
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional group migrations between boron and metal centres within transition metal–borane and –boryl complexes and cleavage of H–H, E–H and E–E′ bonds
Gareth R.
Owen
School of Applied Science, University of South Wales, Upper Glyntaff, Pontypridd, CF37 4AT, UK. E-mail: gareth.owen@southwales.ac.uk; Tel: +44 1443 65 4527
First published on 19th July 2016
Abstract
This feature article examines some of the recent advances in the chemistry of Z-type transition metal–borane and X-type transition metal–boryl complexes. It focuses on the employment of these boron-based functionalities acting as stores and transfer agents for functional groups such as hydrides, alkyl groups and aryl groups which can either be abstracted or delivered to the metal centre. The review also explores the rather novel reactivity involving the cleavage of H–H, E–H and E–E′ bonds (where E and E′ are a range of groups) across the transition metal–boron bond in such complexes. It explores the early examples of the addition of H–H across transition metal–borane bonds and describes the new transformation in the context of other known modes of hydrogen activation including classic oxidative addition and heterolytic cleavage at transition metal centres as well as Frustrated Lewis Pair chemistry. Similar reactivity involving transition metal–boryl complexes are also described particularly those which undergo both boryl-to-borane and borane-to-borohydride transformations. The delivery of hydride to the metal centre in combination with the potential to regenerate the borohydride functional group via a recharging process is explored in the context of providing a new strategy for catalysis. Finally, a light-hearted look at the analogy of the ‘stinging processes’ involving Trofimenko type ligands is taken one step further to determine whether it is indeed in the nature of scorpionate ligands to repeatedly ‘sting’ just as the real life scorpions do.
 Gareth R. Owen | Gareth Owen received his PhD from Imperial College London in 2003. He subsequently worked in the research group of Professor John A. Gladysz, first as a postdoctoral researcher and later as an Alexander von Humboldt Research Fellow. Dr Owen returned to the UK following the award of a Centenary Ramsay Memorial Research Fellowship which was hosted at University of Bristol. He was later awarded a Royal Society Dorothy Hodgkin Research Fellow again at Bristol. Dr Owen is currently working as Senior Lecturer in Inorganic Chemistry at University of South Wales. His main research interests lie in the chemistry of boron based ligands which act as reversible hydrogen atom stores and the investigation of hydrogen shuttle-type transformations for the development of novel transformations. |
Introduction
A bee sting is an effective tool for delivering venom to potential threats to the hive.1 While it is often a misconception that bees can only sting once, their fate is often quite unfortunate when the skin of their victim is too thick (for example in mammals) and the demise of these important creatures ensues. Scorpions on the other hand are much more deadly creatures, with a much more powerful sting (sometimes fatal to humans), they are certainly able to continually deliver stings to many a victim.2 In 1966, Trofimenko coined the term “scorpionates” to represent tripodal ligand systems which bind tightly to transition metal centres.3 Originally, the term was used to depict polypyrazolylborate ligands, however this generally accepted analogy is now used to describe a plethora of multidentate ligand systems which bind to transition metals centres like the claws of a scorpion.4 At the time, even Trofimenko himself could not have envisaged that scorpionate ligands would share a more pertinent characteristic with these arthropods. This review outlines some of these analogous characteristics, more specifically, the scorpionate ligand “stinging the metal centre” and a means of “recharging the sting” so that the scorpionate is able to repeatedly sting.The activation of the sigma bond in H2 is a fundamental process which impacts across the whole field of chemistry. Not only does it serve as an important transformation in its own right, it acts as a model for the activation of other two-centre-two-electron bonds. Oxidative addition is perhaps the first transformation that comes to mind when one thinks about the activation of element–element bonds. The classic transition metal based oxidative addition step is certainly a ubiquitous transformation known to all who have studied this discipline. Oxidative addition has been utilised as a methodology for catalytic hydrogenations for some time now as exemplified by the classic Wilkinson's Catalyst5 where the oxidation state and coordination number of the metal centre increases by two units (Fig. 1, top-left).6
 | ||
| Fig. 1 Various classes of H–H bond activation by transition metals and/or main group species (M – transition metal, LB – Lewis base, LA – Lewis acid). | ||
From the 1980's onwards, the first bifunctional catalysts were developed in which pre-coordinated Lewis basic ligands were found to be intimately involved in the activation of the bond in H2.7 In these systems, the bond is cleaved via a heterolytic mechanism where the metal accepts the “hydridic hydrogen” and the Lewis base component acts as the proton acceptor. Considerable achievements have been made in this area which are still undergoing further substantial development.8
The field of hydrogen activation underwent yet another period of growth with outstanding developments surrounding “Frustrated Lewis Pair” (FLP) activation chemistry.9 The so-called “Frustrated Lewis Pair” activation concept has certainly had a major impact in the field, dramatically altering the perception on the reactivity of main group compounds.10 The headline achievement of FLP chemistry is their ability to carry out transformations only thought possible by transition metals. This includes transformations such as catalytic hydrogenations, small molecule activations and many more.9 FLP concepts have opened significant scope for development in many areas of chemistry. Perhaps it was initially thought that “Lewis pair activations” might serve as an alternative to traditional transition metal based chemistry. However, in the past number of years this standpoint has again shifted. A partnership between the FLP concept and transition metal based reactivity has indeed been demonstrated. This has led to examples of FLP chemistry where the Lewis acidic component is indeed a transition metal centre.11 These are somehow reminiscent of the heterolytic cleavage found in the original bifunctional catalysts without, of course, the element of frustration.
All of these concepts have opened up significant potential particularly in such transformations where the ligand becomes intimately involved within the activation process, so called “ligand cooperation”.12 This is increasingly becoming a popular and highly successful strategy for carrying out new catalytic reactions.12,13 Despite all of the aforementioned achievements, there had not been any focus on heterolytic cleavage involving Lewis acidic (Z-type) ligands until only recently. The principal reason being that the Z-type ligands are still comparatively rare and much less understood than the archetypal L- and X-type ligands.14
The purpose of this feature article is to highlight the new Lewis acid (LA) mediated heterolytic cleavage approach to H2 activation which has been taken by researchers in the field and to draw attention to this transformation as a means of addition across transition metal–borane bonds as a new strategy which has been successfully applied to a number of catalytic systems (Fig. 1, bottom right). In the case of Lewis acid mediated heterolytic cleavage, the distribution of charge during the cleavage is reversed due to the fact that the borane ligand is a Z-type ligand. When the hydrogen species is transferred to the boron, the borane function group becomes a negatively charged borohydride unit. By using Trofimenko's well-known scorpionate analogy, this addition puts the sting back into the scorpionate (vide infra). This review examines these transformations with respect to catalytic potential and somehow demonstrates that it is indeed in the nature of scorpionate ligands to sting again and again.
The “Sting of the Scorpionate” analogy
Trofimenko lived to see that it was indeed in the nature of scorpionate ligands to sting. Originally, the sting was considered as the coordination of the third “arm” of the ligand to a metal centre. However, this perception was altered when it was discovered that the “hydride species” from the borohydride unit could be transferred to the metal centre (i.e. the stinging process). In 1999, Hill reported the formation of the first authenticated transition metal–borane complex15 utilising Reglinski's Tm as ligand precursor (Fig. 2).16 This fascinating new class of compound has generated significant interest in the chemical literature17–22 for two main reasons. Firstly, the metal–borane interaction features a metal-to-boron dative interaction in which the boron functionality acts as a Z-type (σ-acceptor) ligand.23 Secondly, the chemistry demonstrates the potential to utilize the borohydride function as a hydrogen atom store.20,21a,22a,b It is now over a century since the award of the Nobel Prize to Werner for his pioneering work in coordination compounds24 which highlights the significance of these then unknown Z-type ligands. It is over a hundred years since the foundations of coordination chemistry where laid. Hill's seminal work has certainly played its part instigating the current widespread interest in Z-type ligands.15,17–23 This interest along with the emergence of the Frustrated Lewis Pair concept has perhaps sparked developments such as Metal-Only Lewis Pairs (MOLP's).25 The ‘stinging’ process, i.e. the potential to transfer a hydrogen atom from the ligand to the metal centre as shown in Scheme 1, is an interesting process. It was suggested in Hill's original report that this could have potential catalytic applications. Concepts of ‘ligand cooperation’ and ‘borrowing hydrogen’, where a ligand temporarily stores hydrogen atoms, have indeed been successfully developed and applied to a number of catalytic reactions.12,13 Following on from the original sting of the scorpionate article, a significant volume of investigations have been carried out by several distinguished groups in order to try and understand the nature of the sting. This topic has previously been reviewed.21a | ||
| Fig. 2 Trofimenko's ubiquitous trispyrazolylborate ligand (Tp), Reglinski's new generation more flexible scorpionate ligand (Tm) and Hill's metallaboratrane complex derived from Tm (left to right). A two atom bridge is required between the boron and the donor atom for a significant interaction between the metal centre and boron to occur. | ||
 | ||
| Scheme 1 A transition metal centre being ‘stung’ by the scorpionate ligand. Several strategies have been developed which favour hydrogen migration (see ref. 21a for details). | ||
Several strategies to drive hydrogen migration have been achieved. However, at this point the question that remained unanswered was “Do scorpionate ligands share more similarities to the humble bee (i.e. only being able to sting once) or is it possible to administer repeated stings to the metal centre?” It was over a decade after Hill's original report that this question was duly answered by independent work carried out by Owen26 and later by Peters.27 These and subsequent developments are outlined below.
The ligand
There is a constant search for the right combination of metal and ligand to meet the desired properties for specific application. Z-type ligands are a particularly interesting class. A number of Lewis acidic species have now been shown to act as Z-class ligands.28 The most studied functional group is the borane functionality which is focused on within this review. In order to incorporate a borane functional group within a transition metal complex, it needs to be anchored to the metal centre via at least one supporting unit containing an L-type donor group, i.e. κ2-LB (Fig. 3, E = a non-hydrogen substituent at boron). Tethering the borane function utilising an X-type supporting unit is also feasible, however, to date this is unknown to the best of the author's knowledge. Until recently, research encompassing transition metal–borane chemistry interactions was limited to a handful of motifs based on sulfur, nitrogen and phosphine L-type donors (Fig. 4). As outlined below, there has been a significant increase in the number and diversity of such motifs over the past few years. Some of these compounds have demonstrated enhanced catalytic properties as a result of the presence of the borane unit.22a Amongst the most intensively studied ligand systems are those in which the borane is anchored to the metal centre via two supporting groups containing donor functional groups, i.e. κ3-L2B. Those supported by three donor functional groups are also well known (κ4-L3B). The degree of interaction between the transition metal and the borane functional group can vary significantly with transition metal–borane distances found on the Cambridge Structural Database of such complexes ranging from 0.192 Å less than the sum of the covalent radii of transition metal and boron (in a cobalt complex) to 0.463 Å greater than the sum of the covalent radii of transition metal and boron (in a gold complex).29 Furthermore, the nature of the interaction can vary from direct η1-B interaction with the metal centre to η7-BC6 interactions in which the boron atom and all six carbon atoms of an arylborane unit are all interacting with the metal centre.30,31 These η(n+1)-BCn interactions are discussed in more detail below. | ||
| Fig. 3 Generic κ2-LB, κ3-L2B and κ4-L3B coordination modes (top) and the commonly observed interactions of one or more of the carbons on an aryl ring additionally supporting the interaction of the borane unit with the metal centre (bottom). | ||
 | ||
| Fig. 4 Demonstration of the potential fac- and mer-coordination modes of the κ3-L2B ligands (top). Selected ligand components which have been commonly utilised to support transition metal–borane interactions (bottom: the blue atom (E) indicates the atom attached to boron while the red atom indicates the L-donor). | ||
In addition to the varying interaction between the transition metal and the borane unit within these complexes, there is also variation in the specific mode of coordination in the κ3-L2B and κ4-L3B ligands. In the case of the tridentate ligands both fac- and mer-coordination modes have been observed (Fig. 4, top). For those cases considered as mer-coordination modes, the L–M–L angles are typical significantly less than the idealised 180°. The scorpionate descriptor can perhaps be given to the former, however, the latter are better described as pincer type coordination. The difference between the two structural types is related to the potential for the L–M–L bond angles to increase or decrease within the specific ligands.
The significant number of possible variables in terms of flexibility in ligand conformation, resulting coordination geometry at the transition metal centre, nature of the supporting units and degree of metal–boron interaction suggest great potential for tuning these type of complexes for catalytic application. As highlighted in the following sections, this is further enhanced by the fact that along with the transition metal centre, there is potential for reactivity to occur also at the boron centre.
Transfer and exchange of hydride between metal and boron centres
Borohydride to borane and vice versa
Within a few years of the original paper, Hill further demonstrated that it was possible to “tame the stinging process” by providing a means of driving the hydrogen atom between the boron and metal centres (Scheme 2).32 In this case, it was shown that there was a fine balance between the Pt–H and the B–H species, where the latter can be favoured by addition of a strongly donating phosphine ligand. While there have been significant advances in this field, and hydride migration has been observed both from metal-to-boron and from boron-to-metal, this remains the only example in which the migration in both directions has been unambiguously demonstrated within the same system. | ||
| Scheme 2 Reversible transfer of the hydride species from metal-to-boron and from boron-to-metal. | ||
Whilst Hill's example above remains the only one which demonstrates reversibility, several other developments have provided further understanding into the hydrogen migration process. There appears to be several factors which influence whether or not migration of the hydride species from boron occurs.21a These are related to the factors indicated above, i.e. the specific nature of the ligand, the transition metal centre itself, the potential within the coordination sphere to form a transition metal-hydride species (a potential vacant site) or the presence of a co-ligand which has the ability to act as a hydrogen acceptor. Regarding the nature of the ligand, Fig. 4 highlights some of the potential variation of the ligand architecture. The majority of the E–L groups are made up of three-atom bridges allowing the boron centre to form one or more five-membered rings upon interaction with the transition metal centre. Flexibility and potential variation in the L–M–L bond angles is important for effective delivery or abstraction of the hydride to or from the metal centre. This is highlighted in an example reported by Owen in which the hydride species was located at an intermediate point between a ruthenium metal centre and a boron centre (Fig. 5).33 In this case, a pincer type arrangement of the ligand was observed where the N–B–N and S–Ru–S bond angles were 117.4(3)° and 166.13(4)°, respectively.
 | ||
| Fig. 5 A ruthenium complex derived from a scorpionate ligand in which the position of the blue hydrogen atom was located as being at the midpoint between the boron and ruthenium centres. | ||
The nature of the E–L supporting unit was also found to have a particularly important influence on the hydride migration properties of the ligand. This is demonstrated in the different sulfur based ligands shown in Fig. 6.34–37 There are seven examples in which no hydride migration occurs and the non-activated κ3-SSH coordination mode is observed. In the case where the 2-mercaptopyridine supporting unit is used, hydride migration rapidly occurs spontaneously at ambient temperature leading to the rhodium–borane complex (κ3-SSB).38 In this transformation, the former cyclooctadiene ligand (COD) has acted as a hydrogen atom acceptor forming a η3-coordinated cyclooctenyl ring. There are also several related azaindolyl complexes in which the non-activated κ3-NNH coordinated ligand is observed with the same metal and co-ligand.39,40
 | ||
| Fig. 6 Rhodium–cyclooctadiene complexes containing sulfur based scorpionate ligands. Hydride migration is only observed in the one containing the 2-mercaptopyridine heterocycles. | ||
Some insight into the hydride migration process has also been obtained from a computational investigation carried out on a series of iridium complexes containing the anionic ligands, [Tai]− = [HB(7-azaindolyl)3]− and [PhBai]− = [Ph(H)B(7-azaindolyl)2]−.39 In this study, the further reactivity of the complexes, [Ir(κ3-NNH-Tai)(COD)] and [Ir(κ3-NNH-PhBai)(COD)] was explored. As found in the majority of the above cases, spontaneous hydride migration was not observed under ambient conditions. Hydride migration was, however, triggered upon the addition of carbon monoxide to the reaction mixture.39,41 The addition of this strong ligand substituted one of the double bonds of the COD ligand thereby changing its coordination mode from η4 to η2. This change in coordination mode enabled hydride migration to occur. The mechanism involving the transfer of the hydride species from the borohydride functional unit to the iridium centre was modelled computationally. The hydride migration process, involving the reaction steps shown in Fig. 7, was examined. These investigations demonstrated a low energy barrier through the transition state (TS2) for the migration between the boron and iridium centres (ΔG298 = 10.3 kcal mol−1). Furthermore, it was found that there was only a 3.4 kcal mol−1 energy difference between the borohydride species [Ir(κ3-NNH–Tai)(CO)(η2-COD)] (4ii) and the iridium-hydride species [Ir(H)(κ3-NNB-Tai-H)(CO)(η2-COD)] (4iii).
 | ||
| Fig. 7 Computational investigation focusing on the migration of hydrogen from boron-to-iridium centre. Adapted with permission from ref. 39. Copyright 2013 American Chemical Society. | ||
The hydrogen migration approach utilising borohydride (scorpionate-type) ligands highlighted above has featured quite prominently surrounding the synthesis and investigation of metal–borane complexes since they were first reported. An alternative strategy for the synthesis of metal–borane complexes is to directly prepare ligands which already possess the borane functional group. These so-called ambiphilic ligands (since they contain both Lewis Basic and Lewis acidic functional groups) are now well studied. There are a number of different examples of ambiphilic ligands, some of which are described below. By far the most prevalent ligand system which has demonstrated most promise and some truly fascinating reactivity is the one developed by Bourissou.42 This now archetypal ligand motif is comprised of a central triarylborane unit in which two or three of the aryl groups contain a phosphine group in the ortho position (Fig. 8), RDPBR′ and RTPB, respectively. Various derivatives of these ligands have been synthesised where the R-groups at phosphorus have been changed and where the R′ group at boron is an aryl group (e.g. phenyl and mesityl).
 | ||
| Fig. 8 The phosphine–borane ambiphilic ligand motif developed by Bourissou in 2006. The bis-phosphine ligands have been abbreviated as RDPBR′ (where R represents the substituents on the phosphine and the R′ represents the aryl group on boron). The tris-phosphine ligands have been abbreviated as RTPB. | ||
Just like the scorpionate ligands (following a B–H activation step), the bis- and tris-phosphinoborane ligands have been shown to act as “hydride acceptors”. The transfer of hydrogen from metal-to-boron has been demonstrated in a series of studies carried out by Nakazawa and Kameo (Scheme 3).43 For example, the following complexes were prepared, [Ir(H)(κ3-PPB-PhDPBPh)(CO)(PPh3)] and [Rh(κ3-PPH-PhDPBPh + H)(CO)(PPh3)] via addition of the PhDBPPh ligand to the metal precursors [Ir(H)(CO)(PPh3)3] and [Rh(H)(CO)(PPh3)3], respectively. The hydride species remains on the metal in the case of the iridium whilst in the case of the rhodium complex, the hydride was observed to have migrated to the boron centre. Nakazawa and Kameo calculated the energies of the products formed in these reactions along with the corresponding complexes where the position of the hydride was opposed. They found that [Rh(κ3-PPH-PhDPBPh + H)(CO)(PPh3)] was 8.0 kcal mol−1 lower in energy than its hydride migration isomer [Rh(H)(κ3-PPB-PhDPBPh)(CO)(PPh3)] and that [Ir(H)(κ3-PPB-PhDPBPh)(CO)(PPh3)] was 2.4 kcal mol−1 lower in energy than [Ir(κ3-PPH-PhDPBPh + H)(CO)(PPh3)].43b These results further confirm the low energy difference between the B–H and M–H hydride migration isomers in such complexes which has clear implications for their application as potential catalysts.
 | ||
| Scheme 3 Examples showing the transfer of hydrogen from transition metal-to-borane function in a series of studies carried out by Nakazawa and Kameo. | ||
Borane to boryl and vice versa
Along with the transformation between borane (BR3) and borohydride (BR3H) functional groups, the transformation between borane and boryl (BR2) species is also possible. This has been demonstrated in the emergence of a diphosphino-boryl pincer ligand, PBP, which was first reported by Yamashita and Nozaki shown in Scheme 4.44a This example shows the main strategy for installing the PBP pincer ligand via oxidative addition starting from the corresponding borane species, PB(H)P. This ligand has now been utilised in the formation of a wide range of transition metal complexes some of which are outlined below.44,45 | ||
| Scheme 4 The addition of the PB(H)P pro-ligand to iridium. Oxidative addition of the B–H bond led to the generation of a novel PBP boryl pincer ligand. | ||
During their investigations exploring the potential “hydride shuttle” properties and transition metal boron cooperativity, López-Serrano and Rodríguez found that it was possible to carry out the reverse transformation where a hydride species was driven from metal-to-boron centre (Scheme 5).45a They prepared the nickel complex featuring the κ3-PBP pincer ligand and a hydride ligand in the fourth coordination site. Addition of 1,5-cyclooctadiene to the solution resulted in the coordination of the diene ligand and the former nickel-hydride was driven to the boron centre to form the corresponding PB(H)P species. Furthermore, the addition of the diene destabilised the coordination of the newly formed PB(H)P ligand such that only one of the two phosphine donors was coordinated in the observed species. Over time, the free PB(H)P ligand was observed within the reaction along with concomitant formation of [Ni(COD)2]. This transformation perhaps highlights a potential downside of the hydrogen acceptor properties of the boryl and borane functional groups and indicates the importance of anchoring these units to the metal centre via supporting units containing strong donors.
 | ||
| Scheme 5 A means for driving the hydrogen atom from the nickel metal centre to boron thus forming the κ2-PH-PB(H)P ligand. | ||
Transfer and exchange of alkyl and aryl groups between metal and boron centres
A further interesting transformation which has now been observed in a number of cases is the transfer or indeed exchange of alkyl and aryl groups between boron and transition metal centres.46 Early examples of such a transformation were reported by Vedernikov in 2007 (Scheme 6).47a In these transformations a boron-bound methyl group was transferred to the metal centre. The transfer occurred at Pt(IV) centres and was facilitated by addition of alcohols where the alkoxide group was included within the final product by forming a new B–OR bond. This was later developed where the Pt(IV) centres were generated from the Pt(II) via other oxidants.47b,c | ||
| Scheme 6 Transfer of a boron bound methyl group to platinum mediated via oxidation of the platinum centre. | ||
Whilst Vedernikov reported the transfer of methyl groups from boron-to-metal centre, Sadow reported the reverse reactivity. In this case, they reported dialkyl zinc complexes in which one of the alkyl groups was transferred to the boron centre of a neutral ambiphilic ligand, PhB(OxMe2) (Scheme 7).48 Furthermore, if the alkyl group featured a β-hydrogen then a portion of the hydroborate product was formed alongside the alkylborate product as shown in Scheme 7.
 | ||
| Scheme 7 Reactivity involving the transfer of ethyl or hydride fragments from zinc-to-boron reported by Sadow and co-workers. | ||
More recently, Emslie has reported an interesting series of complexes derived from the ambiphilic ligand, TXPB (TXPB = 2,7-di-tert-butyl-5-diphenylboryl-4-diphenylphosphino-9,9-dimethylthioxanthene; as shown in Scheme 8).49 This ligand contains two phenyl groups at the boron centre. Emslie investigated its coordination and subsequent reactivity with [PtMe2(COD)].49a As shown in Scheme 8, the reaction led to the formation of the products resulting from exchange of phenyl and methyl groups between the boron and platinum centres. The exchange of the first methyl group from platinum-to-boron and one phenyl group from boron-to-platinum occurred at ambient temperatures while the second exchange required heating of the samples. Nevertheless it was possible to obtain the product where the two methyl groups formerly located on the platinum centre had exchanged positions with the two phenyl groups formerly located on the boron centre. It was found that the first step in the exchange reaction involved the transfer of the methyl group to boron to form the borate species and this was subsequently followed by transfer of the phenyl group to the metal centre. Evidence for this was obtained via an NMR investigation. Furthermore, it was also possible to trap the borate species by addition of two equivalents of CNXyl (Xyl = 2,6-dimethylphenyl) to form the complex, trans-[PtMe(CNXyl)2(TXPB + Me)] in which the TXPB + Me ligand is coordinated to the platinum centre with a κ1-P coordination mode.
 | ||
| Scheme 8 Exchange of methyl and phenyl groups between platinum and boron centres reported by Emslie. The first step involves the transfer of one of the methyl groups to boron resulting in the formation of a borate species. | ||
A very recent investigation by Ozerov has provided a fascinating insight into the reactivity of transition metal complexes containing arylborane ligands.50 Ozerov investigated the reaction of the iPrDPBPh ligand with the precursor [IrCl(COE)2]2 (where COE = cyclooctene) (Scheme 9). In the case of the reaction involving the iridium complex, which was heated to 100 °C for 5 h, a single product was obtained. This was found to be the product resulting from phenyl migration to the iridium metal centre. Here, the former phenylborane ligand was transformed into an iridium–boryl species, [IrCl(Ph)(iPrDPB)] (where iPrDPB is the iPrDPBPh ligand minus the Ph group on boron). This interesting result led to the reinvestigation of a previous study carried out by Bourissou in which he used the reagents [RhCl(NBD)]2 (NBD = norbornadiene) and iPrDPBPh.42 Reaction of the ligand with an alternative precursor [RhCl(COD)]2, led to a similar mixture of three species to that previously observed, as determined by 31P{1H} NMR spectroscopy. Ozerov assigned these three species as (i) the phenyl migrated X-type boryl complex, (ii) the non-activated Z-type borane complex and (iii) the non-activated Z-type borane complex featuring a ηn+1-BCn interaction involving the phenyl group as shown in Scheme 9. Further reactivity involving these complexes is described below. The ηn+1-BCn interactions of the arylborane units appear to feature prominently in many structures containing the ambiphilic ligands and it is likely that this feature facilitates the migration of the phenyl unit in many of the above examples.
 | ||
| Scheme 9 An investigation into the reactivity of the iPrDPBPh ligand with monovalent iridium and rhodium complexes by Ozerov indicated that it was possible for the phenyl group to migrate from boron-to-rhodium. | ||
Transition metal–borane mediated bond cleavage
Hydrogen–hydrogen and element–hydrogen bond activations
In all of the above reactivity, the transformations originate internally from groups that are attached to either the transition metal or the boron centres. The following sections describe those transformations in which external reactants are activated upon addition to transition metal–borane and –boryl complexes.As highlighted above, significant developments over the past decade have demonstrated that hydride, alkyl and aryl fragments can effectively be stored at boron and/or can potentially be transferred from boron-to-metal centre. Using the analogy described in the introduction, this latter migration would correspond to the sting from the scorpionate ligand. If the delivered fragment is somehow used up by a co-ligand, then is there a means of recharging the boron centre thus providing a potential catalytic strategy? This was indeed demonstrated in 2011 where a metal bound borane functional group was “recharged” to borohydride functional groups via the activation of dihydrogen across a transition metal–borane bond. This development provided a new transformation, namely, the Lewis acid mediated heterolytic cleavage of the sigma bond in H2 as indicated in Scheme 10 (see also Fig. 1). This type of reactivity can also be thought of as Lewis Pair activation where the borane function acts as the Lewis acid component and the transition metal acts as the Lewis base component. This property of the transition metal where it acts as a Lewis base can be considered as atypical. Such developments, of course, have only been possible by the establishment that mid to late transition metals can act as the σ-donating Lewis base component within a coordination complex and undergo σ-acceptor interactions with Lewis acids.15,17–23 This section describes the developments on Lewis acid mediated heterolytic cleavage of H–H as well as the activation of H–E bonds (where E represents any non-hydrogen element or group).
 | ||
| Scheme 10 Lewis base Activation of H2 in which the metal acts as a Lewis base and the ligand acts as a Lewis acid generating new borohydride and metal-hydride functional groups. | ||
Owen and co-workers provided the first example of a Lewis acid mediated activation of H2 at a transition metal centre (Scheme 11).26 In their strategy, the nitrogen based scorpionate ligand containing 7-azaindole heterocycles was utilised. In this system (Scheme 11), the original borohydride hydrogen undergoes an initial sting resulting in the first hydrogen being incorporated into the unsaturated organic ligand, where the former norbornadiene ligand acts as a hydrogen atom acceptor.21 This organic species accepts the hydrogen atom while at the same time undergoes a rearrangement to form the rhodium bound nortricyclyl species.51 The rhodium complex, which at this point contains a strong rhodium–borane interaction [Rh–B = 2.064(4) Å] undergoes reaction with two equivalents of H2 in the presence of a trialkylphosphine. During this reaction, one of the hydrogen atoms is transferred to the nortricyclyl species allowing it to be eliminated from the metal's coordination sphere (via reductive elimination). The three other hydrogen atoms remain on the complex. The transformation leads to a situation where the rhodium–borane bond is broken and one hydrogen atom is located on the boron forming a borohydride functional group. The two other hydrogen atoms are located at the rhodium centre (as a dihydride). This formal addition of H2 across the rhodium–borane bond ‘charges’ the borane functional group back to borohydride. This recharging process provides a methodology for regenerating the sting in the scorpionate ligand and was therefore suggestive that this could have catalytic application. Indeed the complexes were found to be active catalysts for the hydrogenation of olefins under low pressures of H2 (2.5 bar) and low catalytic loadings (0.1 mol%).26
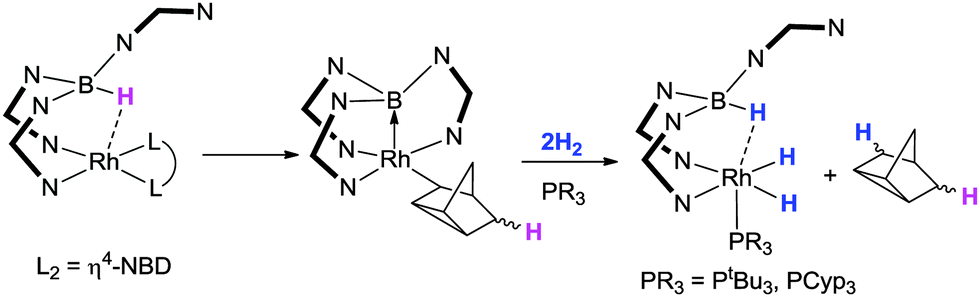 | ||
| Scheme 11 An initial transfer of hydrogen from boron followed by a rhodium–borane Lewis pair activation of H2 reported by Owen and co-workers in 2011 (N–N = 7-azaindolyl). | ||
This initial investigation by Owen was subsequently advanced into an expansive area of research most exemplified by the pioneering work emanating from the Peters' research group. They provided a unique perspective on heterolytic cleavage of hydrogen by utilising the neutral ligand motifs RDPBR′ and RTPB. Soon after the initial investigation, Peters highlighted a similar reactivity in which H2 is added across nickel-borane complex to form nickel-hydride borohydride complex (Scheme 12).27 They prepared the complex [Ni(η2-BC-PhDPBMes)Br] which was reduced by Na/Hg to form the zerovalent complex, [Ni(η3-BC2-PhDPBMes)]. They elegantly demonstrated that this complex was able to cleave the H–H bond in H2 where the atoms are added across the Ni–B bond in the complex (Scheme 12). The resulting nickel hydride borohydride complex, [Ni(H)(κ3-PPH-PhDPBMes + H)] was confirmed via structural characterisation and spectroscopic techniques. Furthermore this transformation was found to be reversible and the complexes [Ni(η3-BC2-PhDPBMes)] and [Ni(H)(κ3-PPH-PhDPBMes + H)] could be interconverted via the addition or removal of the hydrogen atmosphere. The latter complex was shown to promote stoichiometric hydrogenation of olefins reforming the former as shown in Scheme 12. As with the previous example, the nickel complexes were confirmed to be catalytically active for the hydrogenation of olefins.27
 | ||
| Scheme 12 The first of a series of pioneering investigations by Peters looking at the cleavage of H–H across transition metal–borane bonds. Here, the H2 addition product [Ni(H)(κ3-PPH-PhDPBMes + H)] goes on to hydrogenate olefin substrates and regenerate the starting complex. | ||
Peters has since reported a number of further nickel complexes in which similar H–H activations occur. Changing the aryl group at boron had a significant impact on the resulting reactivity of the complex with hydrogen. For example, changing the aryl group from mesityl to phenyl in the above example (to form the PhDPBPh ligand) switched off the reactivity of the resulting complex. In this case the presence of a THF molecule coordinated to the nickel centre was presumed to preclude its reactivity.27 The substituents on the phosphine donors were also important. The addition of hydrogen was found to occur in the di-isopropyl phosphine complex, [Ni(η2-BC-iPrDPBPh)], however, this occurred slowly over period of hours.52 Interestingly, the κ4-PPBP-complexes [Ni(RTPB)] (where R = Ph, iPr) showed no indication of coordination by N2 (which had been the case with related complexes). Whilst there was some indication of weak coordination to H2 under atmospheres of this gas at low temperature, no new product could be isolated.53
In addition to this, they demonstrated that it was also possible to activate E–H bonds (where E = SiH2Ph, SiHPh2) within the nickel–borane systems (Scheme 13).54 The Si–H bond was found to be readily cleaved across the Ni–B bond in their complexes generating new borohydrido-nickel-silyl species. The potential application of the novel Si–H bond breaking process was subsequently tested within the hydrosilylation of para-substituted benzaldehydes.
 | ||
| Scheme 13 Reaction of [Ni(η3-BC2-PhDPBMes)] with H2 and aryl substituted silanes leading to active complexes for hydrosilylation of benzaldehydes. | ||
The E–H bond activation process within the RDPBR′ and RTPB ligand systems have also been explored with iron metal centres.30,55–58 In a series of reports, these researchers have exploited the boron mediated E–H bond activation process for some truly fascinating and high impact transformations (Scheme 14). They have exploited their complexes in order to synthesise a carbon monoxide derived olefin via hydrogenative C–C coupling under mild conditions (Scheme 14a).56 They have also demonstrated that a dinitrogen ligand can be functionalised using [Fe{PhDBPiPr}Br].30 The complex is reduced with Na/Hg in the presence of 1,2-bis(chlorodimethylsilyl)ethane under a nitrogen atmosphere to generate an iron–aminoimide complex. This species then undergoes reaction with H–H or H–SiPhH2 in which a hydrogen adds to the boron centre forming a Fe–H–B bridge and the other, either H or SiPhH2, adds to the nitrogen α to the iron centre as shown in Scheme 14b.
 | ||
| Scheme 14 C–C bond coupling and hydrogenation of two carbon monoxide ligands (top) and stepwise functionalisation and reduction of nitrogen with silanes where one hydrogen originating from the silane is transferred to boron (bottom). | ||
The addition of hydrogen across the iron–borane bond in [Fe(iPrTPB)(N2)] has also been explored (Scheme 15).58 The complex undergoes rapid addition of H2 across the iron–boron bond at room temperature to form the complex [Fe(H)(μ-H)(iPrTPB)(N2)]. As shown in Scheme 15, the bridging hydride is bound to the boron centre of the former iPrTPB ligand (essentially forming a borohydride unit) which then interacts with the iron centre where the coordination mode has changed from κ4-PPBP-iPrTPB to κ4-PPHP-iPrTPB + H. Such a complex provides a rare example of an inverted scorpionate κ4-coordination mode.59 The nitrogen ligand in [Fe(H)(μ-H)(TPB)(N2)] was shown to be labile and was readily replaced by a dihydrogen ligand to form [Fe(H)(μ-H)(TPB)(H2)]. As shown in Scheme 15, the three complexes were interconvertible confirming that the activation of H2 across the iron–boron bond is reversible. It was shown that any of the three complexes could be utilised as effective catalysts for the hydrogenation of alkenes and alkynes. In addition to the cleavage of H–H bonds, [Fe(TPB)(N2)] was also shown to undergo C–H activation in arylacetylenes and formaldehyde amongst other E–H bond activations.
 | ||
| Scheme 15 Demonstrating reversible H–H cleavage reactivity of iron complexes containing the iPrTPB ligand in addition to the activation of C–H bonds. | ||
The Peters research group also explored the potential of iron complexes containing the iPrTPB ligand to act as catalysts for the conversion of nitrogen to ammonia.57 These investigations show great promise and insight into this challenging transformation. The study seems to suggest however that the complex [Fe(H)(μ-H)(iPrTPB)(N2)] inhibits the catalytic transformation in this case.
In addition to nickel and iron transition metal centres, they have also explored the chemistry of cobalt complexes containing the RDPBR′ and RTPB ligand systems.55,60,61 The cobalt complexes exhibited similar E–H bond activations to the other two first row transition metals and have been studied for the application surrounding the conversion of N2 to NH3. Interestingly, even though E–H bond activation was observed in most cases, the specific product formed was influenced by the metal itself and the specific ligand derivative used. For example, the reaction of phenol and thiophenol with the complexes [Fe(iPrDPBPh)]2(μ-N2) and [Co(iPrDPBPh)(N2)] both gave O–H and S–H activated products (Scheme 16). In the case of the iron based systems, the complexes, [Fe(OPh)(iPrDPBPh)] (two equiv.) and [Fe(μ-SPh)(iPrDPBPhH)]2 were formed, respectively (Scheme 16). During the formation of the phenoxide complex, the hydrogen is lost as H2 while the hydrogen is transferred to the boron unit in the corresponding thiophenol reaction forming the borohydride species. In the corresponding reactions involving [Co(iPrDPBPh)(N2)] the hydrogen is lost as H2 in both cases where the borane products [Co(OPh)(iPrDPBPh)] and [Co(SPh)(iPrDPBPh)] are formed. The activation of C–H, N–H and Si–H bonds was also shown to be possible with these complexes via reaction with benzo[h]quinolone, 8-aminoquinoline and diphenylsilane, respectively (Scheme 17). Furthermore, as with the nickel and iron complexes of this type, the corresponding cobalt complexes were found to be active catalysts for the hydrosilylation of ketones and aldehydes. Indeed, [Co(iPrDPBPh)(N2)] was reported to be the most active homogeneous cobalt catalyst reported for this application under the conditions utilised.55
 | ||
| Scheme 16 E–H bond activations utilising iron and cobalt complexes containing the iPrDPBPh ligand showing variation in the reactivity depending on the metal centre. | ||
 | ||
| Scheme 17 N–H, C–H and Si–H bond activations across the cobalt–borane bond. The latter reactivity was further developed into a catalyst for the hydrosilylation of ketones and aldehydes. | ||
Over several years Emslie has developed some interesting ligand motifs where the borane functional group is located at the terminal position of the ligand framework (Fig. 9; see also the TXPB ligand in Scheme 8).62,63 In 2014, Emslie introduced the FcPPB ligand in which they synthesised a range of complexes, investigated their properties and studied their reactivity.63,64 The ligand was added to a zerovalent platinum centre to form the complex [Pt(FcPPB)]. The mode of coordination of this single ligand complex features donation via the two phosphines in addition to a η3-BC2–arylborane interaction similar to that observed in other B-aryl systems (Scheme 18). This complex was subjected to one atmosphere of H2 which led to the formation of the species [PtH(μ-H)(FcPPB)] which was characterised in situ. Both NMR and IR spectroscopic data were consistent with a terminal platinum-hydride and a bridging platinum-borohydride unit. The addition of H2 to the complex was found to be reversible and it was slowly released under an argon atmosphere or under reduced pressure. While it was not possible to obtain structural characterisation of [PtH(μ-H)(FcPPB)], a calculated structure was obtained by DFT calculations and found to be consistent with the spectroscopic interpretation. Unfortunately, the platinum complexes were found to be inactive as catalysts for the hydrogenation of the alkenes and alkynes tested in the presence of mercury. In an attempt to further explore the reactivity of the complex, Emslie tested the reaction of [Pt(FcPPB)] with phenylacetylene. A C–H activation indeed occurred to form the expected product, [Pt(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)(μ-H)(FcPPB)]. This species, however, was only observed in the mixture for minutes before it transformed into a complex containing a vinylborane functional group (Scheme 18).
CPh)(μ-H)(FcPPB)]. This species, however, was only observed in the mixture for minutes before it transformed into a complex containing a vinylborane functional group (Scheme 18).
 | ||
| Fig. 9 The FcPPB and FcPPAl ligands containing terminal Lewis acid functional groups developed by Emslie. | ||
 | ||
| Scheme 18 The reaction of [Pt(FcPPB) with H2 and also with phenylacetylene. | ||
Interestingly, a related alane system has recently been reported by Emslie using the ligand FcPPAl.65 In this case, upon addition of H2, two terminal hydride ligands are formed on the corresponding platinum complex and no significant interaction was observed between the aluminium centre and neither of the terminally coordinated hydrides. Furthermore, the addition of H2 to the complex was not reversible even at elevated temperatures.
Another rather interesting ligand system where the borane functional group is located at the terminal position has also been reported by Figueroa (Scheme 19).66a The borane functional group was installed within the complex from the zerovalent bis isocyanide complex [Pt(CNArDipp2)2] (where ArDipp2 = 2,6-(i-Pr)2C6H3)2] via a 1,1-hydroboration on one of the coordinated isocyanide ligands. The resulting T-shaped complex featured the newly constructed Cy2B(H)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NArDipp2 ligand (Cy2BIM) which coordinated to the platinum centre via a κ2-NB coordination mode and featured a significant platinum-to-boron dative interaction [Pt–B 2.314(6) Å]. As with the above examples, [Pt{κ2-NB Cy2B(H)CNArDipp2}(CNArDipp2)] was capable of cleaving the bond in H2, presumably via the Lewis acid mediated activation mechanism, resulting in the formal addition of dihydrogen across the platinum–boron bond (Scheme 19). In this case, the cleavage of hydrogen was found to be irreversible. It was further demonstrated that [Pt{κ2-NB-Cy2B(H)CNArDipp2}(CNArDipp2)] was capable of activating other E–H bonds (where E = N, O, C) as shown in Scheme 19. Interestingly, Figueroa also demonstrated that the free ligand Cy2BIM was also capable of activating certain bonds in transformations reminiscent of Frustrated Lewis Pair chemistry.66b
NArDipp2 ligand (Cy2BIM) which coordinated to the platinum centre via a κ2-NB coordination mode and featured a significant platinum-to-boron dative interaction [Pt–B 2.314(6) Å]. As with the above examples, [Pt{κ2-NB Cy2B(H)CNArDipp2}(CNArDipp2)] was capable of cleaving the bond in H2, presumably via the Lewis acid mediated activation mechanism, resulting in the formal addition of dihydrogen across the platinum–boron bond (Scheme 19). In this case, the cleavage of hydrogen was found to be irreversible. It was further demonstrated that [Pt{κ2-NB-Cy2B(H)CNArDipp2}(CNArDipp2)] was capable of activating other E–H bonds (where E = N, O, C) as shown in Scheme 19. Interestingly, Figueroa also demonstrated that the free ligand Cy2BIM was also capable of activating certain bonds in transformations reminiscent of Frustrated Lewis Pair chemistry.66b
 | ||
| Scheme 19 The construction of a nitrogen based ligand featuring a terminal borane functional group via addition of dicyclohexylborane with a pre-coordinated isocyanide ligand. The further reactivity of this complex and activation of H–H and H–E bonds. | ||
Computational studies on hydrogen–hydrogen bond cleavage
Computational investigations have been carried out in order to elucidate the possible mechanisms involved in the addition of dihydrogen across the transition metal–boron bond in these complexes.52,67,68 The Peters system (Scheme 12) was modelled in these studies. The η3-BC2–arylborane interaction (i.e. the interaction of the boron and both the ipso and an ortho carbon within the central aromatic ring) with the metal centre also plays an important role in the mechanism and so is not solely influenced by the boron functionality. Nevertheless, these computational studies provide great insight in the role of the borane moiety within the activation of dihydrogen.The first investigation was published by Sakaki soon after Peters' original report.67 This study supported a heterolytic cleavage type mechanism in which the H–H σ-bond was broken via a cooperative process involving both boron and nickel centres. The work showed that the first step in the reaction mechanism involved the coordination of the H2 species to the nickel centre. At this stage the σ-coordinated ligand is positioned trans to the boron. The hydrogen molecule has to approach the open face of the complex since any other approach is blocked by the mesityl group and its interaction with the nickel centre. Furthermore, no significant interaction of the hydrogen molecule directly with the boron centre is possible since it is also blocked by ligand. Upon coordination of the dihydrogen ligand, the BCC interaction with the nickel centre is relaxed, the Ni–B and Ni–C bond distances begin to increase and the mesityl group gradually moves away from the nickel centre. This presumably provides the space for the H2 ligand to move from its original position trans to boron to a point where the boron and one of the hydrogen atoms in the H2 ligand are close enough to interact with each other. In the transition state resulting in the activation of the H2 the relevant bond distances were found to be B–Ha 1.654 Å, Ha–Hb 0.997 Å, Ni–B 2.200 Å, Ni–Ha 1.537 Å and Ni–Hb 1.475 Å. These values indicate a significant interaction across the B–H–H–Ni moiety prior to the cleavage of the H–H bond confirming the cooperation between the electron-rich nickel and the electron-poor boron centres. Sakaki's investigation also ruled out the homolytic cleavage (oxidative addition) at the formal d10 nickel metal centre.
Peters later carried out a computational investigation on a derivative complex which indicated a very similar reaction pathway also ruling out the oxidative addition pathway since it was found to be 9 kcal mol−1 higher in energy. The bond distances found in the B–H–H–Ni moiety prior to the cleavage were B–Ha 1.642 Å, Ha–Hb 1.018 Å, Ni–B 2.203 Å, Ni–Ha 1.589 Å and Ni–Hb 1.526 Å.52
Earlier this year a further computational investigation was carried out by Ke and co-workers.68 Again, this study looked at the same system in which the starting complex featured the η3-BC2 interaction with the nickel centre. In this investigation, Ke probed four possible modes of H2 activation where in this case, the ligand featuring a phenyl group at boron rather than the mesityl group (i.e.PhDPBPh rather than PhDPBMes) was used. The four modes of activation that they studied, (i) cis homolytic, (ii) trans homolytic, (iii) synergetic heterolytic and (iv) dissociative heterolytic activation are shown in Fig. 10. The first two modes of activation looked at homolytic cleavage of the H–H bond by only the metal centre in a typical oxidative addition step. The two modes differ in the orientation of the dihydrogen molecule with respect to the boron when it is activated by the metal centre. It was found that activation in the position cis to the boron was the pathway with lower energy of the two modes. Even though the boron is not directly involved in the activation in this case, it was found that the borane functionality stabilises the transition state for activation by interacting with the filled dz2 orbital of the nickel centre. Their investigation subsequently went on to look at possible steps where there was indeed involvement of the boron within the activation process, i.e. those in which both the metal and boron work in cooperation to cleave the H–H bond. The first of these two processes studied involved a synergetic heterolytic cleavage in which the nickel–boron bond remains intact until that later stages of the activation process. The second examined process involved the potential activation process in which the nickel–boron bond is broken via insertion of the H2 unit prior to activation. This latter process was found to be the higher energy process of the two. Overall, the synergetic heterolytic process was found to be the lower energy process of the four although the cis-homolytic process was found to be only 4 kcal mol−1 higher in energy. The evidence supports the fact that the boron moiety is involved in the activation process and that synergetic heterolytic activation is the most likely mode of activation in these complexes. All three computational investigations are consistent with this conclusion.
 | ||
| Fig. 10 The four possible modes of activation of hydrogen studied by Ke and co-workers. Synergetic heterolytic cleavage (mode 3) was found to be the lowest energy pathway. Adapted with permission from ref. 68. Copyright 2016 American Chemical Society. | ||
Element–element bond cleavage (those not involving hydrogen)
Related E–E′ bond activations (where E or E′ ≠ H) are much less explored than the corresponding H–H and E–H reactions. They had previously been observed some time ago in the scorpionate-type complexes by Parkin and co-workers.69,70 In 2006, they carried out a series of reactions involving the activation of small molecules via a formal 1,2 addition across the metal–borane bond within iron and nickel based metallaboratrane complexes as highlighted in Scheme 20. | ||
| Scheme 20 E–E′ bond activation via formal 1,2-addition reactions across metal–borane bonds. | ||
Tauchert has more recently observed rare E–E′ bond activations in which the C–O bond in various allyl substrates were added across a palladium–boron bond (Scheme 21).71 The complexes utilised for these formal oxidative additions were zerovalent palladium complexes bearing the η2-BC-PhDPBPh ligand where the fourth coordination site at the metal centre was occupied by a pyridine or 2,6-lutidine ligand. The C–O bond activations occurred at room temperature over the course of 1–20 h depending on the allyl species utilised.
 | ||
| Scheme 21 The first example of C–O bond cleavage across a transition metal–borane bond. | ||
Transition metal–boryl mediated hydrogen–hydrogen bond cleavage
Peters has also explored the reactivity of Yamashita and Nozaki's diphosphino-boryl (PBP) pincer ligand (Scheme 4) as a means for obtaining novel H2 activation chemistry.29b,72 They prepared a pseudo square planar cobalt(I) complex which was supported by the meridional coordination of the PBP pincer ligand with a terminally coordinated N2 ligand in the fourth coordination site. This complex was shown to react with H2 to form a complex which was assigned as dihydroborato-cobalt-dihydride on the basis of the spectroscopic evidence as well as DFT calculations (Scheme 22). The transformation from cobalt–boryl to dihydroborato cobalt dihydride results from the activation of two H–H bonds. A theoretical study of this system was later investigated by Paul which suggests that the cleavage of the H–H bond occurs at the cobalt metal centre rather than at both metal and boron centres as found with the nickel–borane systems (vide supra).73 Later studies on derivative ligands also demonstrated the reversible hydrogen atoms storage potential of nickel– and cobalt–boryl functional groups and their application towards olefin hydrogenation.72 | ||
| Scheme 22 Boryl-mediated reversible H2 activation at a cobalt metal centre. | ||
Yamashita later demonstrated a similar motif to that observed by Peters when investigating the coordination and reactivity of a series of ruthenium complexes containing the PBP boryl pincer ligand.44h In this case, the borane ligand precursor was added to [RuH(OAc)(PPh3)2] (Scheme 23). Rather than forming the boryl pincer, which is conceivable via elimination of H2, the ruthenium-hydride species was found to transfer to the boron centre thus forming a borohydride moiety in which the [BH2]− unit was bridged with the metal centre. The resulting product was further reacted with Na[BH4] to form a complex containing a B(μ-H)2Ru(μ-H)2B motif.
 | ||
| Scheme 23 The transfer of a ruthenium-hydride species on to the borane functional group in Yamashita's PB(H)P ligand to form the corresponding borohydride ligand, PB(H2)P and its further reactivity with sodium borohydride. | ||
The newly discovered iridium–boryl pincer complex reported by Ozerov earlier this year was also found to react with H2 (Scheme 24).50 Thermolysis of the complex [IrCl(Ph)(iPrDPB)] in the presence of H2 led to the isolation of the trigonal bipyramidal complex [IrCl(H)(iPrDPB)] where the phenyl group had been lost from the complex as benzene. Under milder conditions, an intermediate species was observed in solution via NMR spectroscopy. The identity of this intermediate species was assigned as [Ir(μ-Cl)(H)2(iPrDPBPh)] where the phenyl group had been transferred back to the boron centre, two new hydride ligands had formed and the chloride ligand was bridging the boron and iridium centres. This intermediate species slowly transformed to a new species at room temperature or within 1 h at 100 °C. This new species was identified as [Ir(Cl)(H)2(iPrDPBH)] again where the phenyl group was lost as Ph–H and three other hydrogen atoms where found on the complex (i.e. where addition of 2 equivalents of H2 had reacted with the original complex). The chemical environments of these three hydrogen atoms were determined to be two terminal iridium-hydrides and one B–H on the basis of the proton NMR spectrum (−2.84 (br, 1H, B–H), −14.70 (br, 1H, Ir–H), −16.89 (br, 1H, Ir–H)). Thus, in this reaction a boryl to borane transformation had occurred where one of the H2 molecules was cleaved by formal addition across the iridium–boryl bond. Finally [Ir(Cl)(H)2(iPrDPBH)] was found to reversibly lose one equivalent of hydrogen to form the trigonal bipyramidal complex [IrCl(H)(iPrDPB)] (Scheme 24).
 | ||
| Scheme 24 Transformations involving iridium–boryl and iridium–borohydride functional groups reported by Ozerov demonstrating reversible H2 activation. | ||
Conclusions
In summary, it has been shown that borane and boryl functional groups have a rich and fascinating reactivity when they are acting as ligands within transition metal complexes. There have been substantial developments in the utilisation of transition metal–borane and transition metal–boryl complexes for the purposes of transfer of hydrides, alkyl groups and aryl groups between the boron and metal centre in recent years. Furthermore, these two boron based groups can be utilised cooperatively with the metal to carry out a wide range of H–H, H–E and E–E′ bond activations. At this stage in the development of the research area, where there are still only a handful of ligand systems, it appears that there is plenty of scope for future development.The combination of the potential to transfer hydrogen between boron and metal centre together with the methodology to formally add H2 across the metal–boron bond provides a powerful methodology. The development of a ‘recharge and sting’ methodology has already shown great promise in terms of future catalytic application. It would appear that Trofimenko's analogy seems to go deeper than perhaps even he could have imagined. The developments by researchers in this field certainly demonstrate that it is possible for scorpionate and other related borane ligands to be recharged and repeatedly sting just like a scorpion.
Acknowledgements
The author would like to thank the Leverhulme Trust for funding (RPG-2015-097).Notes and references
- D. Goulson, Bumblebees: Behaviour, Ecology, and Conservation, Oxford University Press, 2009 Search PubMed.
- G. A. Polis, The Biology of Scorpions, Stanford University Press, 1990 Search PubMed.
- S. Trofimenko, J. Am. Chem. Soc., 1966, 88, 1842 CrossRef CAS.
- (a) S. Trofimenko, Scorpionates: The Coordination of Poly(pyrazolyl)borate Ligands, Imperial College Press, London, 1999 Search PubMed; (b) S. Trofimenko, Polyhedron, 2004, 23, 197 CrossRef CAS; (c) C. Pettinari, Scorpionates II: Chelating Borate Ligands, Imperial College Press, London, 2008 Search PubMed; (d) G. P. A. Yap, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 937 CrossRef CAS PubMed a special issue dedicated to the chemistry of scorpionate ligands has very recently been published; see; (e) C. Pettinari, Eur. J. Inorg. Chem., 2016, 2209 CrossRef CAS , and other articles in this issue.
- (a) J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711 RSC; (b) M. L. H. Green and W. P. Griffith, Platinum Met. Rev., 1998, 42, 168 CAS.
- R. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley-Interscience, 2005 Search PubMed.
- (a) Y. Blum, D. Czarkie, Y. Rahamim and Y. Shvo, Organometallics, 1985, 4, 1459 CrossRef CAS; (b) H. Doucet, T. Ohkuma, K. Murata, T. Yokozawa, M. Kozawa, E. Katayama, A. F. England, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1998, 37, 1703 CrossRef CAS; (c) M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466 CrossRef CAS; (d) C. A. Sandoval, T. Ohkuma, K. Muñiz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490 CrossRef CAS PubMed.
- (a) M. Trincado and H. Grützmacher, Cooperating Ligands in Catalysis, in Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, ed. R. Peters, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, ch. 3 Search PubMed; (b) O. Eisenstein and R. H. Crabtree, New J. Chem., 2013, 37, 21 RSC.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed.
- (a) G. Erker, Dalton Trans., 2011, 40, 7475 RSC; (b) A. T. Normand, C. G. Daniliuc, B. Wibbeling, G. Kehr, P. Le Gendre and G. Erker, Chem. – Eur. J., 2016, 22, 4285 CrossRef CAS PubMed.
- For leading references on this theme see: (a) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588 CrossRef CAS PubMed; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236 CrossRef CAS PubMed.
- (a) R. Peters, Designing Efficient Catalysts for Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015 Search PubMed; (b) A. J. A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS PubMed.
- (a) M. L. H. Green, J. Organomet. Chem., 1995, 500, 127 CrossRef CAS; (b) G. Parkin, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2006, ch. 1, vol. 1 Search PubMed.
- A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 2759 CrossRef CAS.
- (a) M. Garner, J. Reglinski, I. Cassidy, M. D. Spicer and A. R. Kennedy, Chem. Commun., 1996, 1975 RSC; (b) M. D. Spicer and J. Reglinski, Eur. J. Inorg. Chem., 2009, 1553 CrossRef CAS.
- F.-G. Fontaine, J. Boudreau and M.-H. Thibault, Eur. J. Chem., 2008, 5439 CrossRef CAS.
- I. Kuzu, I. Krummenacher, F. Armbruster and F. Breher, Dalton Trans., 2008, 5836 RSC.
- (a) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924 CrossRef CAS PubMed; (b) H. Braunschweig and R. D. Dewhurst, Dalton Trans., 2011, 40, 549 RSC.
- H. Kameo and H. Nakazawa, Chem. – Asian J., 2013, 8, 1720 CrossRef CAS PubMed.
- (a) G. R. Owen, Chem. Soc. Rev., 2012, 41, 3535 RSC; (b) G. R. Owen, Transition Met. Chem., 2010, 35, 221 CrossRef CAS.
- (a) G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065 RSC; (b) M. Devillard, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 730 CrossRef CAS PubMed; (c) A. Amgoune, G. Bouhadir and D. Bourissou, Top. Curr. Chem., 2013, 334, 281 CrossRef PubMed; (d) A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 8163 RSC; (e) G. Bouchadir, A. Amgoune and D. Bourissou, Adv. Organomet. Chem., 2010, 58, 1 CrossRef; (f) A. Maity and T. S. Teets, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.6b00034.
- For an interesting discussion on the formal assignment of Z-type ligands see: (a) A. F. Hill, Organometallics, 2006, 25, 4743 Search PubMed; (b) G. Parkin, Organometallics, 2006, 25, 4744 CrossRef CAS.
- (a) K.-H. Ernst, F. R. W. P. Wild, O. Blacque and H. Berke, Angew. Chem., Int. Ed., 2011, 50, 10780 CrossRef CAS PubMed; (b) A. Werner, Z. Anorg. Allg. Chem., 1893, 2, 267 CrossRef; (c) G. B. Kauffman, in Classics in Coordination Chemistry, Part 1: The Selected Papers of Alfred Werner, Dover, New York, 1968, p. 9 Search PubMed.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS PubMed.
- N. Tsoureas, Y.-Y. Kuo, M. F. Haddow and G. R. Owen, Chem. Commun., 2011, 47, 484 RSC.
- W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080 CrossRef CAS PubMed.
- This review focuses on transition metal–boron chemistry however there have been significant achievements in other Z-type sigma acceptor ligand systems. For leading article see: (a) S. L. Benjamin and G. Reid, Coord. Chem. Rev., 2015, 297–298, 168 CrossRef CAS; (b) D. M. P. Mingos, Coord. Chem. Rev., 2015, 293–294, 2 CrossRef CAS; (c) J. S. Jones, C. R. Wade and F. P. Gabbai, Angew. Chem., Int. Ed., 2014, 53, 8876 CrossRef CAS PubMed; (d) B. E. Cowie, F. A. Tsao and D. J. H. Emslie, Angew. Chem., Int. Ed., 2015, 54, 2165 CrossRef CAS PubMed; (e) J. S. Jones and F. P. Gabbaï, Acc. Chem. Res., 2016, 49, 857 CrossRef CAS PubMed . See also ref. 22a and d.
- A search of the Cambridge Structural Database was carried out looking at the transition metal–borane distances of ambiphilic ligands (April 2016). (a) S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056 CrossRef CAS PubMed; (b) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310 CrossRef CAS PubMed; (c) For the shortest distance in a transition metal–borane complex derived from a scorpionate complex see: G. Nuss, G. Saischek, B. N. Harum, M. Volpe, K. Gatterer, F. Belaj and N. C. Mosch-Zanetti, Inorg. Chem., 2011, 50, 1991 CrossRef CAS PubMed.
- D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 4938 CrossRef CAS PubMed.
- M. P. Boone and D. W. Stephan, Chem. – Eur. J., 2014, 20, 3333 CrossRef CAS PubMed.
- I. R. Crossley, A. F. Hill and A. C. Willis, Dalton Trans., 2008, 201 RSC.
- G. C. Rudolf, A. Hamilton, A. G. Orpen and G. R. Owen, Chem. Commun., 2009, 553 RSC.
- I. R. Crossley, A. F. Hill, E. R. Humphrey and M. K. Smith, Organometallics, 2006, 25, 2242 CrossRef CAS.
- R. J. Blagg, C. J. Adams, J. P. H. Charmant, N. G. Connelly, M. F. Haddow, A. Hamilton, J. Knight, A. G. Orpen and B. M. Ridgway, Dalton Trans., 2009, 8724 RSC.
- M. J. López-Gómez, N. G. Connelly, M. F. Haddow, A. Hamilton and A. G. Orpen, Dalton Trans., 2010, 39, 5221 RSC.
- R. J. Blagg, N. G. Connelly, M. F. Haddow, A. Hamilton, M. Lusi, A. G. Orpen and B. M. Ridgway, Dalton Trans., 2010, 39, 11616 RSC.
- G. Dyson, A. Zech, B. W. Rawe, M. F. Haddow, A. Hamilton and G. R. Owen, Organometallics, 2011, 30, 5844 CrossRef CAS.
- N. Tsoureas, A. Hamilton, M. F. Haddow, J. N. Harvey, A. G. Orpen and G. R. Owen, Organometallics, 2013, 32, 2840 CrossRef CAS.
- (a) N. Tsoureas, G. R. Owen, A. Hamilton and A. G. Orpen, Dalton Trans., 2008, 6039 RSC; (b) N. Tsoureas, T. Bevis, C. P. Butts, A. Hamilton and G. R. Owen, Organometallics, 2009, 28, 5222 CrossRef CAS; (c) G. R. Owen, N. Tsoureas, R. F. Hope, Y.-Y. Kuo and M. F. Haddow, Dalton Trans., 2011, 40, 5906 RSC.
- N. Tsoureas, M. F. Haddow, A. Hamilton and G. R. Owen, Chem. Commun., 2009, 2538 RSC.
- S. Bontemps, H. Gornitzka, G. Bouhadir, K. Miqueu and D. Bourissou, Angew. Chem., Int. Ed., 2006, 45, 1611 CrossRef CAS PubMed.
- (a) H. Kameo, Y. Hashimoto and H. Nakazawa, Organometallics, 2012, 31, 3155 CrossRef CAS; (b) H. Kameo, Y. Hashimoto and H. Nakazawa, Organometallics, 2012, 31, 7476 CrossRef CAS.
- (a) Y. Segawa, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 9201 CrossRef CAS PubMed; (b) Y. Segawa, M. Yamashita and K. Nozaki, Organometallics, 2009, 28, 6234 CrossRef CAS; (c) M. Hasegawa, Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2012, 51, 6956 CrossRef CAS PubMed; (d) H. Ogawa and M. Yamashita, Dalton Trans., 2013, 42, 625 RSC; (e) T. Miyada and M. Yamashita, Organometallics, 2013, 32, 5281 CrossRef CAS; (f) A. F. Hill, S. B. Lee, J. Park, R. Shang and A. C. Willis, Organometallics, 2010, 29, 5661 CrossRef CAS; (g) K. Tanoue and M. Yamashita, Organometallics, 2015, 34, 4011 CrossRef CAS; (h) T. Miyada, E. H. Kwan and M. Yamashita, Organometallics, 2014, 33, 6760 CrossRef CAS.
- (a) N. Curado, C. Maya, J. López-Serrano and A. Rodríguez, Chem. Commun., 2014, 50, 15718 RSC; (b) P. Ríos, N. Curado, J. López-Serrano and A. Rodríguez, Chem. Commun., 2016, 52, 2114 RSC.
- For some early examples see: (a) L. Turculet, J. D. Feldman and T. D. Tilley, Organometallics, 2004, 23, 2488 CrossRef CAS; (b) A. Fischbach, P. R. Bazinet, R. Waterman and T. D Tilley, Organometallics, 2008, 27, 1135 CrossRef CAS; (c) S. Fromel, G. Kehr, R. Frohlich, C. G. Daniliuc and G. Erker, Dalton Trans., 2013, 42, 14531 RSC; (d) B. J. Eleazer, M. D. Smith and D. V. Peryshkov, Organometallics, 2016, 35, 106 CrossRef CAS.
- (a) E. Khaskin, P. Y. Zavlij and A. N. Vedernikov, Angew. Chem., Int. Ed., 2007, 46, 6309 CrossRef CAS PubMed; (b) S. Pal and A. N. Vedernikov, Dalton Trans., 2012, 41, 8116 RSC; (c) For an interesting related transformation see: S. Pal, P. Y. Zavalij and A. N. Vedernikov, Chem. Commun., 2014, 50, 5376 RSC.
- J. Zhu, D. Mukherjee and A. D. Sadow, Chem. Commun., 2012, 48, 464 RSC.
- (a) B. E. Cowie and D. J. H. Emslie, Organometallics, 2015, 34, 2737 CrossRef CAS; (b) B. E. Cowie and D. J. H. Emslie, Organometallics, 2013, 32, 7297 CrossRef CAS.
- W.-C. Shih, W. Gu, M. C. MacInnis, S. D. Timpa, N. Bhuvanesh, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2016, 138, 2086 CrossRef CAS PubMed.
- J. A. Mata, E. Peris, C. Incarvito and R. H. Crabtree, Chem. Commun., 2003, 184 RSC.
- W. H. Harman, T.-P. Lin and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 1081 CrossRef CAS PubMed.
- C. Tsay and J. C. Peters, Chem. Sci., 2012, 3, 1313 RSC.
- S. N. MacMillan, W. H. Harman and J. C. Peters, Chem. Sci., 2014, 5, 590 RSC.
- M. Nesbit, D. L. M. Suss and J. C. Peters, Organometallics, 2015, 34, 4741 CrossRef CAS.
- D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 12580 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef CAS PubMed.
- H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053 CrossRef CAS PubMed.
- (a) B. M. Bridgewater and G. Parkin, Inorg. Chem. Commun., 2000, 3, 534 CrossRef CAS; (b) Y. Rong, J. H. Palmer and G. Parkin, Dalton Trans., 2014, 43, 1397 RSC.
- T. J. Del Castillo, N. B. Thompson, D. L. M. Suess, G. Ung and J. C. Peters, Inorg. Chem., 2015, 54, 9256 CrossRef CAS PubMed.
- W. A. Gunderson, D. L. M. Suess, H. Fong, X. Wang, C. M. Hoffmann, G. E. Cutsail III, J. C. Peters and B. M. Hoffman, J. Am. Chem. Soc., 2014, 136, 14998 CrossRef CAS PubMed.
- D. J. H. Emslie, J. M. Blackwell, J. F. Britten and L. E. Harrington, Organometallics, 2006, 25, 2412 CrossRef CAS.
- B. E. Cowie and D. J. H. Emslie, Chem. – Eur. J., 2014, 20, 16899 CrossRef CAS PubMed.
- B. E. Cowie and D. J. H. Emslie, Organometallics, 2015, 34, 4093 CrossRef CAS.
- B. E. Cowie and D. J. H. Emslie, Angew. Chem., Int. Ed., 2015, 54, 2165 CrossRef CAS PubMed.
- (a) B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 10262 CrossRef CAS PubMed; (b) B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Chem. Commun., 2015, 51, 541 RSC.
- G. Zeng and S. Sakaki, Inorg. Chem., 2013, 52, 2844 CrossRef CAS PubMed.
- Y. Li, C. Hou, J. Jiang, Z. Zhang, C. Zhao, A. J. Page and Z. Ke, ACS Catal., 2016, 6, 1655 CrossRef CAS.
- (a) J. S. Figueroa, J. G. Melnick and G. Parkin, Inorg. Chem., 2006, 45, 7056 CrossRef CAS PubMed; (b) K. Pang, J. M. Tanski and G. Parkin, Chem. Commun., 2008, 1008 RSC.
- see also: M. J. López-Gómez, N. G. Connelly, M. F. Haddow, A. Hamilton and A. G. Orpen, Dalton Trans., 2010, 39, 5221 RSC.
- T. Schindler, M. Lux, M. Peters, L. T. Scharf, H. Osseili, L. Maron and M. E. Tauchert, Organometallics, 2015, 34, 1978 CrossRef CAS.
- T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672 CrossRef CAS PubMed.
- G. Ganguly, T. Malakar and A. Paul, ACS Catal., 2015, 5, 2754 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |