenyne 4 (ref. 2a). | ||
For this new set of calculations, the trimethyl-substituted complexes K and P, corresponding to arenynes 65 and 68 respectively, were considered (Scheme 12). From the 2,4,5-isomer K, a transition state corresponding to the 6-exo-dig cyclization was located at 13.6 kcal mol−1 on the PES. The cyclized Wheland intermediate L has virtually the same energy as K. A direct pathway from L to O by 1,3-H shift could not be modelled. Moreover, presumably due to the steric hindrance brought about by the methyl groups, it was not possible to catch the arene proton with an ester group from L directly. Manifold efforts to do so led to a 1,2-H shift transition state connecting L to M. This transition state lies only 2.1 kcal mol−1 above L. Complex M is appreciably more stable than L by 6.4 kcal mol−1. Protonation of one ester moiety became computationally feasible through a transition state lying 9.6 kcal mol−1 above M. The resulting complex N is formed exergonically (−10.8 kcal mol−1 from M) and it could be connected to the final complex Ovia an oxygen-to-carbon proton transfer. This step requires 9.3 kcal mol−1 of free energy of activation and is strongly exergonic by 22.9 kcal mol−1.
 | ||
| Scheme 12 M06-2X/6-311+G(d,p) free energy profile (kcal mol−1) corresponding to the cyclization of arenynes 65 and 68; geometry of TSPQ and TSP′Q′ (selected distances in Å). | ||
As for the 2,4,6-isomer P, the ipso attack is significantly more demanding in free energy of activation than the aforementioned cyclization step (difference of 3.2 kcal mol−1 between TSPQ and TSKL). The resulting complex Q is more stable than P by 7.0 kcal mol−1. The surprize of this study is that the formation of Q involves a C–C bond formation between the arene and the alkyne and a 1,2-methyl shift through the same transition state. In other words, the expected complex is actually not a stationary point and it collapses directly to Q. Geometrically, the transition state between P and Q corresponds to a 1,2-methyl shift. The C–C bond between the arene and the alkyne is already formed (1.50 Å). A very similar transition state, TSP′Q′, was obtained with the (NHC)GaCl2+ moiety instead of GaCl3. It lies 10.1 kcal mol−1 over P′ and leads to Q′ which is more stable than P′ by 15.8 kcal mol−1. The lower barrier computed for (NHC)GaCl2+ compared to GaCl3 can be explained by the higher electrophilic character of the cationic complex. Going back to GaCl3 as model catalyst, complex Q could be connected to Lvia a 1,2-H shift transition state lying 11.6 kcal mol−1 above Q. This step is markedly endergonic but the overall process is funnelled by the downhill mechanism leading to O.
Although the fundamental role of the substrate backbone esters in metal-catalyzed cyclizations has already been discussed elsewhere,2a new elements further corroborated this hypothesis. First, while the reaction still works well with one ester group (Scheme 13, Z = CO2Et), only degradation took place with no functional group in the tether (Z = CH2). Second, no reaction took place (or only partial degradation of the substrate) with arenynes in which Z = O, NTs or (PhO2S)2C.
 | ||
| Scheme 13 Hydroarylation of compound 69. | ||
Another intriguing transformation potentially based on ipso attack was unveiled when using arenyne 75 (Scheme 14). Treatment with the cationic gallium complexes led to an intractable mixture of unidentified products. With the softer Lewis acid GaCl3, the unusual decadiene 76 was isolated in low yield from the complex mixture. Although this product could not be obtained in a preparative manner, it displays a fascinating structure which was unambiguously confirmed by single crystal X-ray analysis (Fig. 1).§
 | ||
| Scheme 14 Dimerization of compound 75. | ||
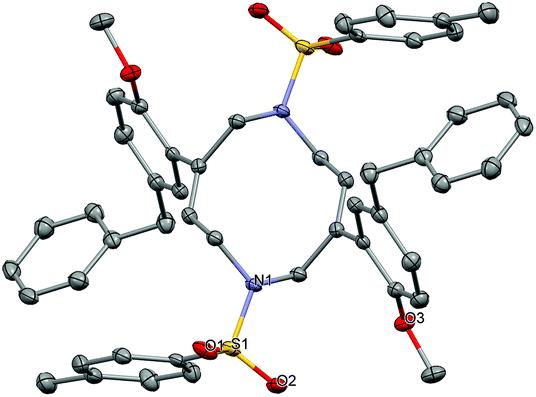 | ||
| Fig. 1 Ortep view of decadiene 76 (thermal ellipsoids at 50% probability, hydrogen atoms omitted for clarity). | ||
We propose that this dimer is formed after ipso attack of the arene moiety to the complexed alkyne fragment of R. Unlike the preceding case, this attack would take place in a 5-exo-dig fashion because the nucleophilicity of the tethered arene carbon is boosted by the ortho methoxy group. This would give rise to a spiro derivative S, which cleavage would yield iminium T. Since this intermediate also displays a nucleophilic vinyl gallium moiety, two units may react in a head-to-tail fashion to give the decadiene scaffold.
The dimerization of the naphthalene-derived arenyne 77 was also observed, yet this time in high yields (Scheme 15). The spiro compound 78 was obtained in up to 99% yield as a single diastereomer using [IPr·GaCl2(TFBN)][SbF6] as precatalyst. The connectivity of the atoms could be ascertained by X-ray analysis, yet the low quality of the crystals did not allow the complete refinement of the structure (Fig. 2). It seems that the last complex of the catalytic cycle U reacted with expected product V to give W. Attack of the naphthalene moiety to the carbocationic center would lead to the spiro σ-intermediate X, which finally undergoes deprotonation and subsequent protodemetallation.
 | ||
| Scheme 15 Dimerization of compound 77. | ||
 | ||
| Fig. 2 Not refined X-ray structure of compound 78. | ||
 | ||
| Scheme 16 Dihydroarylations of arenynes with anisole, 1,2-dimethoxybenzene and 1-(phenylsulfonyl)indole. | ||
Up to 4 C–C bonds were created in a single operational step from compound 86 (Scheme 17). This substrate converted into the expected tricyclic product 87 using the cationic gallium complexes (eqn (1)). With anisole, 86 transformed into the tetrahydroarylation product 88, isolated in 64% yield as a 3:1 dl:meso mixture (eqn (2)).
 | ||
| Scheme 17 Di- and tetrahydroarylation of compound 86 (E = CO2Me; adetermined by HPLC with CD detector). | ||
3. Conclusions
Through this work, we have shown that cationic complexes of gallium can be useful in homogeneous molecular catalysis. They are both oxophilic Lewis acids able to abstract OH groups, and carbophilic Lewis acids able to activate alkynes or alkenes. Since they are easily formed in situ from bench-stable precursors, they are much more practical to use than GaX3 salts. In spite of their putative highly electron-deficient nature, they do not promote extensive polymerization of the substrates or reaction products, even polyunsaturated ones. Among the reactions studied, those related to Friedel–Crafts alkenylation and alkylation showed an interesting dynamic behavior in C–C bond formation and cleavage. Dynamic covalent chemistry is intensively used in various research fields but the Friedel–Crafts alkylation, which is known as “faithfully” reversible compared to acylation,34 has been virtually ignored so far in this domain. These findings have stimulated further research in our group on this aspect.4. Experimental section
All reactions were performed in oven-dried flasks under argon. Unless otherwise stated, commercially available reagents were used as received without further purification. Gallium(III) chloride, bromide and iodide were obtained from Alfa Aesar and used as received. 1,4-Cyclohexadiene was obtained from Sigma-Aldrich and used as received. Anisole was distilled prior to use. 1,2-Dichloroethane was distilled from calcium(II) hydride and degassed by freeze–pump–thaw technique. Wet DCE was prepared by shaking DCE with water in a separatory funnel followed by collection of the organic layer. Unless otherwise stated, products were purified by chromatography over silica gel. TLC plates were visualized by UV light (254 nm) and p-anisaldehyde staining. GC analysis was performed on a Varian 430-GC Instrument, equipped with an autoinjector (CP8410) using a Zebron ZB-1MS or ZB-1 capillary GC column (15 m × 0.25 mm × 0.25 μm). dl:meso ratio was determined by HPLC equipped with a Circular Dichroism detector (PU-2089 JASCO, CD-2095 JASCO) using a chiral column (chiralpak IC, 250 × 4.6 mm). NMR spectra were recorded on AM250, AV300 or AV360 MHz Bruker spectrometers. Chemical shifts are reported in parts per million (ppm). The spectra were referenced to the residual 1H and 13C signals of the solvents as follows: CDCl3 (1H, δ = 7.27 ppm; 13C, δ = 77 ppm), CD2Cl2 (1H, δ = 5.32 ppm). Data are given in the following order: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant (J) in Hz and integration. Infrared spectra were recorded on a FTIR spectrometer (Perkin-Elmer spectrum one, NaCl pellets or Bruker Vertex 70 ATR Pike Germanium) and are reported in cm−1. HRMS were performed on MicrOTOFq Bruker spectrometer by electrospray ionization. MS were recorded on DSQ Thermo Fisher instrument by electronic impact. IPr·GaX31 (X = Cl, Br, I) and [IPr·GaCl2(TFBN)][SbF6] 3 were synthesized following a reported procedure.5a,b,7 The following compounds were previously described: 4,96,97,5a8,359,5a12,3615,2b17,2b18,3722,3828,3930,3932,3946,948,4059,5a62,965,971,4172,4277,986.9 Characterization data of the new compounds 20, 23–26, 47, 49, 50–54, 56, 60, 63, 64, 66–69, 73–76, 78–85, 87, and 88 are provided in the ESI.†
4.1 C–O bond formation from alkynes and alkenes (Scheme 3)
General procedure: IPr·GaX3 (X = Cl, Br or I, 5 mol%, 0.0125 mmol) and AgSbF6 (7 mol%, 6 mg, 0.0175 mmol) were mixed in a screw-cap vial which was evacuated and filled with argon. Wet DCE (0.5 mL) was added and the mixture was stirred at room temperature for 5 min. A solution of phenylacetylene, 12, or 18 (1 equiv., 0.25 mmol) diluted in wet DCE (0.5 mL) was added and the mixture was stirred at 80 °C for 20 h. The mixture was then filtered on a short pad of silica gel, which was rinsed with DCM (5 mL). Unless otherwise stated, solvents were removed under reduced pressure and the yield was determined by 1H NMR analysis after addition of p-anisaldehyde (1 equiv., 30 μL) as internal standard.4.2 C–C bond formation from alcohols (Scheme 4)
A [IPr·GaCl2][SbF6] solution was prepared by adding IPr·GaCl3 (5 mol%, 7 mg, 0.0125 mmol) and AgSbF6 (7 mol%, 6 mg, 0.0175 mmol) to DCE (1 mL) in a screw-cap vial under argon. In a separated tube, anisole (3 equiv., 81.1 mg, 0.0815 mL, 0.75 mmol) was added to a solution of 21 (1 equiv., 52.1 mg, 0.25 mmol) in DCE (0.9 mL). 0.1 mL of the [IPr·GaCl2][SbF6] solution previously prepared was then added and the mixture was stirred at room temperature for 1 h. The reaction was monitored by TLC. After filtration on a Celite pad and evaporation of the volatiles, the crude product was purified by chromatography over silica gel (cyclohexane) to afford compound 22 (80%, 60 mg, 0.201 mmol).4.3 Cyclization of compound 23 (Scheme 4)
A [IPr·GaCl2][SbF6] solution was prepared by adding IPr·GaCl3 (5 mol%, 7 mg, 0.0125 mmol) and AgSbF6 (7 mol%, 6 mg, 0.0175 mmol) to DCE (1 mL) in a screw-cap vial under argon. In a separated tube, compound 23 (1 equiv., 111 mg, 0.25 mmol) was dissolved in DCE (0.9 mL). 0.1 mL of the [IPr·GaCl2][SbF6] solution previously prepared was then added and the mixture was stirred at the indicated temperature for 1–16 h. The mixture was then filtered on a short pad of silica gel, which was rinsed with DCM (5 mL). After evaporation, the crude product was purified by flash chromatography over silica gel (cyclohexane, EtOAc).4.4 C–H bond formation from alcohols (Table 1)
4.5 (Di)hydroarylation of arenynes (Schemes 5–7, 9, 13–17)
General procedure: IPr·GaCl3 (5 mol%, 7 mg, 0.0125 mmol) and AgSbF6 (7 mol%, 6 mg, 0.0175 mmol) were mixed in a screw-cap vial under argon. DCE (0.5 mL) was added and the mixture was stirred at room temperature for 5 min. Arenyne 4, 46, 48, 53, 62, 65, 68–73, 77 or 86 (1 equiv., 0.25 mmol) and the corresponding aromatic nucleophile (0 or 3 equiv., 0.75 mmol) diluted in DCE (0.5 mL) were added and the mixture was stirred at the indicated temperature until completion of the reaction. The mixture was then filtered on a short pad of silica gel, which was rinsed with DCM (5 mL). After evaporation, the crude product was purified by flash chromatography over silica gel (cyclohexane, EtOAc).4.6 Two-step formal transfer hydrogenative cyclization of enyne 8 (Scheme 8)
IPr·GaCl3 (5 mol%, 7 mg, 0.0125 mmol) and AgSbF6 (7 mol%, 6 mg, 0.0175 mmol) were mixed in a screw-cap vial under argon. DCE (0.5 mL) was added and the mixture was stirred at room temperature for 5 min. A solution of the Friedel–Crafts adduct 9 (1 equiv., 0.25 mmol) and 1,4-cyclohexadiene (1.2 equiv., 29 μL, 0.30 mmol) in DCE (0.5 mL) was added and the mixture was stirred at 80 °C for 2 h. The crude mixture was then filtered through a pad of Celite and the volatiles were removed under reduced pressure. The crude product was purified by chromatography over silica gel (cyclohexane, EtOAc) to afford compound 60 as an equimolar mixture of regioisomers (74%, 42 mg, 0.19 mmol).Acknowledgements
This work was supported by UPS, MESR, IUF, and CNRS. We used the computing facility of the CRIAAN, Centre Régional Informatique et d'Applications Numériques de Normandie (project 2006-013). We thank Dr. Christophe Meyer (ESPCI) and Prof. Ognjen Miljanic (University of Houston) for useful discussions.Notes and references
- (a) M. Yamaguchi, S. Matsunaga, M. Shibasaki, B. Michelet, C. Bour and V. Gandon, e-EROS Encycl. Reagents Org. Synth., 2014, 1 CrossRef CAS; (b) M. K. Gupta and T. P. O'Sullivan, RSC Adv., 2013, 3, 25498 RSC; (c) M. Yamaguchi and Y. Nishimura, Chem. Commun., 2008, 35 RSC; (d) R. Amemiya and M. Yamaguchi, in Acid Catalysis in Modern Organic Synthesis, ed. H. Yamamoto and K. Ishihara, Wiley-VCH, Weinheim, 2008, pp. 347–375 Search PubMed; (e) R. Amemiya and M. Yamaguchi, Eur. J. Org. Chem., 2005, 5145 CrossRef CAS; (f) M. Yamaguchi, in Main Group Metals in Organic Synthesis, ed. H. Yamamoto, K. Oshima, Wiley-VCH, Weinheim, 2004, pp. 307–322 Search PubMed; (g) D. C. Barman, Synlett, 2003, 2440 CrossRef CAS.
- (a) B. Michelet, G. Thiery, C. Bour and V. Gandon, J. Org. Chem., 2015, 80, 10925 CrossRef CAS PubMed; (b) H. Li, R. Guillot and V. Gandon, J. Org. Chem., 2010, 75, 8435 CrossRef CAS PubMed.
- Y. Yamamoto, J. Org. Chem., 2007, 72, 7817 CrossRef CAS PubMed.
- (a) Y. Sarazin and J.-F. Carpentier, Chem. Rev., 2015, 115, 3564 CrossRef CAS PubMed; (b) M. R. Lichtenthaler, S. Maurer, R. J. Mangan, F. Stahl, F. Mönkemeyer, J. Hamann and I. Krossing, Chem. – Eur. J., 2015, 21, 157 CrossRef PubMed; (c) M. R. Lichtenthaler, A. Higelin, A. Kraft, S. Hughes, A. Steffani, D. A. Plattner, J. M. Slattery and I. Krossing, Organometallics, 2013, 32, 6725 CrossRef CAS; (d) S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869 CrossRef CAS; (e) S. Dagorne and C. Fliedel, Top. Organomet. Chem., 2013, 41, 125 CrossRef CAS; (f) S. Dagorne and D. A. Atwood, Chem. Rev., 2008, 108, 4037 CrossRef CAS PubMed; (g) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS.
- (a) S. Tang, J. Monot, A. El-Hellani, B. Michelet, R. Guillot, C. Bour and V. Gandon, Chem. – Eur. J., 2012, 18, 10239 CrossRef CAS PubMed; (b) A. El-Hellani, J. Monot, S. Tang, R. Guillot, C. Bour and V. Gandon, Inorg. Chem., 2013, 52, 11493 CrossRef CAS PubMed; (c) A. Hellani, J. Monot, R. Guillot, C. Bour and V. Gandon, Inorg. Chem., 2013, 52, 506 CrossRef PubMed; (d) C. Bour, J. Monot, S. Tang, R. Guillot, J. Farjon and V. Gandon, Organometallics, 2014, 33, 594–599 CrossRef CAS; (e) B. Michelet, J.-R. Colard-Itté, G. Thiery, R. Guillot, C. Bour and V. Gandon, Chem. Commun., 2015, 51, 7401 RSC; (f) C. Bour, R. Guillot and V. Gandon, Chem. – Eur. J., 2015, 21, 6066 CrossRef CAS PubMed.
- C. Bour and V. Gandon, Coord. Chem. Rev., 2014, 279, 43 CrossRef CAS.
- N. Marion, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, L. Fensterbank, M. Malacria and S. P. Nolan, Organometallics, 2007, 26, 3256 CrossRef CAS.
- C. Bour and V. Gandon, Synlett Account, 2015, 26, 1427 CrossRef CAS.
- B. Michelet, C. Bour and V. Gandon, Chem. – Eur. J., 2014, 20, 14488 CrossRef CAS PubMed.
- (a) M. Kutscheroff, Chem. Ber., 1881, 14, 1540 CrossRef; (b) M. Kutscheroff, Chem. Ber., 1884, 17, 13 CrossRef.
- L. Hintermann and A. Labonne, Synthesis, 2007, 1121 CrossRef CAS.
- N. Marion, R. S. Ramón and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448 CrossRef CAS PubMed.
- For gold-catalyzed hydrative cyclization of 1,6-diynes, see: C. Zhang, D.-M. Cui, L.-Y. Yao, B.-S. Wang, Y.-Z. Hu and T. Hayashi, J. Org. Chem., 2008, 73, 7811 CrossRef CAS PubMed.
- For another example of IPr·GaCl2+-catalyzed aldol condensation, see: M. Presset, B. Michelet, R. Guillot, C. Bour, S. Bezzenine-Lafollée and V. Gandon, Chem. Commun., 2015, 51, 5318 RSC.
- S. Antoniotti, E. Genin, V. Michelet and J.-P. Genêt, J. Am. Chem. Soc., 2005, 127, 9976 CrossRef CAS PubMed.
- (a) I. J. S. Fairlamb, S. Grant, S. Tommasi, J. M. Lynam, M. Bandini, H. Dong, Z. Lin and A. C. Whitwood, Adv. Synth. Catal., 2006, 348, 2515 CrossRef CAS; (b) F. Grau, A. Heumann and E. Duñach, Angew. Chem., Int. Ed., 2006, 45, 7285 CrossRef CAS PubMed; (c) B. Cacciuttolo, S. Poulain-Martini and E. Duñach, Eur. J. Org. Chem., 2011, 3710 CrossRef CAS.
- M. Paz Muñoz and G. C. Lloyd-Jones, Eur. J. Org. Chem., 2009, 516 CrossRef.
- M. Niggemann and M. J. Meel, Angew. Chem., Int. Ed., 2010, 49, 3684 CrossRef CAS PubMed.
- C.-F. Xu, M. Xu, L.-Q. Yang and C.-Y. Li, J. Org. Chem., 2012, 77, 3010 CrossRef CAS PubMed.
- Y. Masuyama, M. Hayashi and N. Suzuki, Eur. J. Org. Chem., 2013, 2914 CrossRef CAS.
- W. Huang, P. Zheng, Z. Zhang, R. Liu, Z. Chen and X. Zhou, J. Org. Chem., 2008, 73, 6845 CrossRef CAS PubMed.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Grybo, Angew. Chem., Int. Ed., 2013, 52, 9900 CrossRef CAS PubMed.
- J. M. Herrmann and B. König, Eur. J. Org. Chem., 2013, 7017 CrossRef CAS.
- (a) T. Miyai, M. Ueba and A. Baba, Synlett, 1999, 182 CrossRef CAS; (b) M. Yasuda, Y. Onishi, M. Ueba, T. Miyai and A. Baba, J. Org. Chem., 2001, 66, 7741 CrossRef CAS PubMed; (c) A. Baba, M. Yasuda, Y. Nishimoto, T. Saito and Y. Onishi, Pure Appl. Chem., 2008, 80, 845 CrossRef CAS.
- V. J. Meyer and M. Niggemann, Chem. – Eur. J., 2012, 18, 4687 CrossRef CAS PubMed.
- G. G. K. S. Narayana Kumar and K. K. Laali, Org. Biomol. Chem., 2012, 10, 7347 CAS.
- (a) V. Gevorgyan, J.-X. Liu, M. Rubin, S. Benson and Y. Yamamoto, Tetrahedron Lett., 1999, 40, 8919 CrossRef CAS; (b) V. Gevorgyan, M. Rubin, S. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179 CrossRef CAS PubMed; (c) M. Denancé, M. Guyot and M. Samadi, Steroids, 2006, 71, 599 CrossRef PubMed.
- (a) M. Khaled, Z.-Z. Jiang and L.-Y. Zhang, J. Ethnopharmacol., 2013, 149, 24 CrossRef CAS PubMed; (b) H. Xu, M. Lv and X. Tian, Curr. Med. Chem., 2009, 16, 327 CrossRef CAS PubMed; (c) Y. J. You, Curr. Pharm. Des., 2005, 11, 1695 CrossRef CAS PubMed; (d) M. Gordaliza, P. A. Garcia, J. M. Miguel Del Corral, M. A. Castro and M. A. Gomez-Zurita, Toxicon, 2004, 44, 441 CrossRef CAS PubMed.
- For a seminal report on GaCl3-catalyzed hydroarylation of arenynes, see: (a) H. Inoue, N. Chatani and S. Murai, J. Org. Chem., 2002, 67, 1414 CrossRef CAS PubMed. See also: (b) S. Pascual, C. Bour, P. de Mendoza and A. M. Echavarren, Beilstein J. Org. Chem., 2011, 7, 1520 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634 RSC.
- M. Holler, N. Allenbach, J. Sonet and J. F. Nierengarten, Chem. Commun., 2012, 48, 2576 RSC.
- F. I. Zubkov, E. V. Nikitina, V. V. Kouznetsov and L. Dary Avellaneda Duarte, Eur. J. Org. Chem., 2004, 5064 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Mala'bi, S. Pogodin, S. Cohen and I. Agranat, RSC Adv., 2013, 3, 21797 RSC.
- J. W. Faller and P. P. Fontaine, J. Organomet. Chem., 2006, 691, 1912 CrossRef CAS.
- J. Mandal, S. K. Prasad, D. S. S. Rao and S. Ramakrishnan, J. Am. Chem. Soc., 2014, 136, 2538 CrossRef CAS PubMed.
- D. P. Curran and N. Fairweather, J. Org. Chem., 2003, 68, 2972 CrossRef CAS PubMed.
- J. J. Kennedy-Smith, L. A. Young and F. D. Toste, Org. Lett., 2004, 6, 1325 CrossRef CAS PubMed.
- V. J. Meyer and M. Niggemann, Chem. – Eur. J., 2012, 18, 4687 CrossRef CAS PubMed.
- N. Chatani, H. Inoue, T. Ikeda and S. Murai, J. Org. Chem., 2000, 65, 4913 CrossRef CAS PubMed.
- S. T. A. Shah, K. M. Khan, H. Hussain, M. U. Anwar, M. Fecker and W. Voelter, Tetrahedron, 2005, 61, 6652 CrossRef CAS.
- W. Huang, Q. Shen, J. Wang and X. Zhou, J. Org. Chem., 2008, 73, 1586 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental details, single crystal X-ray analyses, coordinates of the computed structures, copies of NMR spectra. CCDC 1483915 and 1483916. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00470a |
| ‡ DCE is humidified with water in a separatory funnel, collected and used as such. |
| § CCCDC 1483915 and 1483916 contains the supplementary crystallographic data for this paper. |
| This journal is © the Partner Organisations 2016 |