
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the hydrogen transfer catalytic activity of hybrid carbon nanotube-based NHC–iridium catalysts by increasing the oxidation degree of the nanosupport†
Matías
Blanco
a,
Patricia
Álvarez
a,
Clara
Blanco
a,
M. Victoria
Jiménez
*b,
Jesús J.
Pérez-Torrente
 b,
Luis A.
Oro
b,
Javier
Blasco
c,
Vera
Cuartero
d and
Rosa
Menéndez
*a
b,
Luis A.
Oro
b,
Javier
Blasco
c,
Vera
Cuartero
d and
Rosa
Menéndez
*a
aInstituto Nacional del Carbón (INCAR) – CSIC, P.O. Box 73, 33080, Oviedo, Spain. E-mail: rosmenen@incar.csic.es
bDepartment of Inorganic Chemistry, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH-CSIC), University of Zaragoza, 50009, Zaragoza, Spain. E-mail: vijimenez@unizar.es
cInstituto de Ciencia de Materiales de Aragón (ICMA), Departamento de Física de la Materia Condensada, CSIC – Universidad de Zaragoza, 50009 Zaragoza, Spain
dESRF – The European Synchrotron, 71, Avenue des Martyrs, Grenoble, France
First published on 21st March 2016
Abstract
CVD-grown multiwalled carbon nanotubes were purified by applying four different treatments with increasing oxidation severity. The growing severity of the treatment results in progressive oxygen functionalization of the surface along with introduction of an increasing quantity of defects on the carbon nanotube walls. Iridium–N-heterocyclic carbene complexes were covalently anchored to those oxidized surfaces through their surface carboxylic acids via acetyl linkers. The carbon nanotube-based iridium–NHC hybrid materials developed are active in the hydrogen-transfer reduction of cyclohexanone to cyclohexanol with 2-propanol/KOH as hydrogen source but with rather different activity. The catalytic activity of the hybrid catalysts is strongly influenced by the type and amount of oxygenated functionalization resulting from the treatment applied to the support, being the most active and the most oxidized material.
1. Introduction
N-Heterocyclic carbenes (NHCs) have attracted increasing attention as ancillary ligands for the design of transition metal homogeneous catalysts.1 On the one hand, the tunable character of NHC ligands allows for the control of the sterical and electronic properties at the metal centre.2 On the other hand, organometallic catalysts can be anchored on solid supports through the wingtip of the NHC ligand with potential improvement in the catalytic activity by support effects permitting, additionally, the easy recovery and subsequent recyclability of the hybrid catalysts.3,4Carbon nanotubes (CNT),5 among other carbon materials, present excellent electronic, thermal, chemical and mechanical properties6–8 which make them suitable to support molecular transition metal complexes. All the synthetic methods used in their production, i.e. arc discharge, laser vaporization or CVD,9 result in impurities that join every produced CNT batch, which are mainly based on amorphous carbon particles and traces of the metals employed for the growth of the nanotubes.10 Purification methods involve thermal annealing, electrochemical or magnetic treatments, but the most applied procedures are based on different acid/oxidant treatments in order to dissolve the metals and eliminate the carbon impurities.11 It is known that these treatments also affect the carbon walls of the CNTs. In fact, the oxidation surface chemistry of CNTs is reasonably well established,12 and surface oxygen groups are deployed after the purification by oxidation processes, mainly carboxylic acids on the edges, tips and defects and hydroxylic and epoxy groups on the basal planes.13 depending on the grade of oxidation.14–16 Several proposals about the mechanism involving the oxidation of the CNT walls have been reported in the literature, but all of them agree that the severity of the treatments is directly related to the degree of oxidation.
Iridium–NHC complexes are efficient catalyst precursors for the reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds to generate alcohols or amines under mild transfer hydrogenation conditions.17–19 The covalent immobilization of Ir–NHC complexes on CNTs can be achieved by taking advantage of their oxygenated surface chemistry, but their development as hybrid catalysts is under expansion in comparison with other suitable supports.20 In this regard, we have developed synthetic protocols for the covalent immobilization of Ir–NHC complexes to carbon surfaces through the carboxylic acid or the hydroxyl surface groups by using hydroxy-functionalized imidazolium salt. This functionalization approach results in the formation of ester and carbonate functions, respectively, as linkers to the Ir–NHC species. Interestingly, the hybrid materials were found to be efficient hydrogen-transfer catalysts exhibiting good recyclability.
O and CN bonds to generate alcohols or amines under mild transfer hydrogenation conditions.17–19 The covalent immobilization of Ir–NHC complexes on CNTs can be achieved by taking advantage of their oxygenated surface chemistry, but their development as hybrid catalysts is under expansion in comparison with other suitable supports.20 In this regard, we have developed synthetic protocols for the covalent immobilization of Ir–NHC complexes to carbon surfaces through the carboxylic acid or the hydroxyl surface groups by using hydroxy-functionalized imidazolium salt. This functionalization approach results in the formation of ester and carbonate functions, respectively, as linkers to the Ir–NHC species. Interestingly, the hybrid materials were found to be efficient hydrogen-transfer catalysts exhibiting good recyclability.
The aim of this work is the covalent anchoring of Ir–NHC complexes, via acetyl linkers, through the carboxylic acids of different oxidized CNT supports resulting from oxidation treatments of increasing severity. We are aware that only a few N-acyl-substituted NHC–transition metal complexes have been reported to date.21 In addition, it has been found that these complexes usually react quickly and irreversibly with a great variety of mild nucleophiles, including water and alcohols, to give the corresponding protic NHC complexes.21a However, to our delight, we have found that these iridium hybrid catalysts exhibit an outstanding catalytic activity in hydrogen transfer, which is probably a consequence of the stabilizing effect exerted by the carbon nanotube support. Thus, the catalytic activity, stability and recyclability of the resultant hybrid materials in transfer hydrogenation reactions have been studied as a function of the surface chemistry of their corresponding support. In this context, it is worth noting that detailed catalytic studies based on supported catalysts with a gradual oxidation level of their surfaces are scarce.
2. Experimental
Materials
All the chemicals, including starting multiwalled carbon nanotubes, were purchased from Aldrich. Reagent or HPLC grade was employed in all the experiments. Solvents were distilled immediately prior to use from the appropriate drying agents or obtained from a Solvent Purification System (Innovative Technologies). The starting organometallic compound [Ir(μ-OMe)(cod)]2 (cod = 1,5-cyclooctadiene) was prepared according to a standard literature procedure.22Oxidation of the carbon nanotubes
Carbon nanotubes were subjected to four different purification treatments of increasing severity: (a) 0.3 g of raw carbon nanotubes was dispersed in 70 mL of concentrated HCl. The mixture was magnetically stirred at 60 °C for 2 h. The suspension was washed by addition of several portions of 250 mL of water, centrifugation, and elimination of the supernatant until neutral pH was reached. The solid collected was dried at 100 °C until constant weight, and labelled CNT-HCl. (b) 0.3 g of raw carbon nanotubes was dispersed in 25 mL of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ammonium hydroxide (28%) and hydrogen peroxide (30%). The mixture was magnetically stirred at 80 °C for 5 h, and then, the washing procedure described above was applied until neutral pH was reached. The solid was dried and labelled CNT-LT. (c) 0.3 g of raw CNT was dispersed in 70 mL of a solution of nitric acid (3 M). The mixture was magnetically stirred at 60 °C for 15 min, sonicated for 2 h and then diluted with 250 mL of water. After centrifugation, the washing procedure with water was repeated until neutral pH was reached as in the previous preparations. The collected solid was dispersed in 70 mL of hydrogen peroxide (30%), and the mixture was magnetically stirred at 60 °C for 15 min and then sonicated for 2 h. The solid was collected by filtration, washed with water and dried at 100 °C until constant weight. The dried solid obtained was named CNT-MT. (d) 0.3 g of raw CNT was dispersed in 40 mL of a 3:1 mixture of concentrated sulphuric and nitric acids. The mixture was sonicated for 10 min, magnetically stirred at 80 °C for 20 min and then sonicated for another 20 min. Finally, it was diluted with 250 mL of water, and after centrifugation, the standard washing procedure with water was repeated until neutral pH was reached as in the previous preparations. The solid was dried at 100 °C until constant weight and named CNT-ST.
:1 mixture of ammonium hydroxide (28%) and hydrogen peroxide (30%). The mixture was magnetically stirred at 80 °C for 5 h, and then, the washing procedure described above was applied until neutral pH was reached. The solid was dried and labelled CNT-LT. (c) 0.3 g of raw CNT was dispersed in 70 mL of a solution of nitric acid (3 M). The mixture was magnetically stirred at 60 °C for 15 min, sonicated for 2 h and then diluted with 250 mL of water. After centrifugation, the washing procedure with water was repeated until neutral pH was reached as in the previous preparations. The collected solid was dispersed in 70 mL of hydrogen peroxide (30%), and the mixture was magnetically stirred at 60 °C for 15 min and then sonicated for 2 h. The solid was collected by filtration, washed with water and dried at 100 °C until constant weight. The dried solid obtained was named CNT-MT. (d) 0.3 g of raw CNT was dispersed in 40 mL of a 3:1 mixture of concentrated sulphuric and nitric acids. The mixture was sonicated for 10 min, magnetically stirred at 80 °C for 20 min and then sonicated for another 20 min. Finally, it was diluted with 250 mL of water, and after centrifugation, the standard washing procedure with water was repeated until neutral pH was reached as in the previous preparations. The solid was dried at 100 °C until constant weight and named CNT-ST.
Covalent functionalization of the carbon nanotubes
The cationic iridium complexes bearing methyl-imidazol-2-ylidene ligands were anchored to the oxidized carbon nanotubes through acetyl functions by applying a three-step procedure. 100 mg of CNT-X was refluxed in 40 mL of thionyl chloride for 24 h under argon atmosphere. The excess of SOCl2 was removed under vacuum, and the resultant product was washed three times with 20 mL of tetrahydrofuran (THF) and dried for 2 h under vacuum. Then, the solid was dispersed in 15 mL of THF, and N-methylimidazole (2 mL, 25.1 mmol) was added. The mixture was refluxed for 24 h. The solid was filtered and washed with THF (3 × 20 mL), dichloromethane (3 × 20 mL) and diethyl ether (2 × 20 mL). The solids collected were dried at 100 °C in a preheated furnace for 2 h, and the dried samples were labelled CNT-X-MI. The resulting imidazolium functionalized carbon nanotubes (100 mg) were reacted with [Ir(μ-OMe)(cod)]2 (100 mg, 0.150 mmol) in THF (20 mL) under argon atmosphere. The mixture was refluxed for 2 days and then immersed in an ultrasonic bath for 30 min at room temperature. The resultant solids were recovered by centrifugation, washed with THF (5 × 10 mL) and diethyl ether (2 × 5 mL), and dried under vacuum. The hybrid materials were labelled CNT-X-MI-Ir, where X is related to the degree of oxidation of the nanotube (HCl, LT, MT and ST).:2:2:2, 8H, CH2cod). 13C{1H} NMR (298 K, C6D6): δ 186.43 (NCN), 170.63 (CO), 122.24, 118.39 (CHIm), 85.86, 84.70, 52.22, 51.52 (CHcod), 38.45 (CH3Im), 33.89, 33.24, 29.75, 29.67 (CH2cod), 28.12 (COCH3).
:2, 4H, CHcod), 2.38 (m, 4H, CH2cod), 2.19 (s, 3H, CH3Im), 1.70, 1.55 (m, 2:2, 4H, CH2cod). 13C{1H} NMR (298 K, C6D6): δ 182.18 (NCN), 126.0, 120.31 (CHIm), 66.80, 56.64 (CHcod), 32.98 (CH3Im), 32.42, 31.60 (CH2cod).
General procedure for the transfer hydrogenation catalysis
The catalytic transfer hydrogenation reactions were carried out under argon atmosphere in thick glass reaction tubes fitted with a greaseless high-vacuum stopcock. In a typical experiment, the reactor was charged with a solution of cyclohexanone (0.52 mL, 5.0 mmol) in 2-propanol (4.5 mL), an internal standard (mesitylene, 70 μL, 0.5 mmol), a base (0.1 mL, 0.025 mmol of a KOH solution (0.24 M) in 2-propanol) and a catalyst (0.005 mmol, 0.1 mol%). The resulting mixture was stirred at room temperature for 10 min and then placed in a thermostatic oil bath at the required temperature, typically 80 °C. Conversions were determined by gas chromatography analysis under the following conditions: a column temperature of 35 °C (2 min) up to 220 °C at 10 °C min−1 and a flow rate of 1 mL min−1 using ultrapure He as carrier gas. Once the reaction was completed, the hybrid iridium–NHC catalysts were recovered by centrifugation and washed with additional amounts of 2-propanol (3 × 10 mL). Several catalytic cycles were performed with these materials, under the same experimental conditions, without adding any fresh catalyst precursor. The last cycle was carried out in air.Characterization methods
The catalytic reactions were analysed using an Agilent 4890 D system equipped with an HP-INNOWax capillary column (0.4 μm, 25 m × 0.2 mm i.d.) using mesitylene as internal standard. Thermogravimetric analysis (TGA) of the materials was performed using a TA SDT 2960 analyzer thermobalance. The procedure used was as follows: 3 mg of sample was heated in the thermobalance at 10 °C min−1 to 1000 °C using a nitrogen flow of 200 mL min−1. Raman spectra of the parent oxidized nanotubes and the imidazolium-modified materials were recorded from 750 to 3500 cm−1 on a Renishaw 2000 Confocal Raman Microprobe (Rhenishaw Instruments, England) using a 514.5 nm argon ion laser. High-resolution transmission electron microscopy (HRTEM) images were recorded using a JEOL JEM-2100F transmission electron microscope, equipped with a field emission gun (FEG) operating at 200 kV and fitted with an Oxford Instruments microprobe to perform energy-dispersive X-ray spectroscopy (EDX), in order to verify the atomic composition of the catalyst. The samples were prepared by casting a few drops of 1 mg mL−1 ethanol suspensions of the materials over the carbon grids. To minimize exposure of the samples to air, the suspensions were transferred to a lacey carbon grid using a glovebox filled with ultrahigh-purity argon and then to the TEM holder in order to minimize the time required to introduce them into the microscope. The X-ray photoemission spectroscopy (XPS) spectra were recorded using a SPECS system operating under a pressure of 10−7 Pa and equipped with a Mg Kα X-ray source. The functional groups in the nanotube based materials were quantified by deconvolution of the high resolution C1s XPS peak employing Gaussian and Lorentzian functions.23 The binding energy profiles were deconvoluted as follows: undamaged structures of sp2-hybridized carbon (284.5 eV), damaged structures or sp3-hybridized carbons (and C–N groups, 285.5 eV), C–O (and C–N groups, 286.5 eV), CO functional groups (287.7 eV) and COO groups at 288.7 eV. The amount of iridium present in the samples was determined by means of inductively coupled plasma mass spectrometry (ICP-MS) using an Agilent 7700× instrument. The samples were digested following a method described elsewhere.24 Briefly, 30 mg of sample was treated with 5 mL of a mixture of concentrated nitric and hydrochloric acids (3:1 ratio) at 180 °C for 3 h under microwave irradiation.
X-ray absorption measurements at the Ir L3-edge were performed using a Si(111) double crystal monochromator at the BM23 beam line of the ESRF (Grenoble, France) and using a Si(311) double crystal monochromator at the Claess beam line of the Alba (Barcelona, Spain) synchrotron facilities. The spectra collected from both setups were equivalent. The energy resolution ΔE/E was estimated to be about 8 × 10−5 at the Ir L3-edge, and a pellet of Ir metal and cellulose, the complex [IrCl(cod)(MeIm(CH2)3OH)] and the salt IrCl3 were simultaneously measured for energy calibration as references for Ir(0), Ir(I) and Ir(III), respectively. The extended X-ray absorption fine structure (EXAFS) spectra were analysed using the ARTEMIS program,25 which makes use of theoretical phases and backscattering amplitudes calculated from FEFF6.26 The fits were carried out in R space using a Hanning window for filtering purposes.
3. Results and discussion
Preparation and functionalization of carbon nanotubes with different oxidation levels
The carbon nanotube-based support materials, CNT-X, were obtained from commercial CVD-grown multiwalled carbon nanotubes by applying different and gradually increasing oxidation reagents, such as a) hydrochloric acid, b) a mixture of hydrogen peroxide and ammonium hydroxide, c) diluted nitric acid and hydrogen peroxide and d) a mixture of warm concentrated sulphuric and nitric acids.27 It is noticeable that the ongoing oxidation processes on the CNTs generate oxygen functional groups at edges, defects and basal planes of the nanotubes. The quantity of defects observed on the carbon nanotube walls of the oxidized samples is directly proportional to the harshness of the oxidation treatment. In fact, a gradual increment in the D band of the Raman spectra is observed as a result of those treatments. Furthermore, the C/O atomic ratio calculated by XPS data (Table 1) also decreases gradually.| Sample | XPS (atom%) | ICP (wt%) | |||||
|---|---|---|---|---|---|---|---|
| C/O | C | Cl | N | O | S | Ir | |
| CNT-HCl | 28 | 96.6 | 0.0 | 0.0 | 3.4 | 0.0 | |
| CNT-HCl-MI | 23 | 93.7 | 0.5 | 0.9 | 4.0 | 0.8 | |
| CNT-HCl-MI-Ir | 3.1 | ||||||
| CNT-LT | 20 | 95.2 | 0.0 | 0.0 | 4.8 | 0.0 | |
| CNT-LT-MI | 20 | 92.4 | 0.6 | 1.4 | 4.6 | 1.0 | |
| CNT-LT-MI-Ir | 6.1 | ||||||
| CNT-MT | 16 | 94.1 | 0.0 | 0.0 | 5.9 | 0.0 | |
| CNT-MT-MI | 15 | 91.4 | 0.8 | 1.7 | 6.1 | 0.0 | |
| CNT-MT-MI-Ir | 10.0 | ||||||
| CNT-ST | 4 | 80.8 | 0.0 | 0.0 | 19.2 | 0.0 | |
| CNT-ST-MI | 3.5 | 73.9 | 1.3 | 2.3 | 21.0 | 1.5 | |
| CNT-ST-MI-Ir | 12.3 | ||||||
The generated surface groups on the CNT-X materials were analysed by deconvolution of the C 1s band in the XPS spectra (Fig. 1), which also confirm the progressive introduction of oxygen groups following the increase in oxidation severity. Additionally, the diminution in the Csp2 band and the intensification in the Csp3 are both consequences of those oxidation processes. Interestingly, in accordance with this argument, the most oxidized carbon nanotube material, CNT-ST, shows the largest amount of carboxylic acids and hydroxylic groups. Additionally, the large percentage of oxygen in this material, also observed in graphene oxides, is also attributed to an increase in unreactive epoxy or ether groups.28
 | ||
| Fig. 1 XPS C 1s fitting of the parent materials CNT-X: a) CNT-HCl, b) CNT-LT, c) CNT-MT and d) CNT-ST. | ||
Applying a known carbon nanotube functionalization method, the treatment of the parent oxidized nanotubes (CNT-X) with thionyl chloride followed by reaction with N-methylimidazole allow the formation of hanging methylimidazolium salts by alkylation of the heterocycle with the more reactive pendant acyl groups. Operating in such a way, it is possible to obtain the imidazolium functionalized carbon nanotube materials named CNT-X-MI (MI = 1-acyl-3-methylimidazolium, Scheme 1).
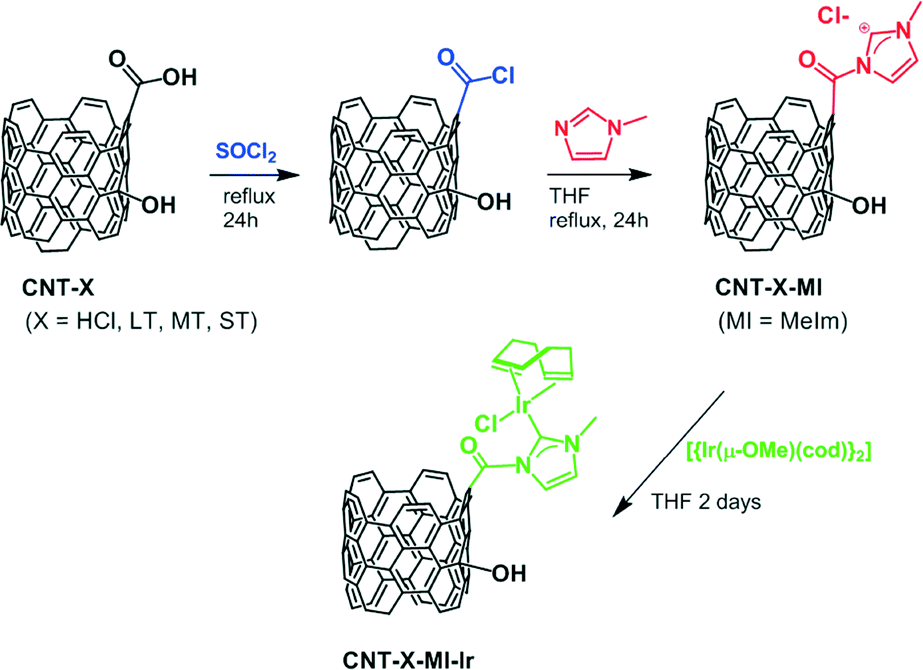 | ||
| Scheme 1 Syntheses of carbon nanotube materials: from CNT-X to CNT-X-MI-Ir. | ||
The high-resolution XPS N 1s bands of the functionalized samples reveal a single band at 401.5 eV that is indicative of a unique functionalization mode. The atomic percentages obtained from XPS data (Table 1) show a steady increase in the amount of nitrogen, which corresponds to increasing functionalization of the carbon nanotube materials with the imidazolium groups. In addition, a higher ID/IG Raman ratio was observed in every sample and also when comparing the parent-oxidized with imidazole-treated materials, that indicates positive functionalization, which is in accordance with the correlation observed in related materials (see the ESI†).29 In addition, the correct N:Cl atomic ratio of ca. 2:1 was obtained which is in agreement with the presence of the imidazole ring (see the ESI†). All these data confirm that the more oxidation treatment applied, the more functionalized support is obtained, which is CNT-ST-MI.
The quantification of the imidazolium groups present in these materials has also been determined by means of thermogravimetric analysis (TGA) as the weight decreases at 400 °C (mainly associated to the elimination of the imidazolium fragments).30 As expected, more pronounced decays in the TGA profiles were observed for the materials coming from the more oxidized CNTs, which is in accordance with a larger number of imidazole units in these materials (see the ESI†).26 In fact, the nitrogen weight percentages determined from the TGA curves compare well with those obtained from the XPS data (see the ESI†).
Synthesis and characterization of carbon nanotube–NHC–iridium hybrid catalysts
The deprotonation of the weak acid proton at the C2 of the supported imidazolium groups by the basic methoxo ligands in the dinuclear complex [Ir(μ-OMe)(cod)]2 (cod = 1,5-cyclooctadiene) generates N-heterocyclic carbene (NHC) anchored iridium complexes (Scheme 1) on the nanotube surfaces, CNT-X-MI-Ir. The insolubility of these materials together with the detection of methanol by gas chromatography in the filtered mother liquids after reaction with [Ir(μ-OMe)(cod)]2 (see the ESI†) confirm the progress of the reactions. The supporting of the metal complexes is substantiated by the high-resolution transmission electron microscopy images of the four types of Ir–NHC hybrid materials derived from gradually oxidized carbon nanotubes (CNT-X-MI-Ir), as shown in Fig. 2. Homogeneous distributions of electron-dense regions were detected with diameters ranging 0.15–0.3 nm all throughout the outer and inner walls (white circles). The metallic spots are in the range of the molecular iridium complexes as confirmed by their EDX spectra (see the ESI†). | ||
| Fig. 2 HRTEM images of the carbon nanotube–NHC–iridium hybrid catalysts: a) CNT-HCl-MI-Ir, b) CNT-LT-MI-Ir, c) CNT-MT-MI-Ir, and d) CNT-ST-MI-Ir. | ||
Although larger iridium particles of 1.2–1.4 nm are also detected, the Ir 4f region of the XPS spectra shows, for all samples, two maxima centred at 62.4 and 66.5 eV, corresponding to Ir 4f7/2 and Ir 4f5/2, according to Ir(I) species.31 Those larger spots, more marked on the unprotected outer walls, could be clusters or nanoparticles possibly formed by electron beam irradiation inside the microscope chamber.32 Similar size distributions were observed for other supported molecular iridium catalysts or even for graphene-based hybrid catalysts.3,4
Additionally, all the materials exhibited the same appearance in terms of number of layers and interlayer distances compared to their parent carbon nanotubes (see the ESI†), which indicates that functionalization has not caused any damage to the nanotube layers.
The amount of iridium in the nanotubes, determined by means of ICP-MS, varies progressively from 3.1% for the less oxidized material, CNT-HCl-MI-Ir, to 12.3% for the most oxidized material, CNT-ST-MI-Ir (6.1% for CNT-LT-MI-Ir and 10.0% for CNT-MT-MI-Ir, Table 1). The nitrogen weight percentages determined from the TGA curves allow for the calculation of the maximum iridium load in the CNT-X-MI-Ir materials assuming that each imidazole-2-ylidene is involved in the formation of an Ir–NHC bond. The ratio between the iridium found and the maximum calculated is 48%, 71% and 98% for CNT-HCl-MI-Ir, CNT-LT-MI-Ir, and CNT-ST-MI-Ir, respectively, although a slight metal overload (106%) was found for CNT-MT-MI-Ir. In this context, it is worth mentioning that the iridium load could be in some way influenced by a poor dispersion capacity of the materials in the polar reaction media, together with the accessibility to the reactive acyl chloride groups.33
Structural features of the carbon nanotube–NHC–iridium hybrid catalysts
In order to gain an insight into the local structure of the iridium atoms in the hybrid catalysts, XAS measurements were performed at room temperature. Fig. 3 presents the κ2-weighted EXAFS signals for all new CNT-X-MI-Ir acetyl hybrid catalysts and the known CNT-ST-1E-Ir ester hybrid catalyst,3,34 compared to [IrCl(cod)(MeIm(CH2)3OH)] as a reference compound. The hybrid catalysts show similar oscillations in the spectra although a higher damping is observed in the signal of CNT-ST-MI-Ir. The main difference between the catalysts and the reference is the clear interference at k ∼ 8–11 Å−1 in the latter compound which is absent in the hybrid catalysts. This interference is ascribed to the Ir–Cl bond in the first coordination shell,2,3 and its absence in the catalysts' spectra suggests that the Cl has been replaced by a light element as was observed in similar catalysts.35Fig. 4a shows the pseudo radial distribution obtained from the Fourier transform (FT) of the previous EXAFS spectra between 3.0 and 13.3 Å−1 using a Hanning window. The hybrid catalysts show a big single peak corresponding to the first coordination shell at R ∼ 1.7 Å (without phase shift correction). The [IrCl(cod)(MeIm(CH2)3OH)] reference compound instead shows an additional shoulder at 2.1 Å in agreement with the contribution of the Ir–Cl bond. The intensity of the main peak is similar for all catalysts with the exception of CNT-ST-MI-Ir whose peak is smaller in agreement with the high damping of the EXAFS signal. Structures beyond 2.2 Å correspond to mixed contributions from the next-neighbour coordination shells. The peaks are significantly weaker and broader suggesting a strong disorder in these shells. Therefore, EXAFS analysis was limited to the first coordination shell, when the study was performed between 1.1 and 2.3 Å in R-space.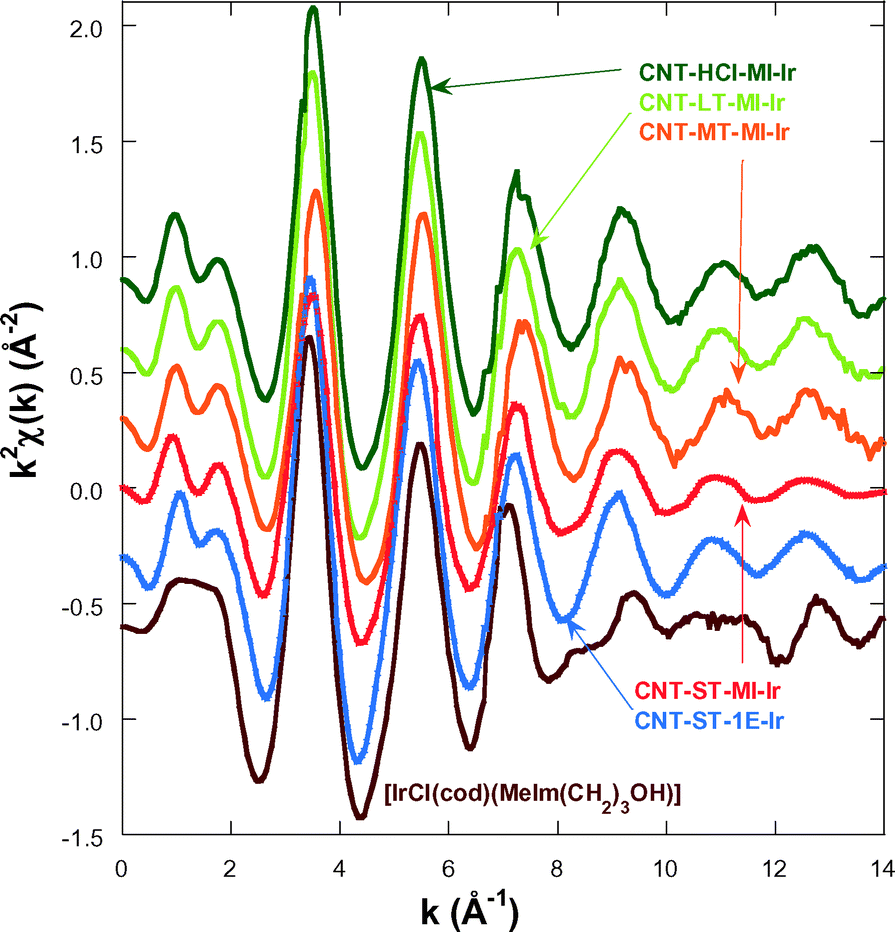 | ||
| Fig. 3 κ2-Weighted EXAFS spectra for the hybrid catalysts and the [IrCl(cod)(MeIm(CH2)3OH)] reference compound. The data are shifted in the vertical scale for the sake of comparison. | ||
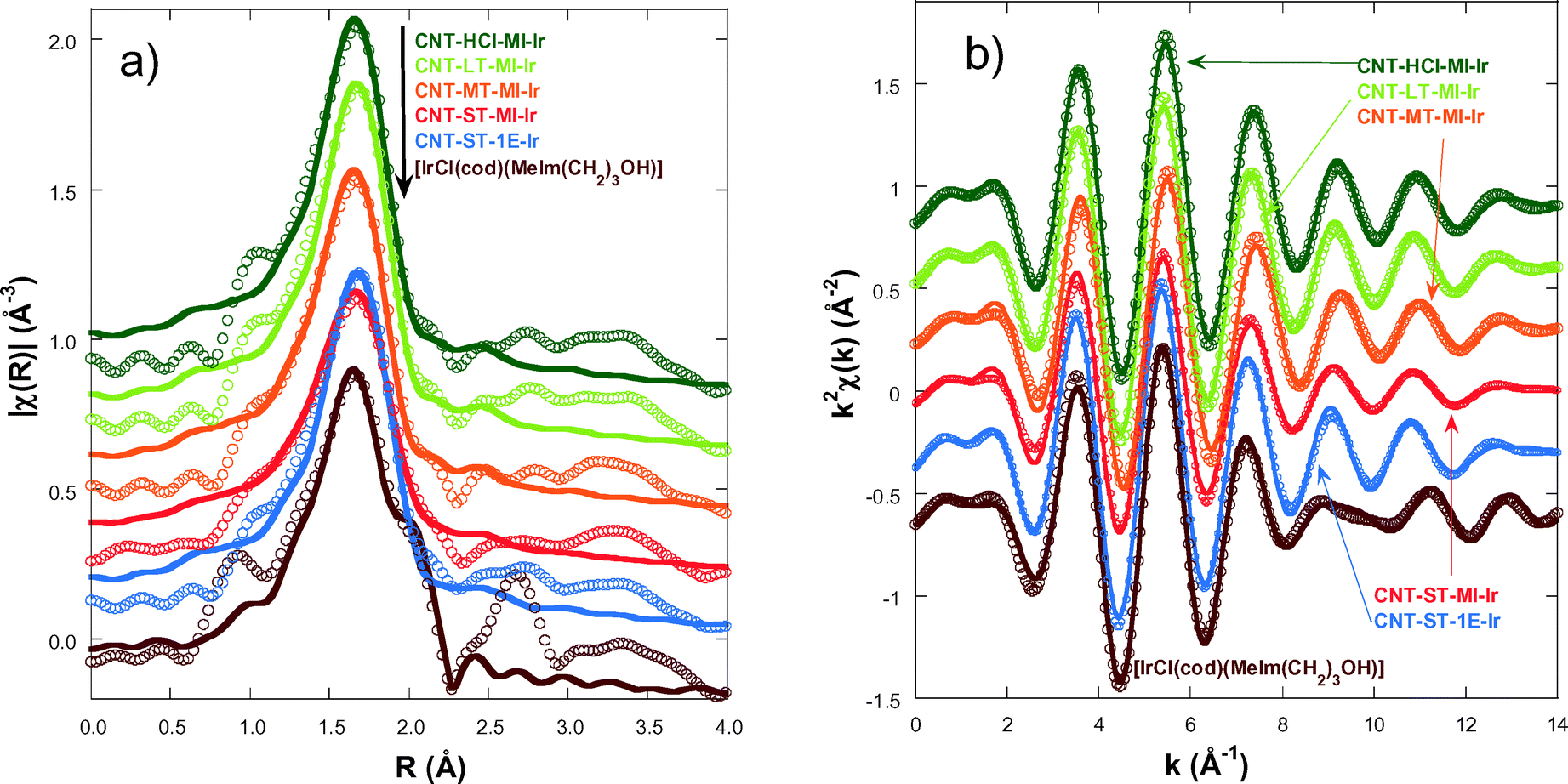 | ||
| Fig. 4 a) Fits (lines) of the Fourier transform signal (points) from the κ2-weighted EXAFS signal (points) of the catalysts and the reference neutral chloride compound [IrCl(cod)(MeIm(CH2)3OH)] until R = 2.3 Å. b) Fits (lines) of the real part of the Fourier-filtered spectra (points) between R = 1–2.3 Å for the same samples in k-space. | ||
The first coordination shell of the Ir atom in the reference compound, [IrCl(cod)(MeIm(CH2)3OH)], as it was previously reported35 is composed of a Cl atom, a C atom of the imidazol-2-ylidene (C1) ring and 4 C atoms of both CC bonds present in the cyclooctadiene ligand (Fig. 5a). Our analysis yields the following distances: Ir–Cl = 2.369(9) Å, Ir–C1 = 2–016(14) Å, Ir–C2,3 = 2.089(13) Å and Ir–C3,4 = 2.120(13) Å. The four latter distances correspond to the interatomic distances to the 1,5-cyclooctadiene carbons, while C1 refers to the carbon atom belonging to the NHC. In contrast, the Cl atom is missing in the Ir coordination shell of the supported catalysts and it is probably substituted with an O atom from the oxidized CNT materials.
 | ||
| Fig. 5 a) Schematic molecular structure of the reference neutral chloride compound [IrCl(cod)(MeIm(CH2)3OH)]. b) Molecular structure of the cation of [Ir(NCCH3)(cod)(MeIm(CH2)3OH)][BF4] (hydrogen atoms have been omitted for clarity). Selected bond lengths (Å) and angles (°): Ir–C(1) 2.031(3), Ir–C(2) 2.127(3), Ir–C(3) 2.134(4), Ir–C(4) 2.199(3), Ir–C(5) 2.186(3), Ir–N(1) 2.032(3), C(1)–Ir–N(1) 92.44(12), C(1)–Ir–C(2) 90.58(13), N(1)–Ir–C(5) 87.55(12), C(2)–Ir–C(4) 90.11(14). | ||
In order to model the Ir local environment in the catalysts, we have replaced the Cl atom by oxygen, preserving the rest of the Ir–C bonds. In fact, the Ir–O distance was matched to the Ir–C1 bond length because this approach strongly stabilizes the fitting procedure and minimizes the number of free parameters. Moreover, it agrees with isostructural compounds like [Ir(NCCH3)(cod)(MeIm(CH2)3OH)][BF4], whose molecular structure, determined by X-ray diffraction (Fig. 5b), shows a N atom of the acetonitrile ligand bonded at 2.032(3) Å while the Ir–C1 bond length is 2.031(3) Å.35 We have observed a strong correlation between the values of Ir–C2,3 and Ir–C4,5; therefore, these interatomic distances were refined with a single parameter (ΔR2) starting from the theoretical value of the reference compound. Therefore, a total of 4 parameters are refined: an average inner potential correction of the threshold (ΔE0), an average Debye–Waller factor (σ2) and two distance parameters, ΔR1 for Ir–C1 and Ir–O and ΔR2 for Ir–C2,3 and Ir–C4,5. The amplitude reduction factor S02 is fixed to 1 in agreement with the value obtained for the reference compounds and previous studies.
Fig. 4a compares the best fit and experimental spectra corresponding to the moduli of the FTs of the κ2-weighted EXAFS shown in Fig. 3. Fig. 4b shows the back-Fourier filtered spectra in k-space corresponding to the first coordination shell together with the best fit results. These best fit results for CNT-X-MI-Ir and CNT-ST-1E-Ir34 are summarized in Table 2. Our analysis confirms a local environment composed of six light elements around the Ir atom in all the hybrid catalysts. The distances of Ir to the carbons in the diolefin range between 2.078 and 2.22 Å, while the shortest distances are between 2.025 and 2.045 Å. Finally, the higher value of the Debye–Waller factor is observed for CNT-MT-MI-Ir and especially for CNT-ST-MI-Ir suggesting a stronger structural disorder in the first coordination shell for these catalysts.
| Catalyst | ΔE0 (eV) | R 1 (Å) | Ir–C2,3 (Å) | Ir–C4,5 (Å) | σ 2 (103, Å2) | R F |
|---|---|---|---|---|---|---|
| a The residual factor accounts for the misfit between the actual data and the theoretical calculations.25 Numbers in parentheses are the errors estimated from different analyses to the best significant digit. R1 stands for Ir–C1 and Ir–O. | ||||||
| CNT-HCl-MI-Ir | 8.9(9) | 2.025(16) | 2.104(13) | 2.190(13) | 1.6(9) | 0.004 |
| CNT-LT-MI-Ir | 9.0(9) | 2.037(16) | 2.114(14) | 2.199(14) | 1.7(9) | 0.006 |
| CNT-MT-MI-Ir | 8.8(9) | 2.033(63) | 2.078(53) | 2.164(53) | 4.0(30) | 0.014 |
| CNT-ST-MI-Ir | 8.8(9) | 2.046(12) | 2.118(11) | 2.204(11) | 5.0(30) | 0.011 |
| CNT-ST-1E-Ir | 9.3(9) | 2.045(13) | 2.135(11) | 2.221(11) | 1.1(9) | 0.005 |
Hydrogen-transfer catalytic activity of the carbon nanotube–NHC–iridium hybrid catalysts
Once the anchoring of Ir–NHC organometallic complexes on the inner and outer surfaces of the nanotubes was confirmed, the hybrid materials were tested as catalysts in the hydrogen-transfer reduction of cyclohexanone to cyclohexanol, using 2-propanol both as hydrogen source and as a non-toxic solvent with a moderate boiling point (Scheme 2). The reaction conditions were those previously optimized for related O-functionalized NHC–iridium(I) homogeneous catalysts such as [IrBr(cod)(MeIm(2-methoxybenzyl))] in previous studies.2 Thus, standard catalyst loads of 0.1 mol% Ir, with 0.5 mol% KOH as a co-catalyst, and a temperature of 80 °C were routinely employed. | ||
| Scheme 2 Transfer hydrogenation of cyclohexanone with 2-propanol. | ||
The hybrid catalysts derived from gradually oxidized carbon nanotubes having anchored Ir–NHC complexes via acetyl linkers (CNT-X-MI-Ir) were active in the transfer hydrogenation of cyclohexanone. Reaction times required reaching a conversion of more than 90%, and the turnover frequencies (TOF), at initial time and at 50% conversion, for all the examined catalysts are summarized in Table 3. The four hybrid materials showed similar kinetic profiles, but appreciable differences in the catalytic activity were found (Fig. 6a). Interestingly, the cyclohexanone reduction was observed immediately after the thermal equilibration of the reactant mixture with no detectable induction period. The hybrid catalyst CNT-ST-MI-Ir is the most active in this series reaching 91% conversion in 1.6 h with a TOF50 of 3000 h−1. CNT-MT-MI-Ir, CNT-LT-MI-Ir and CNT-HCl-MI-Ir were considerably less active with TOF50 values of 789, 428 and 254 h−1, although CNT-MT-MI-Ir showed an initial TOFo of 6500 h−1 which is the highest in the series.
| Catalyst | Time (min) | Conv. (%) | TOF0 (h−1)c | TOF50 (h−1)d |
|---|---|---|---|---|
| a Reaction conditions: catalyst/substrate/KOH ratio of 1/1000/5, 0.1 mol% of catalyst in 2-propanol (5 mL) at 80 °C. b The reactions were monitored by GC using mesitylene as internal standard. c TOFs, turnover frequencies [(mol product/mol catalyst)/time (h)], were calculated at initial time (60 s), TOF0. d TOFs, turnover frequencies [(mol product/mol catalyst)/time (h)], were calculated at 50% conversion, TOF50. | ||||
| CNT-HCl-MI-Ir | 900 | 91 | 1050 | 250 |
| CNT-LT-MI-Ir | 300 | 93 | 1500 | 430 |
| CNT-MT-MI-Ir | 200 | 90 | 6500 | 790 |
| CNT-ST-MI-Ir | 100 | 91 | 5400 | 3000 |
| CNT-ST-1E-Ir | 80 | 94 | 6300 | 3350 |
| CNT-ST-1C-Ir | 210 | 89 | 3000 | 1300 |
| TRCNT-ST-1C-Ir | 100 | 92 | 6000 | 3000 |
| Ir-ImidO | 200 | 94 | 3000 | 1500 |
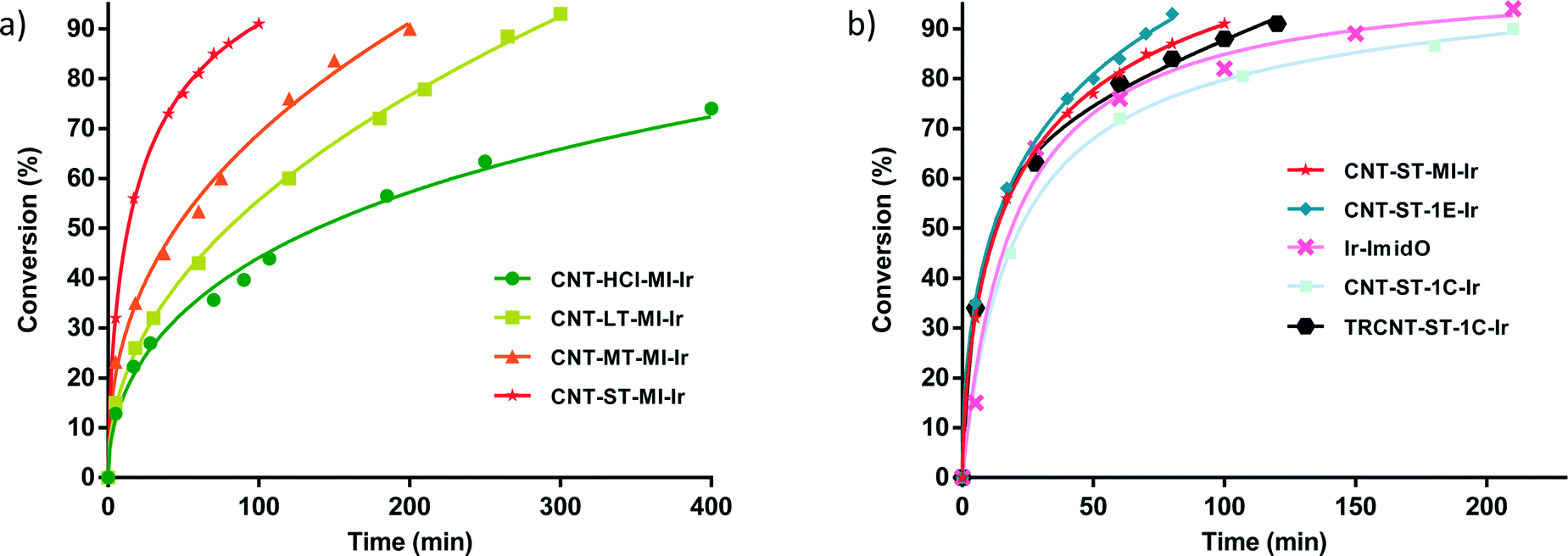 | ||
| Fig. 6 Reaction profiles for the transfer hydrogenation of cyclohexanone (%) with 2-propanol by carbon nanotube based iridium–NHC hybrid catalysts (0.1 mol% Ir) at 80 °C. a) Cyclohexanone reduction by CNT-X-MI-Ir hybrid catalysts. b) Cyclohexanone reduction by CNT-ST-MI-Ir compared with CNT-ST-1E-Ir, CNT-ST-1C-Ir, TRCNT-ST-1C-Ir, and the homogeneous [IrCl(cod)(MeIm(CH2)3OCOCH3] (Ir-ImidO) catalyst. Continuous lines represent a mathematical fit with the experimental data points. | ||
The pattern of catalytic activity shown by these hybrid catalysts CNT-X-MI-Ir correlates with the oxidation degree of the corresponding parent carbon nanotubes which points to a surface effect derived from the intensity of the oxidation treatment. According to the solid characterization techniques, those supports treated with weaker oxidizing purification procedures (HCl, LT and MT treatments) result in supports with an extended sp2 structure with higher C/O ratios, low intrinsic porosity and tips scarcely opened. The low level of hydrophilic functional groups could also imply a lower dispersion capacity in the polar catalytic reaction medium which makes the access of the reactants to the active sites and, in particular, to those settled in the inner cavity of the nanotube difficult. On the other hand, the severe oxidation treatment (ST treatment in particular) leads to a nanotube with a defective structure, plenty of holes, oxygen moieties, structural defects and opened tips.36,37 The defective structures contain suitable functional groups exposed to form active centres situated in the holes and open channels where reactants can arrive more easily in the diffusional stages of the heterogeneous catalytic process. Under these circumstances, the nanotube can also act as a “nano-reactor” offering a confinement effect that can occasionally enhance the catalysis activity.38–40
The molecular complex [IrCl(cod)(MeImCOCH3)] has been revealed as extremely moisture and alcohol sensitive which precluded the comparison of its catalytic activity with that of the carbon nanotube–NHC–iridium hybrid catalysts. [IrCl(cod)(MeImCOCH3)] is formed in the reaction of [{Ir(μ-OMe)(cod)}2] with the corresponding N-acetyl imidazolium salt along with complex [IrCl(cod)(MeImH)] resulting from the methanolysis of the acyl fragment. In fact, [IrCl(cod)(MeImCOCH3)] was cleanly converted to [IrCl(cod)(MeImH)] and methyl acetate upon reaction with methanol (NMR evidence).
It is noteworthy that neither the iridium-free carbon nanotube-based materials nor iridium supported materials without an imidazolium ligand have shown significant catalytic activity, which indirectly supports the covalent anchoring of the Ir–NHC complexes to the carbon nanotube-based materials via acetyl linkers. In this context, it has been recently reported that iridium oxide nanoparticles supported on nanoparticulate cerium oxide exhibit moderate catalytic activity in the transfer hydrogenation of cyclohexanone.41 However, the presence of IrO2 nanoparticles in our NHC–Ir hybrid catalysts is negligible because the fit of the first shell of our EXAFS spectra to a model with a rutile crystal structure (IrO2) renders a poor model. In fact, the EXAFS signal of IrO2 shows some structures in the oscillation between 7 and 9 Å−1 that are not found in the spectra or our catalysts, and the Fourier transform shows an important peak around 3 Å corresponding to the Ir–Ir path that is also absent in our spectra, and the white line (very sensitive to the number of d-holes) is significantly higher for the IrO2 spectrum.42
The catalytic performance of CNT-ST-MI-Ir has been compared with that of the molecular acetoxy-functionalized NHC complex [IrCl(cod)(MeIm(CH2)3OCOCH3] (Ir-ImidO)3 and of related hybrid catalysts having supported Ir–NHC complexes on carbon nanotubes obtained under the most severe oxidation conditions through the flexible 3-alkoxipropyl linker 1. In particular, the hybrid catalysts CNT-ST-1E-Ir3 and CNT-ST-1C-Ir, with ester and carbonate linking surface functions, result from the nanotube functionalization through the carboxylic acid and the hydroxyl surface groups, respectively, whereas TRCNT-ST-1C-Ir comes from the surface hydroxylic group functionalization of thermally reduced nanotubes (Scheme 3).35
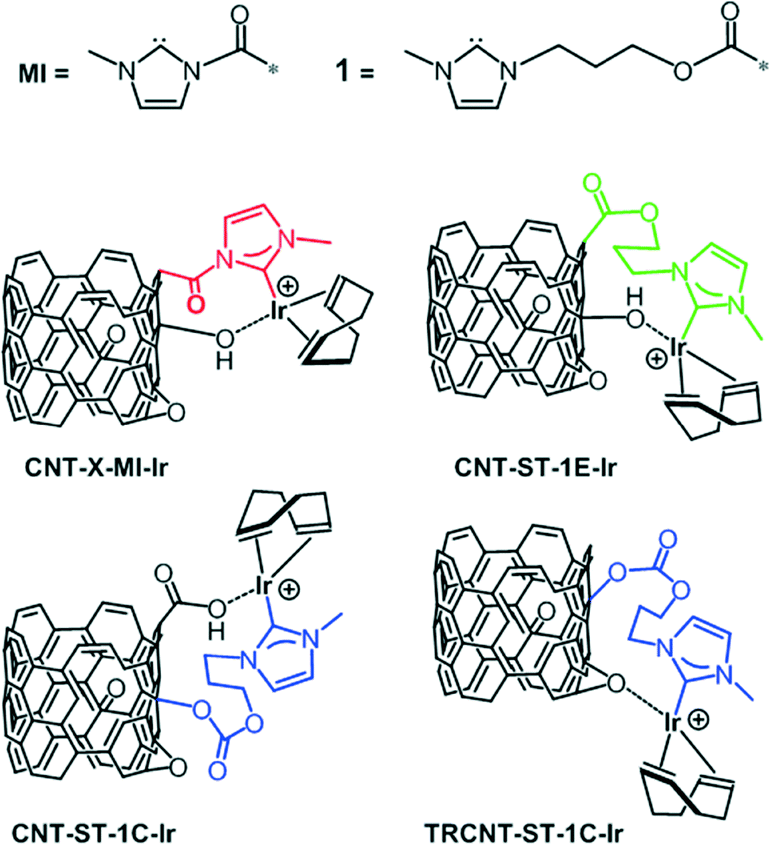 | ||
| Scheme 3 Carbon nanotube based iridium–NHC hybrid catalysts. | ||
As can be observed in Fig. 6b, the hybrid catalyst CNT-ST-1E-Ir is only slightly more active than CNT-ST-MI-Ir reaching 94% conversion in 1.3 h with a TOF50 of 3350 h−1 (Table 3). Thus, the flexibility imparted by the carbon chain linking the Ir–NHC supported complexes in CNT-ST-1E-Ir appears to influence positively the catalytic activity as both hybrid catalysts have a similar degree of oxidation. Interestingly, both hybrid catalysts are more active than the homogeneous catalyst [IrCl(cod)(MeIm(CH2)3OCOCH3] (Ir-ImidO) and the hybrid catalysts prepared by covalent functionalization of the hydroxyl surface groups CNT-ST-1C-Ir and TRCNT-ST-1C-Ir (Fig. 6b).
In contrast with the hybrid catalysts CNT-X-MI-Ir, for which the catalytic performance improves with the level of oxidation of the parent carbon nanotube material, the thermally reduced TRCNT-ST-1C-Ir was found to be more active than CNT-ST-1C-Ir (Fig. 6b).35 The structural characterization of the catalysts based on thermally reduced carbon nanotube materials showed an increase in the amount of Csp2 and a decrease in the oxygen functional groups, as a consequence of the partial restoring of the aromatic structure after the thermal treatment. Having in mind that EXAFS measurements showed a similar first-neighbour coordination shells for all the hybrid Ir–NHC catalysts,35 that difference should be ascribed to the different localization of the iridium centres in the hybrid catalysts. The active centres are located at the basal planes of the nanotubes for the catalyst with –OH functionalization, while for the –COOH functionalized materials, the Ir–NHC complexes are located at the holes and defects, where the approach of the reactants is easier because these centres are more exposed. However, in the case of a cleaner surface, which is provided for the thermal reduction, the diffusional stages can be faster, which enhances the activity of the reduced samples. These observations could be of a great importance because it gives a clear idea of how the functionalization should be in order to obtain more active catalysts.
Recycling studies (Fig. 7) were also carried out with the four hybrid catalysts CNT-X-MI-Ir based on gradually oxidized carbon nanotubes. The black solids obtained after each catalytic run were recovered, washed with fresh 2-propanol (4–5 mL) and subjected to another catalytic cycle. The recycling processes render conversions above 90% after five catalytic runs without any loss of catalytic activity. In addition, the kinetic profiles in the successive experiments are very similar to those plotted in Fig. 6a, even for the last cycle which was performed under air, which demonstrate the air stability of the hybrid catalysts3,35 (see the ESI†).
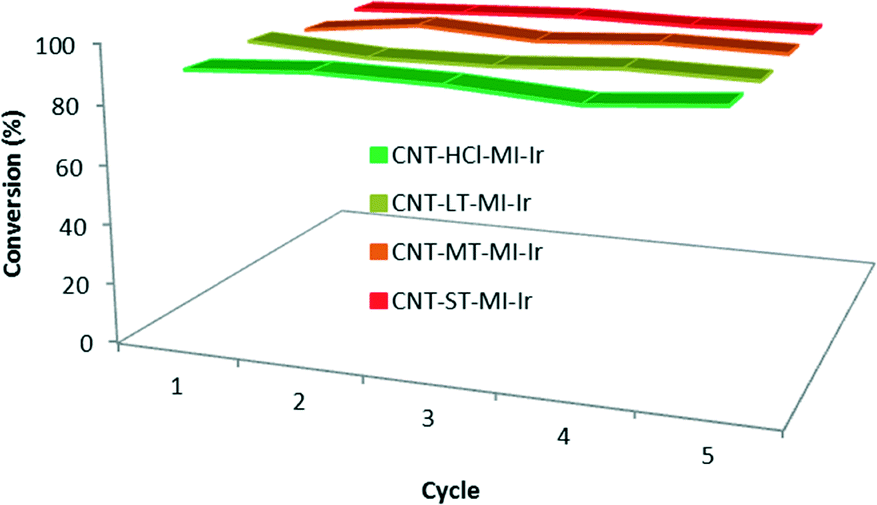 | ||
| Fig. 7 Recyclability of the hybrid catalysts CNT-X-MI-Ir: conversion to cyclohexanol in five catalytic runs with recycling reaction times shown in Table 3. The 5th cycle was performed under air. | ||
Conclusions
Iridium–N-heterocyclic carbene complexes have been supported through the surface carboxylic acid groups of carbon nanotube materials obtained after purification methods of different oxidizing power. The oxidized carbon nanotubes have been functionalized by reaction of the surface acyl chloride groups formed after thionyl chloride treatment with N-methylimidazole, which allow for the covalent anchoring of Ir–NHC complexes via acetyl linkers. The carbon nanotube based iridium–NHC hybrid materials, CNT-X-MI-Ir, were efficient catalysts for the reduction of cyclohexanone by transfer hydrogenation. The most oxidized hybrid catalyst CNT-ST-MI-Ir has shown a superior catalytic performance, observing a gradual increase in the catalytic activity with the oxidation degree of the corresponding parent carbon nanotubes. The supported catalysts can be recycled through successive catalytic runs without any loss of activity.In sharp contrast with the behaviour exhibited by a related N-acyl-substituted Ir–NHC molecular complex, the catalytic activity exhibited by the iridium hybrid catalysts suggests that the acetyl linkers in these materials do not undergo methanolysis under hydrogen transfer conditions which could be a consequence of the protection and stabilizing effect exerted by the nanocarbon walls of the support.
The local structure of the iridium atoms in the hybrid catalyst determined by EXAFS has shown a common local environment for all of them implying a coordination iridium sphere formed by two olefin bonds of the cod ligand, the carbon atom of the NHC carbene ligand, and an O atom from the oxidized carbon matrix as a result of the iridium–support interaction after ionization of the chlorido ligand.
The covalent functionalization through carboxylic acid moieties in the hybrid Ir–NHC catalyst makes possible both a confinement effect due to the porosity of the material and a surface effect based on the potential cooperation of hydroxyl functional groups on the nanotube walls. In this context, it has been found that the catalytic activity is enhanced by the presence of a high concentration of oxygen functionalities in the periphery of the active centers predominantly located at the structural defects including holes or opened tips.
Acknowledgements
The authors thank the Spanish Ministry of Economy and Competitiveness (MINECO/FEDER) (Projects Consolider Ingenio 2010 CSD2009-00050 and CTQ2013-42532-P) and the Diputación General de Aragón (FSE-E07 and FSE-E69) for their financial support. Dr. P. A. thanks MINECO for a Ramón y Cajal contract. M. B. acknowledges his fellowship from MECD (AP2010-0025). The authors also thank Dr. Gloria Subías for fruitful discussion on the EXAFS analysis.Notes and references
- (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (b) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS; (c) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS.
- M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, F. J. Modrego, S. Winterle, C. Cunchillos, F. J. Lahoz and L. A. Oro, Organometallics, 2011, 30, 5493–5508 CrossRef.
- (a) M. Blanco, P. Álvarez, C. Blanco, M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, L. A. Oro and R. Menéndez, ACS Catal., 2013, 3, 1307–1317 CrossRef CAS; (b) M. Blanco, P. Álvarez, C. Blanco, M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, L. A. Oro and R. Menéndez, Carbon, 2015, 83, 21–31 CrossRef CAS.
- (a) S. Sabater, J. A. Mata and E. Peris, Organometallics, 2015, 34, 1186–1190 CrossRef CAS; (b) S. Sabater, J. A. Mata and E. Peris, ACS Catal., 2014, 4, 2038–2047 CrossRef CAS; (c) Q. Zhao, Y. Li, R. Liu, A. Chen, G. Zhang, F. Zhang and X. Fan, J. Mater. Chem. A, 2013, 1, 15039–15045 RSC; (d) Q. Zhao, C. Bai, W. Zhang, Y. Li, G. Zhang, F. Zhang and X. Fan, Ind. Eng. Chem. Res., 2014, 53, 4232–4238 CrossRef CAS; (e) G. Liu, B. Wu, J. Zhang, X. Wang, M. Shao and J. Wang, Inorg. Chem., 2009, 48, 2383–2390 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- C. T. White and T. N. Todorov, Nature, 1998, 393, 240–242 CrossRef CAS.
- J. P. Lu, Phys. Rev. Lett., 1997, 79, 1297–1300 CrossRef CAS.
- J. Zhao and R. H. Xie, J. Nanosci. Nanotechnol., 2003, 3, 459–462 CrossRef CAS PubMed.
- M. Wilson, K. Kannagaram, G. Smith and B. Raguse, in Nanotechnology. Basic Science and Emerging Technologies, CRC Press, London, 2002 Search PubMed.
- S. Porro, S. Musso, M. Vinante, L. Vanzetti, M. Anderle, F. Trotta and A. Tagliaferro, Phys. E, 2007, 37, 58–61 CrossRef CAS.
- A. Aqel, K. M. M. A. El-Nour, R. A. A. Ammar and A. Al-Warthan, Arabian J. Chem., 2012, 5, 1–23 CrossRef CAS.
- G. G. Wildgoose, P. Abiman and R. G. Compton, J. Mater. Chem., 2009, 19, 4875–4886 RSC.
- (a) K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS; (b) V. Datsyuk, M. K. K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- J. Zhang, H. Zou, Q. Qing, Y. Yang, Q. Li, Z. Liu, X. Guo and Z. Du, J. Phys. Chem. B, 2003, 107, 3712–3718 CrossRef CAS.
- F. Avilés, J. V. Cauich-Rodríguez, L. Moo-Tah, A. May-Pat and R. Vargas-Coronado, Carbon, 2009, 47, 2970–2976 CrossRef.
- A. G. Osorio, I. C. L. Silveira, V. L. Bueno and C. P. Bergmann, Appl. Surf. Sci., 2008, 255, 2485–2489 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–495 CrossRef CAS PubMed.
- A. C. Hillier, H. M. Lee, E. D. Stevens and S. P. Nolan, Organometallics, 2001, 20, 4246–4252 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- (a) M. R. Axet, O. Dechy-Cabaret, J. Durand, M. Gouygou and P. Serp, Coord. Chem. Rev., 2016, 308, 236–345 CrossRef CAS; (b) I. Romanenko, D. Gajan, R. Sayah, D. Crozet, E. Jeanneau, C. Lucas, L. Leroux, L. Veyre, A. Lesage, L. Emsley, E. Lacôte and C. Thieuleuxet, Angew. Chem., Int. Ed., 2015, 54, 12937–12941 CrossRef CAS PubMed; (c) Y. Yan, J. Miao, Z. Yang, F.-X. Xiao, H. B. Yang, B. Liu and Y. Yang, Chem. Soc. Rev., 2015, 44, 3295–3346 RSC; (d) L. Zhang, E. Castillejos, P. Serp, W. Sun and J. Durand, Catal. Today, 2014, 235, 33–40 CrossRef CAS; (e) A. Schaetz, M. Zeltner and W. J. Stark, ACS Catal., 2012, 2, 1267–1284 CrossRef CAS.
- (a) G. E. Dobereiner, C. A. Chamberlin, N. D. Schley and R. H. Crabtree, Organometallics, 2010, 29, 5728–5731 CrossRef CAS; (b) B. Bovio, A. Burini and B. R. Pietroni, J. Organomet. Chem., 1993, 452, 287–291 CrossRef CAS; (c) M. Jonek, A. Makhloufi, P. Rech, W. Frank and C. Ganter, J. Organomet. Chem., 2014, 750, 140–149 CrossRef CAS; (d) R. A. Batey, M. Shen and A. J. Lough, Org. Lett., 2002, 4, 1411–1414 CrossRef CAS PubMed.
- R. Usón, L. A. Oro and J. A. Cabeza, Inorg. Synth., 1985, 23, 126–127 CrossRef.
- P. M. A. Sherwood, in Practical Surface Analysis in Auger and X-ray Photoelectron Spectroscopy, ed. D. Briggs and M. P. Seah, Wiley, New York, 1990, vol. 1, p. 574 Search PubMed.
- D. Elgrabli, M. Floriani, S. Abella-Gallart, L. Meunier, C. Gamez, P. Delalain, F. Rogerieux, J. Boczkowski and G. Lacroix, Part. Fibre Toxicol., 2008, 5, 20–33 CrossRef PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- M. Blanco, P. Álvarez, C. Blanco, N. Campos, D. Gómez and R. Menéndez, Diamond Relat. Mater., 2013, 37, 1–7 CrossRef CAS.
- Y. C. Chiang, W. H. Lin and Y. C. Chang, Appl. Surf. Sci., 2011, 257, 2401–2410 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47–99 CrossRef.
- M. J. Park, J. K. Lee, B. S. Lee, Y. W. Lee, I. S. Choi and S. Lee, Chem. Mater., 2006, 18, 1546–1551 CrossRef CAS.
- C. Crotti, E. Farnetti, S. Filipuzzi, M. Stener, E. Zangrando and P. Moras, Dalton Trans., 2007, 133–142 RSC.
- J. Lu, P. Serna, C. Aydin, N. D. Browning and B. C. Gates, J. Am. Chem. Soc., 2011, 133, 16186–16195 CrossRef CAS PubMed.
- D. G. Rees and P. J. Halling, Enzyme Microb. Technol., 2000, 27, 549–559 CrossRef CAS PubMed.
- J. Blasco, V. Cuartero, G. Subías, M. V. Jiménez, J. J. Pérez-Torrente, L. A. Oro, M. Blanco, P. Álvarez, C. Blanco and R. Menéndez, J. Phys.: Conf. Ser., 2016 Search PubMed , accepted.
- M. Blanco, P. Álvarez, C. Blanco, M. V. Jiménez, J. Fernández -Tornos, J. J. Pérez-Torrente, J. Blasco, G. Subías, V. Cuartero, L. A. Oro and R. Menéndez, Carbon, 2016, 96, 66–74 CrossRef CAS.
- F. Su, C. Lu and S. Hu, Colloids Surf., A, 2010, 353, 83–91 CrossRef CAS.
- F. Su, C. Lu and S. Hu, Appl. Surf. Sci., 2008, 254, 7035–7041 CrossRef.
- X. Pan and X. Bao, Acc. Chem. Res., 2011, 44, 553–562 CrossRef CAS PubMed.
- R. M. Malek Abbaslou, J. Soltan and A. K. Dalai, Appl. Catal., A, 2010, 379, 129–134 CrossRef.
- P. Serp and E. Castillejos, ChemCatChem, 2010, 2, 41–47 CrossRef CAS.
- C. Hammond, M. T. Schümperli, S. Conrad and I. Hermans, ChemCatChem, 2013, 5, 2983–2990 CrossRef CAS.
- E. Prouzet, J. Phys.: Condens. Matter, 1995, 7, 8027–8033 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cy01998b |
| This journal is © The Royal Society of Chemistry 2016 |