
Nanocrystalline iron oxide based electroactive materials in lithium ion batteries: the critical role of crystallite size, morphology, and electrode heterostructure on battery relevant electrochemistry
Andrea M.
Bruck†
a,
Christina A.
Cama†
a,
Cara N.
Gannett
bc,
Amy C.
Marschilok
*ad,
Esther S.
Takeuchi
*ade and
Kenneth J.
Takeuchi
*ad
aDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, USA. E-mail: amy.marschilok@stonybrook.edu; esther.takeuchi@stonybrook.edu; kenneth.takeuchi.1@stonybrook.edu
bDepartment of Chemistry, State University of New York at Geneseo, Geneseo, NY 14454, USA
cCenter for Inclusive Education, Stony Brook University, Stony Brook, NY 11794, USA
dDepartment of Materials Science and Engineering, Stony Brook University, Stony Brook, NY 11794, USA
eEnergy Sciences Directorate, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 8th December 2015
Abstract
The importance of crystallite size control and direct synthesis of materials with desirable properties is broadly applicable for the rational design and development of new active materials for energy storage. Recently, the use of nanoparticles and crystallite size control has redefined electrode design strategies, due in part to the large surface area/volume ratios providing more pathways for ion movement within the bulk electrode. This review is structured primarily as a case study, where reports involving a specific densely structured iron oxide, magnetite, Fe3O4, and its use as an electrode in LIBs are used as examples. Due to the high theoretical capacity (924 mA h g−1), and opportunity for implementation of a low cost electrode material, magnetite was selected as the model material for this review. Notably, crystallite size, morphology, and electrode heterostructure can all play a critical role in battery relevant electrochemistry, particularly for crystallographically dense materials such as Fe3O4. Several examples of Fe3O4 based composites are described, incorporating different types of conductive materials such as carbons as part of the structure. Additionally, this review also provides a brief introduction to a newer iron oxide based material with a 2D layered structure, silver ferrite, where crystallite size control was synthetically achieved. By focusing on two specific iron oxide based nanoscale inorganic materials, this review highlights and distinguishes the contributions of electroactive material crystallite size, morphology and electrode heterostructure to electrochemical behavior, facilitating the future development of next generation of battery electrodes.
 Andrea M. Bruck | Andrea Bruck obtained her BS in chemistry from Illinois State University as an Honors Program Scholar with Merit in 2014. She is currently a PhD candidate at Stony Brook University Department of Chemistry under the advisement of Dr Esther Takeuchi, Dr Kenneth Takeuchi, and Dr Amy Marschilok. Her research focuses on lifting the current limitations of Li-ion battery electrodes to better utilize renewable energy sources. In 2015 she was awarded a National Science Foundation Graduate Research Fellowship based on her potential to “strengthen the vitality of the US science and engineering enterprise”. |
 Christina A. Cama | Christina A. Cama received her Bachelor of Science in Chemistry from Hofstra University, graduating with highest honors (Summa Cum Laude). After her first year in the Chemistry Ph.D. program at Stony Brook University, she received a First Year Graduate Teaching Award in recognition of her efforts as a teaching assistant. Christina is pursuing her Ph.D. degree in Chemistry under the guidance of Dr Esther Takeuchi, Dr Kenneth Takeuchi, and Dr Amy Marschilok. Her research includes the synthesis and application of various inorganic materials and she actively participates in collaborations which focus on the complex reactions associated with lithium-ion batteries. |
 Cara N. Gannett | Cara Gannett began her undergraduate career at the State University of New York at Geneseo as an Edgar Fellow, and she is currently a third year undergraduate honors student, working towards earning a B.S. in Chemistry. During the summer of 2015, she conducted research with Professors Amy Marschilok, Esther Takeuchi, and Kenneth Takeuchi, through the Research Experience for Undergraduates (REU) for Health, Energy, and the Environment at Stony Brook University. Cara intends to pursue a Ph.D. in Chemistry after graduating from SUNY Geneseo. |
 Amy C. Marschilok | Amy Marschilok is a Research Professor in the Departments of Materials Science and Engineering and Chemistry at Stony Brook University. She received her B.A. in and Ph.D. in Chemistry from the University at Buffalo. She was previously employed at Greatbatch Inc., where she was recognized as a Visionary of the Year. She received the Leadership Award, Professional Service Category from the Western New York YWCA. She was also recognized as a Woman of Distinction by the Girl Scouts of Western New York. Her research involves synthesis, advanced characterization and electrochemical study of novel materials and systems for energy storage. |
 Esther S. Takeuchi | Esther Takeuchi, SUNY Distinguished Professor in the Departments of Chemistry and Materials Science and Engineering at Stony Brook University, holds a joint appointment at Brookhaven National Laboratory. She received a BA from the University of Pennsylvania and a PhD from Ohio State University. Prior to her academic career, she conducted battery research in industry, particularly for implantable applications. She is a member of National Academy of Engineering, National Inventors Hall of Fame and National Academy of Innovation and received the National Medal of Technology and Innovation. Her research focus is on understanding governing mechanisms related to energy storage. |
 Kenneth J. Takeuchi | Kenneth Takeuchi is a SUNY Distinguished Teaching Professor in the Department of Chemistry at Stony Brook University. Professor Takeuchi holds a BS from the University of Cincinnati and a PhD in chemistry from The Ohio State University. He is a Fellow of the American Chemical Society and the American Association for the Advancement of Science. Professor Takeuchi has received over 20 regional and national awards for excellence in teaching and mentoring. Professor Takeuchi's research involves the synthesis and utility of inorganic materials towards energy storage, with a recent focus on synthetic development for crystallite size control. |
1. Introduction
Non-renewable energy is being consumed at a high enough rate to warrant a modern strategy for managing the remaining energy stores. Towards this end, a key component of the efficient generation and use of energy is energy storage. While there are a variety of energy storage technologies, batteries are a promising solution for both stationary and mobile applications, because both the chemistries and the engineering of batteries can be adjusted to suit the application, and historically batteries have enabled a number of devices. However, critical applications such as electric grid re-design and totally electric vehicles await batteries which can fulfill both energy and safety requirements.Historically, aqueous-electrolyte batteries were prevalent as consumer batteries, but were limited to 1.5 V. Non-aqueous batteries were then introduced because they offered high voltages and larger capacities. Further improvement of the non-aqueous-electrolyte batteries led to the development of the lithium-ion battery (LIB) – first prototyped in 1986.1 Yoshino from the Asahi Kasei Corporation was the first to create a Li1−xCoO2/C cell,1 which employed a transition-metal oxide electrode, a carbonaceous electrode, and a non-aqueous lithium ion-based electrolyte. In 1991, the LIB was first commercialized by SONY; initially used for camcorders and cell phones, it featured twice the energy density of prior batteries which had a profound impact on the portable electronics market.1,2
Applications of LIBs have seen a dramatic growth because of high capacity, high discharge voltage, and cycle durability. The electrochemical performance of LIB systems is continually being optimized for consumer use with no indication of retracting as shown by Fig. 1. Research focuses on improvements to these batteries systems such as maximizing the power density by improving both the voltage and capacity. This capability is necessary to satisfy the current interest in hybrid and electric vehicles (EVs) which require a high power energy source to function. LIBs have shown the greatest promise for EVs but still require increased power and energy density to compete with current fossil fuel sources.3 Much of LIB science and technology research has focused on the individual components of the LIB, including the anode, cathode, electrolyte, and separator. Anode research has centered on graphite-based materials, metallic alloys, intermetallics, as well as Li metal, where each strategy is rooted in specific desirable material attributes.4,5 Cathode research has involved the synthesis and electrochemistry of new materials, many being metal oxides.6,7 This section provides a brief history of inorganic electroactive materials developments significant to the introduction of LIB, followed by a discussion of materials parameters which form the subsequent sections of this review. While many parameters can affect the performance of lithium battery active materials, such as the electrolyte composition,8–10 active material surface area,11 and parasitic reactions including active material dissolution12–14 and surface film formation,15 the bulk of this review is focused on the key contributions of electroactive material crystallite size, morphology and electrode heterostructure to electrochemical behavior.
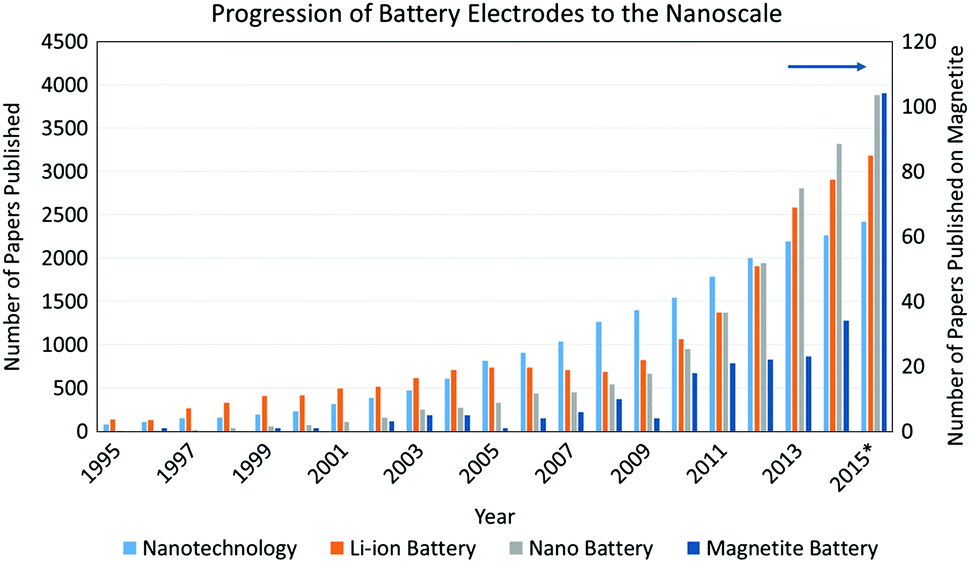 | ||
| Fig. 1 The number of papers published regarding nanotechnology, lithium ion batteries (LIBs), the integration of nanotechnology in LIBs, and magnetite's incorporation into LIBs. * Values for 2015 are projected based on values taken in June 2015. | ||
In general, LIB research has been inspired by the first Li1−xCoO2/C cell. Since its introduction by Goodenough and coworkers in 1980, LiCoO2 remains a commonly used cathode material in lithium ion batteries.16 With the desire to create a lower cost material while maintaining the success seen by LiCoO2, an interest in nickel based lithium oxides developed due to the isostructural properties of the analogous materials. A family of materials, Li[Ni1−xCox]O2, was first studied by Delmas and coworkers.17 Subsequently, a group of layered Li[Ni1−x−yCoxMny]O2 compounds demonstrated good capacity retention and high discharge rates.6,18
As LIB research progressed, metal oxides from several structural families were studied as LIB cathode materials, including spinel (LiM2O4) and olivine (LiMPO4), as well as other layered compounds (LiMO2). Recently, intercalation materials including silicates (Li2MSiO4), borates (LiMBO3), and tavorites (LiMPO4F) were studied as cathode materials.19 Concomitant with an expansion of the range of materials studied, concepts such as surface structure and variation in composition of materials have become increasingly important in battery research.20 Specifically, surface versus interior material differences for properties such as ion insertion and electrical conductivity may prove critical towards understanding fundamental battery chemistry and the rational development of new battery technologies.
At the turn of the 21st century, metal oxide based anodes were reported to produce more than twice the capacity of traditional carbon electrodes upon full utilization. At that time, LIB anodes contained carbon materials coupled with these metal oxides, with metal oxide crystallite sizes in the micrometer range. Impressive capacities were achieved through full reduction of the metal oxide with the formation of metal nanoparticles during the discharge process.21 After this discovery, research in batteries for both anodes and cathodes turned to uncovering the properties of nanoparticles relevant to electrical energy storage.
The use of nanoparticles redefined electrode design strategies, due in part to the large surface area/volume ratios providing more pathways for ion movement within the bulk electrode. Further, nanomaterials reduce the path length of ion movement within an individual crystal, thus, reducing the resistance to Li+ diffusion into the interior of the crystal of an electrode material,3 and a more facile lithium transport within the bulk electrode.20 This is a promising feature of nanomaterials in LIBs; facile discharging and charging results in greater power opportunities, shorter charging times, less heat generation and greater capacities under load-desirable properties for all battery applications, but especially for portable applications such as electric vehicles. Finally, by synthetically manipulating the morphology and porosity of the nanomaterials, the above effects can be mitigated or enhanced.
Combining two or more materials within an electrode to generate a composite electrode provides an opportunity to address multiple issues simultaneously. Composite electrodes can be prepared by a variety of strategies, including: (1) mixing the various components, (2) coating one material with another in a core–shell strategy, (3) layering materials using techniques such as chemical vapor deposition or atomic layer deposition, or (4) developing synthetic strategies where composite materials are formed in situ.22–25 Historically, an important goal for composite electrodes was enhanced electrical conductivity, attained by mixing electrically conductive carbon materials with less conductive electroactive materials. The sizes and morphologies of the electroactive particles and carbon particles can determine the optimal amount of carbon to be used to address electrical conductivity. From percolation theory, the carbon particles should be much smaller than the electroactive particles, to enhance electrical conductivity and facilitate Li+ transport. In practice, the size, morphology and phase of the carbon particles are variable, and variety of carbon particles have been used as additives. Notably, semiconducting and conducting polymers such as polypyrrole and polyaniline have shown to be effective in raising rate capabilities and therefore cathode performance by increasing electrical conductivity without significant decrease in lithium ion flow.7
This review is structured primarily as a case study, where reports involving a specific iron oxide, magnetite, Fe3O4, and its use as an electrode in LIBs are used as examples. As magnetite has a densely packed inverse spinel structure, the use of nanoscale materials and synthetic crystallite size control are particularly important. However, the importance of crystallite size control and direct synthesis of materials with desirable properties is broadly applicable. Therefore, this review also provides an introduction to a newer material with a 2D layered structure, silver ferrite, where crystallite size control was synthetically achieved. Further, the silver ferrite material is synthesized as a nanocomposite, consisting of silver ferrite and maghemite. Due to the newness of these materials, the battery-relevant literature is limited, thus the two introductions in this review represent new strategies towards future structure/function studies involving battery-relevant electrochemistry. In all cases, the role of crystallite size proved to significantly impact electrochemical behavior, a paradigm for the next generation of batteries.
2. Material properties and resulting electrochemistry of magnetite
Lithiation of magnetite
Magnetite is a naturally occurring mineral found in the earth's crust; therefore, it is abundant, environmentally friendly, cheap, and non-toxic. Magnetite is also easy to synthesize and can be prepared through simple, non-hazardous techniques such as through a facile aqueous co-precipitation method. Complete reduction of magnetite involves the transfer of 8 electrons, providing opportunity for an impressive capacity of 925 mA h g−1.26 Magnetite assumes a cubic inverse spinel structure with a space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The formula of magnetite can be written as Fe2+Fe3+2O4,27 where the unit cell contains eight Fe2+ cations in octahedral sites and sixteen Fe3+ cations, distributed evenly among the tetrahedral sites and the octahedral sites as shown in Fig. 2. The oxygen anions form layers parallel to 111 in a cubic close-packed arrangement.
m. The formula of magnetite can be written as Fe2+Fe3+2O4,27 where the unit cell contains eight Fe2+ cations in octahedral sites and sixteen Fe3+ cations, distributed evenly among the tetrahedral sites and the octahedral sites as shown in Fig. 2. The oxygen anions form layers parallel to 111 in a cubic close-packed arrangement.
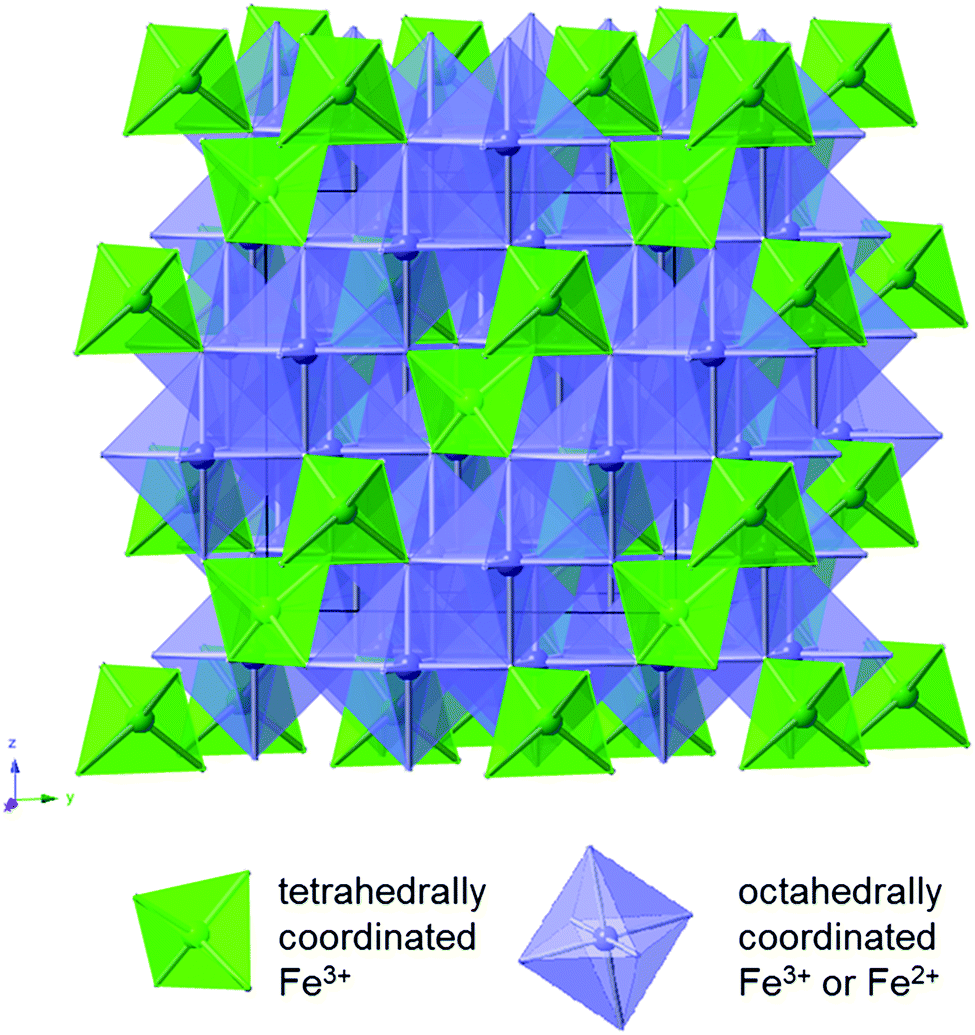 | ||
| Fig. 2 Crystal structure of magnetite (Fe3O4). | ||
Lithiation of magnetite first forms LixFe3O4, where x = 2.28 Upon further lithiation, LixFe3O4 is formed, where x = 2–5. However, in this step, Thackeray and Goodenough first proposed that the iron cations in the tetrahedral sites of Fe3O4 are displaced by lithium ions in a cooperative displacement reaction,29 where a rock salt-type structure is formed with a lattice constant of 8.47 Å.30 The iron ions move into octahedral sites during this displacement.28 As the reduction progresses, Li2O becomes displaced from the lattice and Fe metal is formed which has a body-centered cubic structure. The structural changes that occur when the rock salt is changed to iron metal have been proposed to be at least partially reversible.30 Notably, the initial reactions are insertion reactions where the lithium ion can insert into the Fe3O4 structure with minimal structural distortion, while upon further reduction, a conversion reaction occurs generating Fe metal and Li2O as products of full discharge.
Notably, achieving full conversion and complete utilization of the electroactive metal centers can be challenging for crystallographically dense materials such as Fe3O4. These densely structured materials do not have well defined layers or tunnels for facile lithium ion insertion. Conceptually, it can be envisioned that electrochemical lithiation would proceed from the surface to the interior, which could be visualized as a shrinking core of magnetite surrounded by reduced or partially reduced surface.31 This could affect the functional capacity of these systems due to polarization resulting from kinetic limitations such as slow lithium ion diffusion.32 These systems in particular would significantly benefit from nanosized materials with increased surface area to volume ratios, as discussed in the next section.
Crystallite size effects on magnetite's electrochemical performance
In 2008, Komaba et al. compared the electrochemical performance of magnetite particles that were 400, 100 and 10 nm in size. There was no significant reactivity observed in the 400 nm systems; however, the 100 and 10 nm systems reached initial discharge capacities of 70 mA h g−1 and 130 mA h g−1 after discharging to 1.5 V at 20 mA g−1, respectively. After 30 cycles, the capacity still delivered 96 mA h g−1 which was attributed to the improved performance of the nano-sized sample.33The nano-size effect describes the change in properties of nanoscale materials from their corresponding bulk form. As the size of a crystallite decreases, the surface area increases, thus increasing the surface area-to-volume ratio of the solid. The small size can also improve kinetics since Li+ ion diffusion pathways can take place at smaller distances in nanoscale materials. Thus, decreasing the crystallite size can enhance capacity retention and coulombic efficiency. Despite all of these advantages, working on the nanoscale does have several challenges. For example, due to the large surface area, the nanocrystals can experience side reactions with the electrolyte. These side reactions often cause large irreversible capacity loss. Nanocrystals also have high surface energy due to the incomplete bonding at the surface. To accommodate the high surface energy, nanocrystals tend to aggregate. Aggregation can be detrimental to battery performance as it disrupts the conductive pathways within the cathode, thus causing poor efficiency and capacity retention. It is the aim of battery researchers to overcome these challenges associated with nanomaterials to enhance battery performance.
Various studies in the Takeuchi group have evaluated the Li/Fe3O4 system and have studied the nano-size effect at over a smaller size range and lower voltages than the Komaba studies.33,34 Magnetite particles from 6 to 11 nm were successfully synthesized through a novel co-precipitation technique that varied concentration of the starting materials using triethylamine as a reagent.26,35–38 The Li/Fe3O4 half cells were discharged at constant current between 3.0 and 1.0 V. As crystallite size decreased, the discharge capacity increased by 30–200%.35
A closer look at the discharge profiles of different crystallite sizes of magnetite indicates significant differences in the smoothness of the voltage curves with varying crystallite size, as shown in Fig. 3. Discharge slopes are smoother for the smaller crystallite sizes; however, the larger crystallite size sample exhibits a distinct plateau. Depending on the application, a flat voltage profile with a distinguishable plateau providing more constant energy delivery may be more desirable. The discharge profile is influenced by the large quantity of surface defects present in the nanocrystalline magnetite. These surface defects cause a reduction in the band gap; thus, as Li+ ions are inserting into the magnetite structure, the Fermi energy is gradually changing, rather than abruptly changing as it would in the bulk sample.39
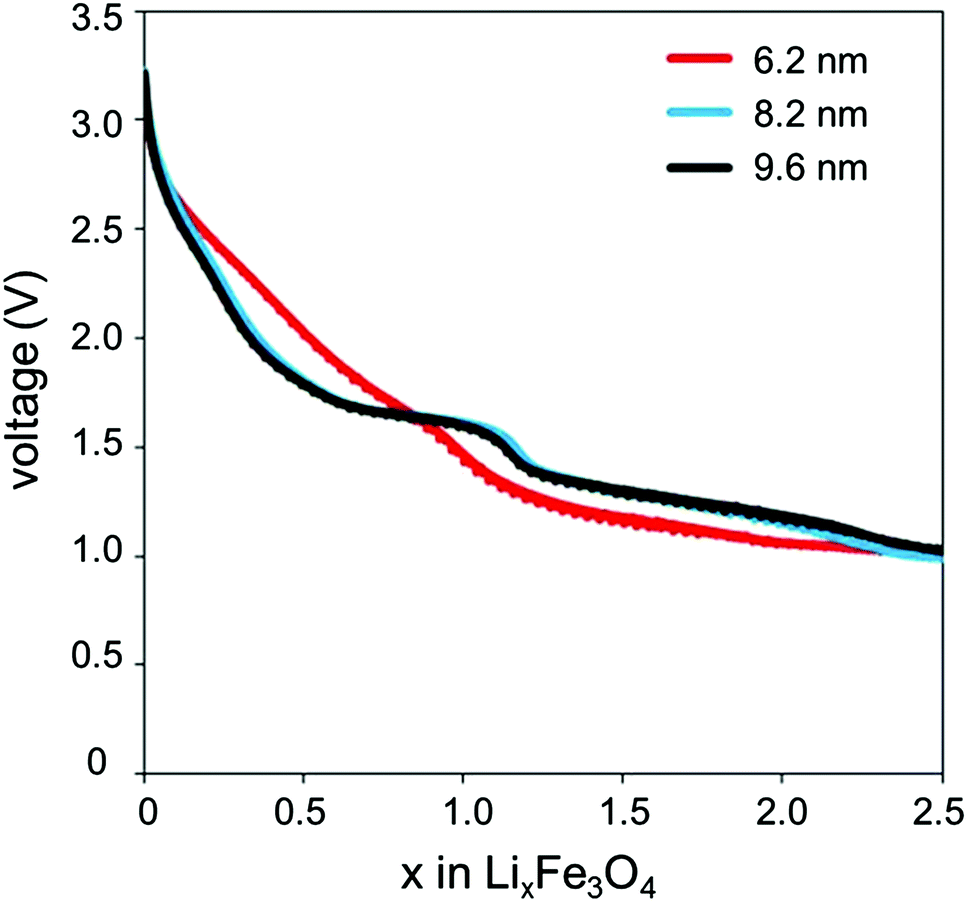 | ||
| Fig. 3 Discharge profiles of lithium cells containing magnetite (Fe3O4) of varying crystallite size prepared by coprecipitation technique. | ||
Saturation magnetization and XAS measurements are useful tools to determine the average oxidation state of materials. Bulk magnetite has a magnetization saturation of about 90 emu g−1, which is higher than the saturation magnetization values for the co-precipitated magnetite.36 Consistent with prior reports,40–43 the saturation magnetization decreased as crystallite size decreased for nanosized Fe3O4, indicating the presence of excess Fe3+ in the smaller crystallite size magnetite. The XAS results in Fig. 4 show bulk magnetite samples with crystallite sizes of 26 nm to have lower pre-edge energies than the 8–10 nm co-precipitation prepared samples, consistent with a higher concentration of Fe3+ in the co-precipitation prepared samples,37 as seen in other studies of nanocrystallite magnetite.44 The saturation magnetization and XAS measurements are both consistent with the assumption of oxidized Fe3+ rich layer on the surface of the Fe3O4 nanoparticles, where the smaller crystallite size materials have a higher average iron oxidation state due to a larger surface area to volume ratio. While an increase in average iron oxidation state would increase the theoretical capacity of the material, calculations predict only ∼10% increase based on the XAS results. Thus, the achieved 30–200% increase in capacity as a function of crystallite size cannot be explained solely by differences in iron oxidation state.
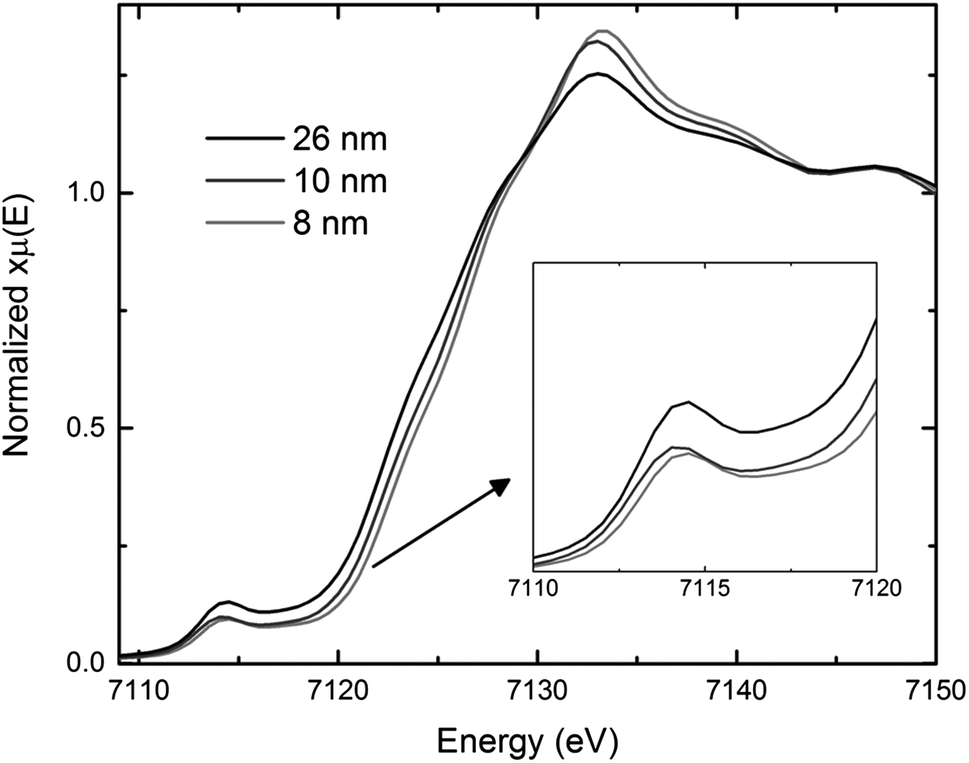 | ||
| Fig. 4 Comparison of X-ray absorption near edge structure (XANES) spectra for 26, 10, and 8 nm magnetite (Fe3O4). | ||
In order to gain additional insight regarding the discharge mechanism of magnetite, a combination of X-ray absorption spectroscopy and X-ray diffraction was used to determine structural changes as a function of electrochemical reduction for Fe3O4 of differing crystallite size (9–26 nm).26 At the onset of electrochemical reduction through 1.7 electron equivalents of discharge, the reduced materials were consistent with a spinel like LixFe3O4 local structure with iron ions in both tetrahedral and octahedral environments. Between 2.8 and 4.0 electron equivalents, a rock-salt phase was observed. Further reduction at 6.0 and 8.0 electron equivalents provided XAS consistent with Fe0 nanoparticle formation, for the smaller crystallite sized material ≤11 nm. Notably, controlling the crystallite size of the Fe3O4 parent material was key to obtaining this first direct empirical evidence of metallic Fe0 formation upon full electrochemical reduction.
The larger surface area of nanoscale materials often causes a high degree of agglomeration, where particle size can be dependent on crystallite size.36 Active material aggregation can contribute to increased charge transfer resistance for the electrode. Recently, the relationship between Fe3O4 crystallite size, Fe3O4 agglomerate size within a battery electrode, and electrochemical performance of Li/Fe3O4 cells was investigated using TEM, Transmission X-ray microscopy (TXM) and X-ray absorption near-edge spectroscopy (XANES).38 TEM results indicated the agglomerate size within processed electrodes containing 8 nm and 28 nm Fe3O4 was the same, as shown in Fig. 5a. Despite the similar degrees of agglomeration, electrodes containing the smaller crystallite size material delivered ∼20–200% higher specific energy, with the larger differences apparent under higher rate discharge. TXM-XANES data indicated that electrodes containing the 8 nm material discharged more evenly than those with the 28 nm material, Fig. 5b, consistent with higher utilization for the smaller crystallite size material.
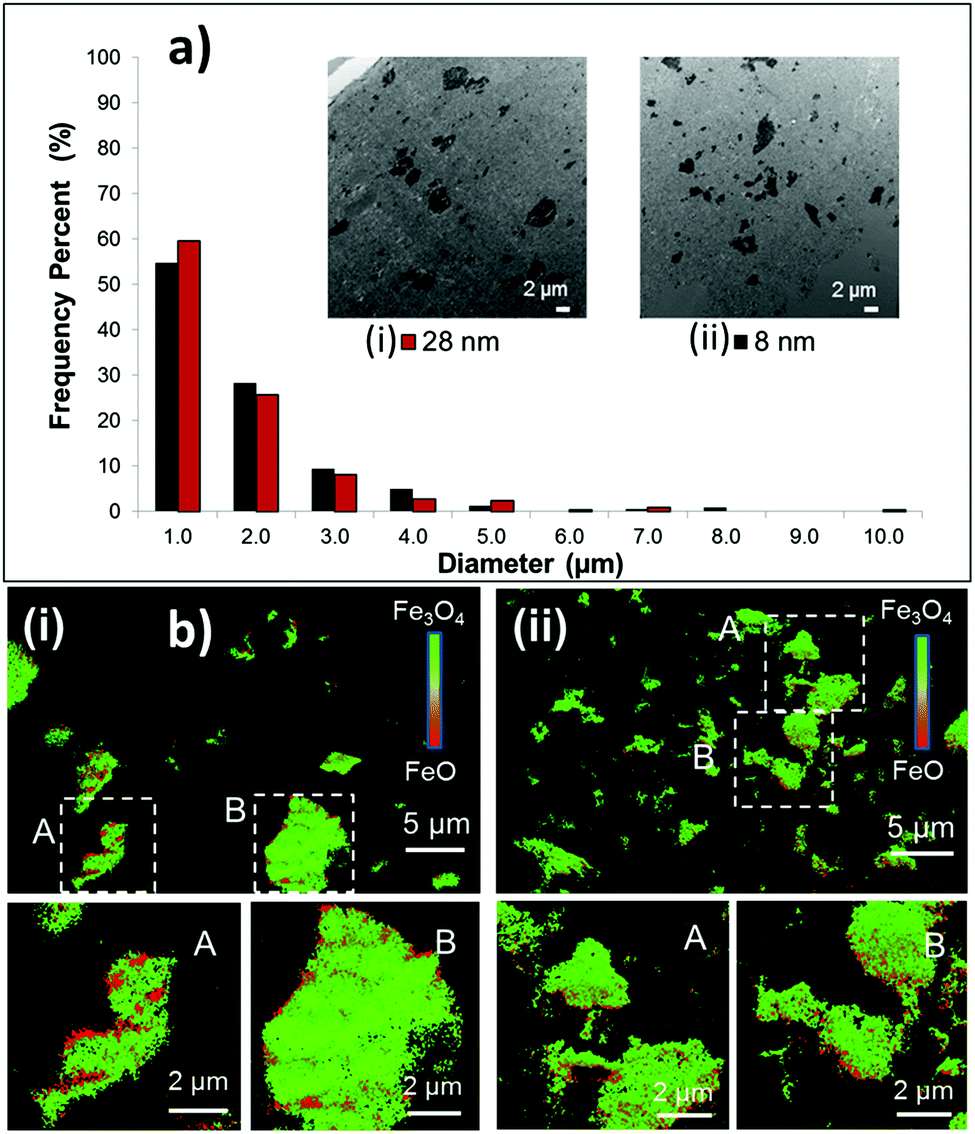 | ||
| Fig. 5 Electrodes prepared with (i) 28 and (ii) 8 nm magnetite (Fe3O4). (a) Aggregate size distributions in undischarged electrodes. Inset: Bright-field transmission electron microscopy (TEM) images of sectioned electrodes fabricated with 8 and 28 nm sized Fe3O4. (b) Transmission X-ray microscopy-X-ray absorption near edge structure (TXM-XANES) images of electrodes discharged to 100 mA h g−1. Selected regions A and B are magnified in each image. | ||
The voltage changes occurring within magnetite electrodes during discharge and open circuit voltage recovery were analyzed through a comparison of the mass transport time-constants associated with different length-scales within the electrode.32 Development of an effective multi-scale mathematical performance model required inclusion of both crystallite size and agglomerate size, indicating that both factors were significant to the mass transport process.31 Notably, even a small number fraction (<10%) of large Fe3O4 aggregates can have a significant impact on delivered capacity.
Magnetite of different morphologies
Magnetite of several types of morphologies have been synthesized and assessed for their battery performance. Table 1 provides a summary of the electrochemical behavior of the different morphological forms of magnetite. Of the morphologies studied, hollow spheres seem to achieve the best performance. In the hollow sphere morphology, magnetite nanoparticles are clustered around empty space. The hollow sphere morphology alleviates the stress caused by volume changes during the conversion mechanism. Furthermore, the hollow sphere is composed of nanoparticles which retain a small diffusion pathway for Li-ion, thus continuing to provide improved kinetics.45–47| Fe3O4 morphology | Size (nm) | Fe3O4 in electrode | Initial capacity, rate, voltage limit | Capacity, rate, # cycles | Ref. |
|---|---|---|---|---|---|
| Hollow spheres | 30–40 | 70% | 1261 mA h g−1, 0.1 C, 0.01–3.0 V | 852 mA h g−1, 1 C, cycles 5–50 | 46 |
| Hexahedra | Length, 200 | 80% | 1304 mA h g−1, 100 mA g−1, 0.01–3.0 V | 644.1 mA h g−1, 100 mA g−1, 40 | 49 |
| Octahedra | Length, 1000–3000 | 80% | 1216 mA g−1, 100 mA g−1, 0.01–3.0 V | 350.5 mA h g−1, 100 mA g−1, 40 | 49 |
| Polyhedra | 120–150 | 50% | 2632 mA h g−1, 50 mA g−1, 0.05–3.0 V | 1100 mA h g−1, 50 mA g−1, 50 | 48 |
| Nanowires | 50 | 60% | 2416 mA h g−1, 50 mA g−1, 0.01–3.0 V | 774 mA h g−1, 50 mA g−1, 50 | 85 |
| Nanowires | 20–50 | 70% | 1240 mA h g−1, 0.1 C, ∼0.1–3.0 V | 820 mA h g−1, 0.1 C, 50 | 86 |
| Submicron spheres composed of nanospheres | 30 | 40% | 1332 mA h g−1, 0.2 C, 0.01–3.0 V | 1100 mA h g−1, 0.2 C, 60 | 57 |
| Hollow spheres organized by nanosheets | 10 | 70% | 1614 mA h g−1, 500 mA g−1, 0.01–3.0 V | 1046 mA h g−1, 500 mA g−1, 100 | 45 |
| Hollow microspheres | 4180 | 70% | 945 mA h g−1, 0.1 C, 0.02–3.0 V | 830 mA h g−1, 0.1 C, 35 | 47 |
| Pyramid | 10 | 100% | 1800 mA h g−1, 40 mA g−1, 0.05–3.0 V | 1300 mA h g−1, 40 mA g−1, 7 | 87 |
There are different ways to prepare magnetite hollow spheres. In one reference, the hollow spheres were synthesized by hydrolyzing a solution of FeCl3 in the presence of polyvinylpyrrolidone (PVP) at 200 °C. As the Fe3O4 forms, the crystals begin to aggregate. The PVP stabilizes the surface of the crystallites and acts a nucleation site. It is suggested that Ostwald rippening causes the inner crystallites to dissolve and reform on the surface of the outer coated crystallites, thus forming a void in the center of the aggregate. SEM images show that the hollow microspheres are composed of nanoparticles between 30 and 40 nm with surface area of 12 m2 g−1. The material was tested against Li/Li+ and exhibited less polarization and better reversibility than the solid Fe3O4 microspheres. Furthermore, the hollow spheres achieved 40% higher capacity and better rate capability than the solid spheres. Electrochemical impedance spectroscopy experiments further suggested that the hollow sphere morphology reduces the charge-transfer resistance, thereby facilitating the transfer of Li+ ions.46
The hollow sphere morphology can also be formed by other morphologies such as nanosheets.45 These hollow spheres exhibited a high initial capacity of 1614 mA h g−1 and retained 65% of the initial capacity after 100 cycles at 500 mA g−1. The solid spheres exhibited about 800 mA h g−1 capacity, thus, higher capacity was achieved by the hollow morphology. Further testing showed that at different rates the hollow morphology achieved higher capacities than the solid spheres. EIS experiments provided further evidence suggesting that the hollow spheres decrease the resistance more than the solid spheres.
Another technique that has been used to prepare hollow spheres is through ionic adsorption of magnetite nanoparticles to a hydrogel such as poly (methacrylic acid-co-ethylene glycol dimethacrylate) (poly(MMA/EGDMA)). The hollow morphology is achieved after annealing the sample at 500 °C to remove the hydrogel. The hollow spheres were composed of Fe3O4 nanoparticles ranging in size from 10–100 nm, according to SEM images. Compared to Fe3O4 nanoparticles, the hollow spheres exhibited better capacity retention over 35 cycles such that the nanoparticles and hollow spheres lost 67% and 12% of their initial capacity, respectively.47
Other studies have prepared magnetite nanoparticles which exhibit different shapes such as octahedra and hexahedra.48,49 A low temperature hydrothermal synthesis was used to synthesize polyhedra which exhibited the different facets (010), (111), (110), (011), (11![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), (11), (01), (001), (101), (100), and (01). This morphology exhibited better performance than the octahedra49 and hexahedra49 synthesized, as summarized in Table 1. After 50 cycles, the polyhedra nanoparticles achieved capacities of about 1100, 800, 600, and 400 mA h g−1 at current rates of 50, 100, 400, and 1000 mA g−1, respectively.48
), (11), (01), (001), (101), (100), and (01). This morphology exhibited better performance than the octahedra49 and hexahedra49 synthesized, as summarized in Table 1. After 50 cycles, the polyhedra nanoparticles achieved capacities of about 1100, 800, 600, and 400 mA h g−1 at current rates of 50, 100, 400, and 1000 mA g−1, respectively.48
These studies suggest that through manipulation of both crystallite size and morphology, electrochemical performance can be significantly improved and altered. Hollow spheres appear to offer the best morphology since that structure facilitates ion transfer as well as the reducing strain produced by the volume change experienced during the conversion reaction of Fe3O4 to Fe0. These features result in higher capacities during cycling at both high and low rates.
3. Electrochemistry of magnetite containing electrode heterostructures
Although magnetite is a semiconductor it is commonly studied combined with an electrically conductive carbon thus enhancing the electronic conductivity of the electrode.50 Carbons of various type have been employed, including carbon black (CB), acetylene black (AB), Super P (SP), graphite, and carbon nanotubes (CNTs). Varying the carbon additive can have profound effects on the electrochemical properties of these electrode systems. For example, a recent study by Deng et al. involved the effects of AB, SP, CNTs and a mixture of AB plus CNTs on the electrochemical performance of magnetite. Notably, a mixture of AB and CNTs provides increased rate capability, larger specific capacity after 30 cycles, and a lower impedance that overall maximizes the performance of the magnetite electrode in a LIB.51 Interest in composite electrodes such as electrically conductive materials plus redox electroactive material has inspired other studies in nanostructured materials and their effect on composite electrode electrochemistry. When carbon is employed as an electrically conductive material in a composite electrode, particularly those discharged under low voltage, it is important to consider the following reaction: 6C + Li → LiC6 in capacity measurements.Research in magnetite electrodes has included carbon nanostructures and magnetite nanostructures in a variety of LIB composite electrodes. One research strategy is to investigate carbon materials such as CB or graphene which may yield a higher theoretical capacity than conventional carbon additives (372 mA h g−1). There are two common approaches for this strategy (1) change the structure of magnetite on the nanoscale then use carbon as a coating or additive, or (2) use a carbon nanostructure such as graphene or graphene oxide as an anode material then deposit magnetite nanoparticles on their surface. In general, combinations of magnetite morphology, magnetite crystallite size, and a variety of carbon additives have been studied to optimize magnetite performance as an LIB electrode.
Studies of magnetite nanostructures where carbon was incorporated into the synthesis to promote electronic conductivity are summarized in Table 2. The morphology, crystallite size of magnetite, and weight percent of magnetite in the active electrode material was provided where reported. The mixture of active material and additional carbon additive was also provided where reported with the identification of the carbon type used. The initial and final capacities reported are also tabulated. Details from a subset of these studies are provided, to illustrate unique or representative syntheses and electrochemistry results.
| Active material composition and morphology | Fe3O4 crystallite size | Fe3O4 in active material | Active material | Carbon additive | Initial capacity, rate, voltage limit | Final capacity, cycle number | Ref. |
|---|---|---|---|---|---|---|---|
| AB = acetylene black, C = carbon, CB = carbon black, G = graphene, GO = graphene oxide, SP = Super-P carbon black, CNT = carbon nanotubes, SWCNT = single walled carbon nanotubes. | |||||||
| Fe3O4@C core–shell nanotubes | 250 nm | 93% | 80% | 10%, SP | 1213 mA h g−1, 100 mA g−1, 0.01–3.0 V; 500 mA g−1, 0.01–3.0 V | 808 mA h g−1, 20 cycles; 560 mA h g−1 100 cycles | 73 |
| Fe3O4@C core–shell nanorods | 200 nm | 96% | 80% | 10%, SP | 2117 mA h g−1, 1 C, 0.01–3.0 V | 807 mA h g−1, 100 cycles | 74 |
| Fe3O4@C core–shell nanorods | 150–200 nm | NR | 75% | 15%, AB | 1126 mA h g−1, 0.1 C, 0.01–3.0 V | 394 mA h g−1, 100 cycles | 75 |
| “Grapecluster”: core–shell Fe3O4@C nanoparticles on CNT | 150 nm | 91% | 65% | 20%, SP | 1450 (w/CNT), 900 (w/o CNT) mA h g−1, 60 mA g−1, 0.01–3.0 V | 693 mA h g−1, 200 cycles | 78 |
| Fe3O4 nanorods embedded in SWCNT | Not reported | Not reported | Not reported | Not reported | 1000 mA h g−1, 0.1 C cycles 1–5, 1 C cycles 6–50, 0.005–3.0 V; 1030 mA h g−1, 0.1 C cycles 1–5, 5 C cycles 6–100, 0.005–3.0 V | 1050 mA h g−1, 50 cycles, 690 mA h g−1, 100 cycles | 88 |
| Porous core–shell Fe3O4@C nanospheres | 100–120 nm | 70% | 75% | 15%, Ketjen black | 1691 mA h g−1, 100 mA g−1, 0.01–3.0 V | 1100 mA h g−1, 60 cycles | 81 |
| Core–shell N-doped Fe3O4@C nanoparticles | 100 nm | 84% | 70% | 15%, CB | 1173 mA h g−1, 500 mA g−1, 0.01–3.0 V | 976 mA h g−1, 50 cycles | 71 |
| Core–shell Fe3O4@C hollow spheroids | 90–30 nm | Very high | 65% | 25%, SP | 1369 mA h g−1, 200 mA g−1, 0.05–3.0 V | 900 mA h g−1, 180 cycles | 80 |
| Hollow Fe3O4 microspheres | 44–79 nm | 100% | 70% | 20%, AB, CNTs 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
1200 mA h g−1, 60 mA g−1, 0.01–3.0 V | 900 mA h g−1, 30 cycles | 51 |
| Onion-like carbon coated Fe3O4 nanoparticles | 30–60 nm | NR | 80% | 10%, CB | 1449 mA h g−1, 100 mA g−1, 0.01–3.0 V | 918 mA h g−1, 300 cycles | 68 |
| Carbon coated Fe3O4 nanospindles | 30 nm | 79% | 70% | 15%, CB | 749 mA h g−1, 0.2 C cycle 1–5, 0.5 C cycle 6–160, ∼0.01–3 V | 530 mA h g−1, 160 cycles | 76 |
| Carbon encapsulated Fe3O4 | 18.2 nm | 68.7% | 80% | 10%, CB | 1499 mA h g−1, 1000 mA g−1, 0.005–3.0 V | 980 mA h g−1, 350 cycles | 89 |
| Carbon coated Fe3O4 dendrites | 20 nm | Very high | 80% | 10%, CB | 1100 mA h g−1, 0.1 C, 0–3.0 V | 405 mA h g−1, 50 cycles | 77 |
| Carbon coated Fe3O4 microcuboids | 20 nm | 98% | 70% | 20%, AB | 1719 mA h g−1, 100 mA g−1, 0.05–3.0 V | 975 mA h g−1, 50 cycles | 82 |
| Carbon coated Fe3O4 nanoparticles on CNTs | 10–15 nm | 90% | 80% | 10% C | 1030 mA h g−1, 50 mA g−1, 0.5–2.5 V | 620 mA h g−1, 50 cycles | 72 |
| Core–shell Fe3O4@C nanoparticles | 12 nm | NR | 70% | 15%, SP | 1310 mA h g−1, 100 mA g−1, 0.1–3.0 V | 859 mA h g−1, 50 cycles | 70 |
| Carbon coated sea urchin-like Fe3O4 nanoclusters | 10.8 nm | 88% | 70% | 15%, SP | 1228 mA h g−1, 100 mA g−1, 0.01–3.0 V | 900 mA h g−1, 10 cycles | 79 |
| Core–shell Fe3O4@C nanoparticles | 10 nm | 93% | 70% | 15%, SP | 1400 mA h g−1, 185 mA g−1, 0.01–3.0 V | 700 mA h g−1, 400 cycles | 69 |
| rGO/Fe3O4 paper | NR | 65% | 85% | 10%, SP-CB | 1100 mA h g−1, 1 C, 0.01–3.0 V | 1140 mA h g−1, 220 cycles | 52 |
| Fe3O4/G nanoscrolls | NR | 50% | 90% | 0% | 1720 mA h g−1, 0.1 C, 0.01–3.0 V | 1010 mA h g−1, 50 cycles | 59 |
| C coated flower-like Fe3O4/G microstructure | NR | NR | 80% | 10%, AB | 1602 mA h g−1, 0.2 C, 0.01–3.0 V | 1439 mA h g−1, 47 cycles | 60 |
| Semi exfoliated reduced graphite oxide, Fe3O4/graphite oxide nanosheets | NR | NR | 100% | 0% | 1616 mA h g−1, 75 mA g−1, 0.01–3.0 V | 1309 mA h g−1, 10 cycles | 58 |
| Fe3O4 nanocrystals on C matrix | 70 nm | NR | 100% | 0% | 863 mA h g−1, 0.1 C, 0.1–3.0 V | 777 mA h g−1, 30 cycles | 66 |
| Fe3O4/G nanosheets | 20–70 nm | NR | 80% | 10%, CB | 1320 mA h g−1, 5 C, 0.01–3.0 V | 650 mA h g−1, 30 cycles | 57 |
| Fe3O4/G nanosheets | 30–50 nm | 70% | 80% | 10%, AB | 1040 mA h g−1, 1000 mA g−1, 0.01–3.0 V | 335 mA h g−1, 300 cycles | 54 |
| Fe3O4/G nanosheets | 40 nm | NR | 80% | 10%, AB | 1334 mA h g−1, 928 mA g−1, 0.01–3.0 V | 504 mA h g−1, 50 cycles | 55 |
| Fe3O4 on C foam-like composite | 13–27 nm | 55% | 80% | 10%, super C65 | 1710 mA h g−1, 100 mA g−1, cyc. 3–150 500 mA g−1, 0.01–3.0 V | 950 mA h g−1, 150 cycles | 64 |
| Fe3O4/G nanosheets | 20 nm | 11% | 80% | 10%, AB | 1976 mA h g−1, 200 mA g−1, 0.001–3.0 V | 1243 mA h g−1, 50 cycles | 53 |
| Fe3O4/G nanosheets | 10–20 nm | 55% | 80% | 10%, SP-Li | 1126 mA h g−1, 0.1 C, 0.01–3.0 V | 500 mA h g−1, 100 cycles | 61 |
| Fe3O4-G nanocomposite | Not reported | 78% | 75% | 15%, SP | 1060 mA h g−1, 100 mA g−1, 0.1–3.0 V | 1048 mA h g−1, 85 cycles | 90 |
| Fe3O4/G composites | 15 nm | 40% | 85% | 10%, AB | 1545 mA h g−1, 0.05 C, 0.001–3.0 V | 675 mA h g−1, 50 cycles | 62 |
| Fe3O4@G foam nanosheet | 7–15 nm | NR | 100% | 0% | 1671 mA h g−1, 1 C, 0.01–3.0 V | 1200 mA h g−1, 500 cycles | 63 |
| Fe3O4/G nanosheet | 10 nm | 42% | 82% | 8%, SP, KS-6 1:1 |
700 mA h g−1, 0.2 C, 0.01–3.0 V | 575 mA h g−1, 50 cycles | 56 |
| G wrapped Fe3O4 | Not reported | 87% | 80% | 10%, SP | 1400 mA h g−1, 35 mA g−1, 0.001–3.0 V | 770 mA h g−1, 100 cycles | 91 |
| G encapsulated Fe3O4 | 12 nm | 62% | 80% | 10%, AB | 1080 mA h g−1, 100 mA g−1, 0–3.0 V | 650 mA h g−1, 100 cycles | 92 |
| Fe3O4 on C foamlike composite | 15–20 nm | 46% | 80% | 10%, SP-CB | 1263 mA h g−1, 200 mA g−1, 0.005–3.0 V | 1008 mA h g−1, 400 cycles | 65 |
| Fe3O4 on CNTs | 5 nm | 41% | 80% | 10%, AB | 840 mA h g−1, 0.1 C, 0.02–3.0 V | 390 mA h g−1, 75 cycles | 67 |
| Fe3O4-CNT composite | Very small | 66.7% | 90% | 0% | 988 mA h g−1, 100 mA g−1, 0.02–3.0 V | 650 mA h g−1, 145 cycles | 93 |
| CNT-Fe3O4 coaxial nanocables | 9.5 nm | Not reported | 80% | 10% AB | 1431 mA h g−1, 200 mA g−1, 0.01–3.0 V, 800 mA h g−1, 2000 mA g−1, 0.01–3.0 V | 1290 mA h g−1, 200 mA g−1, 80 cycles, 690 mA h g−1, 2000 mA g−1, 200 cycles | 94 |
| CNT-reduced GO Fe3O4 nanocomposite | 7 nm | 53% | 80% | 10%, SP | 1277 mA h g−1, 200 mA g−1, 0.00–3.0 V | 680 mA h g−1, 100 cycles | 95 |
| Fe3O4 sheath on aligned CNT | 5–7 nm | 50–60% | N/A | N/A | 1814 mA h g−1, 100 mA g−1, 0.1–3.0 V | 1670 mA h g−1, 100 cycles | 96 |
As magnetite electrodes utilize carbon additives to enhance electronic conductivity, there have been many investigations on optimizing the carbon material's structure. A recent approach has been to use two-dimensional, single-atom thick, hexagonally arranged graphene sheets with the additional benefit that their rigid sp2 hybridized structure reduces agglomeration of the magnetite nanoparticles.52 The most common synthetic approach starts with graphite and turns it to graphene oxide (GO) using a modified Hummers method. Using microwave techniques or an ultrasonic suspension an Fe3+ species is added to the GO and incorporated into the graphene matrix by forming Fe–O–C bonds as shown in Fig. 6a. Zhang et al. reported having magnetite incorporated with polyethylenimine to hold a slightly positive charge and graphene with poly(sodium 4-styrenesulfonate) to hold a slightly negative charge where the electrostatic attraction holds the materials together. The Fe3O4-graphene paper-like material isolated from the preparation showed impressive capacity (1140 mA h g−1) for 220 cycles.52 Another study that added 20 nm magnetite particles on graphene nanosheets with a pore size around 3 nm via sonication reported a retained capacity of 1243 mA h g−1 for 50 cycles when cycled at a 0.2 A g−1 rate.53 Others reported magnetite nanoparticles on graphene sheets providing between 500–650 mA h g−1 after 50 cycles under rates of ∼1 A g−1.54–57
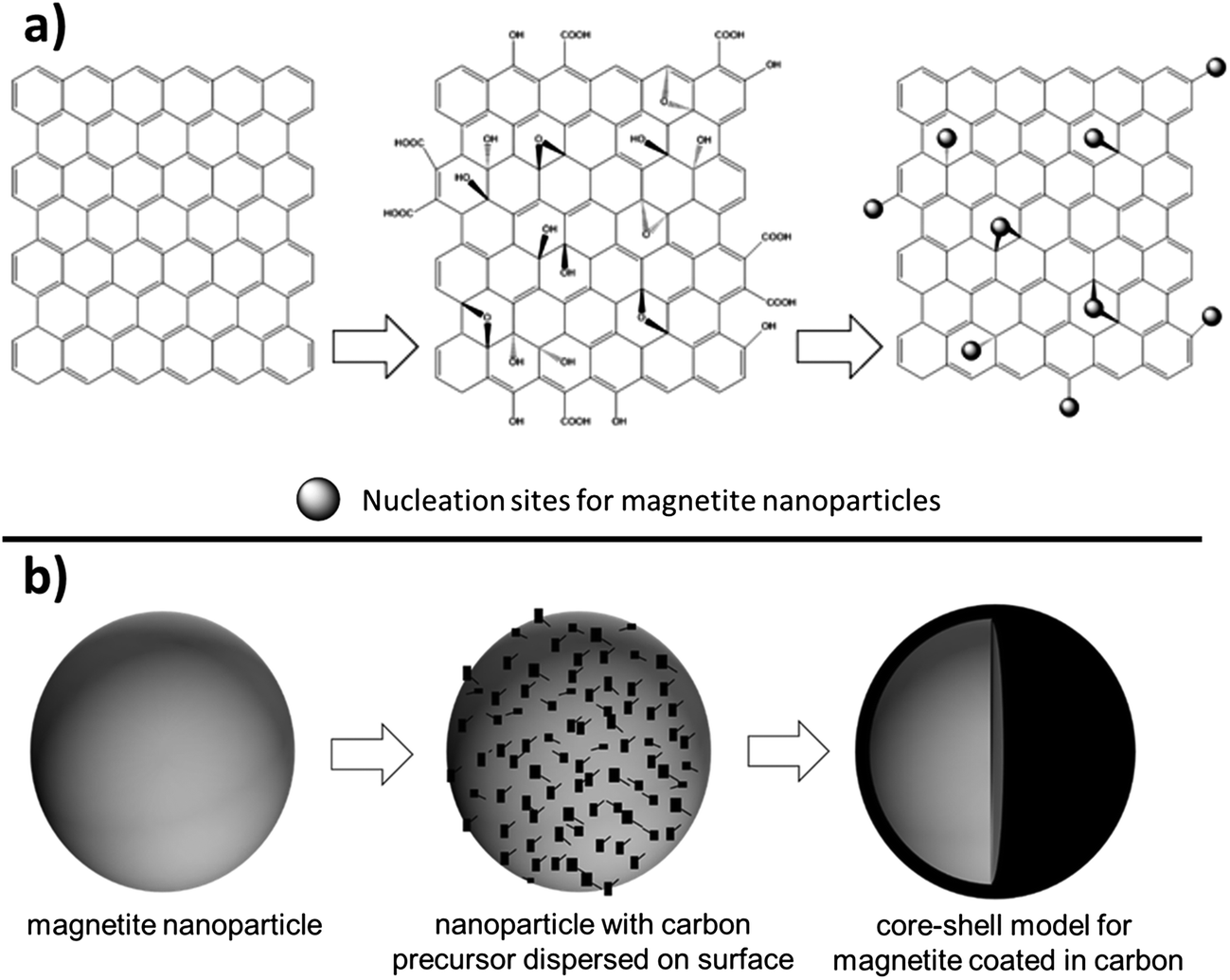 | ||
| Fig. 6 Conceptual models of magnetite (Fe3O4) nanoparticle-carbon heterostructures. (a) Graphene sheet transformed to a graphene oxide composite by Hummers method. The hydroxyl, oxide, and carboxylic acid substituents provide nucleation sites for magnetite to bind to the sheet. (b) Carbon source converted to an evenly dispersed carbon coating on the Fe3O4 surface-referred to as core shell particles. | ||
Another study reported magnetite dispersed on a graphite oxide nanosheet with no binder or carbon conducting additive synthesized using a modified Hummers method. Microwave irradiation was used to disperse an Fe3+ species and the graphite oxide which was then chemically reduced. This study showed that varying electrolyte from DEC to DMC had significant effect on the capacity retention of the material. The best performance with DEC provided 994 mA h g−1 after 50 cycles.58
Zhao et al. developed a new method to modify the structure of the nanosheet by developing GO nanoscrolls with magnetite embedded on the interior by cold quenching GO sheets in liquid nitrogen. Three different wrapping structures were studied with the best electrochemical result showing a capacity of 1010 mA h g−1 after 50 cycles, similar to the nanosheets.59 Another variation of the nanosheets is the assembly of a flower-like structure 3–5 microns across with pedals 60–90 nm thick and approximately 1 micron long. These flower-like materials provided a capacity of 1449 mA h g−1 after 49 cycles.60 Similar studies were conducted with magnetite/GO composites consisting of nanoparticles between 10–20 nm crystallite size where magnetite represented 40–55% of the active material in the electrode, giving sustained capacities of 674 mA h g−1.61,62
Three dimensional structures such as foams and carbon matrices have also been gaining interest in recent years. A study by Hu et al. used nickel foam cut into disks and annealed under hydrogen gas in a quartz tube with small amount of methane used for the graphene matrix, the mixture was cooled to room temperature and finally nitric acid was used to remove excess nickel metal resulting in a graphene foam. The graphene foam, a Fe3+ source, and carbon dioxide were added to a high pressure vessel resulting in the formation of graphene sheets with magnetite particles on the surface. Good reversibility of the foam structure was achieved as illustrated by the capacity of 924 mA h g−1 for 500 cycles, the highest cycle number reported on Table 2.63
Other carbon foams with magnetite particles incorporated into their structure have been tested for their performance as electrode materials with varying results. In a study by Kang et al., they synthesized a mesocellular foam and added the magnetite particles into the cells then studied Al2O3's effect on the material to further stabilize the SEI layer. Their results showed that Al2O3 did aid in side reactions caused at the SEI, but this material still showed considerable fade from 1007 to 533 mA h g−1 after 150 cycles.64 A more successful capacity retention of carbon foam materials was reported by Wu et al. by drying a gelatin doped with an Fe3+ species that was annealed to produce the porous carbon foam with the magnetite nanoparticles. This foam retained a higher capacity of 1008 mA h g−1 over 400 cycles.65
Other forms of carbon matrixes have been derived by synthesizing FeOOH nanoparticles, coating them in alcohol, then carbonizing in vaccum to produce an agglomeration of particles connected by the carbon residue, similar to the porous nanospheres previously mentioned. These batteries were constructed without any additional carbon added and still demonstrated a capacity of 777 mA h g−1 after 30 cycles.66 Another study embedded magnetite nanoparticles (∼5 nm) on CNT's with magnetite representing 41 wt% of the active material, then they used 10% AB as a conductive additive and 10% PVDF binder for their electrode and to found a capacity only slightly higher than graphite after 75 cycles.67
One synthetic technique for generating Fe3O4 based nanostructures is the arc-discharge method. Fe metal is used as the anode and carbon needle as the cathode, the chamber is filled with Ar and H2 then ethanol is introduced and held at a precise pressure for 24 hours resulting in 30–60 nm magnetite nanoparticles with an onion-like carbon coating. The “onion-like” layering is attributed to the defects in the collapsed graphite layer. These defects seemed to accommodate the volume changes during the lithiation/delithiation process and provided high coulombic efficiency (∼86% after 300 cycles).68 A more typical synthesis for nanoparticles is a simple one or two step wet chemical synthesis with an iron oxide precursor isolated. A carbon source can be added (such as sucrose, polymerized dopamine, etc.) or sourced from residual hydrocarbons from synthesis. The resulting particles are then subjected to heat well over combustion of the carbon source and resulting in formation of core–shell structures as depicted conceptually in Fig. 6b. Enhanced capacity, improved cycling capability, and a significant decrease in AC impedence for magnetite based electrodes in both lithium and sodium batteries have been reported as a result of the core–shell synthesis approach.69–72 Frequently, the core–shell Fe3O4 material is combined with another conductive carbon to generate the composite electrode. For example, Park and Myung prepared core–shell nanoparticles where the core magnetite is 10–100 nm and the shell is approximately 1–10 nm and deposited them onto two-dimensional carbon nanotubes.72 Different types of Fe3O4 morphologies have been achieved in concert with core–shell synthesis. For example, in a synthesis reported by Xia et al. Fe2O3 nanotubes were generated then heated above 400 °C for 3 h in inert atmosphere to produce magnetite nanotubes. Glucose was added as a carbon source during synthesis then degraded to a carbon coating producing a core–shell nanotube 200–300 nm long with walls 10–20 nm in thickness. These materials exceeded the capacity of graphite, attributed to shortening of the Li+ ion diffusion path length.73
A similar synthesis was used to generate 200 nm magnetite nanorods from FeOOH nanorods also using glucose as a carbon source. These rods are similar to the nanotubes but lack the hollow structure in the center but still exhibit comparable initial capacity and fade.74 A magnetite core-carbon shell nanorod was prepared by Liu et al. using Fe2O3 nanorods and citric acid as reagents.75 These materials were synthesized and tested similarly, but that resulting electrochemistry differed by approximately 450 mA h g−1 after 100 cycles. Although these materials were discharged at different rates as indicated in Table 2, the FeOOH-glucose derived rods have superior cycling ability based on the reported electrochemical evaluations of the materials.
Similar Fe3O4 nanostructures, ovoid in shape ∼500 nm long and 100–125 nm in diameter, refered to as nanospindles, were generated by Zhang et al. from previously synthesized Fe2O3 nanospindles. Glucose was again used as the carbon source, forming a 2–10 nm thick carbon coating. These core–shell nanospindles showed the ability to deliver over 530 mA h g−1 after 160 cycles with a C/5 (cycles 1–5) or C/2 (cycles 6–160) rate. The authors noted that this carbon layer also protected the integrity of the Fe3O4 nanospindles over multiple cycles by ex situ TEM study after a lithiation/delithiation cycle.76 Partially reducing hematite in an autoclave was shown to produce magnetite dendrites 2.5–6 microns long through the backbone with arms resembling a fractal structure. The intent was to design a morphology with interspaces that could better accommodate the lithiation and delithiation processes. However, this structure showed significant early capacity fade over the initial 15 cycles even in the presence of carbon coating.77
Other approaches intended to mitigate the impacts of lattice expansion and contraction during lithiation and delithiation of Fe3O4 have generated composites described as “clustered” systems. A cluster refer to a group of nanoparticles that have agglomerated to form a hierarchical structure. In many cases, the cluster is a mesoscale composite containing Fe3O4, carbon and/or organic polymers. A “grapelike” cluster was studied and classified by the carbon nanotube backbone and the magnetite nanoparticles acting as “grapes” that are connected to the CNT backbone. The cluster is formed starting from an FeOOH precursor, which is coated in carbon via a glucose precursor, and then converted to magnetite. The resulting grapelike structure was confirmed by SEM and TEM imaging and showed a considerable void volume via porosimetry analysis. The grapelike structure showed improved capacity and rate capability relative to previously synthesized core–shell magnetite nanoparticles.78 Another “urchin-like” nanocluster was synthesized by combining FeCl3·6H2O, pyrrole, and sodium docedyl sulfate in water to form polypyrrole coated FeOOH then calcinated to form 200–500 nm magnetite core-carbon coated spheres with 10 nm pores and an urchin-like surface. The urchin-like clusters were tested against commercially used magnetite and shown to have improved cycling capability attributed to improved capability to accommodate volume change during lithiation/delithiation.79
Another successful morphology that accommodates volume changes during the discharge/charge process is the three dimensional hollow spheroid structure. Silk nanofibers and FeCl3·6H2O were dehydrated to form FeOOH/silk spheroids, then Oswald ripening progressed to form Fe2O3/silk hollow spheroids, when were then reduced to generate hollow, core magnetite structures with a carbon shell. The three dimensional hollow spheroid structures showed improved electrochemical performance relative to their Fe2O3/silk precursors, with 100% coulombic efficiency over 180 cycles and capacity exceeding 900 mA h g−1.80 Core–shell, porous spheres were also generated by a hydrothermal reaction where the carbon shell was generated by calcination of the residual ethanol from the reaction. First, porous Fe2O3 particles were generated then upon calcination the carbon fused the particles to form larger magnetite spheres 100–120 nm in diameter. The structure exhibited the highest capacity over 60 cycles in Table 1, implying a successful pathway to short Li+ paths lengths with high three dimensional electronic conductivity.81
Mesoporous microcuboid structures of Fe3O4 approximately 30 microns in length linked by a carbon matrix have been prepared, derived from FeC2O4·2H2O. An advantage of this approach was the opportunity for large scale syntheses, although the method lacks the ability to provide an even carbon coating, leaving some magnetite surfaces exposed to electrolyte.82 Under high rate discharge at 100 mA g−1, a high initial capacity of 1719 mA h g−1 was demonstrated, with good capacity retention of 975 mA h g−1 after 50 cycles.
4. Example of 2D iron oxide material which also exhibits crystallite size effects
Silver ferrite, AgFeO2
Layered structures or 2D materials can be useful as LIB electrodes as they allow for facile insertion and deinsertion of Li+ ions in two dimensions with the possibility for little structural strain during charge/discharge cycles. Silver ferrite (AgFeO2) is a layered structure consisting of FeO6 octahedra with Ag+ ions occupying the space between these layers which can exist in two distinct but similar phases: 3R, rhombohedral and 2H, hexagonal, as shown in Fig. 7. Silver ferrite can be prepared at low temperature using a coprecipitation method.83 Recently, it was demonstrated that crystallite size control of silver ferrite was achievable using the coprecipitation approach. Notably, the crystallite size control of silver ferrite was accompanied by direct formation of a silver ferrite–maghemite composite, demonstrating a new paradigm for direct synthesis of metal oxide composites.84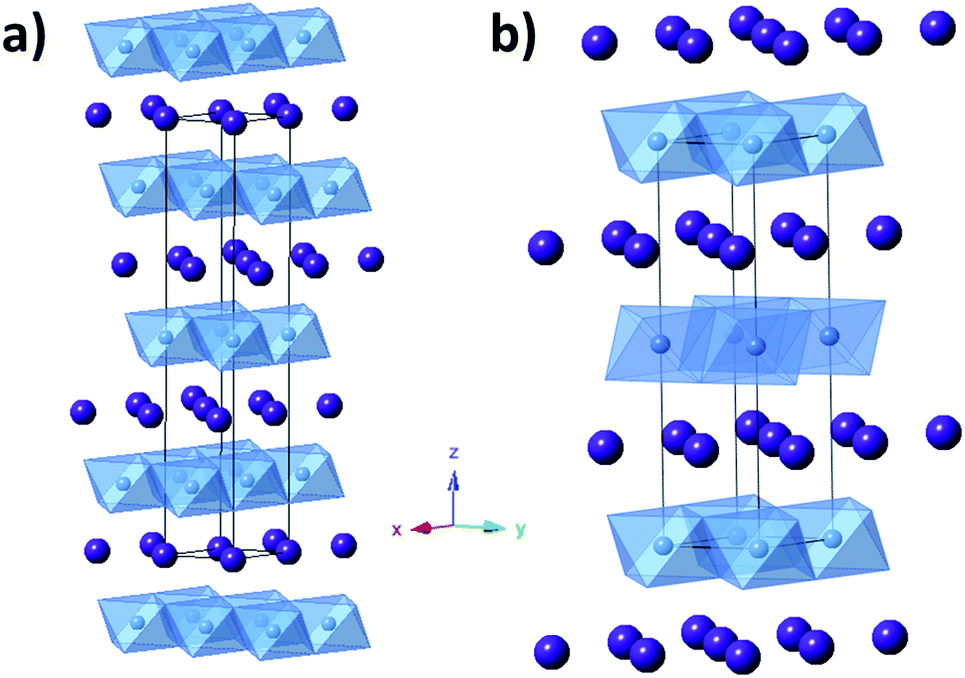 | ||
| Fig. 7 Structure of silver ferrite (AgFeO2). (a) Rhombohedral (3R). (b) Hexagonal (2H). | ||
The composition of the composite was affirmed by a combination of techniques, including X-ray diffraction (XRD), inductively coupled plasma-optical emission spectroscopy (ICP-OES), Raman spectroscopy, and X-ray absorption spectroscopy (XAS). A compositional range of AgxFeO2 where (0.2 ≤ x ≤ 1.0) was achieved. Despite the significant change in silver concentration across the series, the lattice constants did not change and the XRD pattern only exhibited reflections corresponding to the two AgFeO2 polymorphs where the percentage of the 3R phase increased as the silver content decreased. As the Ag/Fe ratio decreased, the crystallite size decreased, Fig. 8a. Further characterization of these materials via Raman spectroscopy indicated the presence of maghemite (γ-Fe2O3) at low silver concentrations. Linear combination fitting of the Raman data showed that at a composition of Ag0.2FeO2, nearly 80% of the material is composed of maghemite, Fig. 8b.84 Even for high maghemite content materials, the maghemite was too amorphous to detect via XRD. The presence of maghemite in the low silver content samples was further supported by XAS, with an increase in intensity of the pre-edge for the Fe K-edge as the silver content decreased, consistent with an increase in a population of Fe3+ in tetrahedral interstitial sites, as are present in the maghemite structure.84
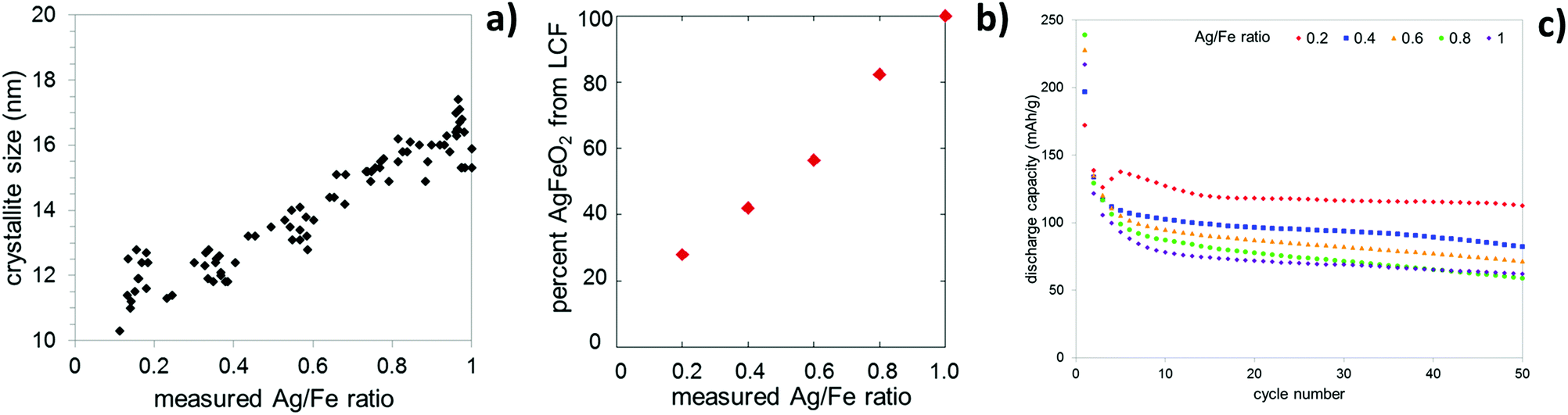 | ||
| Fig. 8 Characterization data for silver ferrite–maghemite (AgFeO2–γ-Fe2O3) composites. (a) Dependence of crystallite size on silver concentration, (b) percent AgFeO2 determined by linear combination fit (LCF) of Raman data versus measured Ag/Fe ratio. (c) Discharge capacity versus cycle number for lithium cells containing silver ferrite–maghemite (AgFeO2–γ-Fe2O3) composite electrodes. | ||
There have been few studies regarding the application of AgFeO2 as a cathode in lithium ion batteries. Initial studies by CV showed that Ag0.98FeO2 is electrochemically active and SEM, EDS, and XRD data proved the formation of silver metal upon discharge.83 Galvanostatic intermittent titration technique (GITT) testing indicated that as the crystallite size decreased, the polarization decreased resulting in higher voltages for the lower silver content samples. The cells were cycled at 0.15 mA cm−2 between 1.5 and 3.5 V. The focus of these initial electrochemical evaluations was the lithium insertion process at higher voltage. As illustrated in Fig. 8c, the discharge capacity during the lithium insertion process is dependent on the Ag(x) composition of the AgxFeO2 material, and in turn, crystallite size. As the crystallite size decreases, the capacity increases. Cycled under the same conditions, maghemite (γ-Fe2O3) achieved a reversible capacity of 60 mA h g−1, and stoichiometric AgFeO2 with a crystallite size of ∼16 nm achieved a reversible capacity of 62 mA h g−1, while the Ag0.2FeO2 composite with a crystallite size of ∼11 nm achieved a reversible capacity of 112 mA h g−1 after 50 cycles. The results highlight the importance of the silver ferrite–maghemite composite and crystallite size on electrochemical performance.
5. Epilogue
Material synthesis and the preparation of nanocomposites are keenly important in determining the function of materials. This review is structured largely as a case study, where the structure/function relationships among magnetite (Fe3O4) crystallite size, nanocomposites of Fe3O4, and battery-relevant electrochemistry are delineated. Following the case study, a brief discussion of the direct synthesis and the electrochemistry of a 2D structured material, specifically, a new nanocomposite based on silver ferrite–maghemite, AgFeO2–γFe2O3, is included. The case study illustrates through a review of the literature how structure/function relationships impacting battery relevant electrochemistry continue to evolve, while the shorter discussions of less studied AgFeO2–γFe2O3 illustrate the growing need for future exploration of structure/function relationships involving battery-relevant electrochemistry through synthetic control of new electroactive materials and composites. As new battery materials and new strategies for battery technologies evolve, the critical role of crystallite size on battery electrochemistry will likely continue as a paradigm for the next generation of batteries.Acknowledgements
The preparation of this manuscript was supported as part of the Center for Mesoscale Transport Properties, an Energy Frontier Research Center supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award #DE-SC0012673. C.N.G. acknowledges support from the National Science Foundation funded Research Experience for Undergraduates Site: Nanotechnology for Health, Energy and the Environment at Stony Brook University. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.References
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800 CrossRef CAS PubMed.
- H. Kumar, S. Rajan and A. K. Shukla, Sci. Prog., 2012, 95, 283–314 CrossRef CAS PubMed.
- S. Flandrois and B. Simon, Carbon, 1999, 37, 165–180 CrossRef CAS.
- L. W. Ji, Z. Lin, M. Alcoutlabi and X. W. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- J. W. Fergus, J. Power Sources, 2010, 195, 939–954 CrossRef CAS.
- M. M. Huie, R. A. DiLeo, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, ACS Appl. Mater. Interfaces, 2015, 7, 11724–11731 CAS.
- R. A. Di Leo, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, Electrochim. Acta, 2013, 109, 27–32 CrossRef CAS.
- R. A. DiLeo, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, J. Electrochem. Soc., 2013, 160, A1399–A1405 CrossRef CAS.
- Y. J. Kim, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, J. Power Sources, 2011, 196, 6781–6787 CrossRef CAS PubMed.
- D. C. Bock, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, J. Power Sources, 2013, 231, 219–225 CrossRef CAS PubMed.
- D. C. Bock, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, Dalton Trans., 2013, 42, 13981–13989 RSC.
- D. C. Bock, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, Phys. Chem. Chem. Phys., 2015, 17, 2034–2042 RSC.
- D. C. Bock, R. V. Tappero, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, ACS Appl. Mater. Interfaces, 2015, 7, 5429–5437 CAS.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783–789 CrossRef CAS.
- A. Rougier, I. Saadoune, P. Gravereau, P. Willmann and C. Delmas, Solid State Ionics, 1996, 90, 83–90 CrossRef CAS.
- S. M. Dou, J. Solid State Electrochem., 2013, 17, 911–926 CrossRef CAS.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., R, 2012, 73, 51–65 CrossRef CAS.
- S.-T. Myung, K. Amine and Y.-K. Sun, J. Power Sources, 2015, 283, 219–236 CrossRef CAS.
- R. Malini, U. Uma, T. Sheela, M. Ganesan and N. G. Renganathan, Ionics, 2009, 15, 301–307 CrossRef CAS.
- L. W. Su, Y. Jing and Z. Zhou, Nanoscale, 2011, 3, 3967–3983 RSC.
- X. R. Wang and G. Yushin, Energy Environ. Sci., 2015, 8, 1889–1904 CAS.
- Z. S. Wu, G. M. Zhou, L. C. Yin, W. Ren, F. Li and H. M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS.
- U. Kasavajjula, C. S. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS.
- M. C. Menard, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, Phys. Chem. Chem. Phys., 2013, 15, 18539–18548 RSC.
- W. D. Nesse, Introduction to Mineralogy, Oxford University Press Inc., Oxford, 2nd edn, 2012 Search PubMed.
- V. Sivakumar, S. Kumar, C. A. Ross and Y. Shao-Horn, IEEE Trans. Magn., 2007, 43, 3121–3123 CrossRef CAS.
- M. M. Thackeray, W. I. F. David and J. B. Goodenough, Mater. Res. Bull., 1982, 17, 785 CrossRef CAS.
- T. Yamada, K. Morita, K. Kume, H. Yoshikawa and K. Awaga, J. Mater. Chem. C, 2014, 2, 5183–5188 RSC.
- K. W. Knehr, N. W. Brady, C. A. Cama, D. C. Bock, Z. Lin, C. N. Lininger, A. C. Marschilok, K. J. Takeuchi, E. S. Takeuchi and A. C. West, J. Electrochem. Soc., 2015, 162, A2817–A2826 CrossRef CAS.
- K. W. Knehr, N. W. Brady, C. N. Lininger, C. A. Cama, D. C. Bock, Z. Lin, A. C. Marschilok, K. J. Takeuchi, E. S. Takeuchi and A. C. West, ECS Trans., 2015, 69, 7–19 CrossRef.
- S. Komaba, T. Mikumo and A. Ogata, Electrochem. Commun., 2008, 10, 1276–1279 CrossRef CAS.
- S. Komaba, T. Mikumo, N. Yabuuchi, A. Ogata, H. Yoshida and Y. Yamada, J. Electrochem. Soc., 2010, 157, A60–A65 CrossRef CAS.
- S. Zhu, A. C. Marschilok, E. S. Takeuchi and K. J. Takeuchi, Electrochem. Solid-State Lett., 2009, 12, A91–A94 CrossRef CAS.
- S. Zhu, A. C. Marschilok, E. S. Takeuchi, G. T. Yee, G. Wang and K. J. Takeuchi, J. Electrochem. Soc., 2010, 157, A1158–A1163 CrossRef CAS.
- M. C. Menard, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, Electrochim. Acta, 2013, 94, 320–326 CrossRef CAS.
- D. C. Bock, A. C. Marschilok, K. J. Takeuchi, E. S. Takeuchi, K. C. Kirshenbaum, J. Wang, W. Zhang, F. Wang, J. Wang, E. S. Takeuchi, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, ACS Appl. Mater. Interfaces, 2015, 7, 13457–13466 CAS.
- C. W. Kwon, M. Quintin, S. Mornet, C. Barbieri, O. Devos, G. Campet and M. H. Delville, J. Electrochem. Soc., 2004, 151, A1445–A1449 CrossRef CAS.
- G. F. Goya, T. S. Berquo, F. C. Fonseca and M. P. Morales, J. Appl. Phys., 2003, 94, 3520–3528 CrossRef CAS.
- A. G. Roca, J. F. Marco, M. d. P. Morales and C. J. Serna, J. Phys. Chem. C, 2007, 111, 18577–18584 CAS.
- J. Santoyo Salazar, L. Perez, O. de Abril, L. Truong Phuoc, D. Ihiawakrim, M. Vazquez, J.-M. Greneche, S. Begin-Colin and G. Pourroy, Chem. Mater., 2011, 23, 1379–1386 CrossRef CAS.
- S. J. Iyengar, M. Joy, C. K. Ghosh, S. Dey, R. K. Kotnala and S. Ghosh, RSC Adv., 2014, 4, 64919–64929 RSC.
- C. Piquer, M. A. Laguna-Marco, A. G. Roca, R. Boada, C. Guglieri and J. Chaboy, J. Phys. Chem. C, 2014, 118, 1332–1346 CAS.
- F. X. Ma, H. Hu, H. B. Wu, C. Y. Xu, Z. Xu, L. Zhen and X. W. David Lou, Adv. Mater., 2015, 27, 4097 CrossRef CAS PubMed.
- Q. Q. Xiong, J. P. Tu, Y. Lu, J. Chen, Y. X. Yu, Y. Q. Qiao, X. L. Wang and C. D. Gu, J. Phys. Chem. C, 2012, 116, 6495–6502 CAS.
- H.-S. Lim, B.-Y. Jung, Y.-K. Sun and K.-D. Suh, Electrochim. Acta, 2012, 75, 123–130 CrossRef CAS.
- D. Su, J. Horvat, P. Munroe, H. Ahn, A. R. Ranjbartoreh and G. Wang, Chem. – Eur. J., 2012, 18, 488 CrossRef CAS PubMed.
- Q. Wang, D. Chen, J. Chen, C. Lai, L. Li and C. Wang, Mater. Lett., 2015, 141, 319–322 CrossRef CAS.
- A. S. Fialkov, Russ. J. Electrochem., 2000, 36, 345–366 CrossRef CAS.
- Y. Deng, Q. Zhang, Z. Shi, L. Han, F. Peng and G. Chen, Electrochim. Acta, 2012, 76, 495–503 CrossRef CAS.
- K. Zhang, W. Zhao, J.-T. Lee, G. Jang, X. Shi and J. H. Park, J. Mater. Chem. A, 2014, 2, 9636–9644 CAS.
- J. Zai, C. Yu, Q. Zou, L. Tao, K. Wang, Q. Han, B. Li, Y. Xiao, X. Qian and R. Qi, RSC Adv., 2012, 2, 4397–4403 RSC.
- J. Zhou, H. Song, L. Ma and X. Chen, RSC Adv., 2011, 1, 782–791 RSC.
- A. Hu, X. Chen, Y. Tang, L. Yang, H. Xiao and B. Fan, Mater. Lett., 2013, 91, 315–318 CrossRef CAS.
- C.-T. Hsieh, J.-Y. Lin and C.-Y. Mo, Electrochim. Acta, 2011, 58, 119–124 CrossRef CAS.
- S. Wang, J. Zhang and C. Chen, J. Power Sources, 2010, 195, 5379–5381 CrossRef CAS.
- R. Ravikumar and S. Gopukumar, J. Power Sources, 2015, 289, 146–153 CrossRef CAS.
- J. Zhao, B. Yang, Z. Zheng, J. Yang, Z. Yang, P. Zhang, W. Ren and X. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 9890–9896 CAS.
- Q. Guo, X. Guo, K. Du, H. Ge and F. Wang, Int. J. Hydrogen Energy, 2015, 40, 1846–1851 CrossRef CAS.
- T. Yoon, J. Kim, J. Kim and J. K. Lee, Energies, 2013, 6, 4830–4840 CrossRef CAS.
- M. Sathish, T. Tomai and I. Honma, J. Power Sources, 2012, 217, 85–91 CrossRef CAS.
- X. Hu, M. Ma, M. Zeng, Y. Sun, L. Chen, Y. Xue, T. Zhang, X. Ai, R. G. Mendes, M. H. Rummeli and L. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 22527–22533 CAS.
- E. Kang, Y. S. Jung, A. S. Cavanagh, G.-H. Kim, S. M. George, A. C. Dillon, J. K. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 2430–2438 CrossRef CAS.
- F. Wu, R. Huang, D. Mu, B. Wu and S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19254–19264 CAS.
- Y. Piao, H. S. Kim, Y.-E. Sung and T. Hyeon, Chem. Commun., 2010, 46, 118–120 RSC.
- J. P. Cheng, J. Yu, D. Shi, D. S. Wang, Y. F. Liu, F. Liu, X. B. Zhang and J. G. Li, Appl. Phys. A: Mater. Sci. Process., 2012, 106, 837–842 CrossRef CAS.
- N. Bi, X. Liu, N. Wu, C. Cui and Y. Sun, RSC Adv., 2015, 5, 32452–32459 RSC.
- S.-D. Seo, D.-H. Lee, H.-W. Shim, S. Lee and D.-W. Kim, J. Am. Ceram. Soc., 2014, 97, 1413–1420 CrossRef CAS.
- H. S. Kim, S. G. Kwon, S. H. Kang, Y. Piao and Y.-E. Sung, Electrochim. Acta, 2014, 136, 47–51 CrossRef CAS.
- C. Lei, F. Han, D. Li, W.-C. Li, Q. Sun, X.-Q. Zhang and A.-H. Lu, Nanoscale, 2013, 5, 1168–1175 RSC.
- D.-Y. Park and S.-T. Myung, ACS Appl. Mater. Interfaces, 2014, 6, 11749–11757 CAS.
- H. Xia, Y. Wan, G. Yuan, Y. Fu and X. Wang, J. Power Sources, 2013, 241, 486–493 CrossRef CAS.
- T. Zhu, J. S. Chen and X. W. Lou, J. Phys. Chem. C, 2011, 115, 9814–9820 CAS.
- H. Liu, G. Wang, J. Wang and D. Wexler, Electrochem. Commun., 2008, 10, 1879–1882 CrossRef CAS.
- W.-M. Zhang, X.-L. Wu, J.-S. Hu, Y.-G. Guo and L.-J. Wan, Adv. Funct. Mater., 2008, 18, 3941–3946 CrossRef CAS.
- M. Zhang, X. Yin, Z. Du, S. Liu, L. Chen, Q. Li, H. Jin, K. Peng and T. Wang, Solid State Sci., 2010, 12, 2024–2029 CrossRef CAS.
- J. Liu, J. Ni, Y. Zhao, H. Wang and L. Gao, J. Mater. Chem. A, 2013, 1, 12879–12884 CAS.
- J. E. Lee, S.-H. Yu, D. J. Lee, D.-C. Lee, S. I. Han, Y.-E. Sung and T. Hyeon, Energy Environ. Sci., 2012, 5, 9528–9533 CAS.
- W. Sheng, G. Zhu, D. L. Kaplan, C. Cao, H. Zhu and Q. Lu, Nanotechnology, 2015, 26, 115603 CrossRef PubMed.
- Y. Luo, X. Zhou, Y. Zhong, M. Yang, J. Wei and Z. Zhou, Electrochim. Acta, 2015, 154, 136–141 CrossRef CAS.
- M. Li, W. Wang, M. Yang, F. Lv, L. Cao, Y. Tang, R. Sun and Z. Lu, RSC Adv., 2015, 5, 7356–7362 RSC.
- K. E. Farley, A. C. Marschilok, E. S. Takeuchi and K. J. Takeuchi, Electrochem. Solid-State Lett., 2012, 15, A23–A27 CrossRef CAS.
- J. L. Durham, K. Kirshenbaum, E. S. Takeuchi, A. C. Marschilok and K. J. Takeuchi, Chem. Commun., 2015, 51, 5120–5123 RSC.
- D. Su, H.-J. Ahn and G. Wang, J. Power Sources, 2013, 244, 742–746 CrossRef CAS.
- T. Muraliganth, A. V. Murugan and A. Manthiram, Chem. Commun., 2009, 7360–7362, 10.1039/b916376j.
- E. S. Takeuchi, A. C. Marschilok, K. J. Takeuchi, A. Ignatov, Z. Zhong and M. Croft, Energy Environ. Sci., 2013, 6, 1465–1463 CAS.
- C. Ban, Z. Wu, D. T. Gillaspie, L. Chen, Y. Yan, J. L. Blackburn and A. C. Dillon, Adv. Energy Mater., 2010, 22, E145–E149 CrossRef CAS PubMed.
- C. He, S. Wu, N. Zhao, C. Shi, E. Liu and J. Li, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS PubMed.
- P. C. Lian, X. F. Zhu, H. F. Xiang, Z. Li, W. S. Yang and H. H. Wang, Electrochim. Acta, 2010, 56, 834–840 CrossRef CAS.
- G. M. Zhou, D. W. Wang, F. Li, L. L. Zhang, N. Li, Z. S. Wu, L. Wen, G. Q. Lu and H. M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS.
- J. Z. Wang, C. Zhong, D. Wexler, N. H. Idris, Z. X. Wang, L. Q. Chen and H. K. Liu, Chem. – Eur. J., 2011, 17, 661–667 CrossRef CAS PubMed.
- Y. He, L. Huang, J. S. Cai, X. M. Zheng and S. G. Sun, Electrochim. Acta, 2010, 55, 1140–1144 CrossRef CAS.
- J. Cheng, B. Wang, C.-M. Park, Y. Wu, H. Huang and F. Nie, Chem. – Eur. J., 2013, 19, 9866–9874 CrossRef CAS PubMed.
- S. Yang, C. Cao, G. Li, Y. Sun, P. Huang, F. Wei and W. Song, Nano Res., 2015, 8, 1339–1347 CrossRef CAS.
- Y. Wu, Y. Wei, J. Wang, K. Jiang and S. Fan, Nano Lett., 2013, 13, 818–823 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |