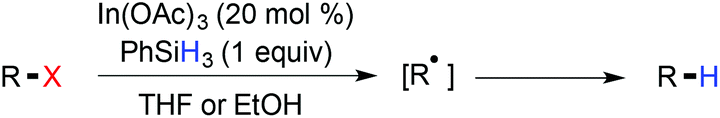DOI:
10.1039/C5OB01949D
(Review Article)
Org. Biomol. Chem., 2016,
14, 21-35
Metal catalyzed defunctionalization reactions
Received
19th September 2015
, Accepted 27th October 2015
First published on 27th October 2015
Abstract
Defunctionalization has a direct impact on the synthesis of value added products (e.g. biomass degradation). In synthetic chemistry it enables the functional group to act as a transient directing group. In this mini review, we have described the chronological development of metal assisted defunctionalization reactions from the stoichiometric to the catalytic stage with their application in synthetic organic chemistry. The proposed catalytic cycles of the transformations have been described to make this review comprehensible.
 Atanu Modak | Atanu Modak was born in West Bengal, India. Following his graduation from Ramakrishna Mission Vidyamandira, Belur (Calcutta University) in 2009, he pursued an M.Sc. in chemistry from Indian Institute of Technology, Kanpur (India) in 2011. Presently he is a PhD student in Prof. D. Maiti's lab from January, 2012. |
 Debabrata Maiti | Prof. Debabrata Maiti received his PhD from Johns Hopkins University (USA) in 2008 under the supervision of Prof. Kenneth D. Karlin. After his postdoctoral studies at Massachusetts Institute of Technology (MIT) with Prof. Stephen L. Buchwald (2008–2010), he joined the department of chemistry of IIT Bombay in 2011. His research interests are focused on the development of new and sustainable synthetic methodologies. |
1. Introduction
Defunctionalization reactions have become an indispensable aspect in the advancement of chemical synthesis despite affording a less functionalized entity from a more functionalized organic material. The key challenge of the defunctionalization reaction lies in the high bond dissociation energy of the C–X (X = carbon or heteroatom) bond. Metal mediated methods emerged as a savior in addressing this issue as the metal destabilizes the C–X bond through single or two electron transfer processes. Moreover, the remarkable electrophilicity of the metal allows them to form the metal carbanion complex which is the key intermediate of defunctionalization as well as cross coupling reactions. In the case of the defunctionalization reaction the metal carbanion intermediate is intercepted by a hydrogen equivalent to result in the required defunctionalized product, whereas in the cross coupling reaction the other coupling partner comes into the picture. Therefore, defunctionalization has an eminent significance in the functionalization reaction in terms of a mechanistic point of view (Scheme 1).
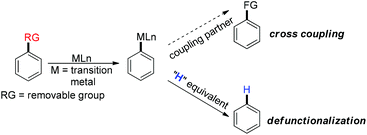 |
| | Scheme 1 Schematic diagram of transition metal mediated defunctionalization. | |
Besides, defunctionalization reactions also have a direct impact in synthetic organic chemistry, facilitating functional groups to be utilized temporarily in a particular synthetic transformation.1,2 In some diverse aspects, defunctionalized substrates are considered more useful than their functionalized form e.g. preparation of useful materials through biomass degradation,3–9 converting environmentally hazardous entities to relatively harmless forms through desulfurization10,11 or dechlorination12–16etc.
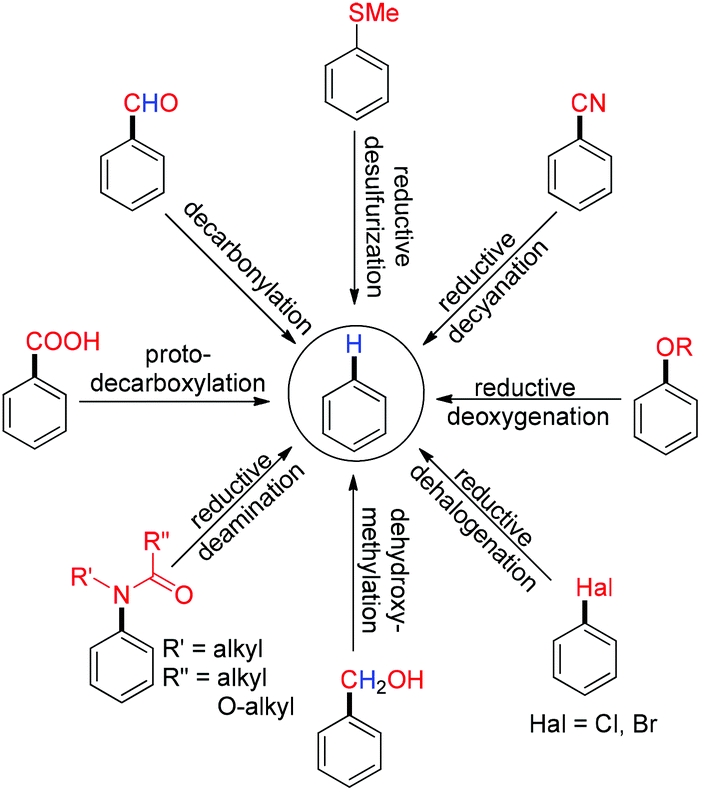
In this mini review, we have highlighted the chronological development of metal assisted defunctionalization reactions from the stoichiometric to the catalytic stage with their application in synthetic organic chemistry (Scheme 2).
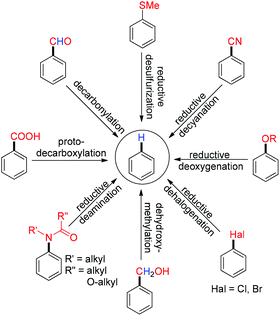 |
| | Scheme 2 Scope of defunctionalization. | |
2. Decarboxylation
Decarboxylation of aromatic carboxylic acids has a great significance in the up-gradation of coal derived substrates, as aromatic carboxylic acids are the major air oxidized products of coal.17 In synthetic chemistry, the carboxylic moiety serves as the directing group through metal-chelation assisted C–H activation or electronically assisted aromatic functionalization. Therefore, an efficient decarboxylation of aromatic carboxylic acids can make the carboxylic group a traceless directing group in aromatic C–H functionalization. A number of metal mediated/catalyzed methods are known for the effective protodecarboxylation of carboxylic acids which occurred through a metal assisted anionic or radical decarboxylation.
2.1 Mercury mediated decarboxylation
Mercury mediated decarboxylation of aromatic carboxylates has been known for a long time. In 1901, Pesci first reported the mercury mediated decarboxylation of an aromatic anhydride to produce an aromatic mono acid.18 Since then a number of groups have worked on mercury as a decarboxylating agent.19–23 Unfortunately, all of these methods involve high reaction temperatures, use of large amounts of toxic metals or a long reaction time. Recently in 2006 Moseley and coworkers reported the mercury mediated decarboxylation of naphthoic anhydride at a faster reaction rate under microwave conditions (Scheme 3).24
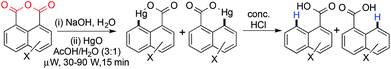 |
| | Scheme 3 Mercury mediated decarboxylation of naphthoic anhydride under microwave conditions. | |
2.2 Copper salt as a decarboxylating agent
After mercury, copper was the second metal which was exploited for effective protodecarboxylation. The first copper mediated decarboxylation was performed in 1930 by Shepard and coworkers using a stoichiometric copper–quinoline combination.25 They successfully decarboxylated halogenated furan carboxylic acid at very high temperature. Later, the decarboxylation method was further developed by the Nilsson,26 Cohen27,28 and Sheppard29 groups for activated aromatic carboxylic acids. According to their study an N-heterocyclic amine type solvent/ligand is essential for stabilization of the copper-decarboxylate intermediate. In 2007, Gooßen and coworkers for the first time reported the copper catalyzed decarboxylation of deactivated carboxylic acids with the help of a bis-N-chelating ligand at 170 °C.30 Decarboxylation of deactivated acids was achieved by introducing a sterically hindered ligand like 4,7-diphenyl-1,10-phenanthroline. A quick version of the protodecarboxylation protocol was also developed by the Goossen group under microwave conditions with a catalytic amount of Cu/phenanthroline.31 According to the mechanistic study and DFT calculation, it was proposed that copper directly inserts into the aryl carboxylate bond and then the hydrolytic cleavage of the copper–carbon bond results in the protodecarboxylated product (Scheme 4).30
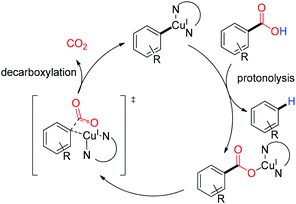 |
| | Scheme 4 Catalytic cycle of the decarboxylation in a copper–phenanthroline system. | |
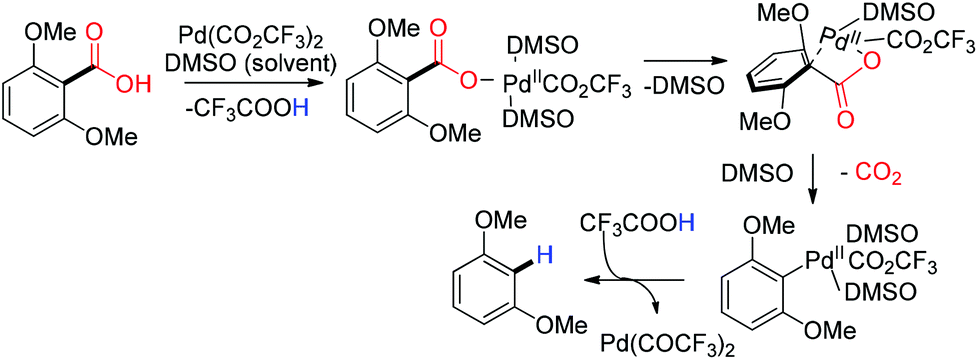
2.3 Palladium catalyzed decarboxylation
Schleyer successfully achieved decarboxylation in the gas phase at a very high temperature with a nickel (180 °C) and palladium (330 °C) catalyst under a hydrogen gas atmosphere.32 Later in 2004 Matsubara and coworkers reported a palladium/charcoal catalyzed hydrothermal condition for decarbonylation and decarboxylation.33 A high deuterium containing entity ensued from a completely non-deuterated substrate due to H–D exchange under their hydrothermal reaction conditions using deuterium oxide as the solvent.33 In 2007 Kozlowski reported a palladium catalyzed method for the decarboxylation of electron rich bis-ortho-substituted aromatic carboxylic acids using trifluoroacetic acid as the proton source.34 In the reaction mechanism, a four-membered palladium species was proposed as an intermediate prior to the decarboxylation (Scheme 5).35
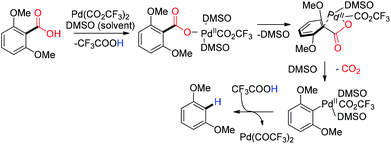 |
| | Scheme 5 Proposed mechanism for palladium catalyzed decarboxylation of bis-ortho-substituted aromatic carboxylic acid. | |
2.4 Ruthenium catalyzed directed decarboxylation
Murai and coworkers in 2001 demonstrated the chelation assisted decarboxylation of esters using the Ru3(CO)12 catalyst through acyl C–O bond fission (Scheme 6).36 According to their proposed reaction mechanism, a heteroatom containing directing group stimulates the metal center for site-selective cleavage of the acyl C–O bond through a tetrahedral intermediate followed by a fast decarbonylation yielding the resultant hydrocarbon (Scheme 6).
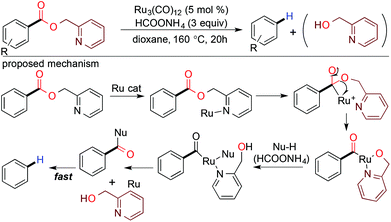 |
| | Scheme 6 Chelation assisted decarboxylation of esters. | |
2.5 Silver catalyzed decarboxylation
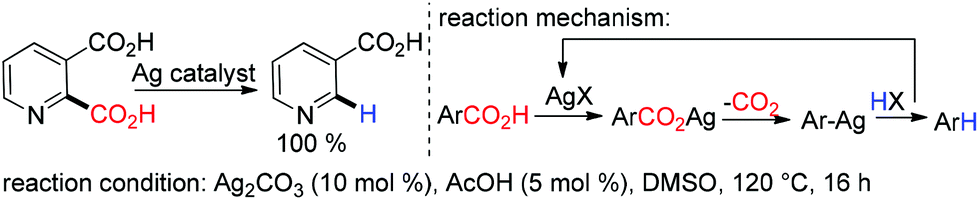
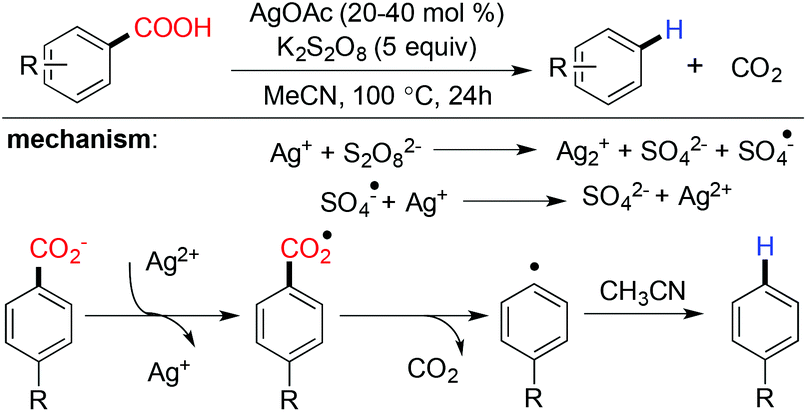
Silver catalyzed protodecarboxylation of heteroaromatic carboxylic acids was explored by Larossa and coworkers in 2009.37 From their substrate scope study it was observed that the α heteroatom is a prerequisite for decarboxylation (Scheme 7). In 2012 they nicely showed that a deutero-substrate can be prepared through deuteron-decarboxylation of the corresponding carboxylic acid using D2O as the D+ source.38 In the same year, Greaney and coworkers established a new approach for protodecarboxylation under oxidative radical conditions using catalytic Ag(I)/K2S2O8 (Scheme 8).39 A variety of benzoic acid derivatives with electron rich and electron donating functional groups were decarboxylated successfully. According to their hypotheses, first Ag(I) is in situ oxidized to Ag(II) in the presence of K2S2O8, later the carboxylate anion is oxidized to the carboxylated radical by in situ generated Ag(II). After that, a successful decarboxylation yielded an aryl radical from the carboxylate radical. Finally, the aryl radical abstracted a hydrogen radical from acetonitrile to deliver the protodecarboxylated product.
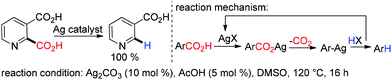 |
| | Scheme 7 Silver catalyzed decarboxylation of hetero-aromatic carboxylic acid. | |
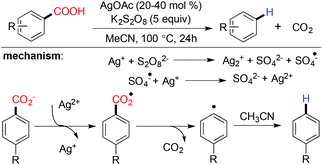 |
| | Scheme 8 Silver catalyzed protodecarboxylation under oxidative radical conditions. | |
2.6 Rhodium catalyzed decarboxylation
A rhodium catalyzed protodecarboxylation was demonstrated by Zhao and coworkers using (cod)Rh(μ-OH)2 as a catalyst in 2010.40 Unfortunately, their substrate scope was limited to ortho-substituted arenecarboxylic acids. Non-activated benzoic acid and ortho-methoxy benzoic acid remained unreactive under their optimized conditions.
2.7 Gold mediated decarboxylation
In 2011, Nolan and coworkers first explored the gold mediated decarboxylation of aromatic carboxylic acids with an N-heterocyclic carbene–gold(I) complex.41 After their initial breakthrough, in 2013 they reported the first gold catalyzed protodecarboxylation of aromatic carboxylic acids.42 Both electron-rich and electron-poor benzoic acid derivatives were successfully employed for decarboxylation. According to the reaction mechanism, protodecarboxylation takes place through a gold carboxylate and gold aryl intermediate (Scheme 9).
 |
| | Scheme 9 Gold catalyzed protodecarboxylation. | |
2.8 Decarboxylation under photochemical conditions
Under photochemical conditions decarboxylation of aliphatic acids was achieved by Bakac and coworkers.43 In the presence of the Fe3+ ion, an aqueous solution of propionic acid was converted to a mixture of hydrocarbons and CO2 by near UV photolysis using anaerobic conditions (Scheme 10).
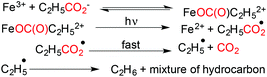 |
| | Scheme 10 Decarboxylation under photochemical conditions. | |
3. Decarbonylation
Transition metal catalyzed decarbonylation of aldehydes has a great importance in synthetic as well as biological chemistry. In a biological system, the cyanobacterial aldehyde decarbonylase enzyme causes the decarbonylation of a fatty aldehyde resulting in the corresponding alkane or alkene and formic acid.44 The synthetic alternative methods of such a reaction can solve the energy crisis producing low boiling point fuel. In terms of the reaction pathway, the decarbonylation of aldehydes in synthetic chemistry differs from nature as it emits carbon monoxide instead of formic acid. On the other hand, the aldehyde group possesses worthwhile features like the ortho directing ability in aromatic systems, increasing the electrophilicity of dienophiles for facile addition reaction, in situ carbon monoxide generation etc. In this regard, the decarbonylation of aldehydes has a potential significance in organic synthesis for utilizing the aldehyde group temporarily in a particular transformation.
3.1 Palladium as a decarbonylating agent
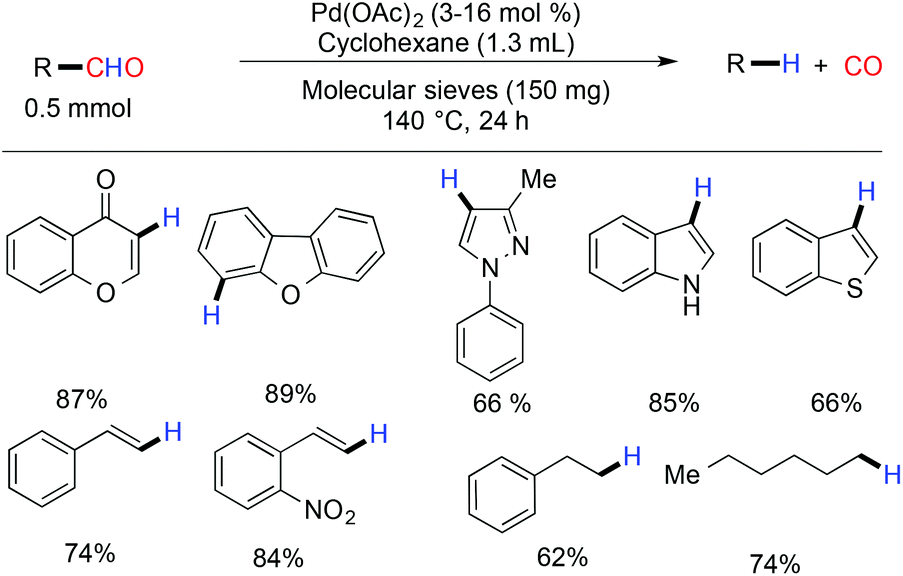
The first decarbonylation of an aldehyde was done in 1960 by Wilt with heterogeneous Pd/charcoal at a high temperature (more than 180 °C).45 A few years later, in 1965 Tsuji and coworkers reported the palladium catalyzed decarbonylation of an acylchloride and an aldehyde, but this protocol is limited due to the high temperature requirement.46 Recently, Maiti and coworkers demonstrated a palladium acetate catalyzed efficient, mild and ligand free protocol for the decarbonylation of aldehydes.47 The reaction exhibited tolerance against a wide range of functional groups such as cyano, ester, keto, nitro and acid. Heterocyclic aldehydes with a single heteroatom and two heteroatoms gave good yields of the corresponding decarbonylated products. It was observed that there was no trace of the β–H eliminated side product formed against simple alkyl aldehydes (Scheme 11). Under the microwave conditions, the decarbonylation of the aldehyde was also reported by the same group.48 A biologically active molecule eulatachromene was synthesized by utilizing the deformylation strategy judiciously (Scheme 12).48 The same strategy was also used by the Bräse group, where they used the rhodium catalyzed method for decarbonylation.49
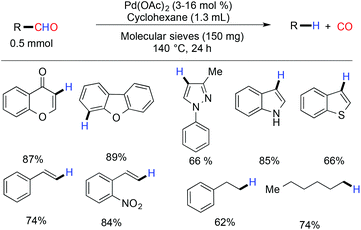 |
| | Scheme 11 Palladium acetate catalyzed decarbonylation. | |
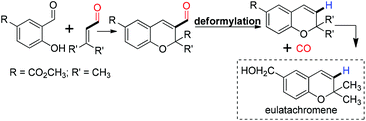 |
| | Scheme 12 Synthesis of eulatachromene. | |
In 2013, Fu and co-workers reported a heterogeneous decarbonylation of biomass derived feedstocks, 5-hydroxymethylfurfural (HMF) to furfuryl alcohol (FFA) with catalytic palladium nanoparticles under mild conditions (Scheme 13). Furfuryl alcohol has a great significance in the preparation of furan based light fuels.4 Later, in 2015 Thomas Rauchfuss and co-workers demonstrated the Pd/C catalyzed decarbonylation of a furfural derivative with the intention to upgrade cellulosic biomass.5
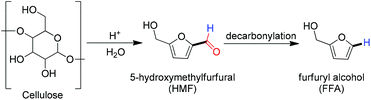 |
| | Scheme 13 Furfuryl alcohol synthesis from cellulose by decarbonylation of aldehyde. | |
3.2 Rhodium as a decarbonylating agent
Rhodium is most abundantly used in decarbonylation reactions compared to other metals. The first report on rhodium as a decarbonylating agent was communicated in 1965 by Tsuji and Ohno, using a stoichiometric amount of Willkinson's catalyst.50 Later, the catalytic decarbonylation of an aldehyde was reported by Doughty and Pignolet in 1978 with Rh(dpp)Cl as the catalyst.51 In 1992 O'Connor demonstrated a rhodium catalyzed efficient method for the decarbonylation of a primary aldehyde, where diphenylphosphoryl azide acted as a carbon monoxide trap.52 Crabtree and coworkers in 1999 discovered yet another efficient way of decarbonylation of primary and aryl aldehydes under mild conditions using the catalytic [Rh(CO)(triphos)][SbF6] complex.53 In 2006, the Madsen group reported a method for the deformylation of aldehydes with the combination of RhCl3·3H2O and the phosphine ligand with a wide substrate scope.54
Following experimental and computational studies they proposed a catalytic cycle for the decarbonylation of aldehydes, where the aldehyde first oxidatively adds to the metal center.55 In the next step, migratory extrusion occurs and then reductive elimination results in the defunctionalized product. Subsequently, the substrate binds with the metal center by the oxygen donor site, replacing the carbon monoxide and repeats the catalytic cycle. The migratory extrusion step is established as the turnover limiting step (Scheme 14). Madsen also applied the decarbonylation strategy to the unprotected aldose using a catalytic amount of Rh(dppp)2Cl and obtained alditol which is one carbon lesser than the starting material.6 This one step degradation of aldose through the decarbonylation reaction has a great significance in glycochemistry. Recently, Kappe and coworkers engineered a continuous-flow protocol for the decarbonylation of aldehydes using a catalytic amount of Rh(OAc)2/dppe.56
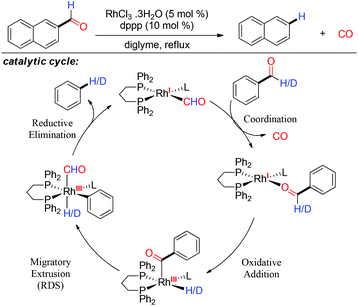 |
| | Scheme 14 Catalytic cycle for rhodium catalyzed decarbonylation of aldehyde. | |
3.3 Decarbonylation using other metals
After palladium and rhodium, ruthenium also proved to be capable for decarbonylation reactions. In 1980, Dolphin and co-workers discovered a ruthenium–porphyrin based complex for stoichiometric decarbonylation.57 An iridium catalyzed decarbonylation method was reported by Tsuji and coworkers in 2008, wherein a variety of functional groups were tolerated under their mild reaction conditions.58
4. Desulfurization
Reductive desulfurization is one of the key steps in manufacturing nonpolluting fuel from natural resources. In view of synthetic organic chemistry, the reductive desulfurization has a great significance owing to temporary advantages coming from the sulfur moiety on its neighboring atoms.59 There are two strategies for reductive desulphurization, one is the direct desulfurization of thio ether and the other being a two-step process where first thio ether gets oxidized to sulfone or sulfoxide followed by the reductive cleavage of the sulfur–carbon bond. The former strategy is more challenging owing to the higher C–S bond dissociation energy in thio ether compared to that in sulfone. The desulfurization of the sulfone moiety has been achieved through simple reduction in the presence of a hydride source. But, in the case of direct desulphurization of thio ether, special conditions are required for activating the C–S bond (Scheme 15). A number of transition metal mediated methodologies have been reported for direct desulfurization of thio ethers. In this review we mainly focused on the desulfurization of (hetero)aromatic thio ethers which occurs through the metal catalyzed reductive cleavage of the C(sp2)–SR bond.
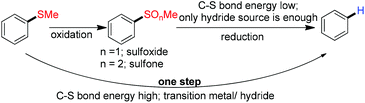 |
| | Scheme 15 Schematic diagram of reductive desulfurization of phenyl methyl thioether. | |
4.1 Nickel as a desulfurizing agent
In 1940 Mozingo and coworkers first reported the desulfurization of thio ether and S-containing amino acids using RANEY® nickel.60 After that, several reports have been published on desulfurization using nickel containing reagents like RANEY® nickel, nickel/borohydride,61,62 nickel-containing complex reducing agent (NiCRA) etc.10 But, all these methods require a high metal/substrate ratio and also render poor chemoselectivity. In 2012, Martin and coworkers demonstrated a general and convenient protocol for cleaving the carbon–sulphur bond of aryl methyl thioethers with a catalytic amount of Ni(COD)2.63 They have also nicely depicted the synthetic applicability of the desulfurization reaction where an aryl sulfide was used as a temporary directing group (Scheme 16).
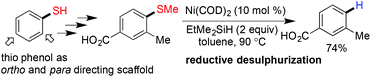 |
| | Scheme 16 Application of reductive desulfurization. | |
4.2 Palladium as a desulfurizing agent
The combination of catalytic palladium/charcoal and a hydride source is capable of reductive desulfurization under heterogeneous conditions. In 1994, hydrazine-Pd/C was used for catalytic desulfurization of methylthioether of pyridimine derivatives.64 Later Graham65 and Nakada66 independently developed the palladium catalyzed reductive desulfurization of alkyl aryl/heterocyclic thio-ethers using triethyl silane as a reducing agent. After their initial report, Nakada and coworkers developed an efficient method for the synthesis of imidazolinium salts by the Pd-catalyzed reduction of their corresponding thioureas with triethylsilane and trialkylsilyl triflate (Scheme 17).67
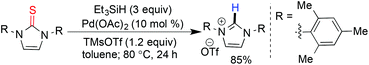 |
| | Scheme 17 Imidazolinium salt preparation through reductive desulfurization. | |
4.3 Rhodium as a desulfurizing agent
Recently in 2013, Willis and co-workers reported a rhodium catalyzed reductive desulfurization of ortho-keto aryl methyl sulfides under mild conditions. The excellent tolerance of the keto group enabled them to use the methyl sulfide moiety as a traceless directing scaffold for hydroacylation of alkanes and alkynes (Scheme 18).68
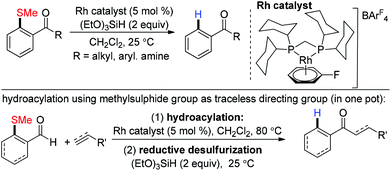 |
| | Scheme 18 Rhodium catalyzed reductive desulfurization of methyl phenyl thio ether. | |
4.4 Other metal mediated desulfurization
Except for palladium and nickel mediated reductive desulfurization protocols, RANEY® copper,69 zinc/HCl,70 red-Al,71,72 Al/HgCl2,73 and Zn/AcOH/Ac2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 74 are known. But, all these protocols are inadequate for broader use due to poor chemoselectivity, limited substrate scope, harsh reaction conditions, excess metal waste etc.
74 are known. But, all these protocols are inadequate for broader use due to poor chemoselectivity, limited substrate scope, harsh reaction conditions, excess metal waste etc.
5. Decyanation
An efficient methodology for removal of the cyano group permits exploration of the expedient feature of the cyano moiety temporarily in the synthesis of complex molecules. The cyano group has been widely used in chelation assisted C–H activation through an end on and side on directing fashion.75–79 Due to its high electron withdrawing ability, it can enhance the α-acidity for the desired organic transformation.80,81 Despite the high C–CN bond dissociation energy,82 decyanation has been achieved with the help of metal mediated reduction. Reductive decyanation with alkali metals is well known but these processes are limited due to the harsh reaction conditions used. In contrast, a transition metal complex/hydride source combination is accepted as a celebrated class for its milder and easily accessible reaction conditions. From a mechanistic point of view, there are two categories of the reaction pathway known for transition metal catalyzed C–CN bond cleavage.83 One of them is the direct insertion of a low valent metal in the C–CN bond through oxidative addition and the other being silyl metal-assisted carbon cyano bond cleavage through an iminoacyl intermediate (Scheme 19).
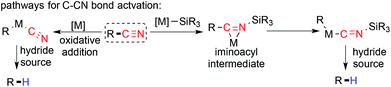 |
| | Scheme 19 Decyanation pathways. | |
5.1 Iron catalyzed reductive decyanation
In 2005, Nakazawa and co-workers developed an iron catalyzed C–CN bond cleavage in the presence of Et3SiH promoted by a photochemical pathway.84 Aliphatic and aromatic nitriles are successfully decyanated in their protocol. According to their proposed mechanistic pathway, Cp(CO)FeMe is generated by the photolysis of Cp(CO)2FeMe, and then Cp(CO)FeMe reacts with Et3SiH resulting in Cp(CO)FeMe(H)(SiEt3). Decyanated species was formed after a successive reductive elimination and yielded active species Cp(CO)Fe(SiEt3) which carried the catalytic cycle forward (Scheme 20).85
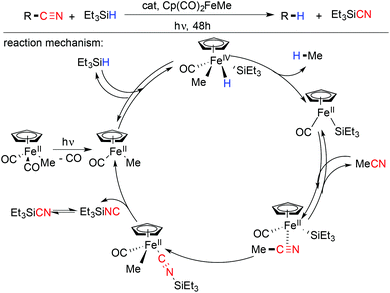 |
| | Scheme 20 Proposed mechanism for Cp(CO)2FeMe catalyzed reductive decyanation. | |
5.2 Rhodium catalyzed reductive decyanation
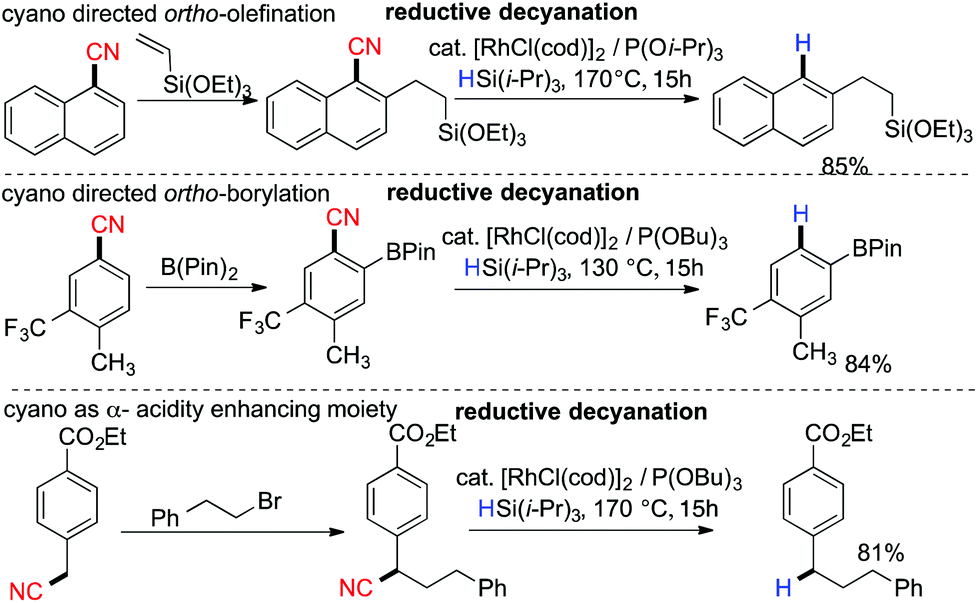
In 2009, Chatani and coworkers reported Rh-catalyzed reductive decyanation of nitriles using hydrosilane as the reducing agent.86 Utilization of hydrosilane as a mild reducing agent allowed a large functional group tolerance towards ethers, amines, amides and esters. In the case of primary alkyl cyanides, the complete suppression of β-hydrogen elimination was achieved using their catalytic system. They also demonstrated the cyano moiety as a transient steering group in regioselective functionalization (Scheme 21). An iminoacyl moiety was proposed as the key intermediate prior to C–CN bond cleavage.87
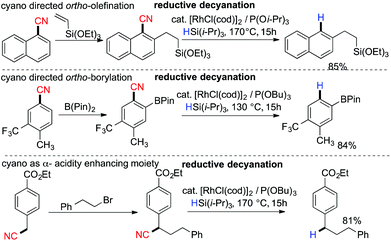 |
| | Scheme 21 Application of reductive decyanation as a traceless steering group. | |
5.3 Nickel catalyzed decyanation
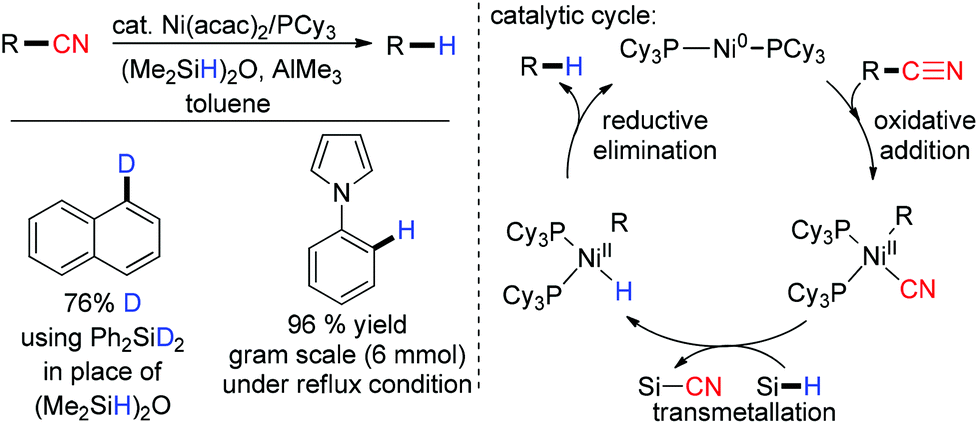
Recently in 2013, Maiti and co-workers studied the Ni-catalyzed decyanation of inert carbon–cyano bonds with (Me2SiH)2O as the hydride source (Scheme 22).88 It was found that alkyl, aryl and heteroaryl nitriles had been decyanated successfully. The direct insertion of the metal between the C–CN bond through oxidative addition was proposed. The resultant species was transmetalated with (Me2SiH)2O and finally reductive elimination yielded the decyanated compound. AlMe3 was used as a Lewis acid which formed an adduct with the nitrile moiety facilitating the oxidative addition of the C–CN bond (Scheme 22).
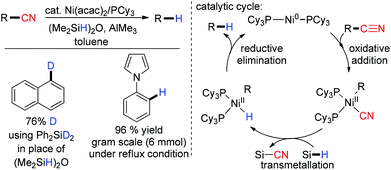 |
| | Scheme 22 Ni(acac)2 catalyzed reductive decyanation. | |
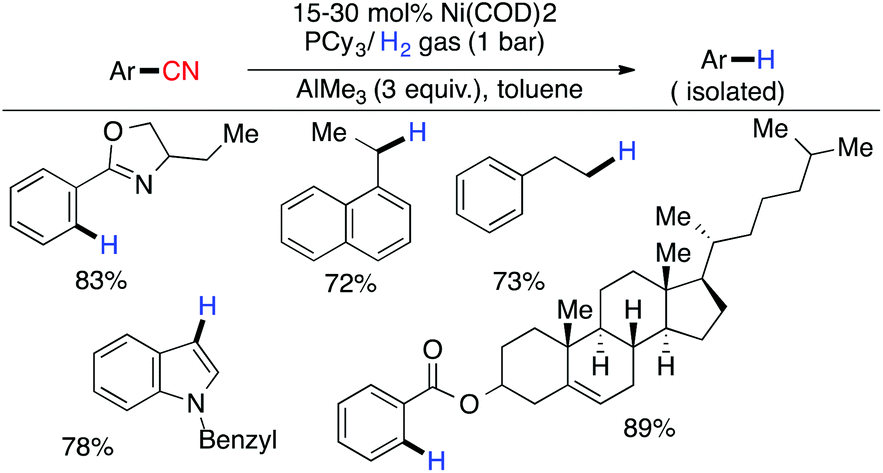
In the same year, Maiti and coworkers reported the Ni-catalyzed hydrogenolysis of inactivated carbon–cyano bonds using catalytic Ni(cod)2/PCy3 and AlMe3 (3 equiv.) under 1 bar pressure of H2 gas.89 The role of AlMe3 was not only to activate the inert C–CN bond for facile oxidative addition of Ni(cod)2 but also to assist in the removal of HCN produced under the reaction conditions (Scheme 23).
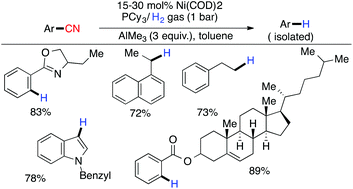 |
| | Scheme 23 Nickel catalyzed hydrogenolysis of an inactivated carbon–cyano bond. | |
Thereafter, Enthaler and coworkers developed a nickel–phosphine precatalyst, which is capable of catalytic hydrodecyanation in the presence of tert-butylmagnesium chloride as the hydride source.90 Later, the same group improved their catalytic system, replacing the hydride source with LiBH4.91
6. Deoxygenation of phenol
Deoxygenation of phenol derivatives through the transition metal catalyzed hydrogenolysis of the aromatic carbon–oxygen bond has a great significance in synthetic chemistry. The electron donating property of the phenol or ether linkage favors the ortho/para functionalization in an aromatic moiety.92 On the other hand, the ether moiety can also act as a directing scaffold through chelation assisted carbometallation.93,94 Thus, deoxygenation can be an interesting strategy for utilizing the beneficial characteristics of the phenol or ether moiety. On the other hand, an efficient deoxygenation protocol can provide a number of deoxygenated analogues of biologically active molecules as the phenol moiety is ubiquitous in natural products. The direct deoxygenation of phenol is quiet difficult due to the high C–O bond strength of phenol. So, the well-known pathway to remove the phenol group is to transform the phenol moiety to electronically poor ester groups like triflates, sulphonates, carbamates etc.95–97 Then, the metal mediated reductive cleavage of the C–O bond results in the deoxygenated product. In other words, the reductive cleavage of the C–O bond in the ether moiety is quite challenging. An efficient method for reductive dealkoxylation or aryloxylation has immense significance in biomass derived fuel production, as ether linkages are quite common in biomass, e.g. lignin.
6.1 Reductive dealkoxylation and dearyloxylation
Nickel has proven itself as the most appropriate metal for the reductive deoxygenation of ether for a long time. In 1935, Duzee and Adkins discovered the deoxygenation/hydrogenolysis of alkyl and aryl ethers in the presence of RANEY® nickel under an H2 atmosphere (150–250 atm).98 But, the disadvantages of this method lie in the uncontrolled hydrogenation of the aromatic ring before and after the hydrogenolysis of ethers. In 2010, Martin and coworkers reported the first metal catalyzed protocol for the reductive scission of inert C–O bonds of aryl methyl ethers.99 This methodology showed tolerance to a broad range of functional groups like silyl, esters, amides, acetals, amines, nitrogen containing heterocycles, etc. They also showed that regioselective demethoxylation can be achieved for a particular methoxy group, in the presence of more than one methoxy group in the substrate (Scheme 24).
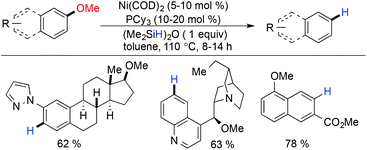 |
| | Scheme 24 Ni(COD)2 catalyzed demethoxylation of arylmethyl ethers. | |
Based on the experimental observation and theoretical calculations, they proposed a reaction mechanism for nickel catalyzed reductive demethoxylation, where at first Ni(COD)2 comproportionates to the key catalytic species Ni(I)−SiEt3 in the presence of PCy3, and Et3SiH. After that Ni(I)−SiEt3 coordinates with 2-methoxy naphthalene in an η2-fashion and following a migratory insertion results in the benzyl nickel species.100 Subsequently, a successive migration of the Ni center releases MeOSiR3. Finally a reversible σ-bond metathesis delivers the demethoxylated product and regenerates the catalytic cycle (Scheme 25).
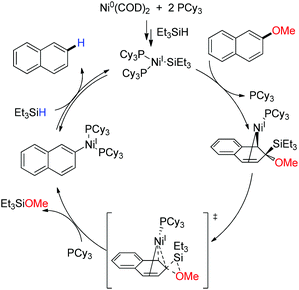 |
| | Scheme 25 Proposed mechanism for Ni(COD)2 catalyzed demethoxylation of arylmethyl ethers. | |
Chatani's group reported the reductive cleavage of aryl-oxygen bonds in alkoxy and pivaloxy arenes using a catalytic Ni(COD)2/PCy3 combination and HSiMe(OMe)2 as a mild reducing agent.101 Under the reaction conditions, the ethoxy and isopropoxy group had been cleaved, but the bulky alkoxy substituent showed lower reactivity. They also judiciously presented the importance of the reductive deoxygenation of aryl pivalates, by using the pivalate moiety as a transient directing group in regioselective aromatic functionalization (Scheme 26).
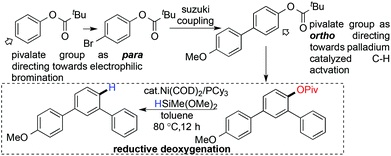 |
| | Scheme 26 Synthetic application of reductive deoxygenation. | |
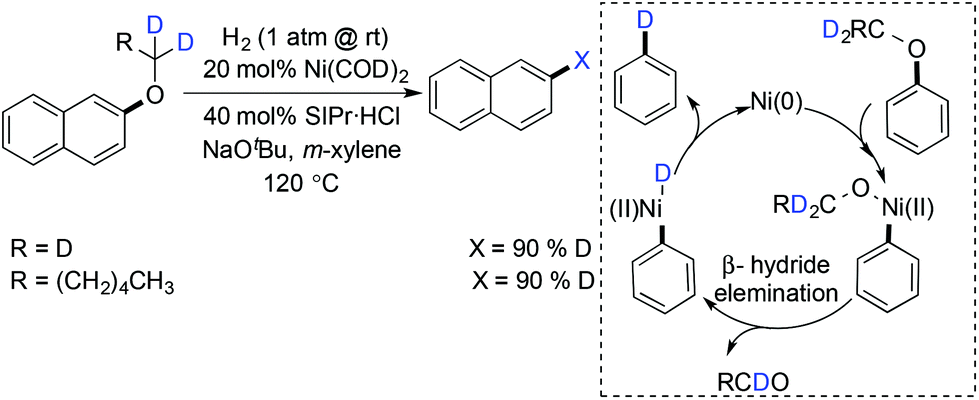
Hartwig and Sergeev in 2011 discovered the first hydrogenolysis of aryl ethers with catalytic Ni(COD)2 and SIPr·HCl as the ligand.7 A strong base, NaOtBu, was used for this transformation, which was thought to help in the C–O bond cleavage or dihydrogen activation forming anionic nickel complexes. The preferential scission of the C–O bond adjacent to the more electron-deficient aryl ring in unsymmetrical diaryl ethers had been attained under their hydrogenolysis conditions. The hydrogenolysis of aryl n-hexyl ethers and alkyl benzyl ethers was achieved successfully (Scheme 27).
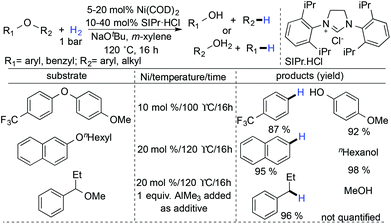 |
| | Scheme 27 Hydrogenolysis of aryl ethers with catalytic Ni(COD)2 and SIPr·HCl as the ligand. | |
A year later, the same group reported a ligand free hydrogenolysis of the C–O bond using Ni(CH2TMS)2(TMEDA) as the catalyst which was proven to be more active than their previously reported catalytic conditions.102 Agapie and coworkers studied the mechanism of the hydrogenolysis reaction of alkyl aryl ethers to gain knowledge about the source of hydrogen.103 The isotope labeling study showed that the α-hydrogen of the alkyl aryl ether was the hydrogen source for reduction in the Ni(COD)2-NHC catalytic system (Scheme 28). Recently, Chatani and coworkers developed external reductant free nickel catalyzed reaction conditions for the reductive deoxygenation of alkyl aryl ethers.104 Their proposed reaction mechanism is similar to the one reported by Agapie.103
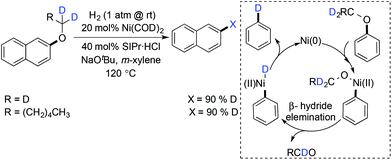 |
| | Scheme 28 Proposed mechanism for hydrogenation of aryl alkyl ether. | |
In 2013 Kawanami and coworkers reported a Rh/C catalyzed hydrogenolysis of diphenyl ether derivatives in the presence of H2 gas (0.5 MPa) in super critical CO2–water medium.105 Wang achieved the selective reductive cleavage of inert aryl C–O bonds by using a catalytic amount of Fe(acac)3 with LiAlH4 as the reducing agent and NaOtBu as the base.106 The experimental studies established that the catalytic reaction occurred through an active heterogeneous species. The same group showed that Co(acac)2 is also capable for the reductive cleavage of inert aryl C–O bonds under similar reaction conditions to the iron catalyzed reaction.107 Recently, Stephenson and coworkers reported the depolymerization of lignin under photolytic conditions using [Ir(ppy)2(dtbbpy)]PF6 as the photocatalyst.108
6.2 Reductive deoxygenation of the sulfonate ester of phenol
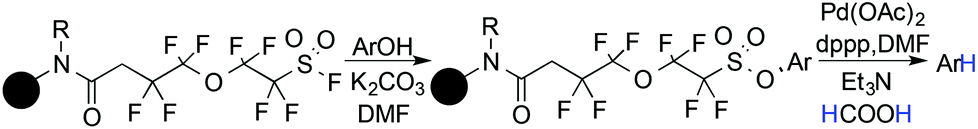
Palladium is the most acknowledged transition metal for the reductive deoxygenation of aryl sulfonate esters in the presence of a hydrogen source. Catalytic palladium on charcoal under a hydrogen atmosphere is one of the well-known reagent combinations for the hydrogenation of aryl nonaflates.109 Afterwards, the reductive deoxygenation of aryl triflates was reported by the Ortar,110 Wulff,111 Chen112 groups independently using trialkylammonium formate as the hydrogen donor in the presence of a catalytic amount of palladium salt/ phosphine ligand.113 Later, in 1989, Saa and his group showed that naphthyl triflate can be deoxygenated in the presence of a catalytic amount of (Ph3P)2PdCl2/dppp in DMF solvent, with Bu3N as the hydride donor.114 Using, triethyl silane as the hydride source, Kotsuki reported the reductive deoxygenation of aryl and enol triflates in the presence of a catalytic amount of a palladium(II) salt and bi-dentate phosphine ligand combination.115 Later Neumeyer successfully employed Katsuki's method in synthesizing pharmacologically important molecules from opioid derivatives.116 Afterwards, Lipshutz found a simple, mild Pd(0) catalyzed deoxygenation protocol for aryl perfluorosulphonates with Me2NH·BH3 as the hydrogenating source.117
Holmes and Pan in 2001 reported a novel method for deoxygenation of phenol with the help of a solid supported perfluoroalkylsulphonyl (PFS) fluoride linker.118 The PFS linker was attached with the phenol, resulting in an aryl sulphonate, and a palladium catalyzed cleavage of the aryl C–O bond followed. Using the same strategy, Revell and Ganesan in 2004 developed a tetrafluoroarylsulphonate linker as a traceless functionality for the solid-phase deoxygenation of phenol (Scheme 29).119
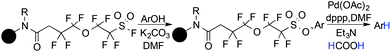 |
| | Scheme 29 Solid supported deoxygenation of phenol. | |
Sajiki and co-workers published a Pd/C catalyzed deoxygenation protocol for aryl triflates and mesylates with a Mg metal–MeOH combination as the hydrogen source.120 A stoichiometric amount of NH4OAc was used for better conversion. According to their mechanistic study it was hypothesized that two consecutive single electron transfer (SET) processes from Pd(0) to aryl sulfonates resulted in Pd(II) and an aryl anion.121 After a protonation by methanol, the aryl anion formed the deoxygenated arene. Palladium(II) transformed to Pd(0) after a successive reduction with magnesium. NH4OAc was speculated to exert a dual activating effect caused by acetic acid and ammonia. Acetic acid helped the magnesium species for better solubility in methanol and ammonia coordinated to Pd/C as the ligand (Scheme 30).
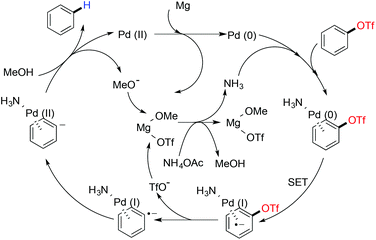 |
| | Scheme 30 Proposed mechanism for palladium catalyzed reductive cleavage of aryl sulphonate. | |
After their initial contribution, Sajiki reported the catalytic Pd/C-diethylamine mediated hydrodeoxygenation of aryl triflates (Scheme 31).122 Unfortunately, their protocol was limited due to the poor tolerance towards functional groups like olefins, benzyl esters, and nitro/aromatic ketones.
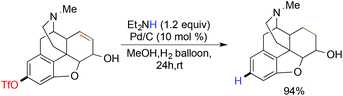 |
| | Scheme 31 Reductive deoxygenation with catalytic Pd/C–diethylamine combination. | |
Recently, Kwong and coworkers developed a palladium catalyzed facile deoxygenation of aryl tosylates using isopropanol as a mild reducing agent.123
In reductive deoxygenation reactions, nickel has also been treated as a celebrated class. In 1975 Kratzl reported a RANEY® nickel assisted deoxygenation of aryl sulphonates.124 Later Sasaki developed the nickel catalyzed deoxygenation of aryl sulphonates.125 A NiBr2(PPh3)2/bidentate phosphine ligand catalyzed method for deoxygenation with a stoichiometric amount of Zn/KI in methanol solvent was reported.126 Recently Lipshutz and coworkers explored nickel/graphite as a catalyst for the reductive deoxygenation of aryl sulphonates using Me2NH·BH3 as the hydride source.127 They have also tuned the reaction under microwave conditions with a shorter reaction time.128
7. Reductive dehalogenation
The reductive removal of the halo moiety has been treated as one of the tempting research areas for a long time. Halo-moieties are used as a blocking group in a particular organic transformation.129–131 Hence, an easy selective removal of this halo moiety allows the synthetic chemist to utilize the halo group as a fertile blocker. On the other hand, reductive dehalogenation has a great significance for environmental cause. Some halogenated, mainly chlorinated arenes are toxic and considered as persistent organic pollutants, but the dehalogenated ones are not.12–16 These halogenated arenes are resistant to biodegradation also. So an efficient artificial degradation through reductive dehalogenation is considered as a savior from these toxic halogenated materials. Transition metals are well known for the activation of the C–halo bond. Therefore, a number of reductive dehalogenation methods are known with a transitional metal and a hydride source combination.
7.1 First row transition metal complexes for reductive dehalogenation
During early stages, reductive dehalogenation was performed by Masamune using a lithium–copper hydride complex.132,133 Afterwards, a copper-hydride species (KCuH2)n was prepared by Negishi, which showed the reductive dehalogenation of aryl halides.134 Later, Tashiro and coworkers reported reductive dehalogenation with the help of a Ni–Al alloy in alkaline aqueous solution.131,135 In 1975, Tyrlik136 and Tufariello137 independently showed a Mg–Ti complex as a dehalogenating reagent, where solvent molecules (THF and H2O, respectively) were speculated as the hydrogen source. An admixture of LiAlH4 and a catalytic/stoichiometric amount of first transition metal chloride salt was used for reductive dehalogenation by Ashby.138 Roth and coworkers reported a Ni(0) catalyzed mild and simple protocol for reductive debromination using NaBH4 as the hydride source.139 In another approach, Ni(0) catalyzed reductive dehalogenation in the presence of Zn and H2O was developed by Colon.140 Using MgH2 as the reducing agent, Ni(II) catalyzed reductive dehalogenation was reported by Carfagna and coworkers.141 In 2001, Lipshutz and co-workers examined the Ni/C catalyzed reductions of aryl chlorides in the presence of Me2NH·BH3 and K2CO3.142 Aryl chlorides containing electron rich and electron donating functional groups and a heterocyclic chloride were successfully dehalogenated under their reaction conditions. They have efficiently converted trichlorobiphenyl to biphenyl through this protocol.
7.2 Palladium as a reductive dehalogenating agent
Palladium emerged as one of the efficient metals for reductive dehalogenation due to its renowned reactivity towards the C–halo bond through oxidative addition. Heck exploited Pd/C as a useful catalyst for reductive dehalogenation in the presence of diethyl ammonium formate.143 Palladium catalyzed reductive dehalogenation was further developed by Helquist144,145 and Blum146 independently. They discovered Pd(PPh3)4 catalyzed reductive dehalogenation of organic halides using sodium methoxide145 or sodium formate144 or benzyl alcohol146 as the hydride source. Reductive dehalogenation using the palladium carbene complex (IPr)Pd(allyl)Cl was explored by Nolan and coworkers under both conventional heating and microwave conditions using isopropanol as the hydrogen equivalent.147 According to their proposed catalytic cycle, Pd(0) was formed in the presence of a base, then oxidative addition of the aryl halide occurred. The Ar–[Pd]–H complex was formed in the presence of isopropanol releasing acetone. Finally, reductive elimination produced the dehalogenated product (Scheme 32).
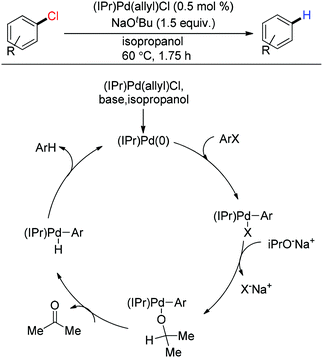 |
| | Scheme 32 Proposed mechanism for palladium catalyzed reductive dehalogenation. | |
In 2010, Jimenez and coworkers presented the Pd/C catalyzed hydrogenation of aryl chlorides and bromide.130 A broad range of functional groups including phenol, ketone, carboxylic acid, amide, nitro, cyano moieties etc. were tolerated under their reaction conditions. Recently, Chelucci and coworkers reported palladium catalyzed mild reaction conditions for the dehalogenation of heterocyclic chlorides and bromide using NaBH4–TMEDA as the hydride source.148 Regioselective hydrodebromination was also achieved in the presence of more than one bromide group (Scheme 33).
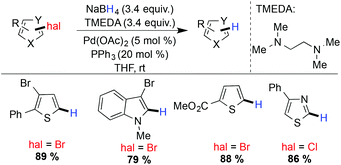 |
| | Scheme 33 Palladium catalyzed reductive dehalogenation of heterocylic chloride. | |
In 2012, Sajiki and coworkers published the Pd/C catalyzed dechlorination of polychlorinated biphenyls in the presence of Mg metal/MeOH at room temperature.149 A broad range of aryl chlorides with a variety of functional groups was tolerated in their reaction protocol. The authors proposed a reaction pathway which was similar to their previously reported reductive deoxygenation of aryl triflates.121
7.3 Other metals in reductive dehalogenation reaction
In 1999, Esteruelas et al. reported the rhodium catalyzed reductive dechlorination of aryl chlorides, using triethylsilane as the hydride source.150 Though a great effort has been made on the reductive dechlorination of polychloroarenes, their substrate scope was rarely addressed to exhibit the functional group compatibility. Miura and coworkers demonstrated an In(OAc)3-catalyzed reduction of organic bromides and iodide with PhSiH3 in ethanol.151 A radical mechanism was proposed for reductive dehalogenation (Scheme 34).
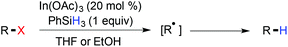 |
| | Scheme 34 Reductive dehalogenation under radical conditions. | |
Corey Stephenson achieved reductive dehalogenation under photochemical conditions in 2009 by using Ru(bpy)3Cl2 as the electron-transfer photoredox catalyst. Benzyl halide and α-halo carbonyl compounds were selectively dehalogenated without affecting any other functional group present in the molecule.152 According to the proposed mechanism, the reaction proceeds through single-electron transfer from the ammonium formate complex to the excited Ru(II)* yielding Ru(I) and the radical cation of the complex. Subsequently, the electron rich Ru(I) complex reduces the alkyl halide to the alkyl radical. In the final step the alkyl radical abstracts the hydrogen radical from either the amine or the formate of the ammonium formate radical cation.
8. Dehydroxymethylation
Dehydroxymethylation is the one pot transformation of a primary alcohol to a one carbon less defunctionalized moiety i.e. removal of the CH2OH group. Dehydroxymethylation permits the synthetic chemist to remove the hydroxymethylene group directly, where the alcohol is oxidized to an aldehyde first followed by decarbonylation of the aldehyde. The successful implementation of one reaction system which can deliver these two processes sequentially is desirable for the development of this strategy. In biological systems also, dehydroxymethylation is a key process in DNA demethylase activity.153
In 2006 Madsen reported the dehydroxymethylation reaction employing two separate catalysts for oxidation and decarbonylation.54 Later, they reported the Ir-catalyzed dehydrogenative decarbonylation of primary alcohols with the liberation of syngas.154 This is achieved with a catalyst generated in situ from [Ir(coe)2Cl]2 and racemic BINAP in a mesitylene solution saturated with water. Lewis acids such as LiCl were found to enhance the rate of transformation.
In 2013, Maiti and co-workers generalized the dehydroxymethylation reaction using one catalyst for the successful transformation where catalytic Pd(OAc)2 was used as both an oxidant and a decarbonylating agent.155 The tolerance to a broad range of functional groups proved the utility of this method. Selective dehydroxymethylation was performed successfully with a natural product derived substrate, bearing the hydroxymethyl group. They have also demonstrated the importance of dehydroxymethylation, by taking advantage of the CH2OH group as an impermanent directing group (Scheme 35).155,156
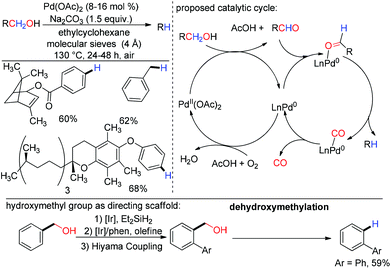 |
| | Scheme 35 Palladium catalyzed dehydroxymethylation. | |
9. Deamination
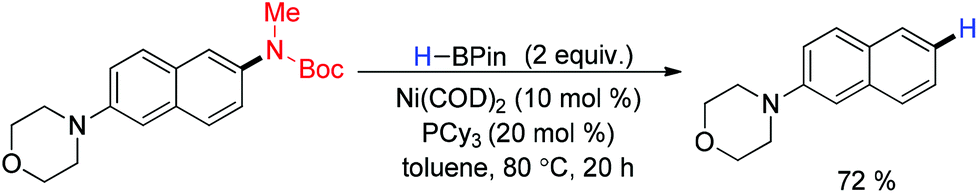
The transition metal catalyzed reductive deamination was achieved through C–N bond cleavage. An electron withdrawing moiety with the nitrogen center was essential for successful implementation. In 2014, Chatani and coworkers have described the Ni-catalyzed reductive aromatic C−N bonds in N-aryl amides and carbamates. A catalytic amount of Ni(cod)2/PCy3 with HB(pin) was required (Scheme 36). Sterically demanding substrates bearing a C−N bond at the 1-position of the naphthalene, as well as those containing an ortho substituent underwent the reductive cleavage reaction to give the desired products. Several other fused aromatic systems also proceeded smoothly under the optimized conditions, whereas benzene derivatives were found to be much less reactive.157
 |
| | Scheme 36 Nickel catalyzed reductive deamination. | |
10. Conclusion
The development of novel strategies for carbon–carbon/heteroatom bond cleavage can lead to exploration of diverse catalytic systems. The development of inexpensive transition-metal catalysts, mild conditions, green and sustainable oxidants, and a wide substrate scope remained the major focus of these studies. Efficient defunctionalization strategies are emerging which will play a vital role in the production of energy viz. bio-fuels are being made from organic sources like biomass. Defunctionalization is also important in the synthesis of drugs and biomimetics, as they form the core in a wide range of pharmaceuticals.
Acknowledgements
We thank SERB-India (EMR/2014/000164) for funding. Financial support received from CSIR-India (fellowship to A. M.) is gratefully acknowledged.
Notes and references
- F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC.
- G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450–2494 CrossRef CAS PubMed.
- A.-L. Marshall and P. J. Alaimo, Chem. – Eur. J., 2010, 16, 4970–4980 CrossRef CAS PubMed.
- Y.-B. Huang, Z. Yang, M.-Y. Chen, J.-J. Dai, Q.-X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1348–1351 CrossRef CAS PubMed.
- J. Mitra, X. Zhou and T. Rauchfuss, Green Chem., 2015, 17, 307–313 RSC.
- R. N. Monrad and R. Madsen, J. Org. Chem., 2007, 72, 9782–9785 CrossRef PubMed.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- S. Becker, Y. Fort, R. Vanderesse and P. Caubere, J. Org. Chem., 1989, 54, 4848–4853 CrossRef CAS.
- J. J. Eisch, L. E. Hallenbeck and M. A. Lucarelli, Fuel, 1985, 64, 440–442 CrossRef CAS.
- S. Agarwal, S. R. Al-Abed and D. D. Dionysiou, Environ. Sci. Technol., 2007, 41, 3722–3727 CrossRef CAS PubMed.
- E. Hadnagy, K. H. Gardner and L. M. Rauch, J. Environ. Sci. Health, Part A, 2007, 42, 685–695 CrossRef CAS PubMed.
- S. Agarwal, S. R. Al-Abed, D. D. Dionysiou and E. Graybill, Environ. Sci. Technol., 2009, 43, 915–921 CrossRef CAS PubMed.
- R. DeVor, K. Carvalho-Knighton, B. Aitken, P. Maloney, E. Holland, L. Talalaj, S. Elsheimer, C. A. Clausen and C. L. Geiger, Chemosphere, 2009, 76, 761–766 CrossRef CAS PubMed.
- A. Ido, S. Ishihara, A. Kume, T. Nakanishi, Y. Monguchi, H. Sajiki and H. Nagase, Chemosphere, 2013, 90, 57–64 CrossRef.
-
R. Hayatsu, R. G. Scott and R. E. Winans, Oxidation in Organic Chemistry 5-D, in Oxidation of Coal, ed. W. Trahanovsky, Elsevier, 2012, ch. IV Search PubMed.
- L. Pesci, Atti Real. Accad. Lincei, 1901, 10 Search PubMed.
- G. J. Leuck, R. P. Perkins and F. C. Whitmore, J. Am. Chem. Soc., 1929, 51, 1831–1836 CrossRef CAS.
- D. Dodd and M. D. Johnson, J. Chem. Soc. B, 1970, 1337–1343 RSC.
- F. C. Whitmore and R. P. Perkins, J. Am. Chem. Soc., 1929, 51, 3352–3353 CrossRef CAS.
- M. S. Newman and M. C. Vander Zwan, J. Org. Chem., 1973, 38, 319–321 CrossRef CAS.
-
G. B. Deacon, S. J. Faulks and G. N. Pain, in Adv. Organomet. Chem, ed. F. G. A. Stone and W. Robert, Academic Press, 1986, vol. 25, pp. 237–276 Search PubMed.
- J. D. Moseley and J. P. Gilday, Tetrahedron, 2006, 62, 4690–4697 CrossRef CAS.
- A. F. Shepard, N. R. Winslow and J. R. Johnson, J. Am. Chem. Soc., 1930, 52, 2083–2090 CrossRef CAS.
- M. Nilsson, Acta Chem. Scand., 1966, 20, 423–426 CrossRef CAS.
- T. Cohen and R. A. Schambach, J. Am. Chem. Soc., 1970, 92, 3189–3190 CrossRef CAS.
- T. Cohen, R. W. Berninger and J. T. Wood, J. Org. Chem., 1978, 43, 837–848 CrossRef CAS.
- A. Cairncross, J. R. Roland, R. M. Henderson and W. A. Sheppard, J. Am. Chem. Soc., 1970, 92, 3187–3189 CrossRef.
- L. J. Gooßen, W. R. Thiel, N. Rodríguez, C. Linder and B. Melzer, Adv. Synth. Catal., 2007, 349, 2241–2246 CrossRef.
- L. J. Goossen, F. Manjolinho, B. A. Khan and N. Rodríguez, J. Org. Chem., 2009, 74, 2620–2623 CrossRef CAS PubMed.
- W. F. Maier, W. Roth, I. Thies and P. V. R. Schleyer, Chem. Ber., 1982, 115, 808–812 CrossRef CAS.
- S. Matsubara, Y. Yokota and K. Oshima, Org. Lett., 2004, 6, 2071–2073 CrossRef CAS PubMed.
- J. S. Dickstein, C. A. Mulrooney, E. M. O'Brien, B. J. Morgan and M. C. Kozlowski, Org. Lett., 2007, 9, 2441–2444 CrossRef CAS PubMed.
- J. S. Dickstein, J. M. Curto, O. Gutierrez, C. A. Mulrooney and M. C. Kozlowski, J. Org. Chem., 2013, 78, 4744–4761 CrossRef CAS PubMed.
- N. Chatani, H. Tatamidani, Y. Ie, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 4849–4850 CrossRef CAS PubMed.
- P. Lu, C. Sanchez, J. Cornella and I. Larrosa, Org. Lett., 2009, 11, 5710–5713 CrossRef CAS.
- R. Grainger, A. Nikmal, J. Cornella and I. Larrosa, Org. Biomol. Chem., 2012, 10, 3172–3174 CAS.
- S. Seo, J. B. Taylor and M. F. Greaney, Chem. Commun., 2012, 48, 8270–8272 RSC.
- Z.-M. Sun, J. Zhang and P. Zhao, Org. Lett., 2010, 12, 992–995 CrossRef CAS PubMed.
- S. Dupuy, F. Lazreg, A. M. Z. Slawin, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2011, 47, 5455–5457 RSC.
- S. Dupuy and S. P. Nolan, Chem. – Eur. J., 2013, 19, 14034–14038 CrossRef CAS PubMed.
- J. M. Carraher, O. Pestovsky and A. Bakac, Dalton Trans., 2012, 41, 5974–5980 RSC.
- T. Patra, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2011, 50, 12140–12142 CrossRef CAS PubMed.
- J. Hawthorne and M. Wilt, J. Org. Chem., 1960, 25, 2215–2216 CrossRef CAS.
- J. Tsuji, K. Ohno and T. Kajimoto, Tetrahedron Lett., 1965, 6, 4565–4568 CrossRef.
- A. Modak, A. Deb, T. Patra, S. Rana, S. Maity and D. Maiti, Chem. Commun., 2012, 48, 4253–4255 RSC.
- Akanksha and D. Maiti, Green Chem., 2012, 14, 2314–2320 RSC.
- M. C. Bröhmer, N. Volz and S. Bräse, Synlett, 2009, 1383–1386 Search PubMed.
- J. Tsuji and K. Ohno, Tetrahedron Lett., 1965, 6, 3969–3971 CrossRef.
- D. H. Doughty and L. H. Pignolet, J. Am. Chem. Soc., 1978, 100, 7083–7085 CrossRef CAS.
- J. M. O'Connor and J. Ma, J. Org. Chem., 1992, 57, 5075–5077 CrossRef.
- C. M. Beck, S. E. Rathmill, Y. J. Park, J. Chen, R. H. Crabtree, L. M. Liable-Sands and A. L. Rheingold, Organometallics, 1999, 18, 5311–5317 CrossRef CAS.
- M. Kreis, A. Palmelund, L. Bunch and R. Madsen, Adv. Synth. Catal., 2006, 348, 2148–2154 CrossRef CAS.
- P. Fristrup, M. Kreis, A. Palmelund, P.-O. Norrby and R. Madsen, J. Am. Chem. Soc., 2008, 130, 5206–5215 CrossRef CAS PubMed.
- B. Gutmann, P. Elsner, T. Glasnov, D. M. Roberge and C. O. Kappe, Angew. Chem., Int. Ed., 2014, 53, 11557–11561 CrossRef CAS PubMed.
- G. Domazetis, B. Tarpey, D. Dolphin and B. R. James, J. Chem. Soc., Chem. Commun., 1980, 939–940 RSC.
- T. Iwai, T. Fujihara and Y. Tsuji, Chem. Commun., 2008, 6215–6217 RSC.
- J. Rentner, M. Kljajic, L. Offner and R. Breinbauer, Tetrahedron, 2014, 70, 8983–9027 CrossRef CAS.
- R. Mozingo, D. E. Wolf, S. A. Harris and K. Folkers, J. Am. Chem. Soc., 1943, 65, 1013–1016 CrossRef CAS.
- A. Zumbrunn, Synthesis, 1998, 1357–1361 CrossRef CAS.
- I. Kawasaki, N. Sakaguchi, A. Khadeer, M. Yamashita and S. Ohta, Tetrahedron, 2006, 62, 10182–10192 CrossRef CAS.
- N. Barbero and R. Martin, Org. Lett., 2012, 14, 796–799 CrossRef CAS PubMed.
- S. Niwas, P. Chand, V. P. Pathak and J. A. Montgomery, J. Med. Chem., 1994, 37, 2477–2480 CrossRef CAS PubMed.
- T. H. Graham, W. Liu and D.-M. Shen, Org. Lett., 2011, 13, 6232–6235 CrossRef CAS PubMed.
- T. Matsumura, T. Niwa and M. Nakada, Tetrahedron Lett., 2012, 53, 4313–4316 CrossRef CAS.
- T. Matsumura and M. Nakada, Tetrahedron Lett., 2014, 55, 1412–1415 CrossRef CAS.
- J. F. Hooper, R. D. Young, A. S. Weller and M. C. Willis, Chem. – Eur. J., 2013, 19, 3125–3130 CrossRef CAS PubMed.
- W. Pfleiderer, Tetrahedron, 1988, 44, 3373–3378 CrossRef CAS.
- J. J. Baldwin, E. L. Engelhardt, R. Hirschmann, G. S. Ponticello, J. G. Atkinson, B. K. Wasson, C. S. Sweet and A. Scriabine, J. Med. Chem., 1980, 23, 65–70 CrossRef CAS PubMed.
- H. Yoshioka, Y. Matsuya, T. Choshi, E. Sugino and S. Hibino, Chem. Pharm. Bull., 1996, 44, 709–714 CrossRef CAS.
- T. Choshi, A. Tonari, H. Yoshioka, K. Harada, E. Sugino and S. Hibino, J. Org. Chem., 1993, 58, 7952–7954 CrossRef CAS.
- T. Sugimoto, N. Nishioka, S. Murata and S. Matsuura, Heterocycles, 1987, 26 Search PubMed.
- C. Yamazaki, H. Arima and S. Udagawa, J. Heterocycl. Chem., 1996, 33, 41–44 CrossRef CAS.
- B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786–2791 CrossRef CAS PubMed.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- M. C. Reddy and M. Jeganmohan, Chem. Commun., 2015, 51, 10738–10741 RSC.
- M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed.
- M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed.
- T. D. Krizan and J. C. Martin, J. Am. Chem. Soc., 1983, 105, 6155–6157 CrossRef CAS.
- J.-M. Mattalia, C. Marchi-Delapierre, H. Hazimeh and M. Chanon, ARKIVOC, 2006, 90–118 CAS.
- M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300–307 RSC.
- H. Kinuta, H. Takahashi, M. Tobisu, S. Mori and N. Chatani, Bull. Chem. Soc. Jpn., 2014, 87, 655–669 CrossRef CAS.
- H. Nakazawa, K. Kamata and M. Itazaki, Chem. Commun., 2005, 4004–4006 RSC.
- H. Nakazawa, M. Itazaki, K. Kamata and K. Ueda, Chem. – Asian J., 2007, 2, 882–888 CrossRef CAS PubMed.
- M. Tobisu, R. Nakamura, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2009, 131, 3174–3175 CrossRef CAS PubMed.
- M. Tobisu, R. Nakamura, Y. Kita and N. Chatani, Bull. Korean Chem. Soc., 2010, 31, 582–587 CrossRef CAS.
- T. Patra, S. Agasti, Akanksha and D. Maiti, Chem. Commun., 2013, 49, 69–71 RSC.
- T. Patra, S. Agasti, A. Modak and D. Maiti, Chem. Commun., 2013, 49, 8362–8364 RSC.
- M. Weidauer, C. I. Someya, E. Irran and S. Enthaler, Asian J. Org. Chem., 2013, 2, 150–156 CrossRef CAS.
- S. Enthaler, M. Weidauer, E. Irran, J. D. Epping, R. Kretschmer and C. I. Someya, J. Organomet. Chem., 2013, 745–746, 262–274 CrossRef CAS.
- D.-G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 7710–7712 CrossRef CAS PubMed.
- Á. Iglesias, R. Álvarez, Á. R. de Lera and K. Muñiz, Angew. Chem., Int. Ed., 2012, 51, 2225–2228 CrossRef PubMed.
- J. Oyamada, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 10720–10723 CrossRef CAS PubMed.
- E. Vowinkel and C. Wolff, Chem. Ber., 1974, 107, 907–914 CrossRef CAS.
- J. M. Herrmann and B. König, Eur. J. Org. Chem., 2013, 7017–7027 CrossRef CAS.
- P. Sebok, T. Timar, T. Eszenyi and T. Patonay, J. Org. Chem., 1994, 59, 6318–6321 CrossRef CAS.
- E. M. v. Duzee and H. Adkins, J. Am. Chem. Soc., 1935, 57, 147–151 CrossRef.
- P. Álvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352–17353 CrossRef.
- J. Cornella, E. Gómez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997–2009 CrossRef CAS PubMed.
- M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2011, 47, 2946–2948 RSC.
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed.
- P. Kelley, S. Lin, G. Edouard, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2012, 134, 5480–5483 CrossRef CAS PubMed.
- M. Tobisu, T. Morioka, A. Ohtsuki and N. Chatani, Chem. Sci., 2015, 6, 3410–3414 RSC.
- M. Chatterjee, A. Chatterjee, T. Ishizaka and H. Kawanami, Catal. Sci. Technol., 2015, 5, 1532–1539 CAS.
- Y. Ren, M. Yan, J. Wang, Z. C. Zhang and K. Yao, Angew. Chem., Int. Ed., 2013, 52, 12674–12678 CrossRef CAS PubMed.
- Y.-L. Ren, M. Tian, X.-Z. Tian, Q. Wang, H. Shang, J. Wang and Z. C. Zhang, Chem. Lett., 2014, 52, 36–39 CAS.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- L. R. Subramanian, A. G. Martinez, A. H. Fernandez and R. M. Alvarez, Synthesis, 1984, 481–485 CrossRef CAS.
- S. Cacchi, P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1986, 27, 5541–5544 CrossRef CAS.
- G. A. Peterson, F.-A. Kunng, J. S. McCallum and W. D. Wulff, Tetrahedron Lett., 1987, 28, 1381–1384 CrossRef CAS.
- Q.-Y. Chen, Y.-B. He and Z.-Y. Yang, J. Chem. Soc., Chem. Commun., 1986, 1452–1453 RSC.
- P. Ganji and H. Ibrahim, J. Org. Chem., 2012, 77, 511–518 CrossRef CAS PubMed.
- J. M. Saa, M. Dopico, G. Martorell and A. Garcia-Raso, J. Org. Chem., 1990, 55, 991–995 CrossRef CAS.
- H. Kotsuki, P. K. Datta, H. Hayakawa and H. Suenaga, Synthesis, 1995, 1348–1350 CrossRef CAS.
- C. D. Hupp and J. L. Neumeyer, Tetrahedron Lett., 2010, 51, 2359–2361 CrossRef CAS PubMed.
- B. H. Lipshutz, D. J. Buzard and R. W. Vivian, Tetrahedron Lett., 1999, 40, 6871–6874 CrossRef CAS.
- Y. Pan and C. P. Holmes, Org. Lett., 2001, 3, 2769–2771 CrossRef CAS PubMed.
- J. D. Revell and A. Ganesan, Chem. Commun., 2004, 1916–1917 RSC.
- H. Sajiki, A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa and K. Hirota, Org. Lett., 2006, 8, 987–990 CrossRef CAS PubMed.
- A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. – Eur. J., 2007, 13, 1432–1441 CrossRef CAS.
- A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa, Y. Monguchi and H. Sajiki, Tetrahedron, 2007, 63, 1270–1280 CrossRef CAS.
- W. K. Chow, C. M. So, C. P. Lau and F. Y. Kwong, Org. Chem. Front., 2014, 1, 464–467 RSC.
- W. Lonsky, H. Traitler and K. Kratzl, J. Chem. Soc., Perkin Trans. 1, 1975, 169–170 RSC.
- K. Sasaki, M. Sakai, Y. Sakakibara and K. Takagi, Chem. Lett., 1991, 20, 2017–2018 CrossRef.
- K. Sasaki, T. Kubo, M. Sakai and Y. Kuroda, Chem. Lett., 1997, 26, 617–618 CrossRef.
- B. H. Lipshutz, B. A. Frieman, T. Butler and V. Kogan, Angew. Chem., Int. Ed., 2006, 45, 800–803 CrossRef CAS PubMed.
- T. A. Butler, E. C. Swift and B. H. Lipshutz, Org. Biomol. Chem., 2008, 6, 19–25 CAS.
- A. Dobbs, J. Org. Chem., 2001, 66, 638–641 CrossRef CAS PubMed.
- A. Ramanathan and L. S. Jimenez, Synthesis, 2010, 217–220 CAS.
- M. Tashiro, G. Fukata and K. Oe, Org. Prep. Proced. Int., 1975, 7, 183–188 CrossRef CAS.
- S. Masamune, P. A. Rossy and G. S. Bates, J. Am. Chem. Soc., 1973, 95, 6452–6454 CrossRef CAS.
- S. Masamune, G. S. Bates and P. E. Georghiou, J. Am. Chem. Soc., 1974, 96, 3686–3688 CrossRef CAS.
- T. Yoshida and E.-i. Negishi, J. Chem. Soc., Chem. Commun., 1974, 762–763 RSC.
- G.-B. Liu, T. Tsukinoki, T. Kanda, Y. Mitoma and M. Tashiro, Tetrahedron Lett., 1998, 39, 5991–5994 CrossRef CAS.
- S. Tyrlik and I. Wolochowicz, J. Chem. Soc., Chem. Commun., 1975, 781–782 RSC.
- T. R. Nelsen and J. J. Tufariello, J. Org. Chem., 1975, 40, 3159–3160 CrossRef CAS.
- E. C. Ashby and J. J. Lin, J. Org. Chem., 1978, 43, 1263–1265 CrossRef CAS.
- S.-T. Lin and J. A. Roth, J. Org. Chem., 1979, 44, 309–310 CrossRef CAS.
- I. Colon, J. Org. Chem., 1982, 47, 2622–2625 CrossRef CAS.
- C. Carfagna, A. Musco, R. Pontellini and G. Terzoni, J. Mol. Catal., 1989, 57, 23–28 CrossRef CAS.
- B. H. Lipshutz, T. Tomioka and K. Sato, Synlett, 2001, 0970–0973 CrossRef CAS.
- N. A. Cortese and R. F. Heck, J. Org. Chem., 1977, 42, 3491–3494 CrossRef CAS.
- P. Helquist, Tetrahedron Lett., 1978, 19, 1913–1914 CrossRef.
- A. Zask and P. Helquist, J. Org. Chem., 1978, 43, 1619–1620 CrossRef CAS.
- A. Zoran, Y. Sasson and J. Blum, J. Mol. Catal., 1984, 27, 349–353 CrossRef CAS.
- O. Navarro, H. Kaur, P. Mahjoor and S. P. Nolan, J. Org. Chem., 2004, 69, 3173–3180 CrossRef CAS PubMed.
- G. Chelucci, S. Baldino and A. Ruiu, J. Org. Chem., 2012, 77, 9921–9925 CrossRef CAS PubMed.
- S. Ishihara, A. Ido, Y. Monguchi, H. Nagase and H. Sajiki, J. Hazard. Mater., 2012, 229–230, 15–19 CrossRef CAS PubMed.
- M. A. Esteruelas, J. Herrero, F. M. López, M. Martín and L. A. Oro, Organometallics, 1999, 18, 1110–1112 CrossRef CAS.
- K. Miura, M. Tomita, Y. Yamada and A. Hosomi, J. Org. Chem., 2007, 72, 787–792 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- K. I. Ladwein and M. Jung, Angew. Chem., Int. Ed., 2011, 50, 12143–12145 CrossRef CAS PubMed.
- E. P. K. Olsen and R. Madsen, Chem. – Eur. J., 2012, 18, 16023–16029 CrossRef CAS PubMed.
- A. Modak, T. Naveen and D. Maiti, Chem. Commun., 2013, 49, 252–254 RSC.
- E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092–17095 CrossRef CAS PubMed.
- M. Tobisu, K. Nakamura and N. Chatani, J. Am. Chem. Soc., 2014, 136, 5587–5590 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence