
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light- or oxidation-triggered CO release from [MnI(CO)3(κ3-L)] complexes: reaction intermediates and a new synthetic route to [MnIII/IV2(μ-O)2(L)2] compounds†
Ulf
Sachs
,
Gerrit
Schaper
,
Daniela
Winkler
,
Daniel
Kratzert
and
Philipp
Kurz
*
Institut für Anorganische und Analytische Chemie, Albert-Ludwigs-Universität Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: philipp.kurz@ac.uni-freiburg.de
First published on 7th October 2016
Abstract
The reaction products and intermediates of the three CO-releasing manganese(I) coordination compounds [Mn(tpm)(CO)3]+, [Mn(bpza)(CO)3] and [Mn(tpa)(CO)3]+ were analysed by combining IR-spectroscopy, electrochemical measurements and single-crystal XRD. The intermediate formation of manganese(I) biscarbonyl compounds and the rather facile oxidation of these species were identified as key reaction steps that accompany CO liberation. For the use of [MnI(CO)3] complexes as light-triggered CO sources, the results indicate that in this case photo- and redox-chemistry seem to be strongly coupled which could be important and potentially even useful in the pharmacological context. Additionally, one has to be aware of the fact that [MnII(κ3-L)(solv)3]2+ complexes, the primary reaction products after CO substitution, are able to bind to proteins, which was demonstrated using bovine serum albumin as a model. And finally it could be shown that the CO-release reactions can be used as a new synthetic route to prepare multinuclear μ-oxido-bridged manganese complexes: the mixed-valence compound [MnIII/IV2(μ-O)2(tpa)2]3+ could be prepared in a single step from [Mn(tpa)(CO)3]+via photo- or electrochemically induced CO substitution.
Introduction
As the role of CO as a physiological trigger molecule in mammals gets more and more acknowledged and therefore well investigated,1 CO-releasing molecules (CORMs) become increasingly popular as possible drugs, especially for inflammatory diseases.2 An attractive family of CORMs liberate CO upon illumination (so-called photo-CORMs) and numerous studies of such substances can be found in the literature reporting the amount and kinetics of the CO release, the targeting of specific body tissues and the wavelength needed to trigger CO release.3–5In this work, we focus on [MnI(CO)3(κ3-L)] compounds, manganese(I) complexes whose photoCORM-activities have been studied for many years. However, details concerning the reaction sequence leading to CO release, the role of manganese redox chemistry in the process or the Mn-containing products of the photoreaction are still scarce. This is surprising, as this information could be crucial for a therapeutic use of these substances. On the other hand, we also know from a previous, initial study on this subject, that a reaction monitoring of CO release by [MnI(CO)3(κ3-L)] is complex as various intermediates and end products might be involved.6
For our investigation, we picked two common, well studied photo-CORMs (Scheme 1), the cationic complex [Mn(tpm)(CO)3]+ (1, tpm = tris(1-pyrazolyl)methane) and the neutral species [Mn(bpza)(CO)3] (2, bpza = bis(1-pyrazolyl)acetic acid). Based on the results of our previous study, we additionally wanted to evaluate the possibility to use photo-CORM chemistry for the synthesis of multinuclear manganese complexes as we previously suspected dinuclear compounds containing the common [Mn2(μ-O(H))2]-core7 as likely reaction products. In this way, the CO-release reactions of the often easily accessible [MnI(CO)3(κ3-L)] complexes could be used for the synthesis of new Mn2-complexes containing various pre-coordinated ligands L. If carried out in the presence of water, the resulting compounds could be of the general type [Mn2(μ-O(H))2L2], which are of broad interest, e.g. as model systems for known manganese based metalloenzymes like Mn-catalase, arginase8,9 or the OEC in PSII.7,10 To probe this concept, we additionally investigated the light-triggered CO substitution for the MnI-carbonyl complex 3, which is also already well investigated as photoCORM11 and for its therapeutic use,12 bearing the potentially tetradentate trispyridylmethylamin (tpa) ligand, for which μ-oxido bridged MnIII/MnIV complexes of high catalase activity have already been reported.13
 | ||
| Scheme 1 Structures of the CO-releasing manganese(I) carbonyl complexes [MnI(CO)3(tpm)]+ (1), [MnI(CO)3(bpza)] (2) and [MnI(CO)3(tpa)]+ (3) investigated in the presented study. | ||
Results and discussion
Tridentate ligand systems: tpm and bpza
Already more than 25 years ago, Elias, Wieghardt and co-workers reported the pronounced light-sensitivity of MnI tricarbonyl complexes like [MnI(CO)3([9]aneS3)]+, which decompose in solution via CO-loss reactions upon illumination.14 A number of previous studies has demonstrated that the CO release of the related compounds [MnI(CO)3(tpm)]+ (1) and [MnI(CO)3(bpza)] (2) can be initiated in a similar fashion by illumination with light of the near ultraviolet region. In our own work,6 it could be demonstrated by the use of IR- and EPR-spectroscopy that this reaction proceeds via (1) the release of a single, first CO ligand leading to a biscarbonyl intermediate (“[MnI(CO)2(tpm)(solv)]”) followed by (2) the oxidation of the manganese centre to MnII and the release of the remaining CO ligands (Scheme 2). So far, the order of the events involved in step (2) and the exact composition of the Mn ligand sphere after the reaction is unknown. However, our observations indicated that both photoexcitation and Mn-oxidation might be involved in the reaction scheme leading to a complete removal of the CO ligands. | ||
| Scheme 2 Overview of reactions leading to the light- and/or oxidation-triggered release of CO ligands from [MnI(CO)3(tpm)]+ (1) or [MnI(CO)3(bpza)] (2). The already previously known initial reactions following the first excitation by UV light are framed by the dashed box. Results from this study led to the identification of new reactions involving electrochemical oxidations (left and right of the box) as well as the oxidized reaction products [MnII(L)2] (for 1), [MnII2(L)4] (for 2) and MnOx (bottom, the latter only after exposure to oxygen). | ||
The typical development of the IR spectrum of a [MnI(CO)3(κ3-L)] photo-CORM during light-triggered CO release is shown in Fig. 1 for the example of complex 1 (see also ESI, Fig. S1,† for additional CO-release reactions monitored by IR). As observed before, the photoreactions result in a decrease of the typical CO bands of the MnI triscarbonyl between 1930 and 2070 cm−1 and the appearance of a new feature at ∼1880 cm−1 assigned to the manganese(I) biscarbonyl intermediate.6
 | ||
| Fig. 1 IR monitoring of the CO release of 1 initialized by UV irradiation. Conditions: 10 mM solution of [MnI(CO)3(bpza)] in MeCN, λirr = 365 nm. | ||
The maximum intensity of this [MnI(CO)2]-band depends on the solvent system, which seems reasonable as the current model for the reaction sequence formulates the intermediate as “[MnI(CO)2(L)(solv)]” in order to maintain the coordination number of 6 (which is most common for MnI complexes) – and thus contains a solvent molecule in the coordination sphere. In acetonitrile, we observe that the [MnI(CO)2(L)(MeCN)]-complexes derived from 1 or 2 are stable in the dark (see ESI, Fig. S2†). In EtOH/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixtures, CO release is slower (see ESI, Fig. S1†) and the biscarbonyl intermediates are also less stable, as the IR-bands at 1880 cm−1 vanish over hours in the case of 1, but within minutes for 2 if irradiations are interrupted (see ESI, Fig. S2†). Taken together, the IR-monitoring of the CO release induced by irradiation indicates that the CO release of the [MnI(CO)2]-intermediate also requires illumination (at least in acetonitrile), but as these photoreactions are at least as fast as the first light-triggered CO release from 1 or 2, the MnI-biscarbonyl complexes can only be generated as intermediate fractions during the photoreactions. This general instability of the Mn(I) biscarbonyl complexes is not surprising, because (in contrast to the situation for the other group 7 metals Tc and ref. 15 and 16) no solution-stable [Mn(CO)2]-compound has been reported so far to the best of our knowledge.
:1) mixtures, CO release is slower (see ESI, Fig. S1†) and the biscarbonyl intermediates are also less stable, as the IR-bands at 1880 cm−1 vanish over hours in the case of 1, but within minutes for 2 if irradiations are interrupted (see ESI, Fig. S2†). Taken together, the IR-monitoring of the CO release induced by irradiation indicates that the CO release of the [MnI(CO)2]-intermediate also requires illumination (at least in acetonitrile), but as these photoreactions are at least as fast as the first light-triggered CO release from 1 or 2, the MnI-biscarbonyl complexes can only be generated as intermediate fractions during the photoreactions. This general instability of the Mn(I) biscarbonyl complexes is not surprising, because (in contrast to the situation for the other group 7 metals Tc and ref. 15 and 16) no solution-stable [Mn(CO)2]-compound has been reported so far to the best of our knowledge.
After extended UV-illumination, we finally observe complete CO release for all complex/solvent combinations and no IR bands are detected any more in the entire spectral region between 1800 and 2100 cm−1 (see end spectra in Fig. 1 and ESI, Fig. S1†). If the irradiation experiments take place in the absence of oxygen (degassed solutions) the reaction ultimately leads to a colourless, paramagnetic product as indicated by the comparison of the 1H-NMR spectra before and after irradiation for 24 h (see ESI, Fig. S3†). We tentatively assign this spectrum to arise from a [MnII(κ3-L)(solv)3] complex.
If air is allowed to come into contact with the pre-illuminated solutions, some manganese oxide precipitates. After removing the brown solid by filtration, colourless crystals could be obtained by slow evaporation of the solvent. A single crystal XRD measurement revealed that these crystals are made up of [MnII(κ3-L)2] complexes. In both cases, we find Mn(II) ions that are six-fold coordinated by ligand spheres containing only donor atoms from tpm or bpza ligands. In the case of [MnII(tpm)2]2+ (Fig. 2), the coordination is very similar to the already reported structure of [MnII(trispyrazolylborate)2]:17 the Mn-centre is slightly distorted octahedral and the Mn2+ ion is placed on an inversion centre of the structure. As a result, all trans ligands bind at 180° angles from each other, while the N–Mn–N angles for each tpm “hemisphere” are smaller (80°–82°), and the N–Mn–N′ angles between the two different tpm are larger than rectangular (97°–99°). The Mn–N bond lengths are between 2.22 and 2.25 Å.
 | ||
| Fig. 2 Molecular structure of [Mn(tpm)2]2+ with ellipsoids at the 50% probability level. The two PF6− counter-ions were omitted. | ||
In the case of bpza, the molecular structure is a ligand-bridged two centred complex ([Mn2(bpza)4]) in which both MnII-centres bind to one bpza exclusively and share two additional bpza-ligands with a second MnII, which is similar to a recently published structure of a ligand bridged [MnII2Br4(azpy)3] by Kottelat et al.18 To one of these bridging ligands they bind via the two nitrogen atoms of the pyrazol-groups and to the other one via the oxygen atoms of the carboxylic acid. The structure has an inversion centre lying between the two MnII. The two MnII-centres are coordinated in a distorted octahedral fashion, with trans-angles of 161°–168° and cis-angles of 79–96°. The two oxygen that bind to the same MnII are binding cis, in this way it's placing the π-donating O-donors trans to the π-accepting aromatic rings. The interaction between the free electron pairs of the oxygen donors is minimized as the acetate groups are slightly twisted. The Mn–N bond lengths (2.23–2.31 Å) are slightly longer than those found for the tpm complex which might be due to the fact that the electron density at MnII is slightly higher here because of the two negative ligand charges. The distances and angles around the MnII centre are also very similar to the known structure of [MnII2(Hphth)2(phen)4](Hphth)2 which has a similar coordination sphere.19
The results presented in the previous paragraphs thus support a CO-release reaction sequence where CO loss from complexes 1 or 2 in the presence of water is accompanied by manganese oxidation from the original MnI state to MnII and beyond, as indicated by the isolation of the two “carbonyl-free” MnII complexes (Fig. 2 and 3) and also manganese oxides from the reaction mixture. To find out more about the interplay of CO loss and Mn oxidation, we now carried out an additional set of experiments in which we probed the redox properties of the intermediate [MnI(CO)2] species and also studied a purely oxidation-induced CO release in the absence of UV irradiation.
 | ||
| Fig. 3 Molecular structure of [Mn2(bpza)4] with ellipsoids at the 50% probability level. Four water molecules embedded in the crystal structure were omitted. | ||
First, a solution containing complex 1 and the electrolyte salt [Bu4N][PF6] in acetonitrile was illuminated for 30 min. by UV light (λirr = 365 nm) to generate a sample containing significant amounts of the MnI-biscarbonyl intermediate. This solution was then quickly transferred to a standard electrochemical cell and a CV was measured immediately (Fig. 4, left). Two irreversible oxidation waves are visible here: a small event at ∼+50 mV and a much larger one at ∼+1000 mV vs. Fc/Fc+. This is in contrast to a CV of a sample of 1 without UV illumination which only shows the oxidation event at ∼+1000 mV (see ESI, Fig. S4†).
 | ||
| Fig. 4 Left: Cyclic voltammogram (scan rate: 100 mV s−1) of a 10 mM solution of 1 in MeCN with [Bu4N][PF6] (0.1 M) as electrolyte. The solution was irradiated (365 nm, 30 min) before the CV. Potentials are calculated vs. Fc/Fc+. Right: Linear sweep voltammogram of the same solution recorded during IR spectro-electrochemistry (potentials vs. q-Pt). | ||
Next, an illuminated solution of 1 was filled into a modified thin-layer liquid IR cell with a platinum net as working electrode (see ESI, Fig. S5† for the set-up) in order to monitor changes in carbonyl coordination induced by electrochemical oxidation. As no proper reference electrode could be incorporated into the cell construction, IR spectro-electrochemistry was carried out in the form of slow linear sweep voltammograms (1 mV s−1, see Fig. 4, right, for the detected LSV trace) during which IR spectra were recorded at 30 s intervals (and thus for every 30 mV of potential difference). When the potential of the platinum net in this set-up reached a pseudo-potential of ∼+0.8 V vs. Pt, the current density started to rise and the band in the IR associated with the [MnI(CO)2] form of 1 at ∼1880 cm−1 vanished instantly. Additionally, the band at ∼1960 cm−1 decreased slightly in intensity. The remaining spectrum is identical with that of the initial complex 1 and is virtually unchanged until the potential has been raised further by more than 600 mV to ∼+1.4 V vs. Pt. Here, large currents are detected in the LSV and these are accompanied in the IR spectra by the rapid loss of all three CO bands without the appearance of any additional intermediate bands (Fig. 5).
 | ||
| Fig. 5 IR monitoring of the CO release caused by electrochemical oxidation for a pre-illuminated solution of 1 (10 mM) in MeCN with (Bu4N)(PF6) (0.1 M) as electrolyte. The spectral changes for four potential regions are shown separately, but all spectra belong to the same IR spectro-electrochemistry experiment. The indicated electrode potentials are given versus the platinum pseudo-reference of the thin-layer IR cell. See Fig. 4, right, for the corresponding LSV trace. | ||
The comparison of the LSV for the spectro-electrochemical experiment with the CV measured in a proper three-electrode setup (Fig. 4) allows a “calibration” of the pseudo-reference potentials inside the IR cell: the first, small event (∼+0.8 V vs. Pt) of the IR spectro-electrochemistry corresponds to the one at +50 mV vs. Fc/Fc+, while the second one (+1.4 V vs. Pt) with large oxidation currents can be found in the CV at +1000 mV vs. Fc/Fc+.
In combination with the potential-dependent IR spectra it is now nicely possible to assign these events in the CV: the process at the lower potential of +50 mV vs. Fc/Fc+ is the oxidation of [MnI(CO)2(tpm)(MeCN)]+. This is accompanied by the loss of the two remaining CO ligands which is also detectable in the IR by the disappearance of the band at 1880 cm−1 and a slight decrease of the 1960 cm−1 signal (Fig. S6†). The second oxidation at +1000 mV vs. Fc/Fc+ must then be the electrochemical oxidation of the starting complex 1. Both trigger the immediate loss of all coordinated COs as no new carbonyl complexes could be detected as products for either step by IR. In the light of the MnII compounds which were isolated as reaction products before, we furthermore think it likely that the observed oxidation process is metal-centred (MnI → MnII). The detected large potential difference of ∼800 mV between the two oxidation events also fits these assignments: as [MnI(CO)2(tpm)(MeCN)]+ contains only two of the strongly π-accepting CO ligands, more electron density resides on the MnI centre which is in consequence much easier to oxidize than in the case of a [MnI(CO)3] complex. Additionally, the detected low oxidation potential for the biscarbonyl form explains why oxidation by O2 (or even H+) might occur for the [MnI(CO)2(tpm)(MeCN)] intermediate, but not for the starting complex 1. We would like to add that these good electron donor properties of the MnI-biscarbonyl intermediates might also contribute to the antioxidant properties of manganese photo-CORMs and add to the already established antioxidant effect originating from the released CO.12 The (spectro-) electrochemical results observed in the case of starting complex 2 are very similar (see ESI, Fig. S4, S7 and S8†), so that we conclude that the reaction sequences described above for 1 are equally valid for [MnI(CO)3(bpza)] (2).
In summary, the combination of the results from photochemical reactions, electrochemistry and an online monitoring of the CO bands by IR spectroscopy provides very detailed information about the reaction intermediates of both the light- and redox-triggered CO-release reactions by 1 and 2 and also the redox properties of the involved species. Scheme 2 shows an overview of the detected reaction steps and intermediates, which greatly extends the knowledge from our previous investigation (marked in the scheme by a dashed box). As already well established, [MnI(CO)3(κ3-L)] complexes can be excited by UV-irradiation which leads to the release of a first CO molecule, observable especially well by the appearance of a new CO band at ∼1880 cm−1 in the IR-spectrum. The thus detected biscarbonyl species is stable in the presence of the strongly coordinating ligand acetonitrile, most likely in the form of the complex [MnI(CO)2(κ3-L)(MeCN)]. The release of the remaining two CO ligands can then either be triggered by oxidation (electrochemically or by O2) or again by light-excitation. The loss of the CO ligand significantly lowers the necessary potential for the MnI → MnII oxidation by more than 600 mV which in part explains the instability of the MnI biscarbonyl species in solution. No MnII carbonyl intermediates could be detected at any stage – instead manganese oxides and “carbonyl free” MnII complexes bearing just tpm and bpza ligands were isolated as end products of the CO-release processes. Finally, and in complete agreement with the other observations, the release of all three CO ligands can also be initiated electrochemically by electrochemical oxidation in acetonitrile at ∼+1000 mV vs. Fc/Fc+. Here, a metal-centred oxidation would first result in a [MnII(CO)3] intermediate. Because of the resulting reduced electron density at the MnII metal centre, π-back-bonding to CO apparently becomes too weak and all three ligands leave the complex too fast to allow for the detection of any intermediates by IR in the set-ups used here. As will be shown in the next section, these efficient light- or oxidation-induced CO-substitution reactions can also be used for the synthesis of dinuclear Mn2-complexes.
CORM-chemistry is especially interesting in the context of a possible pharmaceutical application. As an (of course very crude) model for a “cellular environment”, we also monitored the UV-triggered CO-release of 1 in the presence of bovine serum albumin (BSA) using IR spectroscopy. CORM–BSA adducts have been reported for ruthenium and iridium-CORMs and these adducts have also been investigated as in vivo CORM carriers.20,21 In our experiments, no significant differences to a solution without BSA could be observed in the spectra when the irradiation the complexes were carried out in an anaerobic H2O/MeCN (4:3) solution containing one amino acid monomer equivalent of BSA per Mn complex shown for complex 1 in Fig. 6. Therefore the actual CO release process and its intermediates are neither affected by the much higher concentration of H2O nor by the presence of the BSA protein. On the other hand, it seems that the soluble Mn-containing products of the CO-release process are interacting with the protein, because extended irradiation of the solution using λirr = 365 nm results in the precipitation of the protein from the previously homogeneous mixture containing 1 and BSA (Fig. S9a†). If this experiment is carried out in the presence of air, a dark manganese oxide precipitate again forms and the transparent BSA solution is restored (Fig. S9a†). We also observed that BSA precipitates (a) when BSA is added to a pre-illuminated solution of 1 in anaerobic H2O/MeCN (Fig. S9b†) and (b) when equivalent amounts of Ca2+ are added to BSA in H2O/MeCN (Fig. S9c†). Analogous experiments were also carried out for tenfold lower CORM concentrations (and thus a tenfold higher BSA:CORM ratio), but these did not lead to any different observations (Fig. S10†).
 | ||
| Fig. 6 IR monitoring of the CO release of a 10 mM solution of 1 initialized by UV irradiation with (black) and without (gray) BSA (1 eq. amino acid monomer) in H2O/MeCN 4:3, λirr = 365 nm. | ||
In combination, we firstly conclude from these results that the presence of a model protein like BSA does not affect the CO-release reactions outlined in Scheme 2 above in any major way. Secondly, our results nicely demonstrate how important it is to consider the “fate” of a photoCORM like 1 after it has delivered its CO ligands to the environment: in our case, one of the products – very likely the dicationic [MnII(L)(solv)3]2+ form – is apparently able interact with the protein. The exact nature of this interaction (non-specific binding vs. defined coordination to e.g. histidine side chains) could not be specified by our exploratory experiments and we note that the observed protein precipitation could also be caused by a simple ionic strength effect as known for the case of Ca2+ ions.22
Tetradentate ligand system: tpa
Even though multiple attempts were made, we were not able to isolate other molecular products from the CO-release reactions of 1 or 2 than the two dinuclear [MnII(L)2] complexes shown in Fig. 2 and 3 above. We suspected that this might be related to the very stable ligand environment (especially in the case of [MnII(tpm)2]2+), where the MnII centre is completely encapsulated by the tridentate ligands.We thus turned to a different ligand system to probe whether the identified reaction sequences for CO release from manganese(I) triscarbonyl complexes could also be used to synthesize molecular compounds with a [Mn2(μ-O(H))2]-core and manganese oxidation states of ≥+II, which are commonly found in Mn metalloenzymes.8,23 Our ligand of choice was trispyridylmethylamine (tpa), because μ-oxido bridged MnIII/MnIV complexes with tpa ligands had already been reported.24 However, in these studies the complexes had been prepared by the common route of adding the tpa ligand to (rather ill-defined) solutions of MnIII precursors.24
The starting material, the complex [Mn(CO)3(κ3-tpa)][PF6] (3), is easily accessible via the commonly used reaction of [Mn(CO)5Br] with tpa. The molecular structure of 3 determined by XRD is shown in Fig. 7 and does not significantly differ from published data with bromine as counter ion.11
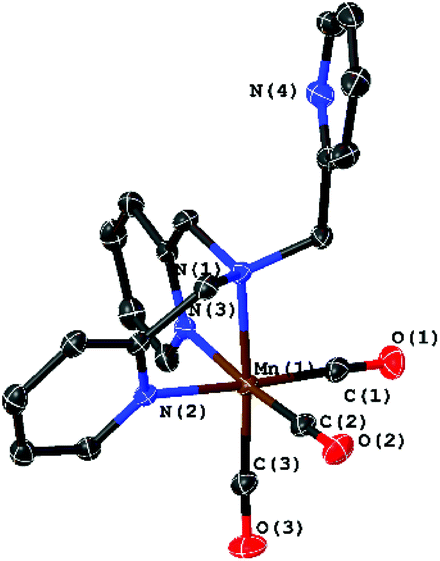 | ||
| Fig. 7 Molecular structure of [Mn(CO)3(κ3-tpa)]+ with ellipsoids at the 50% probability level. The PF6 counter ion was omitted. | ||
The transient IR spectra recorded during UV irradiation in acetonitrile (Fig. 8) and in EtOH/H2O (9:1) look similar to the ones already shown for 1 and 2 (see ESI, Fig. S1†). After a first CO is liberated it is (as shown above for the cases of the tridentate ligands) first replaced by a solvent molecule, thus likely generating the complex [MnI(CO)2(κ3-tpa)(MeCN)] if the reaction is carried out in an acetonitrile solution and this species can again also be detected electrochemically (see ESI, Fig. S2†). However, after some time the solvent gets replaced by the so-far non-coordinated fourth arm of the tetradentate ligand to form [MnI(CO)2(κ4-tpa)] and this reaction sequence has been shown earlier in a detailed investigation by Schatzschneider et al.11 The κ3-tpa intermediate can be detected via an IR shoulder at 1870 cm−1 in addition to the main biscarbonyl signal at 1850 cm−1. This signal decreases in the dark as can be seen in Fig. 9 but is not very strong to begin with, because it decreases already during the irradiation. For the irradiation in EtOH/H2O (9:1) the IR spectra are similar with this small band at 1910 cm−1 (Fig. S11†). The solubility of the complex is worse in EtOH/H2O and solutions of 10 mM were not possible, so statements to kinetic comparison cannot be made.
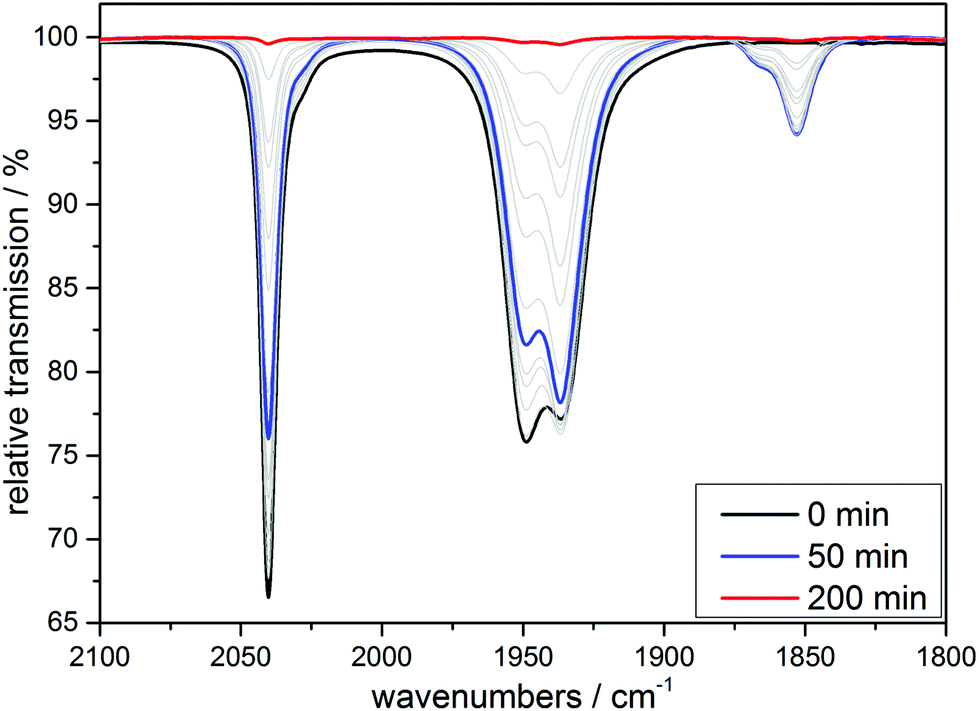 | ||
| Fig. 8 IR monitoring of the CO release via UV irradiation (λirr = 365 nm) of a 10 mM solution of [MnI(CO)3(tpa)][PF6] in MeCN. | ||
 | ||
| Fig. 9 IR-monitoring (in the dark) of the decreasing shoulder at 1870 cm−1 observable after irradiation, which indicates the replacing of a coordinated solvent molecule by the fourth arm of the tpa ligand. | ||
The CO release for 3 in MeCN is slower compared to 1: the irradiation of a 10 mM solution of 3 for 24 h in the presence of air leads to a dark green solution, while unlike before no MnOx precipitates form. From this mixture, dark brownish-green crystals could be obtained by slow evaporation of the solvent.
From single crystal XRD we were able to identify the product as the μ-oxido bridged, mixed valence complex [MnIII,IV2(μ-O)2(κ4-tpa)2]3+ shown in Fig. 10. The observed geometry of the molecule is similar to the one published by Towle et al.,24 as can be seen by a comparison of metal–metal and metal–ligand distances listed in Table 1. The environment of the two manganese ions is clearly different making it possible to clearly assign the different oxidation-states (MnIII and MnIV) to the manganese centres. The Mn–N bonds of MnIV (Mn1A in Fig. 10) are on average 0.1 Å shorter than the bonds for MnIII. As expected for a high-spin d4 system, the coordination sphere around MnIII also shows a pronounced Jahn–Teller-distortion with axial Mn–N distances that are at least ∼0.1 Å longer than the other Mn–ligand interactions.
 | ||
| Fig. 10 Molecular structure of the asymmetric [MnIII,IV2(μ-O)2(κ4-tpa)2]3+ derived by UV-irradiation of [MnI(CO)3(κ3-tpa)]+, with ellipsoids at the 50% probability level. From an analysis of the bond lengths, Mn1A can be identified as the MnIV, Mn2A as the MnIV centre. The three PF6 counter ions were omitted. | ||
| Literature (Towle et al.)24 [Mn2(μ-O)2(κ4-tpa)] [S2O6]1.5 | UV irradiation derived [Mn2(μ-O)2(κ4-tpa)] [PF6]3 | Electrochemically derived [Mn2(μ-O)2(κ4-tpa)] [PF6]3 | |||
|---|---|---|---|---|---|
| Mn1 | Mn2A | Mn1 | |||
| Mn2 | 2.643 | Mn1A | 2.630(5) | Mn1′ | 2.625(6) |
| O1 | 1.835 | O1A | 1.844(16) | O1 | 1.799(13) |
| O2 | 1.839 | O2A | 1.833(16) | O1′ | 1.821(12) |
| N1 | 2.114 | N5A | 2.115(15) | N1 | 2.085(15) |
| N2 | 2.207 | N6A | 2.203(2) | N2 | 1.936(5) |
| N3 | 2.044 | N7A | 2.052(19) | N3 | 2.044(15) |
| N4 | 2.259 | N8A | 2.210(2) | N4 | 2.102(17) |
| Mn2 | Mn1A | Mn1′ | |||
| Mn1 | 2.643 | Mn2A | 2.630(5) | Mn1 | 2.625(6) |
| O1 | 1.782 | O1A | 1.772(16) | O1′ | 1.799(13) |
| O2 | 1.771 | O2A | 1.792(16) | O1 | 1.821(12) |
| N5 | 2.081 | N1A | 2.068(18) | N1′ | 2.085(15) |
| N6 | 2.014 | N2A | 2.033(19) | N2′ | 1.936(5) |
| N7 | 2.027 | N3A | 2.034(19) | N3′ | 2.044(15) |
| N8 | 2.011 | N4A | 2.019(2) | N4′ | 2.102(17) |
Importantly (and supporting our earlier interpretation of the data for the reactions of 1 and 2), [MnIII,IV2(μ-O)2(κ4-tpa)2]3+ can not only be isolated as the reaction product of irradiation experiments in the presence of air but also from an electrochemical oxidation of 3 at +1.12 V vs. Fc/Fc+ in MeCN in the dark. Crystals could again be isolated from the reaction mixture, which were analysed using XRD. In this case, a different unit cell and also a variation of the packing of the molecules in the solid state was found, so that the two manganese centres are indistinguishable here (see Table 1, right column). On the other hand, we are confident that the underlying coordination compounds are identical in all cases, as average bond lengths and the general molecular geometries (e.g. Mn–Mn-distances) are very similar.
From the presented results it is again possible to formulate reaction sequences and intermediates for the MnI CORM 3, shown in Scheme 3. Many steps are similar to the reactions of 1 and 2 (compare Scheme 2), but the presence of a tetradentate ligand leads here to a [MnII(L)(solv)2]-intermediate, which is apparently more stable so that air oxidation results in a MnIII,IV dimer while avoiding MnOx formation. Encouraged by the successful preparation of [MnIII,IV2(μ-O)2(κ4-tpa)]3+ from 3 using the electrochemical route, bulk oxidations were also carried out for 1 and 2, but these lead only to undefined reaction mixtures. It thus seems that a tetradentate ligand system (or probably the presence of additional ligands like the otherwise often-used acetate25) are beneficial in order to prepare dinuclear [MnIII,IV2(μ-O)2(L)2] compounds directly from manganese(I) triscarbonyl precursors.
 | ||
| Scheme 3 Schematic reaction pathway of the CO release of [MnI(CO)3(tpa)]+. The well-known irradiation path is shown in the dashed box, the new proposed path of electrochemical oxidation left and right of the box and the dinuclear complex [Mn2(μ-O)2(κ3-tpa)2]3+ as reaction product after exposure to oxygen at the bottom. | ||
Conclusions
By using the combined analytical data from IR spectroscopy, electrochemistry and X-ray crystallography, it was possible to develop a detailed picture of the reaction pathways eventually leading to the complete release of all CO ligands from the three well-known photo-CORMs [Mn(tpm)(CO)3][PF6], [Mn(bpza)(CO)3] and [Mn(tpa)(CO)3][PF6]. In all cases, the excitation of these molecules by UV light triggers the release of a first CO ligand and the formation of manganese(I) biscarbonyl complexes as key intermediates. These can then liberate the remaining two COs after absorbing at least one more UV photon. Alternatively, the oxidation of the [MnI(CO)2] species is already possible at very mild potentials and this process also leads to the release of the remaining carbonyl ligands. As final reaction products, it was possible to isolate “carbonyl-free” MnII complexes where the MnII centre is still bound to the initially coordinated multidentate ligands. Additionally, experiments in the presence of the “model protein” BSA make it likely that complexes like [MnII(L)(solv)3]2+ are able to interact with biomolecules in aqueous solution. For the possible therapeutic use of MnI photo-CORMs, these results indicate that the “fate” of the Mn precursor after CO substitution and manganese-centred oxidations to MnII have to be considered. Furthermore, the detected manganese-centred oxidation reactions offer a plausible explanation in which way manganese(I) triscarbonyl CORMs can also act as “metal-centred antioxidants” in addition to the well-known antioxidant effect of the liberated CO.By applying strongly oxidising potentials, we were able to show that CO release can also be induced by electrochemical oxidation of the manganese centre to MnII. In this case, all three carbonyl ligands are liberated at once. As the Mn oxidation potential can be varied by the use of different ligands, this process might also be useful for therapy, e.g. to selectively release CO in an oxidative cell environment.
Finally, the detailed understanding of light- and oxidation induced CO substitution could also be used synthetically. As a first example, the combination of either UV illumination or electrochemical oxidation of [Mn(tpa)(CO)3]+ in the presence of O2 resulted in the formation of the dinuclear manganese(III/IV) complex [Mn2(μ-O)2κ3-tpa)2]3+. This method thus offers a new, easy and clean synthetic route to prepare dinuclear manganese complexes starting from the well-defined MnI carbonyl compounds as precursors and avoiding lengthy purification steps. The route is thus a rare example of a fairly simple preparation procedure in the otherwise often difficult field of synthetic MnIII/IV coordination chemistry.
Experimental
Syntheses
The ligands tris(2-pyridylmethyl)amine (tpa), tris(1-pyrazolyl)methane (tpm) and bis(1-pyrazolyl)acetic acid (bpza) and the corresponding MnI triscarbonyl complexes were synthesized according to published procedures: tpm,26 bpza,27 tpa,28 [Mn(CO)3(tpm)][PF6] (1·PF6)29 and [Mn(CO)3(bpza)] (2).27 The preparation of [Mn(CO)3(tpa)][PF6] (3·PF6) was carried out in an analogous way as that of 1·PF6.29 Bovine serum albumin (BSA) was purchased from Sigma Aldrich (lyophilized powder, >96%, agarose gel electrophoresis).To isolate the reaction products of the CO release of 1 PF6 and 2, 10 ml of solutions containing 100 mg of the CORM were irradiated in a large open cuvette for 7 days. The formed manganese oxides were removed by centrifugation and filtration. The resulting product was recrystallized from MeCN (1) and EtOH/H2O (9:1) (2) leading to colourless crystals with yields of 47.6% (61 mg,) for [Mn(tpm)2][PF6]2 and 43.6% (62 mg) for [Mn2(bpza)4]. The reaction product of the light-triggered CO-release of 3 was isolated by reducing the amount of MeCN solvent. Recrystallization from EtOH then lead to brownish green crystals of [Mn2(μ-O)2κ4-tpa)2][PF6]3 in a yield of ∼25%, which were suitable for XRD measurements. IR-spectra of the products show no sign of remaining CO ligands or any of the free multidentade ligand (Fig. S12–S17†). IR (ATR) of [Mn(tpm)2][PF6]2: νmax (cm−1) = 3165 w, 3138 m, 3040 w, 1522 m, 1442 m, 1405 s, 1247 m, 1222 m, 1096 m, 1057 m, 984 m, 914 w, 825 vs, 787 s, 761 s, 737 m, 656 m, 610 m, 603 m, 554 s. IR (ATR) of [Mn2(bpza)4]: νmax (cm−1) = br 3350 w, 3131 w, 3100 w, 2992 w, 1665 w, 1646 s, 1517 w, 1450 m, 1403 s, 1346 m, 1285 s, 1241 m, 1095 m, 1055 s, 980 m, 924 m, 852 m, 808 m, 787 s, 753 s, 678 m, 658 s, 616 m. IR (ATR) of [Mn2(μ-O)2(κ4-tpa)2][PF6]3νmax (cm−1) = 3633 vw, 1605 m, 1572 w, 1483 m, 1438 s, 1290 m, 1156 m, 1190 m, 1057 m, 1015 m, 935 w, 826 vs, 761 vs, 662 s, 552 s.
For the electrochemical synthesis route, 5 ml of a 10 mM solution of 3 in MeCN with [Bu4N][PF6] (0.1 M) as electrolyte were transferred into a electrochemical cell with a three electrode setup. A Pt-net was used as working electrode, a Pt-wire as counter electrode and an Ag/Ag+ electrode as reference. The solution was stirred and the oxidation was carried out at +1.12 V vs. Fc/Fc+ for 48 h. Crystals were obtained without further purification by slowly evaporating the solvent.
UV irradiation experiments
Solutions of the complexes (5 ml; 10 mM) were irradiated using a Herolab UV-hand lamp (λirr = 365 nm) at 5 cm distance. An open quartz cuvette (20 × 10 × 40 mm) with a glass plate as a cover was used as reaction vessel. For IR measurement, irradiation was interrupted, 0.1 ml was transferred into a liquid IR cell (see below) and then filled back into the cuvette after the IR-measurement for further irradiation.For BSA experiments, BSA (ca. 1 eq. amino acid monomer, powder) was added to 3.5 ml of a solution of 1 (10 mM) in H2O/MeCN (4:3). In addition to IR measurements that were proceeded as described above, photographs were taken to document the precipitation of the protein during irradiation.
IR spectroscopy
For IR-spectroscopic measurements of the complexes (10 mM in MeCN and MeOH/H2O 9:1) a Specac Omni-Cell™ with CaF2 windows and a 0.1 mm spacer was used in a Nicolet™ iS™10 FT-IR spectrometer. Spectra of the solid products of the UV-irradiation were measured by using a Nicolet™ diamond ATR unit.
Analytical electrochemistry
Cyclic voltammograms (CVs) of 5 mM solutions of the complexes 1, 2 and 3 were measured using a Princeton Applied Research VersaSTAT 4 potentiostat and a Micro-Cell Kit. The solutions were measured before and after irradiation (λirr = 365 nm, 30 min). The working electrode was a platinum disc electrode (d = 2 mm) and the counter electrode a platinum wire. The electrolyte was [Bu4N][PF6] (0.1 M) in MeCN as solvent and an Ag/Ag+ (0.01 M) reference system was used. At the end of each measurement series, ferrocene was added to calibrate the potential of the reference electrode.Spectroelectrochemistry
The IR spectro-electrochemistry experiments were carried out using a modified Specac Omni-Cell™ (see ESI, Fig. S5†) with a platinum net as working electrode in the path of the IR beam, a platinum net as counter electrode and a platinum wire as quasi reference electrode. LSVs were measured with a scan rate of 1 mV s−1 and the measurements of the IR-spectra were started at 30 s intervals (4 scans each, taking ∼15 s). The 5–10 mM solutions of 1, 2 and 3 in MeCN (also containing [Bu4N][PF6] (0.1 M) as electrolyte) were irradiated before for 30 min (λirr = 365 nm) to maximize the amount of the intermediate biscarbonyl complexes.Bulk electrolysis of 3 was also carried out in the Princeton Applied Research Micro-Cell Kit. The working electrode was a platinum net electrode and the counter electrode was a platinum wire. As electrolyte [Bu4N][PF6] (0.1 M) was used in MeCN as solvent and Ag/Ag+ (0.01 M) was used as reference. Chronoamperometric data can be seen in ESI Fig. S18.† Crystals for XRD measurement were obtained by slowly letting the solvent evaporate and reducing the amount of MeCN to ca. 1 ml.
XRD data for [Mn(tpm)2][PF6]2, [Mn2(bpza)4], [Mn(κ3-tpa)(CO)3][PF6], UV-irradiation derived [Mn2(μ-O)2(κ4-tpa)2][PF6]3 and electrochemically derived [Mn2(μ-O)2(κ4-tpa)2][PF6]3 were collected on a Bruker Apex2 equipped with a CCD area detector, a Mo-Kα X-ray source (λ = 0.70173 Å) and a nitrogen cooling device at 100 K. Crystals were mounted in inert polyether oil on MiTeGen loops. The structures were solved with SHELXT30 and refined with SHELXL31 using Shelxle.32 Data and figures were created using the Olex233 software. Counter ion disorder was modelled using DSR.34 All crystallographic data has been deposited at the Cambridge Crystallographic Data Centre as supplementary publication: CCDC 1475432 for [Mn(κ3-tpa)(CO)3][PF6], CCDC 1475436 for [Mn(tpm)2][PF6]2, CCDC 1475441 for [Mn2(bpza)4], CCDC 1475447 for UV-irradiation derived [Mn2(μ-O)2(κ4-tpa)2][PF6]3 and CCDC 1475452 for the electrochemically derived [Mn2(μ-O)2(κ4-tpa)2][PF6]3.
Acknowledgements
We would like to thank Dr Valentin Radtke from our institute for his advice concerning electrochemical measurements and for making the modified Specac Omni-Cell™ for IR spectro-electrochemistry available. Funding for this project was generously provided by the German Science Foundation (DFG, project KU2885/1-1).Notes and references
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- C. C. Romao, W. A. Blattler, J. D. Seixas and J. L. G. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- W. Huber, R. Linder, J. Niesel, U. Schatzschneider, B. Spingler and P. C. Kunz, Eur. J. Inorg. Chem., 2012, 3140–3146 CrossRef CAS.
- P. Govender, S. Pai, U. Schatzschneider and G. S. Smith, Inorg. Chem., 2013, 52, 5470–5478 CrossRef CAS PubMed.
- C. Bischof, T. Joshi, A. Dimri, L. Spiccia and U. Schatzschneider, Inorg. Chem., 2013, 52, 9297–9308 CrossRef CAS PubMed.
- H.-M. Berends and P. Kurz, Inorg. Chim. Acta, 2012, 380, 141–147 CrossRef CAS.
- S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS PubMed.
- L. Di Costanzo, M. E. Pique and D. W. Christianson, J. Am. Chem. Soc., 2007, 129, 6388–6389 CrossRef CAS PubMed.
- M. T. Sossong Jr., V. S. Khangulov, C. R. Cavalli, R. D. Soprano, C. G. Dismukes and E. D. Ash, J. Biol. Inorg. Chem., 1997, 2, 433–443 CrossRef.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS PubMed.
- C. Nagel, S. McLean, R. K. Poole, H. Braunschweig, T. Kramer and U. Schatzschneider, Dalton Trans., 2014, 43, 9986 RSC.
- M. Tinajero-Trejo, N. Rana, C. Nagel, H. E. Jesse, T. W. Smith, L. K. Wareham, M. Hippler, U. Schatzschneider and R. K. Poole, Antioxid. Redox Signaling, 2016, 24, 765–780 CrossRef CAS PubMed.
- Y. Nishida, T. Akamatsu, K. Tsuchiya and M. Sakamoto, Polyhedron, 1994, 13, 2251–2254 CrossRef CAS.
- H. Elias, G. Schmidt, H. J. Kueppers, M. Saher, K. Wieghardt, B. Nuber and J. Weiss, Inorg. Chem., 1989, 28, 3021–3024 CrossRef CAS.
- L. Kromer, B. Spingler and R. Alberto, J. Organomet. Chem., 2007, 692, 1372–1376 CrossRef CAS.
- F. Zobi, B. Spingler and R. Alberto, Dalton Trans., 2008, 5287 RSC.
- T. Kitano, Y. Sohrin, Y. Hata, H. Wada, T. Hori and K. Ueda, Bull. Chem. Soc. Jpn., 2003, 76, 1365–1373 CrossRef CAS.
- E. Kottelat, A. Ruggi and F. Zobi, Dalton Trans., 2016, 45, 6920–6927 RSC.
- C. Ma, W. Wang, X. Zhang, C. Chen, Q. Liu, H. Zhu, D. Liao and L. Li, Eur. J. Inorg. Chem., 2004, 3522–3532 CrossRef CAS.
- (a) T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernardes, C. C. Romão and M. J. Romão, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed; (b) A. A. Petruk, A. Vergara, D. Marasco, D. Bikiel, F. Doctorovich, D. A. Estrin and A. Merlino, Inorg. Chem., 2014, 53, 10456–10462 CrossRef CAS PubMed; (c) J. D. Seixas, M. F. A. Santos, A. Mukhopadhyay, A. C. Coelho, P. M. Reis, L. F. Veiros, A. R. Marques, N. Penacho, A. M. L. Goncalves, M. J. Romao, J. L. G. Bernardes, T. Santos-Silva and C. C. Romao, Dalton Trans., 2015, 44, 5058–5075 RSC.
- M. Chaves-Ferreira, I. S. Albuquerque, D. Matak-Vinkovic, A. C. Coelho, S. M. Carvalho, L. M. Saraiva, C. C. Romão and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2014, 54, 1172–1175 CrossRef PubMed.
- A. Salis and B. W. Ninham, Chem. Soc. Rev., 2014, 43, 7358–7377 RSC.
- S. Kamachi, K. Wada, M. Tamoi, S. Shigeoka and T. Tada, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2014, 70, 288–293 CrossRef CAS PubMed.
- D. K. Towle, C. A. Botsford and D. J. Hodgson, Inorg. Chim. Acta, 1988, 141, 167–168 CrossRef CAS.
- U. Bossek, K. Wieghardt, B. Nuber and J. Weiss, Inorg. Chim. Acta, 1989, 165, 123–129 CrossRef CAS.
- S. Trofimenko, J. Am. Chem. Soc., 1970, 92, 5118–5126 CrossRef CAS.
- N. Burzlaff, I. Hegelmann and B. Weibert, J. Organomet. Chem., 2001, 626, 16–23 CrossRef CAS.
- H. Wang, Y. Lu, E. Mijangos and A. Thapper, Chin. J. Chem., 2014, 32, 467–473 CrossRef CAS.
- D. L. Reger, T. C. Grattan, K. J. Brown, C. A. Little, J. S. J. Lamba, A. L. Rheingold and R. D. Sommer, J. Organomet. Chem., 2000, 607, 120–128 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 3–8 CrossRef PubMed.
- C. B. Hubschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 1281–1284 CrossRef PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 59–75 CrossRef CAS PubMed.
- D. Kratzert, J. J. Holstein and I. Krossing, J. Appl. Crystallogr., 2015, 933–938 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional IR spectra, CV/LSV curves. CCDC 1475432, 1475436, 1475441, 1475447 and 1475452. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02020h |
| This journal is © The Royal Society of Chemistry 2016 |