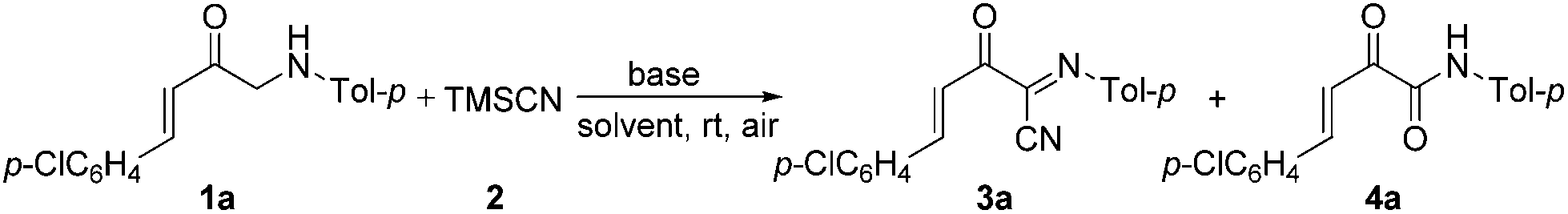DBU-mediated metal-free oxidative cyanation of α-amino carbonyl compounds: using molecular oxygen as the oxidant†
Received
29th September 2015
, Accepted 5th November 2015
First published on 5th November 2015
Abstract
A novel DBU-mediated oxidative cyanation of α-amino carbonyl compounds by using air as the sole oxidant was developed under mild metal-free conditions for the first time. The reaction involves a tandem oxidation/Strecker reaction/oxidation process and provides a new and efficient method for the construction of α-iminonitriles in good to high yields in a single step.
Introduction
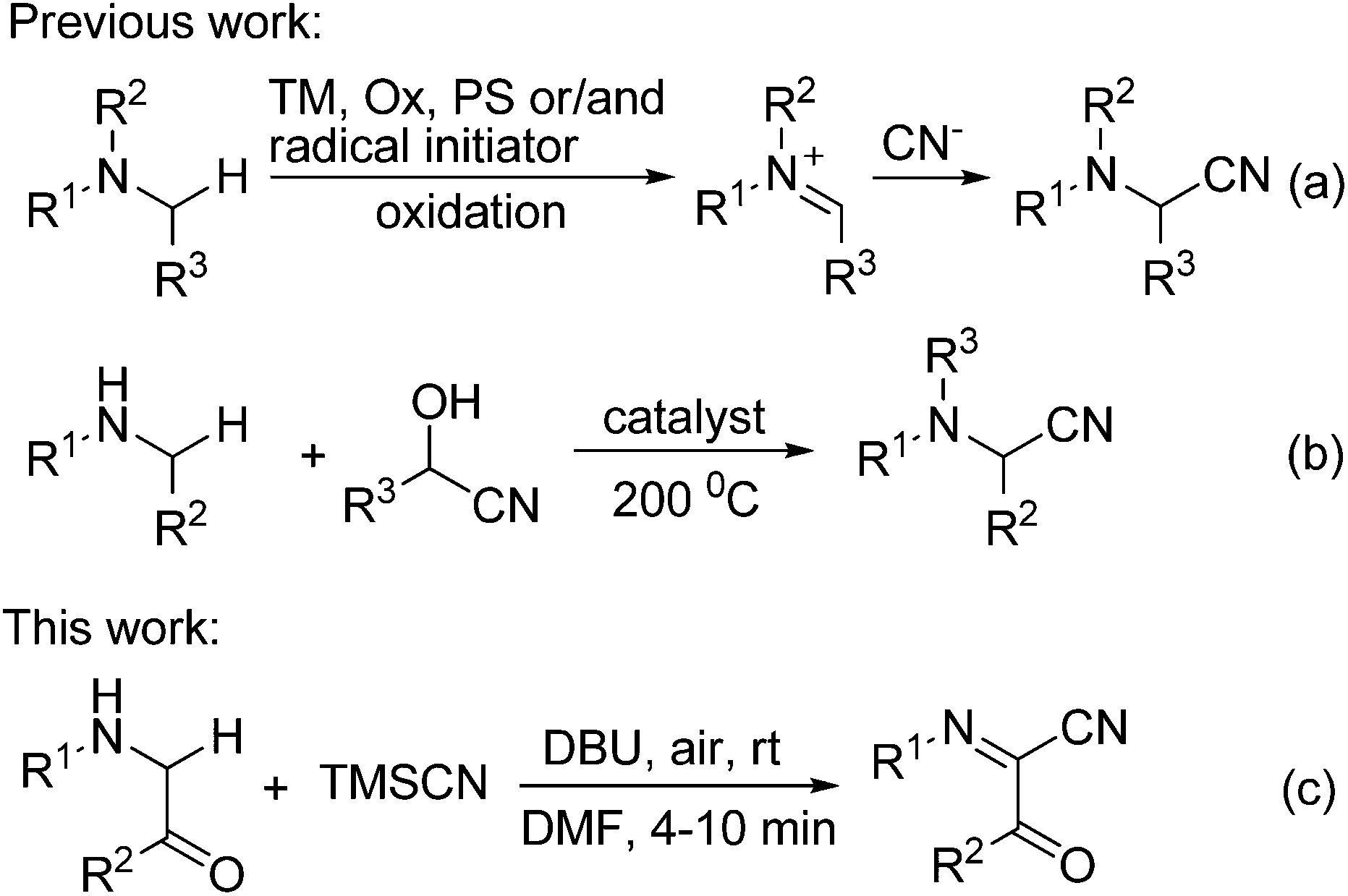
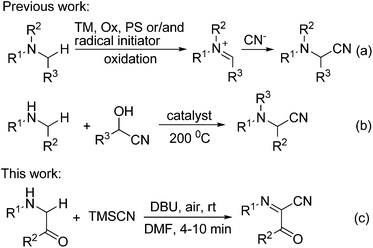
α-Aminonitriles are an important class of versatile intermediates for a wide range of natural products, pharmaceuticals, functional materials, and agricultural chemicals.1 In addition, nucleophilic additions to the nitrile group can provide access to valuable α-amino aldehydes, ketones, and alcohols, as well as 1,2-diamines.2 Accordingly, the development of novel and efficient synthetic methods for α-aminonitriles has been a major topic in synthetic organic chemistry. Among the different methods available for the preparation of these compounds,3–7 the oxidative cyanation of sp3 C–H bonds adjacent to the nitrogen atom represents one of the most straightforward and convenient methods for the synthesis of α-aminonitriles.4–7 However, in all these reactions reported except for the electrochemical methods,5 transition metal (TM) catalysts/mediators, chemical oxidants (Ox), photosensitizers (PS, in light induced reaction) and/or radical initiators are generally required (Scheme 1a).6 Recently, Seidel and co-workers developed a conceptually new strategy for the direct α-cyanation of amines in a redox-neutral fashion, but the reaction was carried out under microwave conditions at a rather high temperature (200 °C, Scheme 1b).7 Obviously, the development of oxidative cyanation of sp3 C–H bonds adjacent to the nitrogen atom in the absence of transition metals, chemical oxidants, photosensitizers and radical initiators under very mild reaction conditions remains a formidable challenge. Herein, we report the first DBU-mediated oxidative cyanation of sp3 C–H bonds adjacent to the nitrogen atom without using any redox-active catalysts, chemical oxidants, photosensitizers and radical initiators (Scheme 1c). Moreover, the reaction takes place under rather mild conditions with molecular oxygen as a green oxidant and provides a new and efficient method for the construction of functionalized α-iminonitriles in a single step.
 |
| | Scheme 1 Oxidative cyanation of sp3 C–H bonds adjacent to the nitrogen atom. | |
Results and discussion
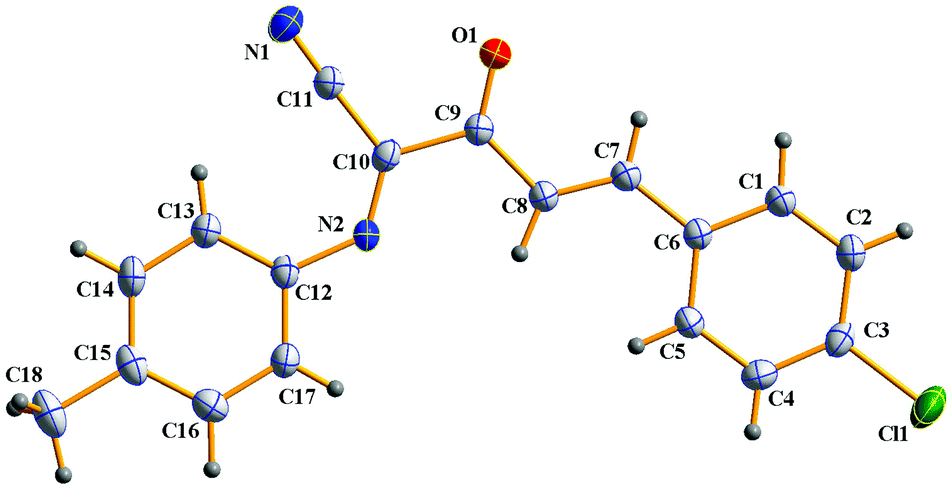
In addition, α-amino carbonyl compounds are ubiquitous subunits in biologically active natural products, biomolecules and therapeutic agents.8 Thus, it is reasonable that considerable efforts have been devoted to the development of novel methods for their assembly. In this field, α-C–H functionalization of the preexisting α-amino carbonyl structures has attracted considerable attention due to its direction and site-specificity.9–11 Among these reactions, the generation of electrophilic iminium intermediates through oxidation of sp3 C–H bonds adjacent to the nitrogen atom of α-amino carbonyl compounds, followed by the attack of various nucleophiles, has recently been recognized as a powerful means for preparation of α-amino carbonyl compounds.11 To the best of our knowledge, however, the direct oxidative α-cyanation of α-amino carbonyl compounds has not been exploited. As part of our continuing research on the formation of carbon–carbon and carbon–nitrogen bonds based on oxidative coupling reactions,12 we recently developed an efficient method for C–C formation via a base-promoted intramolecular CDC of α-amino carbonyl compounds under very mild metal-free conditions.13 These results and our continuous interest in the carbocyanation reaction14 prompted us to investigate the α-cyanation of N-aryl α-amino ketones under basic conditions using molecular oxygen as the oxidant under metal-free conditions. As a result, it was found that the oxidative Strecker reaction of N-aryl α′-amino-α,β-unsaturated ketone 1a (0.2 mmol) with trimethylsilyl cyanide (TMSCN) 2a (0.3 mmol) could proceed rapidly to give (1E,3E)-4-(4-chlorophenyl)-2-oxo-N-p-tolylbut-3-enimidoyl cyanide 3a in 72% yield in the presence of DBU (0.3 mmol, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene) in DMF (1.5 mL) at room temperature in open air for only 4 min along with the oxidation product 4a in 5% yield (entry 3). This reaction is highly chemoselective leaving the cinnamoyl moiety of 1a intact. Further increasing the amounts of DBU leads to a lower yield of 3a (entry 4). Other bases such as DBN (1,5-diazabicyclo[4.3.0]non-5-ene), NaOH, DABCO (1,4-diazabicyclo[2.2.2]octane), Et3N and Cs2CO3 were less (entries 5 and 6) or not effective (entries 7–9). The solvent, DMF, was a much better choice than the other solvents examined, including CH3CN, THF, CH2Cl2 and toluene (entries 10–13). The structure of 3a was determined based on its spectroscopic and analytical data and confirmed by X-ray crystal structure analysis (Fig. 1).15
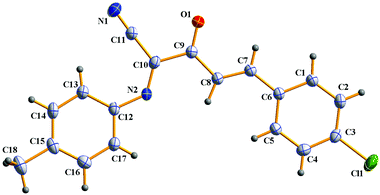 |
| | Fig. 1 ORTEP drawing of 3a. | |
Under the optimal conditions (Table 1, entry 3), the scope and generality of the reaction was next examined. As described in Scheme 2, all of the selected N-aryl α′-amino-α,β-unsaturated ketone substrates 1a–i, bearing phenyl, electron-deficient and electron-rich aryl, heteroaryl and 2-naphthyl groups at the β-position of the enone moiety, reacted smoothly with trimethylsilyl cyanide 2 to give the corresponding (1E,3E)-2-oxo-N,4-diarylbut-3-enimidoyl cyanides 3a–i in good to high yields at room temperature in open air for 4–10 min. In addition, the reaction of substrate 1j bearing a (E)-phenylvinyl group (R) gave the desired product 3j in 75% yield (Scheme 2). On the other hand, various N-aryl groups of 1 were also well-tolerated and the corresponding (1E,3E)-2-oxo-N,4-diarylbut-3-enimidoyl cyanides 3k–n were prepared in good to high yields (Scheme 2). No reaction was observed when α-amino carbonyl compounds 1o and 1p were used as substrates (Scheme 2). In addition, the reaction of α-amino carbonyl compound 1q or 1r with trimethylsilyl cyanide 2 led to a complex mixture under identical conditions to above (Scheme 2), which may be due to the lower pKa value of the hydrogen at the nitrogen.
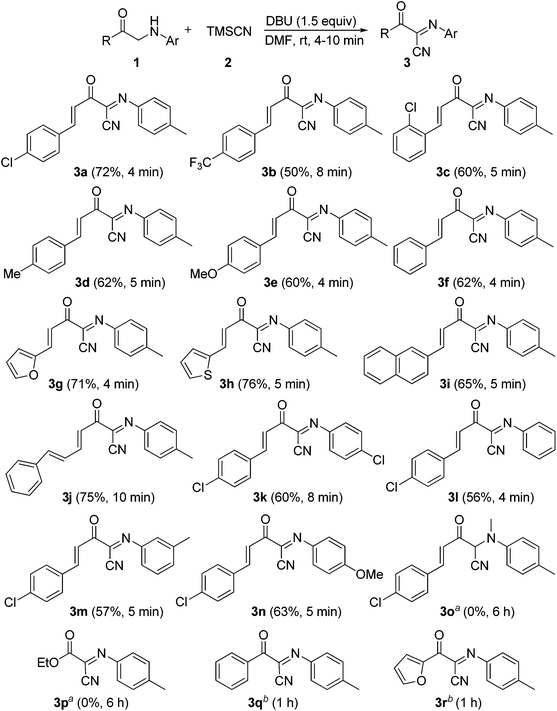 |
| | Scheme 2 DBU-promoted oxidative cyanation of 1 with 2. Reaction conditions: 1 (0.2 mmol), 2 (0.30 mmol), DBU (0.30 mmol), DMF (1.5 mL), rt, 4–10 min. Isolated yield. a1o and 1p were recovered in 95% and 94% yields, respectively. bA complex mixture was produced. | |
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
Base (equiv.) |
Solvent |
Time (min) |
3a Yieldb (%) |
4a Yieldb (%) |
|
Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), base (0.1–0.4 mmol), solvent (1.5 mL), at room temperature for 4–30 min.
Estimated by 1H NMR spectroscopy using dimethyl phthalate as an internal standard.
Isolated yield.
A complex mixture was obtained.
|
| 1 |
DBU (0.5) |
DMF |
5 |
38 |
14 |
| 2 |
DBU (1.0) |
DMF |
4 |
65 |
10 |
| 3c |
DBU (1.5) |
DMF |
4 |
72 |
5 |
| 4c |
DBU (2.0) |
DMF |
4 |
66 |
5 |
| 5 |
DBN (2.0) |
DMF |
4 |
35 |
10 |
| 6 |
NaOH (1.5) |
DMF |
8 |
41 |
7 |
| 7d |
DABCO (1.5) |
DMF |
30 |
— |
— |
| 8d |
Et3N (1.5) |
DMF |
30 |
— |
— |
| 9d |
Cs2CO3 (1.5) |
DMF |
30 |
— |
— |
| 10 |
DBU (1.5) |
MeCN |
5 |
64 |
12 |
| 11 |
DBU (1.5) |
THF |
8 |
61 |
15 |
| 12 |
DBU (1.5) |
CH2Cl2 |
8 |
35 |
18 |
| 13 |
DBU (1.5) |
Toluene |
6 |
61 |
17 |
To further probe the mechanisms for the formation of 3, some control experiments were designed and investigated. As a result, it was found that the yield of product 3a decreased greatly from 72 to 16% under the standard conditions when TEMPO (1.0 equiv.; TEMPO = 2,2,6,6-tetramethylpiperidinooxy) was used as a radical inhibitor (Scheme 3a). No desired product 3a was produced when the reaction of 1a with TMSCN 2 was carried out under a nitrogen atmosphere (Scheme 3b). These results indicate that molecular oxygen as the oxidant (from air) is necessary for the oxidative cyanation reaction and a radical mechanism may be involved in this oxidative coupling process. No reaction was observed under otherwise identical conditions but in the absence of DBU (Scheme 3c), which demonstrates that DBU is also crucial for the above oxidative cyanation reaction. In addition, the imine 5a could be formed in 11% yield (along with a complex mixture) in the absence of TMSCN under otherwise identical conditions to above (Scheme 3d). According to the results of this transformation, we predicted that the imine 5a may be a key intermediate in the reaction. Therefore, the reaction of imine 5a with TMSCN 2 was further investigated. As expected, under otherwise identical conditions to above, the Strecker reaction of imine 5a with TMSCN 2 can smoothly proceed to give the product 3a in 60% yield (Scheme 3e).
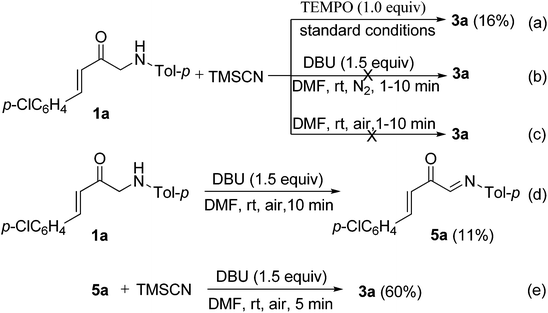 |
| | Scheme 3 Control experiments for mechanistic studies. | |
Based on the above experimental results and related reports,4–7,11,13,16 a possible mechanism for the formation of 3 is proposed (Scheme 4). Initially, the deprotonation of 1 into the enolate anion I followed by a single-electron transfer (SET) process between I and triplet oxygen forms the superoxide anion radical and the radical intermediate II/III. Then, the intermediate IV, generated by the reaction of intermediate III with the superoxide anion radical, undergoes an elimination of the hydroperoxide anion to give the imine intermediate V, which rapidly reacts with trimethylsilyl cyanide 2 to generate the intermediate VI. Finally, (1E,3E)-2-oxo-N-4-diarylbut-3-enimidoyl cyanide 3 was produced via a sequential deprotonation, oxidation and elimination of the TMS-peroxide anion process (VI → 3, Scheme 4).
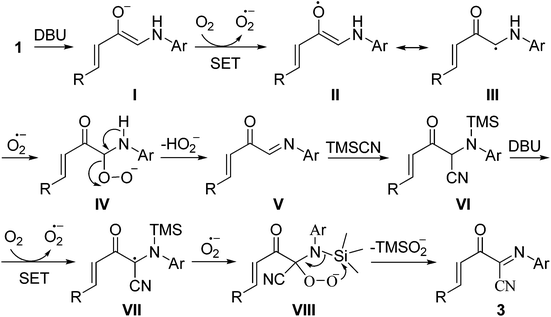 |
| | Scheme 4 Proposed mechanism for the formation of 3. | |
In order to explore the synthetic potential of these functionalized (1E,3E)-2-oxo-N-4-diarylbut-3-enimidoyl cyanides, the cyclization reaction of 3 with benzene-1,2-diamine was examined (Scheme 5). It was found that the cyclization reaction of 3a (0.2 mmol) with benzene-1,2-diamine (0.24 mmol) could easily proceed to give 2-cyano quinoxaline 6a in 92% yield in AcOH (2.0 mL) at 120 °C for 10 min in the presence of NaOAc (0.24 mmol). Similarly, 2-cyano quinoxaline derivatives 6f and 6h were obtained in 91% and 95% yields from (1E,3E)-2-oxo-N,4-diarylbut-3-enimidoyl cyanides 3f and 3h, respectively (Scheme 5).
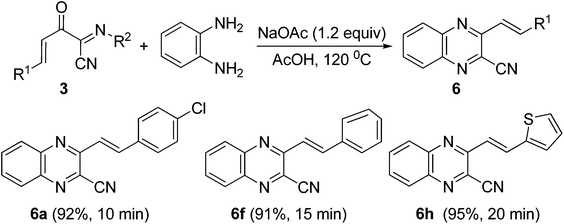 |
| | Scheme 5 Synthesis of 2-cyano quinoxalines 6. | |
Conclusions
In conclusion, we have developed a novel DBU-mediated oxidative cyanation of α-amino carbonyl compounds by using air as the sole oxidant under metal-free conditions for the first time. The reaction involves a tandem oxidation/Strecker reaction/oxidation process and provides a new and efficient method for the construction of α-iminonitriles in a single step. The advantages of these methods include: (1) mild reaction conditions and short reaction time; (2) simplicity and safety of operation; (3) any transition metals, synthetic oxidants, photosensitizers and radical initiators are not required and only cheap DBU and air are necessary. These advantages make this protocol very practical. Further studies are in progress.
Experimental
General methods
All reagents were commercial and were used without further purification. Chromatography was carried out on flash silica gel (300–400 mesh). All reactions were monitored by TLC, which was performed on precoated aluminum sheets of silica gel 60 (F254). Unless noted, the 1H NMR spectra were recorded at 400 MHz and 500 MHz in CDCl3 and the 13C NMR spectra were recorded at 125 MHz in CDCl3 with TMS as the internal standard. All coupling constants (J values) were reported in Hertz (Hz). High-resolution mass spectra (HRMS) were obtained using a Bruker microTOF II focus spectrometer (ESI).
General procedure for the preparation of 3 (3a as example)
To a solution of (E)-4-(4-chlorophenyl)-1-(p-tolylamino)but-3-en-2-one 1a (0.2 mmol, 57.1 mg) and TMSCN 2 (0.30 mmol, 0.040 mL) in DMF (1.5 mL) was added DBU (0.3 mmol, 0.045 mL). Then the reaction mixture was stirred at room temperature in open air for 5 min. After 1a was consumed (monitored by TLC), the reaction mixture was poured into water (50 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to yield the corresponding crude product, which was purified by chromatography (silica gel, petroleum ether/acetone = 10/1, v/v) to give 3a (44.4 mg, 72%) as a yellow solid.
(1E,3E)-4-(4-Chlorophenyl)-2-oxo-N-(p-tolyl)but-3-enimidoyl cyanide (3a).
Yellow solid; m.p. 107–109 °C; 1H NMR (CDCl3, 500 MHz) δ: 7.91 (d, J = 16.0 Hz, 1H), 7.83 (d, J = 16.0 Hz, 1H), 7.63 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 8.5 Hz, 2H), 7.42 (d, J = 8.5 Hz, 2H), 7.35 (d, J = 8.5 Hz, 2H), 2.45 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ: 183.5, 145.4, 143.3, 142.2, 137.4, 135.8, 132.8, 130.3, 130.2, 129.4, 123.0, 118.4, 110.6, 21.6; IR (KBr) ν: 2921, 2215, 1618, 1600, 1556, 1445, 1319, 1108, 1060 cm−1; HRMS (ESI-TOF) calcd for C18H13ClN2ONa+ ([M + Na]+): 331.0609, found: 331.0620.
(1E,3E)-2-Oxo-N-(p-tolyl)-4-(4-(trifluoromethyl)phenyl)but-3-enimidoyl cyanide (3b).
Yellow solid; m.p. 135–137 °C; 1H NMR (500 MHz, CDCl3) δ: 7.94 (d, J = 4.0 Hz, 2H), 7.79 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 8.5 Hz, 2H), 7.53 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 2.46 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.5, 144.7, 143.2, 142.5, 137.6, 135.4, 132.6 (q, JC–F = 32.7 Hz), 130.3, 129.1, 126.0 (q, JC–F = 3.6 Hz), 123.7 (q, JC–F = 270.7 Hz), 123.2, 120.2, 110.6, 21.6; IR (KBr) ν: 2926, 2211, 1608, 1607, 1550, 1324, 1117, 1066 cm−1; HRMS (ESI-TOF) calcd for C19H14F3N2O+ ([M + H]+): 343.1053, found: 343.1049.
(1E,3E)-4-(2-Chlorophenyl)-2-oxo-N-(p-tolyl)but-3-enimidoyl cyanide (3c).
Yellow solid; m.p. 118–120 °C; 1H NMR (500 MHz, CDCl3) δ: 8.39 (d, J = 16.0 Hz, 1H), 7.85–7.81 (m, 2H), 7.50 (d, J = 8.5 Hz, 2H), 7.46 (dd, J = 8.0 Hz, 1.0 Hz, 1H), 7.39–7.32 (m, 4H), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.4, 143.2, 142.5, 142.3, 136.2, 135.6, 132.5, 132.0, 130.4, 130.3, 128.0, 127.1, 123.0, 120.3, 110.6, 21.5; IR (KBr) ν: 3032, 2217, 1663, 1595, 1328, 1272, 1208 cm−1; HRMS (ESI-TOF) calcd for C18H14ClN2O+ ([M + H]+): 309.0789, found: 309.0798.
(1E,3E)-2-Oxo-N-4-dip-tolylbut-3-enimidoyl cyanide (3d).
Yellow solid; m.p. 133–135 °C; 1H NMR (500 MHz, CDCl3) δ: 7.96 (d, J = 16.0 Hz, 1H), 7.81 (d, J = 16.0 Hz, 1H), 7.60 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 8.5 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.25 (d, J = 8.5 Hz, 2H), 2.45 (s, 3H), 2.41 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.6, 147.2, 143.5, 142.3, 141.8, 136.2, 131.6, 130.2, 129.8, 129.2, 122.8, 116.8, 110.7, 21.7, 21.5; HRMS (ESI-TOF) calcd for C19H17N2O+ ([M + H]+): 289.1335, found: 289.1337.
(1E,3E)-4-(4-Methoxyphenyl)-2-oxo-N-p-tolylbut-3-enimidoyl cyanide (3e).
Yellow solid; m.p. 112–114 °C; 1H NMR (500 MHz, CDCl3) δ: 7.94 (d, J = 16.0 Hz, 1H), 7.72 (d, J = 16.0 Hz, 1H), 7.66 (d, J = 8.5 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 9.0 Hz, 2H), 3.87 (s, 3H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.4, 162.4, 146.9, 143.5, 141.7, 136.4, 131.1, 130.2, 127.1, 122.7, 115.4, 114.5, 110.7, 55.5, 21.5; IR (KBr) ν: 2960, 2930, 2217, 1730, 1663, 1258, 1170, 1020 cm−1; HRMS (ESI-TOF) calcd for C19H17N2O2+ ([M + H]+): 305.1285, found: 305.1286.
(1E,3E)-2-Oxo-4-phenyl-N-(p-tolyl)but-3-enimidoyl cyanide (3f).
Yellow solid; m.p. 90–92 °C; 1H NMR (CDCl3, 500 MHz) δ: 7.97 (d, J = 16.0 Hz, 1H), 7.85 (d, J = 16.0 Hz, 1H), 7.69 (d, J = 7.5 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.44–7.46 (m, 3H), 7.34 (d, J = 8.0 Hz, 2H), 2.45 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ: 183.6, 146.9, 143.4, 142.0, 135.9, 134.3, 131.4, 130.2, 129.0, 129.0, 122.9, 117.9, 110.6, 21.5; IR (KBr) ν: 2962, 2938, 2218, 1738, 1670, 1261, 1176, 1032 cm−1; HRMS (ESI-TOF) calcd for C18H15N2O+ ([M + H]+): 275.1179, found: 275.1179.
(1E,3E)-4-(Furan-2-yl)-2-oxo-N-(p-tolyl)but-3-enimidoyl cyanide (3g).
Yellow solid; m.p. 146–148 °C; 1H NMR (500 MHz, CDCl3) δ: 7.70 (s, 2H), 7.58 (s, 1H), 7.49 (d, J = 8.5 Hz, 2H), 7.34 (d, J = 8.5 Hz, 2H), 6.84 (d, J = 3.5 Hz, 1H), 6.56 (dd, J = 3.5, 1.6 Hz, 1H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.5, 151.4, 146.0, 143.4, 141.9, 135.8, 132.2, 130.2, 122.9, 118.3, 115.5, 113.1, 110.7, 21.5; HRMS (ESI-TOF) calcd for C16H13N2O2+ ([M + H]+): 265.0972, found: 265.0977.
(1E,3E)-2-Oxo-4-(thiophen-2-yl)-N-(p-tolyl)but-3-enimidoyl cyanide (3h).
Yellow solid; m.p. 133–135 °C; 1H NMR (CDCl3, 500 MHz) δ: 2.44 (s, 3H), 7.12 (dd, J = 3.5, 5.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 2.0 Hz, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 5.0 Hz, 1H), 7.60 (d, J = 16.0 Hz, 1H), 8.07 (d, J = 15.5 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ: 183.2, 143.4, 141.9, 140.0, 139.1, 135.9, 133.5, 130.5, 130.2, 128.6, 122.9, 116.8, 110.7, 21.5; IR (KBr) ν: 2926, 2214, 1665, 1582, 1420, 1213, 1001 cm−1; HRMS (ESI-TOF) calcd for C16H13N2OS+ ([M + H]+): 281.0743, found: 281.0743.
(1E,3E)-4-(Naphthalen-2-yl)-2-oxo-N-p-tolylbut-3-enimidoyl cyanide (3i).
Yellow solid; m.p. 176–178 °C; 1H NMR (500 MHz, CDCl3) δ: 8.13 (d, J = 15.5 Hz, 1H), 8.09 (s, 1H), 7.95 (d, J = 16.0 Hz, 1H), 7.92–7.82 (m, 4H), 7.57–7.53 (m, 2H), 7.51 (d, J = 8.5 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.5, 147.0, 143.4, 141.9, 136.1, 134.7, 133.2, 131.9, 130.2, 128.8, 127.8, 127.7, 126.9, 123.7, 122.9, 118.0, 110.7, 21.5. Two carbons are not visible due to overlapping peaks; IR (KBr) ν: 2920, 2217, 1666, 1598, 1567, 1363, 1302, 1200, 1002 cm−1; HRMS (ESI-TOF) calcd for C22H17N2O+ ([M + H]+): 325.1335, found: 325.1336.
(1E,3E,5E)-2-Oxo-6-phenyl-N-(p-tolyl)hexa-3,5-dienimidoyl cyanide (3j).
Yellow solid; m.p. 160–162 °C; 1H NMR (500 MHz, CDCl3) δ: 7.74 (dd, J = 15.5, 11.0 Hz, 1H), 7.52 (d, J = 7.5 Hz, 2H), 7.47 (d, J = 8.5 Hz, 2H), 7.41–7.35 (m, 4H), 7.33 (d, J = 8.0 Hz, 2H), 7.12 (d, J = 15.5 Hz, 1H), 7.07–7.02 (m, 1H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.6, 146.9, 144.3, 143.4, 141.9, 135.9, 135.7, 130.2, 129.8, 128.9, 127.6, 126.7, 122.9, 121.3, 110.7, 21.5; HRMS (ESI-TOF) calcd for C20H17N2O+ ([M + H]+): 301.1335, found: 301.1327.
(1E,3E)-N,4-Bis(4-chlorophenyl)-2-oxobut-3-enimidoyl cyanide (3k).
Yellow solid; m.p. 148–150 °C; 1H NMR (CDCl3, 500 MHz) δ: 7.42 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.0 Hz, 2H), 7.78 (d, J = 16.0 Hz, 1H), 7.92 (d, J = 16.0 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ: 183.0, 146.0, 144.2, 137.7, 137.6, 136.8, 132.6, 130.2, 129.9, 129.2, 123.7, 118.0, 110.0; IR (KBr) ν: 2923, 2208, 1668, 1607, 1487, 1335, 1092 cm−1; HRMS (ESI-TOF) calcd for C17H11Cl2N2O+ ([M + H]+): 329.0243, found: 329.0257.
(1E,3E)-4-(4-Chlorophenyl)-2-oxo-N-phenylbut-3-enimidoyl cyanide (3l).
Yellow solid; m.p. 165–167 °C; 1H NMR (500 MHz, CDCl3) δ: 7.93 (d, J = 16.0 Hz, 1H), 7.82 (d, J = 16.0 Hz, 1H), 7.64 (d, J = 8.5 Hz, 2H), 7.57–7.54 (m, 2H), 7.50–7.47 (m, 3H), 7.42 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 183.2, 146.0, 145.8, 137.6, 137.5, 132.6, 130.6, 130.2, 129.6, 129.4, 122.0, 118.1, 110.1; IR (KBr) ν: 2220, 1728, 1674, 1604, 1564, 1332.67, 1208, 993 cm−1; HRMS (ESI-TOF) calcd for C17H12ClN2O+ ([M + H]+): 295.0633, found: 295.0626.
(1E,3E)-4-(4-Chlorophenyl)-2-oxo-N-(m-tolyl)but-3-enimidoyl cyanide (3m).
Yellow solid; m.p. 132–134 °C; 1H NMR (500 MHz, CDCl3) δ: 7.92 (d, J = 16.0 Hz, 1H), 7.81 (d, J = 16.0 Hz, 1H), 7.63 (d, J = 8.5 Hz, 2H), 7.45–7.40 (m, 3H), 7.29 (d, J = 5.5 Hz, 3H), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.3, 146.0, 145.7, 139.7, 137.5, 137.2, 132.7, 131.5, 130.2, 129.4, 129.3, 122.8, 118.9, 118.2, 110.2, 21.3; IR (KBr) ν: 2922, 2217, 1667, 1598, 1563, 1329, 994 cm−1; HRMS (ESI-TOF) calcd for C18H14ClN2O+ ([M + H]+): 309.0789, found: 309.0791.
(1E,3E)-4-(4-Chlorophenyl)-N-(4-methoxyphenyl)-2-oxobut-3-enimidoyl cyanide (3n).
Yellow solid; m.p. 153–155 °C; 1H NMR (500 MHz, CDCl3) δ: 7.88 (d, J = 16.0 Hz, 1H), 7.84 (d, J = 16.0 Hz, 1H), 7.79 (d, J = 9.0 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 9.0 Hz, 2H), 3.92 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 183.9, 162.9, 144.8, 138.2, 137.2, 132.9, 132.3, 130.1, 129.3, 126.8, 118.5, 114.9, 111.5, 55.8; IR (KBr) ν: 2917, 2208, 1667, 1608, 1535, 1312, 1271, 1167, 993 cm−1; HRMS (ESI-TOF) calcd for C18H14ClN2O2+ ([M + H]+): 325.0738, found: 325.0733.
General procedure for the preparation of 6 (6a as example)
To a solution of (1E,3E)-4-(4-chlorophenyl)-2-oxo-N-(p-tolyl)but-3-enimidoyl cyanide 3a (0.20 mmol, 61.6 mg) in AcOH (2 mL) were added benzene-1,2-diamine (0.24 mmol, 25.9 mg) and NaOAc (0.24 mmol, 19.7 mg). Then the reaction mixture was stirred at 120 °C for 10 min. After 3a was consumed (monitored by TLC), the reaction mixture was poured into water (50 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to yield the corresponding crude product, which was purified by chromatography (silica gel, petroleum ether/acetone = 10/2, v/v) to give 6a (53.5 mg, 92%) as a yellow solid.
(E)-3-(4-Chlorostyryl)quinoxaline-2-carbonitrile (6a).
Yellow solid; m.p. 173–175 °C; 1H NMR (500 MHz, CDCl3) δ: 8.13 (d, J = 15.5 Hz, 1H), 8.08 (t, J = 8.5 Hz, 2H), 7.89 (t, J = 8.0 Hz, 1H), 7.79 (t, J = 8.0 Hz, 1H), 7.62 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 15.5 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 150.2, 142.7, 140.9, 138.4, 135.8, 133.8, 133.5, 130.9, 129.6, 129.3, 129.2, 129.2, 129.1, 120.8, 115.6; IR (KBr) ν: 2221, 1615, 1535, 1456, 1355, 1228, 951, 860, 750, 718 cm−1; HRMS (ESI-TOF) calcd for C17H11ClN3+ ([M + H]+): 292.0636, found: 292.0636.
(E)-3-Styrylquinoxaline-2-carbonitrile (6f).
Yellow solid; m.p. 172–174 °C; 1H NMR (500 MHz, CDCl3) δ: 8.18 (d, J = 15.5 Hz, 1H), 8.08 (q, J = 8.5 Hz, 2H), 7.88 (t, J = 8.0 Hz, 1H), 7.77 (t, J = 7.5 Hz, 1H), 7.69 (d, J = 7.5 Hz, 2H), 7.64 (d, J = 16.0 Hz, 1H), 7.44–7.37 (m, 3H); 13C NMR (125 MHz, CDCl3) δ: 150.5, 142.7, 140.8, 139.9, 135.3, 133.4, 130.7, 130.0, 129.5, 129.2, 129.1, 128.9, 128.0, 120.2, 115.6; HRMS (ESI-TOF) calcd for C17H12N3+ ([M + H]+): 258.1026, found: 258.1031.
(E)-3-(2-(Thiophen-2-yl)vinyl)quinoxaline-2-carbonitrile (6h).
Yellow solid; m.p. 184–186 °C; 1H NMR (500 MHz, CDCl3) δ: 8.28 (d, J = 15.0 Hz, 1H), 8.04 (d, J = 9.0 Hz, 2H), 7.85 (t, J = 7.5 Hz, 1H), 7.75 (t, J = 7.5 Hz, 1H), 7.39 (d, J = 15.0 Hz, 1H), 7.39–7.36 (m, 2H), 7.09 (t, J = 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ: 150.4, 142.8, 140.9, 140.7, 133.4, 132.3, 130.6, 130.4, 129.5, 129.1, 128.8, 128.2, 128.1, 119.2, 115.5; IR (KBr) ν: 2219, 1606, 1510.40, 1355, 1203, 958, 864, 756, 708 cm−1; HRMS (ESI-TOF) calcd for C15H10N3S+ ([M + H]+): 264.0590, found: 264.0591.
Acknowledgements
Financial support of this research by the National Natural Sciences Foundation of China (21472017 and 21172032), the Natural Sciences Foundation of Jilin Province (20150101065JC) and the Fundamental Research Funds for the Central Universities is greatly acknowledged.
Notes and references
-
(a) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef PubMed;
(b) D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359 RSC.
-
(a)
Z. Rappoport, The Chemistry of the Cyano Group, Interscience Publishers, London, 1970 Search PubMed;
(b)
R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, VCH, New York, 1989 Search PubMed.
-
(a) T. Li, J. Liang, A. Ambrogelly, T. Brennan, G. Gloor, G. Huisman, J. Lalonde, A. Lekhal, B. Mijts, S. Muley, L. Newman, M. Tobin, G. Wong, A. Zaks and X. Zhang, J. Am. Chem. Soc., 2012, 134, 6467 CrossRef CAS PubMed;
(b) F. Chen, X. Huang, Y. Cui and N. Jiao, Chem. – Eur. J., 2013, 19, 11199 CrossRef CAS PubMed;
(c) L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2009, 131, 4070 CrossRef PubMed;
(d) T. Sakai, T. Soeta, K. Endo, S. Fujinami and Y. Ukaji, Org. Lett., 2013, 15, 2422 CrossRef CAS PubMed.
- For selected recent reviews, see:
(a) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed;
(b) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC;
(c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed;
(d) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed.
- Selected articles on the electrochemical cyanation reaction of amines:
(a) F. Louafi, J.-P. Hurvois, A. Chibani and T. Roisnel, J. Org. Chem., 2010, 75, 5721 CrossRef CAS PubMed;
(b) T. Tajima and A. Nakajima, J. Am. Chem. Soc., 2008, 130, 10496 CrossRef CAS PubMed.
-
(a) E. Boess, C. Schmitz and M. Klussmann, J. Am. Chem. Soc., 2012, 134, 5317 CrossRef CAS PubMed;
(b) P. Liu, Y. Liu, E. L.-M. Wong, S. Xiang and C.-M. Che, Chem. Sci., 2011, 2, 2187 RSC;
(c) J. M. Allen and T. H. Lambert, J. Am. Chem. Soc., 2011, 133, 1260 CrossRef CAS PubMed;
(d) G. Zhang, Y. Ma, G. Cheng, D. Liu and R. Wang, Org. Lett., 2014, 16, 656 CrossRef CAS PubMed;
(e) S.-I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931 CrossRef CAS PubMed;
(f) S.-I. Murahashi, N. Komiya, H. Terai and T. Nakae, J. Am. Chem. Soc., 2003, 125, 15312 CrossRef CAS PubMed;
(g) Y. Zhang, H. Peng, M. Zhang, Y. Cheng and C. Zhu, Chem. Commun., 2011, 47, 2354 RSC;
(h) A. Wagner and A. R. Ofial, J. Org. Chem., 2015, 80, 2848 CrossRef CAS PubMed;
(i) C. Yan, Y. Liu and Q. Wang, RSC Adv., 2014, 4, 60075 RSC;
(j) M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 12709 RSC;
(k) D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94 CrossRef CAS PubMed;
(l) L. Liu, Z. Wang, X. Fu and C. Yan, Org. Lett., 2012, 14, 5692 CrossRef CAS PubMed;
(m) S. Kamijo, T. Hoshikawa and M. Inoue, Org. Lett., 2011, 13, 5928 CrossRef CAS PubMed;
(n) K. Alagiri and K. R. Prabhu, Org. Biomol. Chem., 2012, 10, 835 RSC;
(o) S. Singhal, S. L. Jain and B. Sain, Chem. Commun., 2009, 2371 RSC;
(p) D. P. Hari and B. König, Org. Lett., 2011, 13, 3852 CrossRef CAS PubMed;
(q) Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341 RSC.
-
(a) L. Ma, W. Chen and D. Seidel, J. Am. Chem. Soc., 2012, 134, 15305 CrossRef CAS PubMed;
(b) D. Das, M. T. Richers, L. Ma and D. Seidel, Org. Lett., 2011, 13, 6584 CrossRef CAS PubMed.
-
(a) J. M. Concellon and H. Rodriguez-Solla, Curr. Org. Chem., 2008, 12, 524 CrossRef CAS;
(b) F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367 CrossRef CAS PubMed;
(c)
Chemistry and Biochemistry of the Amino Acids, ed. G. C. Barrett, Chapman and Hall, London, 1985 Search PubMed.
- For selected reviews and papers on the arylation reaction:
(a) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082 CrossRef CAS PubMed;
(b) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234 CrossRef CAS PubMed;
(c) M. Miura and M. Nomura, Top. Curr. Chem., 2002, 219, 211 CrossRef CAS;
(d) J.-C. Wu, R.-J. Song, Z.-Q. Wang, X.-C. Huang, Y.-X. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2012, 51, 3453 CrossRef CAS PubMed.
- For selected reviews on the alkylation reaction using alkyl halides, see:
(a) T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656 CrossRef CAS PubMed;
(b) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS PubMed.
- For selected reports, see:
(a) L. Zhao and C.-J. Li, Angew. Chem., Int. Ed., 2008, 47, 7075 CrossRef CAS PubMed;
(b) K. Li, G. Tan, J. Huang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12942 CrossRef CAS PubMed;
(c) C. Huo, Y. Yuan, M. Wu, X. Jia, X. Wang, F. Chen and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 13544 CrossRef CAS PubMed;
(d) J. Xie and Z.-Z. Huang, Angew. Chem., Int. Ed., 2010, 49, 10181 CrossRef CAS PubMed;
(e) B. Yang, T.-T. Yang, X. A. Li, J.-J. Wang and S.-D. Yang, Org. Lett., 2013, 15, 5024 CrossRef CAS PubMed.
- For selected recent reports, see:
(a) Y.-J. Li, X. Li, S.-X. Zhang, Y.-L. Zhao and Q. Liu, Chem. Commun., 2015, 51, 11564 RSC;
(b) Y. Dong, B. Liu, P. Chen, Q. Liu and M. Wang, Angew. Chem., Int. Ed., 2014, 53, 3442 CrossRef CAS PubMed;
(c) H. Wang, Y.-L. Zhao, L. Li, S.-S. Li and Q. Liu, Adv. Synth. Catal., 2014, 356, 3157 CrossRef CAS;
(d) H. Zhang, W. Pu, T. Xiong, Y. Li, X. Zhou, K. Sun, Q. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2013, 52, 2529 CrossRef CAS PubMed;
(e) T. Xiong, Y. Li, X. Bi, Y. Lv and Q. Zhang, Angew. Chem., Int. Ed., 2011, 50, 7140 CrossRef CAS PubMed.
- L. Li, Y.-L. Zhao, Q. Wang, T. Lin and Q. Liu, Org. Lett., 2015, 17, 370 CrossRef CAS PubMed.
- J. Meng, Y.-J. Li, Y.-L. Zhao, X.-B. Bu and Q. Liu, Chem. Commun., 2014, 50, 12490 RSC.
- CCDC 1028181 (3a) contains the supplementary crystallographic data for this paper.
- H. Kaise, J. Shimokawa and T. Fukuyama, Org. Lett., 2014, 16, 727 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1028181. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob01690h |
|
| This journal is © The Royal Society of Chemistry 2016 |