 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe curious case of zeolite–clay/binder interactions and their consequences for catalyst preparation†
Gareth T.
Whiting‡
*,
Abhishek Dutta
Chowdhury‡
,
Ramon
Oord
,
Pasi
Paalanen
and
Bert M.
Weckhuysen
*
Inorganic Chemistry and Catalysis Group, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: g.t.whiting@uu.nl; b.m.weckhuysen@uu.nl
First published on 2nd December 2015
Abstract
Zeolite-based catalyst bodies are commonly employed in a range of important industrial processes. Depending on the binder and shaping method chosen, vast differences in the reactivity, selectivity and stability are obtained. Here, three highly complementary micro-spectroscopic techniques were employed to study zeolite ZSM-5–binder interactions in SiO2-, Al2O3-, SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3- (2:1 mix) and kaolinite-bound catalyst pellets. We establish how their preparation influences the zeolite–clay/binder interactions. Using thiophene as an acid-catalyzed staining reaction, light absorbing oligomers produced in each sample were followed. To our surprise, kaolinite decreased the overall reactivity of the sample due to the phase change of the binder, creating a hard impenetrable outer layer. Aluminum migration to the zeolite was observed when Al2O3 was selected as a binder, creating additional Brønsted acid sites, which favored the formation of ring-opened thiophene oligomers compared to the larger oligomer species produced when SiO2 was used as a binder. In the latter case, the interaction of the Si–OH groups in the binder with thiophene was revealed to have a large impact in creating such large oligomer species. Furthermore, the combination of a SiO2:Al2O3 mix as a binder enhanced the reactivity, possibly due to the creation of additional Brønsted acid sites between the two binder components during pellet preparation. It is evident that, independent of the shaping method, the intimate contact between the zeolite and binder heavily impacts the reactivity and product selectivity, with the type of binder playing a vital role.
:Al2O3- (2:1 mix) and kaolinite-bound catalyst pellets. We establish how their preparation influences the zeolite–clay/binder interactions. Using thiophene as an acid-catalyzed staining reaction, light absorbing oligomers produced in each sample were followed. To our surprise, kaolinite decreased the overall reactivity of the sample due to the phase change of the binder, creating a hard impenetrable outer layer. Aluminum migration to the zeolite was observed when Al2O3 was selected as a binder, creating additional Brønsted acid sites, which favored the formation of ring-opened thiophene oligomers compared to the larger oligomer species produced when SiO2 was used as a binder. In the latter case, the interaction of the Si–OH groups in the binder with thiophene was revealed to have a large impact in creating such large oligomer species. Furthermore, the combination of a SiO2:Al2O3 mix as a binder enhanced the reactivity, possibly due to the creation of additional Brønsted acid sites between the two binder components during pellet preparation. It is evident that, independent of the shaping method, the intimate contact between the zeolite and binder heavily impacts the reactivity and product selectivity, with the type of binder playing a vital role.
1. Introduction
The use of catalyst materials in the petrochemical and bulk chemical synthesis industries is vast, and plays a vital role in supplying many of our everyday needs.1–3 In industrial processes such as hydrocarbon and/or aromatic isomerization- and alkylation-type reactions, zeolites provide the main active phase of the catalyst materials.4,5 However, in order for the zeolite to function at its optimum level, it is often dispersed non-homogeneously in a binder or matrix, i.e. SiO2, Al2O3 and/or clay, and shaped to the desired needs. Such catalysts are commonly referred to as ‘catalyst bodies’ or ‘technical bodies’, and take the form of extrudates, pellets or granules.6 Binder/matrix components offer numerous advantages to the zeolite (as highlighted in a perspective article by Hargreaves et al.7), but their overriding role is to increase the mechanical strength in order to resist attrition loss within industrial reactor conditions. However, our preconceived ideas that the zeolite's physicochemical properties and catalytic performance will be fully maintained during/after the preparation of catalyst bodies (i.e. shaping with binders, heat treatment, etc.) would be naive, with the binder known to interact with the zeolite and also known to play a vital role in the catalyst's performance.4,7–9Until recently, limited academic research has been reported on such industrial catalysts, but a spate in the development of advanced characterization techniques able to probe the multi-dimensional parameters of these materials with high spatiotemporal resolution (and also non-invasively) has provided significant insights into the structure–performance relationships of these closely guarded materials.10–20 Particularly in the field of Fluid Catalytic Cracking (FCC) particles and zeolite-based binder-bound extrudates and pellets for e.g. methanol-to-hydrocarbon (MTH) catalysis, combinations of chemical imaging and structural imaging techniques, together with standard physicochemical characterization of the materials, have enhanced our knowledge into how these complex materials function.17,18,21–23 For instance, staining probe molecule reactions (i.e. acid-catalyzed fluorostyrene oligomerization), coupled with visible micro-spectroscopy, confocal microscopy and X-ray absorption micro-spectroscopy, have shown that the distribution of the zeolite, the presence of metal poisons, and the type/amount of binder present, all significantly influence the catalytic performance and, in particular, the catalyst’s ability to resist deactivation.12,16,20,24,25 Overall, the information gained on binder effects from such advanced characterization studies of industrially relevant catalyst materials is key to improving the catalyst body design, which could play a vital role in the future of these catalysts. This is particularly prominent, given the ever stricter and challenging societal, environmental and economical demands enforced on catalytic industrial reaction processes.
The use of inexpensive inorganic binders, such as SiO2, Al2O3 and clay, for zeolite-based catalyst bodies is common in industrial processes, such as xylene isomerization and benzene alkylation (among others), but several reports note that the zeolite–binder interactions can indeed influence the reactivity, selectivity and stability of the overall catalyst material. When employing clay (i.e. bentonite, kaolin and attapulgite) as a binder with zeolites, high-temperatures are used in the pelletization process in order to eliminate the activity of the clay and destroy the surface area, whilst creating meso- and/or macro-porosity.26 However, it has been reported that a substantial decrease in the inter- and intra-crystalline acidity of the H-zeolite occurs during pelletization due to the neutralization of acid sites with mobile alkaline or alkaline-earth cations from the clay binder.27 Clay has also been shown to decrease weak acidity and increase high strength acid site density,28 as well as influence product selectivity29 and improve coke stability (by trapping coke precursors in the kaolinite binder).30 Al2O3 as a binder for zeolite-based catalyst bodies has also been known to influence product selectivity and overall activity due to the migration of aluminum from the binder to the zeolite framework, creating additional Brønsted acid sites.17,31,32 It is also used as a heat sink and a trap for metal poisons in petroleum feedstocks, hence retaining zeolite acidity.33 A similar trend occurs for SiO2-bound zeolite materials, whereby a more unexpected solid-state ion exchange takes place, with Michels et al. and Lee et al. reporting a loss of Brønsted acidity due to dealumination from the framework of the zeolite.34,35 Gélin et al. also witnessed the opposite trend, with Si species from the SiO2–Al2O3 matrix migrating to the zeolite framework, healing defects and preserving crystallinity.36 It is clear that, although some binders are often considered as inert, the intimate contact between the zeolite and binder in catalyst bodies can produce remarkable consequences or advantages and their interactions therefore require a greater degree of attention, if improvements in the catalyst design are to be made.
In our previous work, we have focused on SiO2- and Al2O3-bound zeolite-based catalysts in the form of extrudates.17 However, in this work, we set out to synthesize four ZSM-5–binder-bound pellets, using either SiO2, Al2O3, SiO2:Al2O3 (mix 2:1) or kaolinite as a binder, and study whether any zeolite–binder interactions have taken place during their physical mixing, and how these parameters influence the overall catalytic properties of the catalysts prepared. To do this, a staining probe reaction, namely thiophene oligomerization, was employed along with several highly complementary non-invasive in situ micro-spectroscopic techniques: UV-Visible, confocal fluorescence and IR micro-spectroscopy. Using these three techniques, we are able to follow variations in product selectivity as well as in the location and distribution of the light absorbing oligomers formed in each sample and correlate these to any zeolite–clay/binder effects.
2. Experimental
2.1. Materials
Four batches of SiO2- (purchased from Aldrich, Davisil™), Al2O3- (Catapal®), SiO2/Al2O3- (2:1 mix of Davisil™:Catapal®) and kaolinite- (purchased from Sigma Aldrich) bound ZSM-5 samples were prepared by a pelletization procedure: NH4–ZSM-5 powder (purchased from Zeolyst CBV 8014) with a SiO2:Al2O3 ratio of 80 was thoroughly mixed with the relevant binder at a ZSM-5:binder wt% ratio of 30:70 (to ensure noticeable binder effects) before pressing into pellets. Each pellet underwent heat treatment under a N2 atmosphere (to form H–ZSM-5) from 30 °C to 600 °C (at 15 °C min−1) and it was then held at this temperature for 3 h, before cooling to 30 °C. Sample notations are as follows: ZSM-5–a: where ‘a’ corresponds to either: Si (SiO2); Al (Al2O3); SiAl (SiO2–Al2O3 mix, 2:1); Kao (kaolinite).
2.2. Ar physisorption
Ar physisorption on all samples was performed with an automated gas sorption system Micromeritics TriStar 3000. Prior to the measurements at 77 K, all samples were dried at 300 °C for 20 h. The surface areas were calculated using the BET model, whilst the external surface area, micropore surface area and micropore volume were determined using the t-plot method (Table 1 and Fig. 1).| Sample | BET surface area (m2 g−1) | t-Plot micropore area (m2 g−1) | Pore volume (cm3 g−1) | t-Plot micropore volume (m2 g−1) |
|---|---|---|---|---|
| a Predicted values calculated from the pellet composition and the experimental values of the pure binder and pure zeolite components. | ||||
| ZSM-5 | 371 | 306 | 0.18 | 0.12 |
| ZSM-5–Si | 252 (296)a | 90 (110)a | 0.46 (0.80)a | 0.03 (0.04)a |
| ZSM-5–Al | 269 (316) | 93 (97) | 0.30 (0.33) | 0.03 (0.04) |
| ZSM-5–SiAl | 272 | 106 | 0.35 | 0.04 (0.04) |
| ZSM-5–Kao | 176 (113) | 150 (97) | 0.09 (0.06) | 0.06 (0.04) |
| SiO2 | 264 | 26 | 1.06 | 0 |
| Al2O3 | 293 | 33 | 0.39 | 0 |
| Kaolinite | 3 | 7 | 0.01 | 0 |
 | ||
| Fig. 1 Pore volume distributions, determined by the Barrett–Joyner–Halenda adsorption method with Ar, for each selected binder (dashed lines) and its corresponding zeolite–clay/binder pellets (solid lines): ZSM-5 (black diamonds); ZSM-5–Si (red triangles); ZSM-5–Al (green circles); ZSM-5–SiAl (blue squares); ZSM-5–Kao (orange crosses). | ||
2.3. X-ray powder diffraction
All samples were measured with a Bruker D8 Discover diffractometer equipped with a 1-dimensional LynxEye XE energy dispersive detector, in transmission capillary geometry with a Mo (Kα1 0.709 Å) source. The sample powders were loaded into 980 μm inner diameter quartz capillaries (1000 μm outer diameter) so that the sample bed length was about ≈20 mm. The measurement parameters used were 2θ = 2.5–24° with a step size of 0.008 and an exposure time of 3.5 s per step. The Mo source was run with a power of 2500 W (50 kV × 50 mA) (Fig. 2). | ||
| Fig. 2 Powder X-ray diffraction (XRD) patterns of the following ZSM-5-bound pellets (from top to bottom): ZSM-5 (black); ZSM-5–Si (red); ZSM-5–Al (green); ZSM-5–SiAl (blue); and ZSM-5–Kao (orange). The inset corresponds to the main zeolite diffraction peaks, noting no noticeable shifts in 2θ between the samples under study. | ||
| Sample | Rietveld Rwp values | Crystallite size (LVol-IB) (nm) | Microstrain | NH3-TPD min. peak temperature (°C) | NH3-TPD max. peak temperature (°C) |
|---|---|---|---|---|---|
| ZSM-5 | 3.1 | 89.1 | 0.00093 | 191 | 385 |
| ZSM-5–Si | 2.7 | 77.6 | 0.00063 | 175 | 367 |
| ZSM-5–Al | 3.0 | 79.7 | 0.00117 | 192, 290 | 386 |
| ZSM-5–SiAl | 2.6 | 85.8 | 0.00094 | 183 | 365 |
| ZSM-5–Kao | 2.5 | 81.8 | 0.00096 | 175 | 384 |
2.4. NH3-TPD
The acidity of the samples was measured by temperature-programmed desorption (TPD) with ammonia as the probe molecule using Micrometrics AutoChem 2920 apparatus. Prior to the adsorption of ammonia, 100 mg of the sample placed in a quartz tube reactor was preheated at 550 °C under 50 mL min−1 of He flow for 15 min (rate 10 °C min−1). Adsorption of ammonia (10 vol% in He) was performed in a pulse wise manner at 100 °C. In order to remove the physisorbed ammonia, the sample was flushed with a He stream (25 mL min−1) at 100 °C for 1 h. The ammonia desorption was carried out under a He flow in the temperature range 100–700 °C at a heating rate of 5 °C min−1. The amount of adsorbed ammonia was monitored with a thermal conductivity detector (TCD) (Fig. 3 and Table 2). | ||
| Fig. 3 NH3-temperature programmed desorption (TPD) profiles of the following ZSM-5-bound pellets: ZSM-5 (black); ZSM-5–Si (red); ZSM-5–Al (green); ZSM-5–SiAl (blue); and ZSM-5–Kao (orange). | ||
2.5. UV-Vis absorption micro-spectroscopy
The UV-Vis micro-spectroscopy measurements were performed with a CRAIC 20/30 PV™ UV-Vis-NIR micro-spectrophotometer using a 15× objective. A 75 W xenon lamp was used for illumination. The pellets were placed on the heating stage of an in situ Linkam cell (THMS600) equipped with a temperature controller (Linkam TMS94), where they were impregnated with 8 μL of thiophene. During each experiment, after complete adsorption of 8 μL of thiophene under atmospheric conditions, the pellets were heated (from 30 °C up to 120 °C at a rate of 15 °C min−1, and then held at this temperature for a total of 1000 s).2.6. Confocal fluorescence microscopy
A Nikon Eclipse 90i confocal microscope with a 100 × 0.73 NA dry objective was used for the fluorescence microscopy investigations. Excitation light was provided by focusing two specific laser lines, 488 and 561 nm, simultaneously on the desired sample, located in an open in situ cell (Linkam Instruments, FTIR 600). The microscope was equipped with a Nikon A1 scan head, accommodating the optics, which couple fiber optics for excitation and emission light with the microscope. A spectral analyzer in the Nikon A1 system was equipped with 32 photomultiplier tubes (PMTs) set to collect emission light in the region of 480–730 nm, with a resolution of 6 nm. 3D confocal fluorescence microscopy images were recorded at a similar focal depth in each sample using an identical laser power. Prior to microscopic analysis, to each sample, 8 μL of the desired probe molecule was impregnated at 30 °C, before ramping the temperature (at 15 °C min−1) to 120 °C, holding at this temperature for 15 min, and cooling to 30 °C.2.7. Infrared micro-spectroscopy
In situ infrared micro-spectroscopy experiments were performed using a Perkin Elmer Spotlight 400 FT-IR microscope with an aperture size of 100 × 100 μm. Each zeolite–clay/binder-bound pellet was placed under the microscope and spectra were obtained in reflectance mode before, during and after thiophene oligomerization (from 30 °C to 120 °C at 15 °C min−1).3. Results and discussion
3.1. UV-Vis absorption micro-spectroscopy
In order to probe possible zeolite–clay/binder interactions created during the catalyst preparation procedure (i.e. pelletization and/or heat treatment), a suitable reaction was chosen. The oligomerization of thiophene has previously been utilized by our research group for such purposes on zeolite–binder-bound catalyst extrudates, and offers an excellent means to gain insight into characteristics such as how the nature and/or number of Brønsted acid sites within the samples influence the catalytic reactivity and intermediate/product selectivity.17,18 A particular highlight of the chosen reaction is that the majority of oligomer species produced on the acid sites within the samples are light absorbing (Scheme 1). Therefore, UV-Vis absorption spectroscopy allows us to obtain, in situ, the formation of absorption bands and hence the nature and relative amount of oligomer species formed, as a function of reaction time. Scheme 1 provides a general overview of the possible light absorbing species produced (measured using Time-Dependent Density Functional Theory (TD-DFT)), either during an oligomerization reaction pathway, or during a ring-opening pathway (leading to the formation of thiol-like species), with the pathway chosen determined by the strength and number of Brønsted acid sites of the zeolite.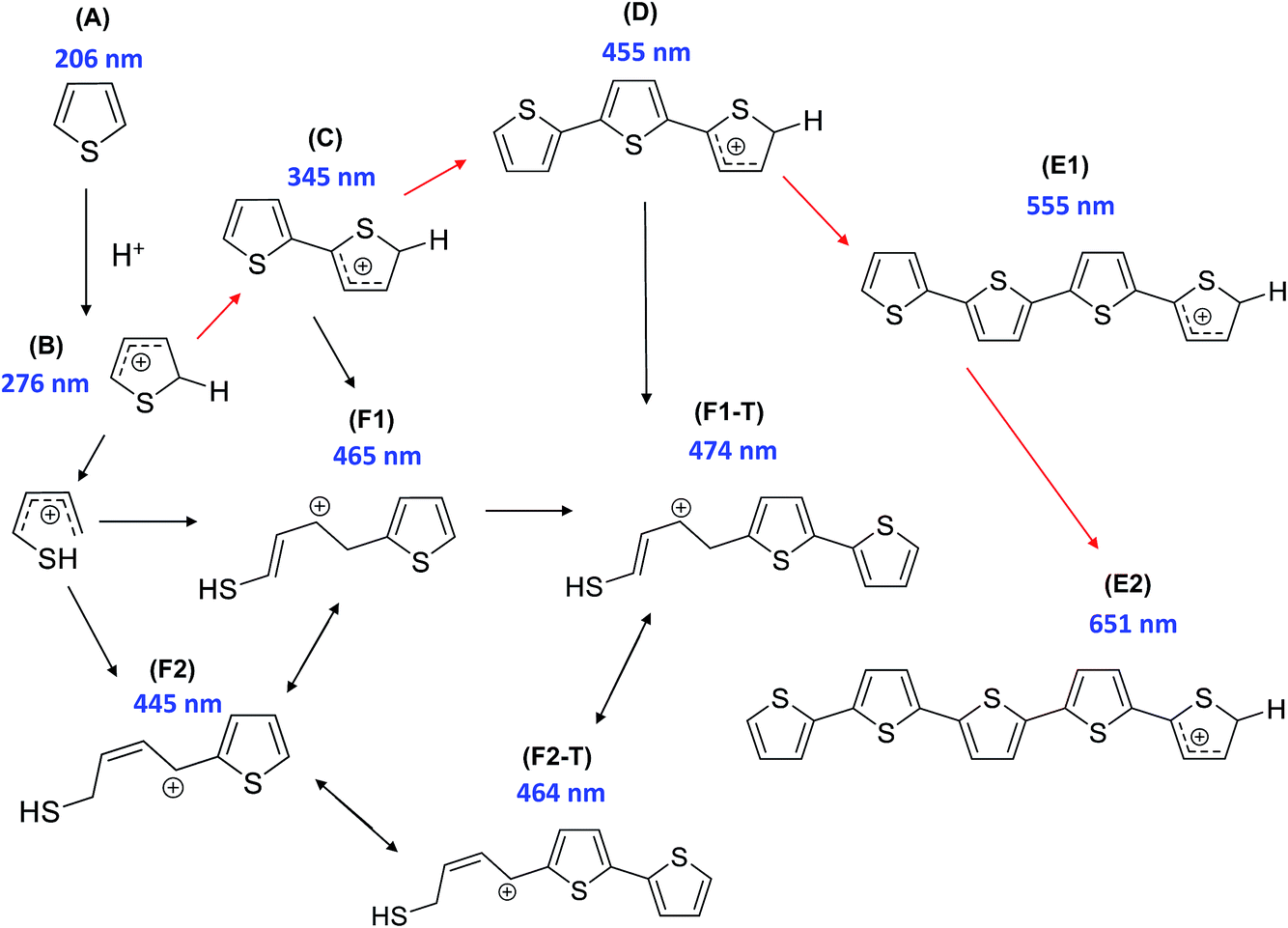 | ||
| Scheme 1 Proposed reaction pathway of thiophene oligomerization on zeolite Brønsted acid sites. Thiophene monomer (A) undergoes protonation on the Brønsted acid sites of H–ZSM-5 to form the protonated monomer (B). Two different reaction pathways are possible at this stage: opening of the thiophene ring and subsequent reaction with a second monomer to form a thiol-like carbocation (F1/F1-T/F2/F2-T), or, alternatively, dimerization (to C) can occur followed by further oligomerization (to D) or opening of the thiophene ring, forming a thiol-like carbocation (F1/F1-T/F2/F2-T). The trimeric carbocationic species (D) can undergo further oligomerization to form more extended/conjugated species (E1/E2). [Reproduced from ref. 17]. | ||
Given the complex nature of the overall UV-Vis absorption spectra of all the samples (Fig. S1 and S2†), absorption spectra of each sample were compared at various reaction times during the oligomerization reaction (Fig. 4). Fig. 4(a) displays the absorption spectra for pellets of ZSM-5, ZSM-5–Si, ZSM-5–Al, ZSM-5–SiAl and ZSM-5–Kao after 10 s of reaction time. Even at this early stage of the reaction, visible differences between each catalyst's absorption spectrum are apparent. The absorption band formed at 275 nm can be assigned to α-protonated thiophene monomers (Scheme 1B) on zeolite Brønsted acid sites.37 The band at 310 nm is attributed to a protonated dimeric carbocation, formed by the interaction of two thiophene molecules linked together in the zeolite pores (C).37 The more intense absorption bands at 410, 500, and 550 nm are assigned to molecules formed via the oligomerization route, namely: a protonated trimeric carbocation species, i.e. C12H13S3+; a protonated trimeric carbocation species, i.e. C12H9S3+; and a tetrameric carbocation species (E1), respectively.37 Finally, the band at 445 nm can be assigned to a thiol-like species (F2) formed via a ring-opening reaction pathway.
 | ||
| Fig. 4 UV-Vis absorption spectra of ZSM-5 (black line), ZSM-5–Si (red), ZSM-5–Al (green), ZSM-5–SiAl (blue), and ZSM-5–Kao (orange) pellets reacted with thiophene after: (a) 10 s; (b) 50 s; (c) 100 s; (d) 200 s; (e) 400 s; and (f) 800 s of reaction time. | ||
Taking each sample into consideration, a feature that stands out in the spectra at 10 s of reaction time (Fig. 4(a)) is the surprising lack of intense absorption bands for the kaolinite-bound sample, ZSM-5–Kao. This suggests a clear inactivity when ZSM-5 crystals are bound to clay, compared with ZSM-5 bound to an amorphous binder. As NH3-TPD showed (Fig. 3), the sample bound to kaolinite displayed the lowest amount of weak and strong Brønsted acid sites, which could explain the low thiophene oligomerization reactivity, but this is not likely considering the similar profile for ZSM-5–Si (which shows a far higher reactivity, and even has slightly weaker acid sites due to a slight shift to lower temperatures in the NH3 desorption peaks). X-ray diffraction of the ZSM-5–Kao material (Fig. 2) showed a clear phase change from kaolinite (Fig. S3†) to a combination of muscovite and orthoclase upon preparation of the ZSM-5–Kao pellet. This feature is due to the loss of hydroxyl groups from kaolinite upon heat treatment of the pellet, which propagates the phase change of the clay,38 and could play a vital role in the inactivity of the sample. Further evidence to support this claim is observed in Fig. S2(a),† where the heat-treated ZSM-5 powder pelletized with non-heat-treated kaolinite reveals a far higher reactivity towards thiophene oligomerization (due to the unchanged phase of kaolinite). Looking towards the Mohs hardness properties of the newly developed binder in ZSM-5–Kao, an increase from 2.5 (kaolinite) to 6 (orthoclase) gives an indication that thiophene impregnation into the pellet could be hindered. Ar physisorption results provide further support that the phase change of the material is detrimental to the textural properties of the catalyst, with limited porosity available in ZSM-5–Kao, and even a striking elevation in the expected micropore area and micropore volume values (Table 1). Given the temperature of the heat treatment of the pellets was 600 °C, it is highly likely that upon the loss of hydroxyl groups and H2O, partial steaming of the zeolite takes place, exposing a greater micropore area. Also of importance is to take into account solid-state interfacial reactions between the clay material and the zeolite, leading to a decrease in the number of Brønsted acid sites.
As expected, the ZSM-5 pellet obtains a higher absorbance at 275 nm compared to the other pellets, considering the larger number of Brønsted acid sites available (Fig. 3) to protonate thiophene monomers. An indication into the acid site strength between each pellet is also evident when comparing the intensity ratio of the oligomers absorbing light at 310, 410, 500 and 550 nm. A high absorbance at 310 nm is displayed for both the ZSM-5 pellet and more so for the ZSM-5–Al pellet, compared to its absence in ZSM-5–Si and weak absorbance in ZSM-5–SiAl. This suggests that the higher amount of acid sites and/or stronger acid sites on both the ZSM-5 and ZSM-5–Al pellets (Fig. 3) favor the oligomerization of the protonated thiophene monomers to form the dimeric carbocation (C).
The 550 nm band accounts for a higher degree of oligomerization, however, as the band is tentatively assigned to a tetrameric carbocation species (Scheme 1), it is unlikely to fit within the ZSM-5 pores and it is therefore probable that it forms on the outer surface of the ZSM-5 crystals. Given that this large species forms at room temperature, it is also feasible that this is a cationic radical species, which can form on Lewis acid sites (extra framework aluminum, EFAL) in zeolites.39
The presence of a 445 nm absorption band for the ZSM-5–Al and ZSM-5–SiAl (weak) pellets, clearly absent for all the other pellets at this reaction time (10 s), also suggests that the Al2O3-bound pellets possess additional acid sites of a different acid strength (most likely Lewis acid sites), which favor the ring-opening pathway and hence the formation of thiol-like species. This was observed in our previous studies on ZSM-5-based Al2O3-bound extrudates, where Al migration occurred and favored the formation of such species. It is important to note that Al2O3 alone is inactive for the reaction (Fig. S2†) and therefore this suggests that Al migration has indeed created additional acid sites in the ZSM-5–Al and ZSM-5–SiAl pelletized samples. An indication that this has taken place for ZSM-5–Al is presented using Rietveld analysis of the ZSM-5 and ZSM-5–Al X-ray diffraction patterns (Table 2) as well as the NH3-TPD results (Fig. 3). When comparing the microstrain values between the two samples (0.00093 and 0.00117, respectively), there is a clear increase for the Al2O3-bound sample, which could suggest the incorporation of Al in the zeolite framework. This indication is enhanced further by the NH3-TPD profile of ZSM-5–Al, which not only presents two peaks corresponding to weak (192 °C) and strong (386 °C) acid sites, but also a new peak (compared to ZSM-5) centered at 290 °C, suggesting Al migration creates additional acid sites of moderate acid strength, also reported by Zhang et al.40
The decrease in absorbance of the 275 nm band and the rise in the 310 nm band for all the pellets (except ZSM-5–Kao) after 50 s (Fig. 4(b)) and 100 s (Fig. 4(c)) show that the protonated thiophene monomers are interacting within the zeolite pores, leading to the formation of the dimeric carbocationic species. This is accompanied by an overall increase in absorbance across the whole wavelength range as expected, due to the increase in the formation of carbocationic species as the reaction time increases. However, it is noticeable that the ratio of the 550 nm band (tetramer oligomer species, E1) to the 410 nm band (trimeric oligomer species, D) is decreasing, accompanied by a rise in the 445 (except for ZSM-5–Si) and 480 nm bands. This suggests that alternative reaction pathways are now taking place, with an increase in the formation of thiol-like species (F2 and F1-T) and trimeric species (D), at the expense of the formation of larger oligomers (E1).
However, the lower ratio of the 550 nm absorbance band can be explained in Fig. 4(d) at a reaction time of 200 s, whereby an increase in a broad band >550 nm is now visible, suggesting that these species are oligomerizing to even larger species over time. Again, the increase in the absorbance of the 445, 460 and 480 nm bands shows that ring-opening is now one of the main reaction pathways for all the catalysts (except ZSM-5–Kao), possibly due to the lack of strong acid sites at this point of the reaction, which are involved in forming larger oligomer species. It is also important to note the much higher overall absorbance of the ZSM-5–SiAl pellet compared with ZSM-5, ZSM-5–Si and ZSM-5–Al (which are all relatively similar), suggesting a binder effect in this sample. A possible explanation could be the interaction between the two components of the binder (i.e. Al migration to SiO2, or Si migration to Al2O3), creating additional acid sites to perform the thiophene oligomerization reaction. This is again backed by the NH3-TPD profile of ZSM-5–SiAl (Fig. 3), which shows a far higher ratio of stronger acid sites to weaker sites compared to ZSM-5 and a greater relative amount of both weak and strong acid sites compared to ZSM-5–Si. However, further studies are needed to develop a better understanding of how this mixed binder promotes such a high product formation.
Similar reactivity and selectivity trends continue towards the termination of the reaction at 400 s (Fig. 4(e)) and 800 s (Fig. 4(f)), with a decrease in the absorbance of both the 275 and 310 nm bands and a continued rise in bands >400 nm, particularly towards the formation of thiol-like species and large oligomers. The appearance of both 580 and 620 nm bands and the broadness of the UV-Vis spectrum background (particularly in the case of ZSM-5–Si) suggests the formation of complex larger molecules and/or coke in each pellet. Regarding ZSM-5–Si, it is important to note at this stage, that a SiO2 pellet produces a very broad band in the >580 nm region (Fig. S2(c)†), which explains the high absorbance in the ZSM-5–Si spectra. The reason for SiO2 reacting with thiophene is, at this stage, not clear, as SiO2 is not expected to provide acid sites with enough strength to convert thiophene monomers.
Confocal fluorescence microscopy corresponds well with UV-Vis micro-spectroscopy and is an excellent method to provide visible insight into the oligomer species formed and how they are distributed over each ZSM-5–clay/binder-bound pellet. Fig. 5 presents the 3D confocal fluorescence microscopy volumes of each individual sample excited with both 488 and 561 nm lasers simultaneously, after the completion of the thiophene oligomerization reaction. These two laser line wavelengths were chosen to gain an understanding into the relative amount and nature of ‘smaller’ to ‘larger’ oligomer species formed. As expected for the ZSM-5 pellet (Fig. 5(a)), a homogeneous distribution of fluorescent oligomers are obtained, with an orange fluorescence indicating the formation both <3-thiophene ring oligomers/thiol-like species and >3-thiophene ring oligomers, with a higher relative amount of the latter (Fig. S4†).
 | ||
| Fig. 5 3D confocal fluorescence microscopy images of the following ZSM-5–binder-bound pellets after reaction of thiophene at 120 °C: (a) ZSM-5; (b) ZSM-5–Si; (c) ZSM-5–Al; (d) ZSM-5–SiAl; and (e) ZSM-5–Kao. The XYZ axis values correspond to: 127 × 127 × 27 μm. | ||
ZSM-5–Al (Fig. 5(c)) and ZSM-5–SiAl (Fig. 5(d)) show similar results to that of ZSM-5, with a more heterogeneous distribution of ZSM-5 crystals/agglomerates due to the binder distribution. However, ZSM-5–Si reveals a higher fluorescence intensity than that for all the other pellets, showing its higher ratio of larger oligomers/coke species (Fig. S4†), as evidenced by UV-Vis absorption (Fig. 4(f)), and this is again explained by the unexpected reactivity of SiO2 (Fig. S2(c)†). The lack of reactivity for the ZSM-5–Kao pellet leads to an almost blank image, with only a few visible fluorescent ZSM-5 crystals, confirming that the binder has a detrimental effect to the active zeolite phase by blocking access to the active zeolite beneath the hard outer layer.
In order to develop a greater understanding of the chemical species formed in each ZSM-5–binder-bound pellet upon thiophene oligomerization, in situ FT-IR micro-spectroscopy was performed. Given the high concentration of moisture present in the ZSM-5 pellet (Fig. S5†), it was difficult to draw detailed conclusions from the whole range of FT-IR spectra before, during and after thiophene oligomerization. Therefore, we will focus on the results of the other pellets (which have only 30 wt% ZSM-5 present).
Fig. 6(a) and (b) present the FT-IR spectra of ZSM-5–Si before, during and after thiophene oligomerization, allowing a correlation to those timescales in the UV-Vis absorption spectra (Fig. 4). Although it is not possible to observe a band at 3610 cm−1 corresponding to SiOHAl groups (Brønsted acid sites) in the fresh pellet and after thiophene impregnation, it is already known (and expected) that this band will decrease in intensity or become perturbed upon the adsorption of thiophene monomers.41 Two other prominent features expected in the fresh ZSM-5–Si pellet are visible at 3746 cm−1 and between 1250 and 950 cm−1, corresponding to the Si–OH groups of SiO2 and zeolite overtones, respectively.
 | ||
| Fig. 6 In situ FT-IR spectra of (a and b) ZSM-5–Si and (c and d) ZSM-5–Al before (black), during (red) and after (blue) reaction with thiophene. | ||
Upon thiophene addition and reaction, numerous bands appear over the whole FT-IR range (4000–750 cm−1). Focusing first on the 4000–2250 cm−1 region (Fig. 6(a)), the most noticeable change, which helps explain the reactivity of SiO2, is the disappearance of the 3746 cm−1 band, indicating that thiophene monomers not only interact with ZSM-5 acid sites, but also with the binder surface. The presence of a broad band around 3620 cm−1 has previously been attributed to the hydrogen bonding of Si–OH groups with π-electron donor molecules, enforcing this theory.41,42 The bands present at 3110, 3076 (unsaturated ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching vibrations) and 1408 cm−1 (symmetric ring stretching vibrations of CC)43 are assigned to thiophene rings hydrogen bonded to the Brønsted acid sites and Si–OH groups, corresponding well with the 275 nm UV absorption band observed previously (Fig. 4(a)). The bands visible at 1588 and 760 cm−1 also refer to the thiophene ring, as C–H in-plane bending vibrations and neighboring aromatic C–H, respectively.
C–H stretching vibrations) and 1408 cm−1 (symmetric ring stretching vibrations of CC)43 are assigned to thiophene rings hydrogen bonded to the Brønsted acid sites and Si–OH groups, corresponding well with the 275 nm UV absorption band observed previously (Fig. 4(a)). The bands visible at 1588 and 760 cm−1 also refer to the thiophene ring, as C–H in-plane bending vibrations and neighboring aromatic C–H, respectively.
However, there are not only bands due to intact/adsorbed thiophene present, but also additional bands even at a low reaction temperature, suggesting several species are formed. The bands observed at 2970, 2932 and 2880 cm−1 are attributed to CH2 groups in proximity to a double bond, i.e. asymmetric *CH2–(CHCH2) stretching vibrations, or a neighboring S atom (CH2–S–).41 Therefore, these are attributed to thiol-like species with further bands at 1806–1770 and 930 cm−1 also indicating their formation (CC–H stretch and bending, respectively). However, Geobaldo et al. propose an alternative theory, suggesting that the bands present at 1800–1600 cm−1 and 2980–2800 cm−1 correspond with carbocationic oligomer species, also reported in our UV-Vis absorption spectra (Fig. 4).37 Both proposals correspond well with our results in this work, however, we can prove the presence of thiol-like species given that perturbed S–H bands are present at 2714, 2584 and 2374 cm−1.41
Towards the termination of the reaction, it is clear that no bands corresponding to adsorbed thiophene monomers are present (i.e. 3110 and 3076 cm−1). This is accompanied by the re-appearance of the Si–OH groups (3746 cm−1), suggesting full conversion of thiophene to its respective intermediate/products, recognized by bands still present at 2970, 2932, 930 cm−1. Another noticeable feature in the region of 1250–900 cm−1 is the shift towards lower wavenumbers for the zeolite overtone bands upon thiophene reaction. Given the higher ratio of larger oligomers/coke species formed in ZSM-5–Si compared to in the other samples (also displayed by the higher orange fluorescence intensity), it could be that such large species formed (inside the pores) are distorting the zeolite framework, but further studies with in situ XRD (Rietveld analysis) are needed to confirm this finding.
The in situ FT-IR spectra of ZSM-5–Al (Fig. 6(c) and (d)) show similar features but also distinct differences compared to the ZSM-5–Si spectra. The decrease in intensity of the 3746 cm−1 band of fresh ZSM-5–Al is expected considering the significant decrease in Si–OH groups compared to ZSM-5–Si, but it is also apparent that bands present upon thiophene impregnation are much lower in intensity in the 4000–2250 cm−1 region. This could be due to the presence of H2O in the sample (as with ZSM-5), but yet it is still possible to distinguish vital additional bands. The presence of bands at 3110, 3076, 2970, 2930, 2710 and 2950 cm−1 all agree with the proposed formation of both oligomer and thiol-like species. However, additional bands at 1610 and 1380 cm−1, as well as the large increase and broadening of the 1410 cm−1 band, confirm perturbed vibration of thiophene molecules, which are strongly interacting with EFAL species.41 This again enforces our previous proposal that Al migration has occurred from the Al2O3 binder to ZSM-5, creating additional acid sites that are now interacting with thiophene. As with ZSM-5–Si, the re-appearance of the 3746 cm−1 band and absence of the 3110 and 3076 cm−1 bands towards the termination of the reaction again suggest the full conversion of thiophene monomers.
As in the case of ZSM-5–Si, very distinct and clear bands are formed with ZSM-5–SiAl upon its impregnation with thiophene (Fig. 7(a) and (b)). The increase in intensity of all relevant bands related to thiophene and intermediate/product species could relate to the high absorbance obtained in the UV-Vis spectra (Fig. 4). This indicates that a far greater absorption of thiophene is present in this sample compared with the other pellets. Other than the main bands relating to oligomer and thiol-like species, similar bands to ZSM-5–Al are also present (1620 cm−1), again suggesting the interaction of thiophene with EFAL species created from Al migration in ZSM-5–SiAl.
 | ||
| Fig. 7 In situ FT-IR spectra of (a and b) ZSM-5–SiAl and (c and d) ZSM-5–Kao, before (black), during (red) and after (blue) reaction with thiophene. | ||
Finally, the in situ FT-IR spectra of ZSM-5–Kao confirm the limited reactivity of the catalyst pellet. Taking the bands present in the region of 4000–3800 cm−1 as an ‘internal standard’ between samples (relating to the adsorption of thiophene), it is apparent that a far lower concentration of thiophene is adsorbed on ZSM-5–Kao, confirming the earlier proposal that the phase change upon pelletization/heat treatment creates a hard layer hindering the access of thiophene to the zeolite active phase. Nevertheless, the vibrational bands present around 2968, 2928 and 2872 cm−1 suggest the formation of oligomers and thiol-like species, possibly due to some accessible zeolite on the surface, but the absence of observed bands in the 2800–750 cm−1 region do not allow further interpretation.
4. Conclusions
In relation to our previous studies on ZSM-5–binder extrudates, four ZSM-5–binder-bound pellets were prepared using SiO2, Al2O3, SiO2:Al2O3 (2:1 mix) and kaolinite as binder materials, in order to determine how zeolite–clay/binder interactions upon physical mixing influence the catalytic performance. Using complementary micro-spectroscopic techniques, coupled with thiophene oligomerization as a staining probe reaction, it is evident that, independent of the shaping method (i.e. pellet or extrudate), the intimate contact between the zeolite and clay/binder in catalyst bodies heavily impacts their reactivity and product selectivity, with the type of binder playing a vital role. Kaolinite was found to limit the reactivity of ZSM-5 within the pellet due to its phase change upon heat treatment, creating a hard impenetrable outer layer, which hindered thiophene accessibility to the ZSM-5 crystals. Aluminum migration, previously witnessed for ZSM-5–binder-bound extrudates, also occurs in the pellet formation of ZSM-5–Al2O3-bound samples, suggesting that it is difficult to limit this process occurring when these components are in close proximity. Given the higher selectivity towards the ring opening of thiophene and its oligomers, compared with ZSM-5 and ZSM-5–Si, it should be taken into account that aluminum migration can alter product selectivity when chosen as a binder. The high reactivity towards larger oligomer species for ZSM-5–Si, clearly visible with confocal fluorescence microscopy, was due to the interaction of the SiO2 binder with thiophene (observed using IR spectroscopy), which, given the lower acid site strength and number compared to ZSM-5 and ZSM-5–Al, was unexpected. Finally, the combination of a SiO2:Al2O3 mixed binder enhanced the reactivity of ZSM-5, which could be due to the interaction of the two binder components creating additional acid sites, but further studies are needed to resolve this finding. Evidently, the combination of UV-Vis, confocal fluorescence and IR micro-spectroscopy with staining probe reactions is highly complementary, providing new insights into zeolite-based catalyst bodies and proving that the choice of binder is crucial to the catalytic performance, whether it be detrimental or advantageous.
Acknowledgements
The authors would like to thank Lennart Webber (Utrecht University, UU) for the Ar physisorption analysis, and Joris Goetze (UU) for fruitful discussions. This work was financially supported by a NWO personal ‘Veni’ grant awarded to G. T. W. and a Top Grant of NWO to B. M. W.References
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS
.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed
- A. Corma, M. J. Díaz-Cabañas, J. Martínez-Triguero, F. Rey and J. Rius, Nature, 2002, 418, 514–517 CrossRef CAS PubMed
- A. de Lucas, J. L. Valverde, P. Sánchez, F. Dorado and M. J. Ramos, Ind. Eng. Chem. Res., 2004, 43, 8217–8225 CrossRef CAS
- C. Perego and P. Ingallina, Catal. Today, 2002, 73, 3–22 CrossRef CAS
- S. Mitchell, N.-L. Michels and J. Pérez-Ramírez, Chem. Soc. Rev., 2013, 42, 6094–6112 RSC
- J. S. J. Hargreaves and A. L. Munnoch, Catal. Sci. Technol., 2013, 3, 1165 CAS
- P. Sánchez, F. Dorado, A. Fúnez, V. Jiménez, M. J. Ramos and J. L. Valverde, J. Mol. Catal. A: Chem., 2007, 273, 109–113 CrossRef
- E. T. C. Vogt, G. T. Whiting, A. Dutta Chowdhury and B. M. Weckhuysen, Adv. Catal., 2015, 58, 143–314 Search PubMed
- F. Meirer, D. T. Morris, S. Kalirai, Y. Liu, J. C. Andrews and B. M. Weckhuysen, J. Am. Chem. Soc., 2015, 137, 102–105 CrossRef CAS PubMed
- Z. Ristanović, J. P. Hofmann, G. de Cremer, A. V. Kubarev, M. Rohnke, F. Meirer, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, J. Am. Chem. Soc., 2015, 137, 6559–6568 CrossRef PubMed
- F. Meirer, S. Kalirai, D. Morris, S. Soparawalla, Y. Liu, G. Mesu, J. C. Andrews and B. M. Weckhuysen, Sci. Adv., 2015, 1, 1400199 Search PubMed
- F. Meirer, S. Kalirai, J. Nelson Weker, Y. Liu, J. C. Andrews and B. M. Weckhuysen, Chem. Commun., 2015, 51, 8097–8100 RSC
- S. Kalirai, U. Boesenberg, G. Falkenberg, F. Meirer and B. M. Weckhuysen, ChemCatChem, 2015, 7, 3674–3682 CrossRef CAS PubMed
- J. Ruiz-Martínez, A. M. Beale, U. Deka, M. G. O'Brien, P. D. Quinn, J. F. W. Mosselmans and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 5983–5987 CrossRef PubMed
- Z. Ristanović, M. M. Kerssens, A. V. Kubarev, F. C. Hendriks, P. Dedecker, J. Hofkens, M. B. J. Roeffaers and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2015, 54, 1836–1840 CrossRef PubMed
- G. T. Whiting, F. Meirer, M. M. Mertens, A.-J. Bons, B. M. Weiss, P. A. Stevens, E. de Smit and B. M. Weckhuysen, ChemCatChem, 2015, 7, 1312–1321 CrossRef CAS
- G. T. Whiting, F. Meirer, D. Valencia, M. M. Mertens, A.-J. Bons, B. M. Weiss, P. A. Stevens, E. de Smit and B. Weckhuysen, Phys. Chem. Chem. Phys., 2014, 16, 21531–21542 RSC
- S. Mitchell, N.-L. Michels, K. Kunze and J. Pérez-Ramírez, Nat. Chem., 2012, 4, 825–831 CrossRef CAS PubMed
- I. L. C. Buurmans, J. Ruiz-Martínez, W. V. Knowles, D. van der Beek, J. A. Bergwerff, E. T. C. Vogt and B. M. Weckhuysen, Nat. Chem., 2011, 3, 862–867 CrossRef CAS PubMed
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC
- I. L. C. Buurmans, F. Soulimani, J. Ruiz-Martínez, H. E. van der Bij and B. M. Weckhuysen, Microporous Mesoporous Mater., 2013, 166, 86–92 CrossRef CAS
- I. L. C. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873–886 CrossRef CAS PubMed
- M. Ibáñez, M. Gamero, J. Ruiz-Martínez, B. M. Weckhuysen, A. T. Aguayo, J. Bilbao and P. Castaño, Catal. Sci. Technol., 2016, 6, 296–306 Search PubMed
- P. Castaño, J. Ruiz-Martínez, E. Epelde, A. G. Gayubo and B. M. Weckhuysen, ChemCatChem, 2013, 5, 2827–2831 CrossRef
- R. V. Jasra, B. Tyagi, Y. M. Badheka, V. N. Choudary and T. S. G. Bhat, Ind. Eng. Chem. Res., 2003, 42, 3263–3272 CrossRef CAS
- V. R. Choudhary, P. Devadas, A. K. Kinage and M. Guisnet, Appl. Catal., A, 1997, 162, 223–233 CrossRef CAS
- P. Cañizares, A. Durán, F. Dorado and M. Carmona, Appl. Clay Sci., 2000, 16, 273–287 CrossRef
- F. Dorado, R. Romero and P. Cañizares, Appl. Catal., A, 2002, 236, 235–243 CrossRef CAS
- J. M. Fougerit, N. S. Gnep and M. Guisnet, Microporous Mesoporous Mater., 1999, 29, 79–89 CrossRef CAS
- M. W. Kasture, P. S. Niphadkar, V. V. Bokade and P. N. Joshi, Catal. Commun., 2007, 8, 1003–1008 CrossRef CAS
- G. de la Puente, E. Falabella Sousa-Aguiar, A. Figueiredo Costa and U. Sedran, Appl. Catal., A, 2003, 242, 381–391 CrossRef CAS
- F. V. Pinto, A. S. Escobar, B. G. de Oliveira, Y. L. Lam, H. S. Cerqueira, B. Louis, J. P. Tessonnier, D. S. Su and M. M. Pereira, Appl. Catal., A, 2010, 388, 15–21 CrossRef CAS
- N.-L. Michels, S. Mitchell and J. Pérez-Ramírez, ACS Catal., 2014, 4, 2409–2417 CrossRef CAS
- K.-Y. Lee, H.-K. Lee and S.-K. Ihm, Top. Catal., 2010, 53, 247–253 CrossRef CAS
- P. Gélin and T. Des Courières, Appl. Catal., 1991, 72, 179–192 CrossRef
- F. Geobaldo, T. Palomino, S. Bordiga, C. Fisica and V. Pietro Giuria, Phys. Chem. Chem. Phys., 1999, 1, 561–569 RSC
- M. Alkan, Ç. Hopa, Z. Yilmaz and H. Güler, Microporous Mesoporous Mater., 2005, 86, 176–184 CrossRef CAS
- X. Liu, K.-K. Iu, J. K. Thomas, H. He and J. Klinowski, J. Am. Chem. Soc., 1994, 116, 11811–11818 CrossRef CAS
- Y. Zhang, Y. Zhou, A. Qiu, Y. Wang, Y. Xu and P. Wu, Ind. Eng. Chem. Res., 2006, 45, 2213–2219 CrossRef CAS
- C. L. Garcia and J. A. Lercher, J. Phys. Chem., 1992, 96, 2669–2675 CrossRef CAS
-
H. Knozinger, The Hydrogen Bond, ed. P. Schuster, G. Zundel and C. Sandorfy, North Holland, Amsterdam, 1976, vol. 3, p. 1275 Search PubMed
- Z. Wu, C. Li, Z. Wei, P. Ying and Q. Xin, J. Phys. Chem. B, 2002, 106, 979–987 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5fd00200a |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |