
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and solid state structure of a metalloid tin cluster [Sn10(trip8)]†
J.
Wiederkehr
,
C.
Wölper
and
S.
Schulz
 *
*
Faculty of Chemistry, Inorganic Chemistry, University of Duisburg-Essen, Universitätsstr. 5-7, S07S03C30, 45114 Essen, Germany. E-mail: stephan.schulz@uni-due.de; Fax: +49-201-1833830; Tel: +49-201-1834635
First published on 25th August 2016
Abstract
(trip2Sn)2 (trip = 2,4,6-i-Pr3C6H2) reacts with Mg(I) reductants (LMg)2 (L = HC[C(Me)N(dipp)]2) and (L′Mg)2 (L′ = HC[C(Me)N(mes)]2) with Sn–C bond cleavage and formation of the novel metalloid tin cluster Sn10trip8 1 or elemental tin. 1, which contains Sn atoms in the formal oxidation states 0, +I and +II, and the side products LMgtrip (2) and L′Mgtrip (3) were characterized spectroscopically and by single crystal X-ray diffraction.
Metal clusters have been investigated for decades since the understanding of the nature of metal–metal bonds is of fundamental interest in chemistry as well as due to the promising technical applications of nanometer sized metal clusters.1 Transition metals are particularly suited for the synthesis of clusters due to the presence of (n + 1)s, (n + 1)p and nd orbitals, but main group metal clusters have also received growing interest and their number steadily increased within the last decades.2 The development of the co-condensation technique by the Schnöckel group resulted in the synthesis of unforeseen main group metal clusters including so called metalloid clusters, in which the number of metal atoms exceeds the number of ligands.3 These clusters contain “naked” metal atoms which are only bonded to other metal atoms and hence adopt the formal oxidation state 0. Even though the topology of the metal atoms in bulk metals and in metalloid clusters differs, metalloid clusters can be considered as intermediates of the bulk metal formation.4
Group 14 (metalloid) clusters have been intensely studied in recent years. They were typically synthesized by reduction reaction of metal complexes RMX3 (R = organic substituent, X = halide) as was shown for instance for octasilacubanes [RSi]8,5 or starting from metastable metal(I) halide solutions as was demonstrated by Schnepf et al. for metalloid germanium and tin clusters including Ge18[Si(SiMe3)3]6, the biggest group 14 metal cluster structurally characterized, to date.4,6–8 The vast majority of clusters of the heavier group 14 elements fall into three categories: ligand-free Zintl anions [Ex]y− (y = 2, 3 or 4, I),9 from which E52− or E9n− (n = 2, 3, 4) are well known, ligand-stabilized neutral clusters ExRx (II) and metalloid clusters of the general type ExRy (x > y, III).10 Moreover, Zintl-type anions such as E94− ions were exo-functionalized with alkyl groups or post-transition metals and the resulting [RGe9]3−, [R2Ge9]2−, [RGe9Ge9R]4−, and [RSn9]3− clusters represent a link between “metalloid” clusters and traditional Zintl ions.11Scheme 1 shows selected tin clusters of each category.
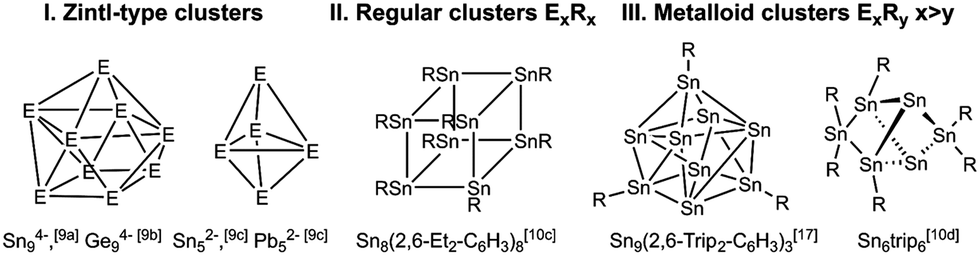 | ||
| Scheme 1 Different types of group 14 cluster compounds. | ||
Metalloid tin clusters SnmRn (m > n) are accessible by controlled disproportionation reaction of monovalent tin halides SnX (X = Cl, Br),4a whose synthesis is generally accompanied by the formation of small amounts of SnX2, in the presence of sterically demanding organic ligands. However, despite the fascinating results, these reactions are often hard to control. For instance, the reaction of Sn(I)Br with LiR′ (R′ = Si(SiMe3)3) under slightly different conditions yielded SnR′3−, Sn3R′4, [Sn4SiR′4(SiMe3)2], Sn9R′22−, Sn10R′6 and Sn10R′42−, respectively.8c,12–16 Another synthetic route is the reductive coupling of RSnCl or RSnCl3 by strong reductants such as alkali metals, KC8, or NaC10H8, i.e. the reaction of [Sn[N(2,6-i-Pr2-C6H3)(SiMe3)](μ-Cl)2] with KC8 yielded a Sn15 cluster.17 Fischer et al. showed that the reaction of SnCl2 with LGa (L = HC[C(Me)N(dipp)]2; dipp = 2,6-i-Pr2-C6H3), a redox-active subvalent Ga(I) species with additional σ-donor properties,18 yielded metalloid clusters [{(L)ClGa}2Sn7] and [{(L)ClGa}4Sn17], to date the largest metalloid tin cluster.19 Unfortunately, the cluster yields are typically low due to the often preferred formation of elemental tin and the reactions can be hardly monitored by in situ NMR spectroscopy due to the poor solubility of the starting reagents in organic solvents.
Herein we introduce organodistannenes R2Sn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SnR2, in which the tin atoms adopt the formal oxidation state +II, as promising starting reagents for the synthesis of metalloid Sn clusters in reactions with strong Mg(I) reductants. Sn10trip81 was obtained from the reaction of (trip2Sn)2 (trip = 2,4,6-i-Pr3C6H2) with (LMg)2 at 95 °C in toluene as dark red crystals after workup. In contrast, the reaction of the somewhat stronger Mg(I) reducing agent (L′Mg)2 (L′ = HC[C(Me)N(mes)]2, mes = 2,4,6-Me3-C6H2) only yielded elemental tin. The reactions proceeded with Sn–C bond breakage and subsequent elimination of HC[C(Me)N(2,6-i-Pr2-C6H3)]2Mg-2,4,6-i-Pr3C6H2 (LMgtrip 2) or HC[C(Me)N(2,4,6-Me3-C6H2)]2Mg-2,4,6-i-Pr3C6H2 (L′Mgtrip 3). 1–3 were characterized by heteronuclear NMR and IR spectroscopy and X-ray crystallography (Scheme 2).
SnR2, in which the tin atoms adopt the formal oxidation state +II, as promising starting reagents for the synthesis of metalloid Sn clusters in reactions with strong Mg(I) reductants. Sn10trip81 was obtained from the reaction of (trip2Sn)2 (trip = 2,4,6-i-Pr3C6H2) with (LMg)2 at 95 °C in toluene as dark red crystals after workup. In contrast, the reaction of the somewhat stronger Mg(I) reducing agent (L′Mg)2 (L′ = HC[C(Me)N(mes)]2, mes = 2,4,6-Me3-C6H2) only yielded elemental tin. The reactions proceeded with Sn–C bond breakage and subsequent elimination of HC[C(Me)N(2,6-i-Pr2-C6H3)]2Mg-2,4,6-i-Pr3C6H2 (LMgtrip 2) or HC[C(Me)N(2,4,6-Me3-C6H2)]2Mg-2,4,6-i-Pr3C6H2 (L′Mgtrip 3). 1–3 were characterized by heteronuclear NMR and IR spectroscopy and X-ray crystallography (Scheme 2).
 | ||
| Scheme 2 Reduction reaction of trip2Sn = Sntrip2 with (LMg)2 yielding the Sn10 cluster 1. | ||
In accordance with the red to purple colour of 1, the UV/VIS spectrum shows an absorption band at about 540 nm (Fig. S9, ESI†). The 119Sn NMR spectrum (Fig S3, ESI†) of 1 shows three resonances at 134.7, 236.7 and 358.9 ppm, pointing to three magnetically inequivalent Sn atoms. The resonances are shifted to lower field compared to that of (trip2Sn)2 (427 ppm). 119Sn NMR resonances are typically not observed for metalloid Sn clusters. Only Power et al. reported on 119Sn NMR values in the clusters Sn7(2,6-dipp2-C6H3)2 (419.5, 529.7 ppm)10e and Sn8(2,6-mes2-C6H3)4 (483.1, 751.7 ppm),10a which are shifted to higher field compared to those observed for 1. The 1H NMR spectrum of 1 is rather complex, showing six different resonances for the methine proton of the i-Pr groups and several doublets for the Me groups (Fig. S1, ESI†). Temperature-dependent 1H NMR spectra (Fig. S7 and S8, ESI†) in the range from −80 to +110 °C point to dynamic behaviour of 1 in solution, but an assignment of the resonances is not possible. The reaction of trip2SnSntrip2 with (LMg)2 was further investigated by in situ1H NMR spectroscopy. No reaction was observed at ambient temperature even after long reaction times (1d), while the reaction slowly proceeded at 95 °C. Resonances of LMg, 1 and 2 were observed after 6 h (Fig. S5, ESI†), while the reaction was finished after 24 h (Fig. S6, ESI†).
Single crystals of 1–3 were obtained from solutions in n-hexane (1, Fig. 1) and pentane (2, 3) upon storage at −30 °C for 1 d (1, 2) and 6 d (3).20 The Sn10 core in 1 contains four “naked” Sn(0) atoms (Sn4, Sn5 and equivalents #1: −x, −y + 1, −z), which form a rhombus structure. This core is capped by a chain of three Sn atoms on each side. Alternatively, the structure of 1 can be described as four edge-sharing five membered rings or as two clamped butterfly-type four-membered rings. Two Sn atoms (Sn2, Sn2#1) carry two ligands (L), hence adopting the formal oxidation state +II, while the remaining four Sn atoms (Sn1, Sn1#1, Sn3, Sn3#1) only carry one ligand and can thus be regarded as Sn(I) atoms.
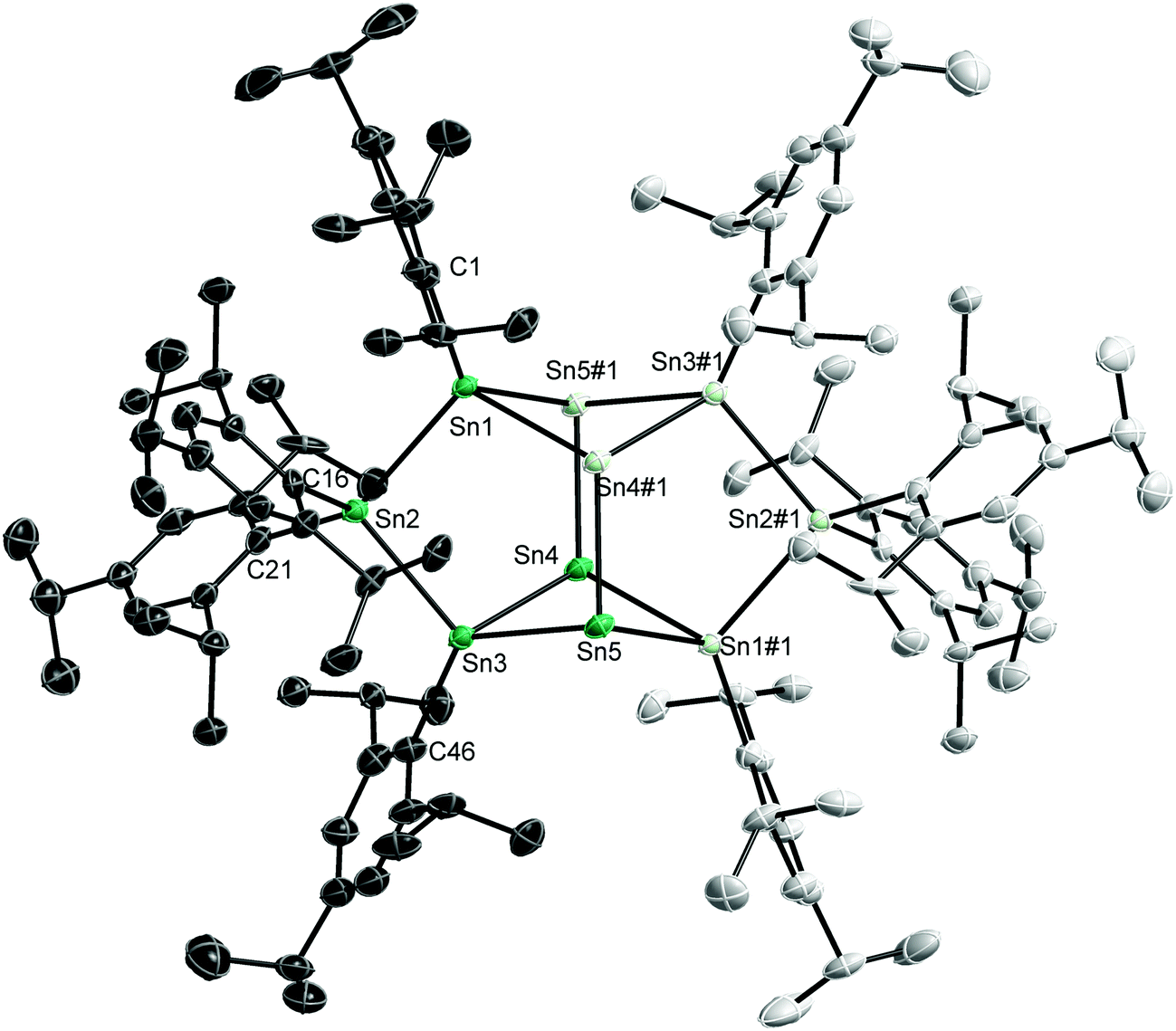 | ||
| Fig. 1 Solid state structure of 1; non-H-atoms are shown as thermal ellipsoids at 50% probability levels. H atoms and solvent molecules are omitted for clarity. Light coloured atoms are symmetry equivalent (#1: −x, −y + 1, −z). Only the ipso C atoms of the trip ligands are labelled. For other atom names please refer to the cif-file. Selected bond lengths [Å] and angles [°]: Sn(1)–Sn(2) 2.8579(9), Sn(2)–Sn(3) 2.8578(8), Sn(3)–Sn(4) 2.8069(9), Sn(3)–Sn(5) 2.8854(9), Sn(4)–Sn(1)#1 2.8243(9), Sn(4)–Sn(5) 3.3544(9), Sn(4)–Sn(4)#1 3.2637(12), Sn(4)–Sn(5)#1 2.8756(8), Sn(5)–Sn(1)#1 2.8883(9), Sn(5)–Sn(5)#1 5.3283(11), Sn(1)–C(1) 2.208(8), Sn(2)–C(31) 2.199(8), Sn(2)–C(16) 2.199(8), Sn(3)–C(46) 2.192(8); Sn(4)#1–Sn(1)–Sn(2) 99.15(2), Sn(4)#1–Sn(1)–Sn(5)#1 71.91(2), Sn(2)–Sn(1)–Sn(5)#1 114.86(3), Sn(3)–Sn(2)–Sn(1) 98.03(2), Sn(4)–Sn(3)–Sn(2) 101.40(2), Sn(4)–Sn(3)–Sn(5) 72.20(2), Sn(2)–Sn(3)–Sn(5) 114.56(3), Sn(3)–Sn(4)–Sn(1)#1 100.43(3), Sn(3)–Sn(4)–Sn(5)#1 120.17(3), Sn(1)#1–Sn(4)–Sn(5)#1 121.80(3), Sn(3)–Sn(4)–Sn(4)#1 81.55(3), Sn(1)#1–Sn(4)–Sn(4)#1 82.75(2), Sn(5)#1–Sn(4)–Sn(4)#1 65.89(2), Sn(4)#1–Sn(5)–Sn(3) 87.39(2), Sn(4)#1–Sn(5)–Sn(1)#1 88.93(2), Sn(3)–Sn(5)–Sn(1)#1 97.09(3). | ||
The structure of the Sn10 core of 1 differs from those reported for Sn10 clusters, i.e. [Sn10R′4]2−,6 [Sn10SiR′4(SiMe3)2]2−,14c [Sn10R′5]−,14b Sn10R′6 (R′ = Si(SiMe3)3),8c which adopt distorted centaur polyhedral arrangements with a cubic side and an icosahedral side in accordance with analogous Ge10 and Pb10 clusters such as (Na6[Ge10Fe(CO4)8]18THF)8b and (Pb10[Si(SiMe3)3]6).21 The structure of 1 also differs from that reported for the largest structurally characterized tin cluster, Sn15[N(2,6-i-Pr2-C6H3)(SiMe3)]6, which contains a body-centred arrangement of the 15 tin atoms.17 However, edge-sharing five membered rings and butterfly-type four membered rings were previously observed in metalloid germanium clusters such as Ge18R67 and Ge14R53−.8a,d
The Sn–Sn bond lengths within the distorted butterfly-type ring of 1 clearly differ. The Sn3–Sn4 (2.8069(9) Å) and Sn(4)–Sn(1)#1 bonds (2.8243(9) Å) are the shortest Sn–Sn bonds observed in 1, while the other two (Sn(3)–Sn(5) 2.8854(9), Sn1–Sn5#1 2.8884(9) Å) are the longest ones. The Sn–Sn bonds between the Sn(II) and Sn(I) atoms (Sn(1)–Sn(2) 2.8579(9), Sn(2)–Sn(3) 2.8578(8) Å) fall in between. The trans-annular Sn4–Sn5 bond (3.3544(9) Å) within the butterfly-type ring as well as the Sn(4)–Sn(4)#1 bond between these rings (3.2637(12) Å) clearly exceed the sum of the covalent radii (2.80 Å)22 but are shorter than the sum of the van-der Waals radii (4.34 Å).23 In contrast, the Sn(5)–Sn(5)#1 distance (5.3283(13) Å) is far too long to be considered as attractive interaction. The Sn–Sn bond lengths in Sn10 centaur polyhedra typically range from 2.90 to 2.95 Å for the cubic part and 2.95 to 3.05 Å for the icosahedral part,6,8c,14b,c while Sn–Sn bond distances between 2.7992(4) and 3.5729(4) Å were reported for the related Sn6trip6 cluster.10d Comparable Sn–Sn bond lengths were observed in other Sn8, Sn9, Sn15 and Sn17 clusters.10a,15,17,19 The interatomic distances in metallic tin are 3.022 and 3.181 Å.24
Mg(I) compounds are promising reductants for the synthesis of metalloid Sn clusters. They readily react with trip2SnSntrip2, which contain strong tin-carbon bonds, clearly underlining the strong reducing potential of Mg(I) compounds, which can be further modified by use of different substituents (L, L′). The mes-substituted Mg(I) compound (L′Mg)2 shows a somewhat higher reducing potential compared to the dipp-substituted derivative (LMg)2. In contrast, the gallanediyl LGa, which reacts with SnCl2 with formation of tin clusters,19 failed to react with trip2SnSntrip2, clearly proving its weaker reducing properties compared to Mg(I) reagents. Since the distannene and the Mg(I) compounds are both soluble in common organic solvents, a homogeneous reaction route is possible, which provides a better reaction control since the formation of elemental tin can easily be observed. In addition, the reaction can be monitored by in situ NMR spectroscopy, which may further help to identify the reaction pathway. We are currently investigating the reduction potential of other soluble reductants toward organotin compounds.
J. Wiederkehr acknowledges financial support (Kekulé-Scholarship) by the Fonds der Chemischen Industrie (FCI) and S. Schulz acknowledges financial support by the University of Duisburg-Essen.
Notes and references
- G. Schmid, M. Baumle, M. Geerkens, I. Heim, C. Osemann and T. Sawitowski, Chem. Soc. Rev., 1999, 28, 179 RSC; Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594 RSC.
- S. T. Liddle, Molecular Metal Metal Bonds, Wiley VCH, Weinheim, 2015 Search PubMed.
- A. Schnepf and H. Schnöckel, Angew. Chem., 2002, 114, 3682 ( Angew. Chem., Int. Ed. , 2002 , 41 , 3532 ) CrossRef; H. Schnöckel, Dalton Trans., 2008, 4344 RSC; H. Schnöckel, Chem. Rev., 2010, 110, 4125 CrossRef PubMed; H. Schnöckel, A. Schnepf, R. L. Whetten, C. Schenk and P. Henke, Z. Anorg. Allg. Chem., 2011, 637, 15 CrossRef.
- (a) A. Schnepf, Chem. Soc. Rev., 2007, 36, 745 RSC; (b) A. Schnepf, New J. Chem., 2010, 34, 2079 RSC.
- H. Matsumoto, K. Higuchi, Y. Hoshino, H. Koike, Y. Naoi and Y. Nagai, Chem. Commun., 1988, 1083 RSC; H. Matsumoto, K. Higuchi, S. Kyushin and M. Goto, Angew. Chem., 1992, 104, 1410 ( Angew. Chem., Int. Ed. , 1992 , 31 , 1354 ) CrossRef CAS.
- C. Schrenk, F. Winter, R. Pöttgen and A. Schnepf, Chem. – Eur. J., 2015, 21, 2992 CrossRef CAS PubMed.
- O. Kysliak, C. Schrenk and A. Schnepf, Angew. Chem., 2016, 128, 3270 ( Angew. Chem., Int. Ed. , 2016 , 55 , 3216 ) CrossRef.
- (a) C. Schenk and A. Schnepf, Chem. Commun., 2008, 4643 RSC; (b) A. Schnepf and C. Schenk, Angew. Chem., 2006, 118, 5499 ( Angew. Chem., Int. Ed. , 2006 , 45 , 5373 ) CrossRef; (c) C. Schrenk, I. Schellenberg, R. Pöttgen and A. Schnepf, Dalton Trans., 2010, 39, 1872 RSC; (d) C. Schenk, A. Kracke, K. Fink, A. Kubas, W. Klopper, M. Neumaier, H. Schnöckel and A. Schnepf, J. Am. Chem. Soc., 2011, 133, 2518 CrossRef CAS PubMed.
- J. D. Corbett and P. A. Edwards, J. Am. Chem. Soc., 1977, 99, 3313 CrossRef CAS; L. Xu and S. C. Sevov, J. Am. Chem. Soc., 1999, 121, 9245 CrossRef; J. D. Corbett, Angew. Chem., 2000, 112, 682 ( Angew. Chem., Int. Ed. , 2000 , 39 , 670 ) CrossRef; S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., 2011, 123, 3712 ( Angew. Chem., Int. Ed. , 2011 , 50 , 3630 ) CrossRef.
- (a) B. E. Eichler and P. P. Power, Angew. Chem., 2001, 113, 818 ( Angew. Chem., Int. Ed. , 2001 , 40 , 796 ) CrossRef; (b) A. F. Richards, B. E. Eichler, M. Brynda, M. M. Olmstead and P. P. Power, Angew. Chem., 2005, 117, 2602 ( Angew. Chem., Int. Ed. , 2005 , 44 , 2546 ) CrossRef; (c) L. R. Sita, Acc. Chem. Res., 1994, 27, 191 CrossRef CAS; (d) C. P. Sindlinger and L. Wesemann, Chem. Sci., 2014, 5, 2739 RSC; (e) E. Rivard, J. Steiner, J. C. Fettinger, J. R. Giuliani, M. P. Augustine and P. P. Power, Chem. Commun., 2007, 4919 RSC.
- D. J. Chapman and S. C. Sevov, Inorg. Chem., 2008, 47, 6009 CrossRef CAS PubMed; M. W. Hull and S. C. Sevov, J. Am. Chem. Soc., 2009, 131, 9026 CrossRef PubMed; F. S. Kocak, P. Y. Zavalij, Y. F. Lam and B. W. Eichhorn, Chem. Commun., 2009, 4197 RSC; F. S. Kocak, P. Zavalij and B. Eichhorn, Chem. – Eur. J., 2011, 17, 4858 CrossRef PubMed.
- C. Schrenk and A. Schnepf, Chem. Commun., 2010, 46, 6756–6758 RSC.
- C. Schrenk, A. Kubas, K. Fink and A. Schnepf, Angew. Chem., 2011, 123, 7411 ( Angew. Chem., Int. Ed. , 2011 , 50 , 7273 ) CrossRef.
- (a) C. Schrenk, M. Neumaier and A. Schnepf, Inorg. Chem., 2012, 51, 3989 CrossRef CAS PubMed; (b) C. Schrenk, J. Helmlinger and A. Schnepf, Z. Anorg. Allg. Chem., 2012, 638, 589 CrossRef CAS; (c) C. Schrenk and A. Schnepf, Main Group Met. Chem., 2013, 36, 161 CrossRef CAS.
- C. Schrenk, F. Winter, R. Pöttgen and A. Schnepf, Inorg. Chem., 2012, 51, 8583 CrossRef CAS PubMed.
- C. Schrenk, B. Gerke, R. Pöttgen, A. Clayborne and A. Schnepf, Chem. – Eur. J., 2015, 21, 8222 CrossRef CAS PubMed; A. Schnepf, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 662 CrossRef.
- M. Brynda, R. Herber, P. B. Hitchcock, M. F. Lappert, I. Nowik, P. P. Power, A. V. Protchenko, A. Ruzicka and J. Steiner, Angew. Chem., 2006, 118, 4439 ( Angew. Chem., Int. Ed. , 2006 , 45 , 4333 ) CrossRef.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991 RSC; N. J. Hardman and P. P. Power, ACS Symp. Ser., 2002, 822, 2 CrossRef CAS; C.-H. Chen, M.-L. Tsai and M.-D. Su, Organometallics, 2006, 25, 2766 CrossRef; M. S. Hill, P. B. Hitchcock and R. Pontavornpinyo, Dalton Trans., 2005, 273 RSC; N. J. Hardman, P. P. Power, J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, J. Chem. Soc., Chem. Commun., 2001, 1866 RSC; C. Gemel, T. Steinke, M. Cokoja, A. Kempter and R. A. Fischer, Eur. J. Inorg. Chem., 2004, 4161 CrossRef.
- G. Prabusankar, A. Kempter, C. Gemel, M.-K. Schröter and R. A. Fischer, Angew. Chem., 2008, 120, 7344 ( Angew. Chem., Int. Ed. , 2008 , 47 , 7234 ) CrossRef.
- Structural details are given in the ESI†.
- S. Yao, K. W. Klinkhammer and Y. Xiong, Angew. Chem., 2004, 116, 6328 ( Angew. Chem., Int. Ed. , 2004 , 43 , 6202 ) CrossRef.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186 CrossRef PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806 CrossRef CAS PubMed.
- J. A. Lee and G. V. Raynor, Proc. Phys. Soc., London, Sect. B, 1954, 67, 737 CrossRef , ICSD entry 40038.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization of 1–3. CCDC 1480548 (1), 1499559 (2) and 1489510 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc06770k |
| This journal is © The Royal Society of Chemistry 2016 |