
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of regioisomerism on the solid-state fluorescence of bis(piperidyl)anthracenes: structurally simple but bright AIE luminogens†
Shunsuke
Sasaki
a,
Kazunobu
Igawa
b and
Gen-ichi
Konishi
*ac
aDepartment of Organic and Polymeric Materials, Tokyo Institute of Technology, Tokyo 152-8552, Japan. E-mail: konishi.g.aa@m.titech.ac.jp; Fax: +81-3-5734-2321
bInstitute for Materials Chemistry and Engineering and Department of Molecular and Material Sciences, Kyushu University, Fukuoka 816-8580, Japan
cPRESTO, Japan Science and Technology Agency (JST), Japan
First published on 11th May 2015
Abstract
A series of regioisomers of piperidylanthracenes (PA) and bis(piperidyl)anthracenes (BPA) were synthesized and their photophysical properties examined in solution, suspension, and the solid state. Aggregation-induced emission (AIE) was observed only for 1,4-BPA and 9,10-BPA, in which two piperidyl groups are substituted at the anthracene moiety in the para-position with respect to each other. Compared to previously reported AIE luminogens, these easily obtainable para-substituted BPAs, exhibited several unique and beneficial features, such as simple structures, bright solid-state fluorescence (Φfl = 0.49 and 0.86 for 1,4-BPA and 9,10-BPA, respectively), tunable fluorescence emission, and large Stokes shifts. Results from diffuse-reflectance and fluorescence lifetime measurements demonstrated that PAs and BPAs intrinsically possess an efficient non-radiative transition pathway in the solid state, and that 1,4-BPA and 9,10-BPA may overcome this pathway. The X-ray crystallographic analysis of 1,4-BPA revealed that undesirable interchromophoric interactions can be minimised, while TD-DFT calculations suggested that the enhanced Stokes shift of 1,4-BPA arises from severe electronic repulsion between neighbouring piperidyl moieties, which presumably results in the absence of self-absorption.
Introduction
Most organic fluorophores suffer from substantial fluorescence quenching in the solid state, an effect usually referred to as aggregation-caused quenching (ACQ). For decades, this characteristic feature has often been considered a bottleneck for practical applications of such organic compounds in e.g. electronics and bioimaging devices.1,2 However, traditional concepts for organic fluorophores have recently changed dramatically, as various studies have described simple and yet accurate systems for the measurement of absolute fluorescence quantum yields (Φfl).3,4 The fluorescence quantum yield is generally considered a quantitative index for solid-state fluorescence,5 and it provides deeper insight into possible new design strategies for efficient solid-state fluorophores, thereby promoting the development of fluorophores that exhibit aggregation-induced emission (AIE)1,2 and efficient solid-state fluorescence.6,7 Especially AIE luminogens have found many applications, not only in organic light-emitting diodes (OLEDs),8,9 but also in environment-responsive fluorescent probes,1,10 which are sensitive to aggregation and self-assembly.Despite the development of a wide variety of AIE luminogens1,2 and efficient solid-state fluorophores,6,7 the majority of functional molecules used in OLEDs8,9 and fluorescence imaging11 are based on tetraphenylethenes (TPEs),12 stilbenes,13,14 and siloles.15 These chromophores are preferentially used, because they exhibit simple structures and are susceptible to further modifications, which may be necessary for certain applications.16 Conversely, most other fluorophores exhibit more complex and complicated structures. Most recently, Tang et al. have addressed this issue and developed simple, small, and easily accessible AIE luminogens based on tetraphenylpyrazines (TPPs).17 Although both TPE and TPP are more readily available than other AIE luminogens, they exhibit Φfl values that remain unsatisfactory for most applications12,17 (Chart 1), and further modifications are required in order to attain efficient solid-state fluorescence.18
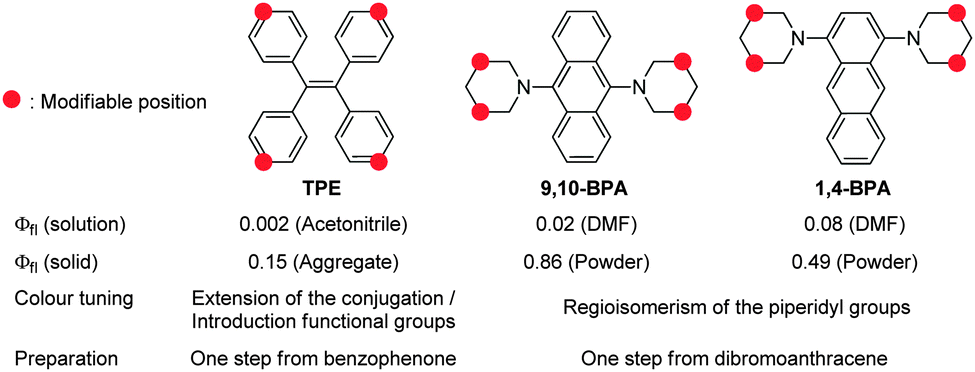 | ||
| Chart 1 Comparison between TPE12 and AIE-active BPAs. | ||
In order to develop simple, easily accessible, and versatile AIE luminogens, we focused on the solid-state fluorescence behaviour of basic dyes.19 Bis(dialkylamino)anthracene is a promising candidate in this context, as it does not exhibit fluorescence in solution.20 This is probably due to fast internal conversion21 and can be suppressed by aggregation. However, it is exactly this fluorescence property that sets bis(dialkylamino)anthracene apart from other fluorescent analogues such as e.g. diphenylanthracene.3
In this study, we report the synthesis of various regioisomers of piperidylanthracenes (PAs) and bis(piperidyl)anthracenes (BPAs), and we discuss the effect of regioisomerism on their solid-state fluorescence properties (Chart 2). Our results revealed that only 1,4-BPA and 9,10-BPA exhibit AIE behaviour, and that their fluorescence colours are tuneable without the introduction of additional substituents (Chart 1). Moreover, 9,10-BPA, which can be easily obtained via a one-step reaction from inexpensive 9,10-dibromoanthracene, also exhibits highly efficient solid-state fluorescence (Φfl = 0.86; neat powder). As BPAs are prepared with relative ease, and as they exhibit considerable levels of AIE, efficient solid-state fluorescence, characteristic tunability of the fluorescence colour, as well as simple and small structures, they are ideally suited to serve as core fluorophores for various functional molecules.
 | ||
| Chart 2 Nomenclature of piperidyl-substituted PA and BPA derivatives used in this study. | ||
However, BPAs are not only attractive with respect to the aforementioned practical aspects, but they are also interesting from a fundamental perspective, as they display efficient solid-state fluorescence as well as a large fluctuation in their Stokes shifts. This is particularly remarkable, as the molecular mass of BPAs is relatively low and they do not contain aryl–aryl bonds, alkene moieties, or bulky substituents.1,2,6 Therefore, the fluorescence behaviour of BPAs will also be discussed from a mechanistic viewpoint.
Results and discussion
Synthesis
A series of PA and BPA derivatives was readily obtained from the Pd-catalysed C–N coupling of mono- and dibromoanthracene, respectively.22,23 This route was given preference over a previously reported approach starting from 9,10-bis(dimethylamino)anthracene,20,24 as the synthetic intermediate 9,10-diaminoanthracene generated therein is unstable and susceptible to rapid oxidation.25 Most likely, this instability is responsible for the shortage of reports on the emission properties of bis(dialkylamino)anthracenes.26–30 Fortunately, recent advances in Pd-catalysed C–N coupling reactions provide synthetic routes to PAs and BPAs that bypass the formation of this unstable intermediate. Furthermore, 1,4-BPA and 9,10-BPA thus prepared do not exhibit any signs of deterioration upon exposure to air, which was attributed to the highly twisted aryl–nitrogen bonds, which suppresses oxidation via the formation of a quinoidal intermediate.25 Thermogravimetric analysis (TGA) measurements revealed decomposition temperatures (Td, corresponding to 5% weight loss under a nitrogen atmosphere) of 245 and 253 °C for 1,4-BPA and 9,10-BPA, respectively (Fig. S17, ESI†). These Td values are higher than that of TPE (Td = 213 °C) and comparable to that of TPP (Td = 275 °C),17 suggesting that 1,4-BPA and 9,10-BPA should possess a sufficiently high thermal stability in order to serve as AIE luminogens. Detailed synthetic procedures and characterization data are summarized in the experimental section.Aggregation-induced emission (AIE)
We measured the fluorescence properties of PAs and BPAs in solution (DMF), in colloidal suspension (THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:9; v/v), and in the solid state (neat powder, Fig. 1). The formation of aggregates was supported by UV-vis spectroscopy19,31 and confirmed by dynamic light scattering (DLS) measurements. For PA and BPA, the DLS measurements revealed typical hydrodynamic radii (Rh) of ca. 50–100 nm (Fig. S19–S26, ESI†). More detailed information about these DLS measurements is contained in the experimental section. Table 1 summarises the fluorescence maxima (λfl) and fluorescence quantum yields (Φfl) for each state. Most PAs and BPAs suffer severely from ACQ, and only 1,4-BPA and 9,10-BPA, which contain piperidyl groups at the para-position of the anthracene moiety with respect to each other, exhibit AIE behaviour. Especially 9,10-BPA demonstrates noteworthy levels of AIE, as it lacks fluorescence in solution (Φfl ≈ 0.02), but exhibits efficient solid-state fluorescence (Φfl ≈ 0.86), which is comparable to previously reported efficient fluorophores.6,7,18,32,33 Interestingly, 1,4-BPA reduces its fluorescence quantum yield as a function of increasing solvent polarity, and significantly enhanced fluorescence is observed in colloidal suspension (Table 1). This polarity-reduced fluorescence and the AIE characteristics are clearly reflected in the fluorescence behaviour when water is added to a THF solution of 1,4-BPA (Fig. 2a and c). Then, the fluorescence intensity plot is quite similar to AIE luminogens, whose fluorescence is quenched substantially by the addition of polar solvents.34 Moreover, the fluorescence colour of PAs and BPAs varies, depending on the position of the substituents. For example, green fluorescence (λfl = 514–519 nm) was observed for 9,10-BPA, while orange fluorescence (λfl = 591–596 nm) was observed for 1,4-BPA. Such a drastic colour change induced by regioisomerism is quite unique compared to conventional design paradigms, where the fluorescence colour is modulated via the introduction of donor or acceptor groups.33 This intriguing feature may be applied to the multicolour imaging of living cells, which imposes strict uniformity requirements on the chemical structures of the fluorophores.11,35 In addition, large Stokes shifts were observed for both 1,4-BPA (7400 cm−1) and 9,10-BPA (5500 cm−1) in the solid state, which renders these compounds attractive for applications in far-field fluorescence microscopy.36 In fact, these Stokes shifts are at least comparable, if not larger than those reported for recently developed fluorescent probes with large Stokes shifts.37
:H2O = 1:9; v/v), and in the solid state (neat powder, Fig. 1). The formation of aggregates was supported by UV-vis spectroscopy19,31 and confirmed by dynamic light scattering (DLS) measurements. For PA and BPA, the DLS measurements revealed typical hydrodynamic radii (Rh) of ca. 50–100 nm (Fig. S19–S26, ESI†). More detailed information about these DLS measurements is contained in the experimental section. Table 1 summarises the fluorescence maxima (λfl) and fluorescence quantum yields (Φfl) for each state. Most PAs and BPAs suffer severely from ACQ, and only 1,4-BPA and 9,10-BPA, which contain piperidyl groups at the para-position of the anthracene moiety with respect to each other, exhibit AIE behaviour. Especially 9,10-BPA demonstrates noteworthy levels of AIE, as it lacks fluorescence in solution (Φfl ≈ 0.02), but exhibits efficient solid-state fluorescence (Φfl ≈ 0.86), which is comparable to previously reported efficient fluorophores.6,7,18,32,33 Interestingly, 1,4-BPA reduces its fluorescence quantum yield as a function of increasing solvent polarity, and significantly enhanced fluorescence is observed in colloidal suspension (Table 1). This polarity-reduced fluorescence and the AIE characteristics are clearly reflected in the fluorescence behaviour when water is added to a THF solution of 1,4-BPA (Fig. 2a and c). Then, the fluorescence intensity plot is quite similar to AIE luminogens, whose fluorescence is quenched substantially by the addition of polar solvents.34 Moreover, the fluorescence colour of PAs and BPAs varies, depending on the position of the substituents. For example, green fluorescence (λfl = 514–519 nm) was observed for 9,10-BPA, while orange fluorescence (λfl = 591–596 nm) was observed for 1,4-BPA. Such a drastic colour change induced by regioisomerism is quite unique compared to conventional design paradigms, where the fluorescence colour is modulated via the introduction of donor or acceptor groups.33 This intriguing feature may be applied to the multicolour imaging of living cells, which imposes strict uniformity requirements on the chemical structures of the fluorophores.11,35 In addition, large Stokes shifts were observed for both 1,4-BPA (7400 cm−1) and 9,10-BPA (5500 cm−1) in the solid state, which renders these compounds attractive for applications in far-field fluorescence microscopy.36 In fact, these Stokes shifts are at least comparable, if not larger than those reported for recently developed fluorescent probes with large Stokes shifts.37
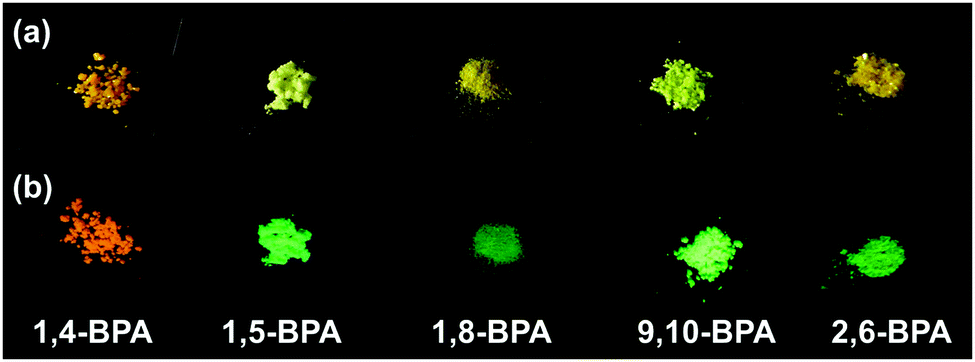 | ||
| Fig. 1 Photographic images of BPAs exposed to (a) ambient light and (b) UV light (λ = 365 nm). | ||
| Entry | λ fl [nm] | Φ fl | ||||||
|---|---|---|---|---|---|---|---|---|
| Solution | Suspensionb | Solidc | Solution | Suspensionb | Solidc | |||
| THF | DMF | THF | DMF | |||||
|
a The excitation wavelength corresponds to either the maximum wavelength of the absorption spectrum, or the diffused-reflectance spectrum.
b THF:H2O = 1:9 (v/v); H2O was added in order to obtain colloidal suspensions, and the formation of aggregates was confirmed by UV-vis spectroscopy19,31 and DLS measurements.
c Data obtained from neat powder.
|
||||||||
| 1-PA | 521 | 550 | 519 | 500 | 0.91 | 0.85 | 0.49 | 0.52 |
| 2-PA | 504 | 525 | 488 | 523 | 0.91 | 0.92 | 0.34 | 0.27 |
| 9-PA | 478 | 495 | 490 | 519 | 0.01 | 0.01 | 0.05 | 0.71 |
| 1,4-BPA | 598 | 631 | 596 | 591 | 0.25 | 0.08 | 0.45 | 0.49 |
| 1,5-BPA | 504 | 533 | 504 | 518 | 0.77 | 0.87 | 0.12 | 0.34 |
| 1,8-BPA | 514 | 540 | 514 | 531 | 0.71 | 0.80 | 0.15 | 0.12 |
| 2,6-BPA | 515 | 528 | 504 | 532 | 0.88 | 0.92 | 0.15 | 0.17 |
| 9,10-BPA | 528 | 537 | 514 | 519 | 0.02 | 0.02 | 0.79 | 0.86 |
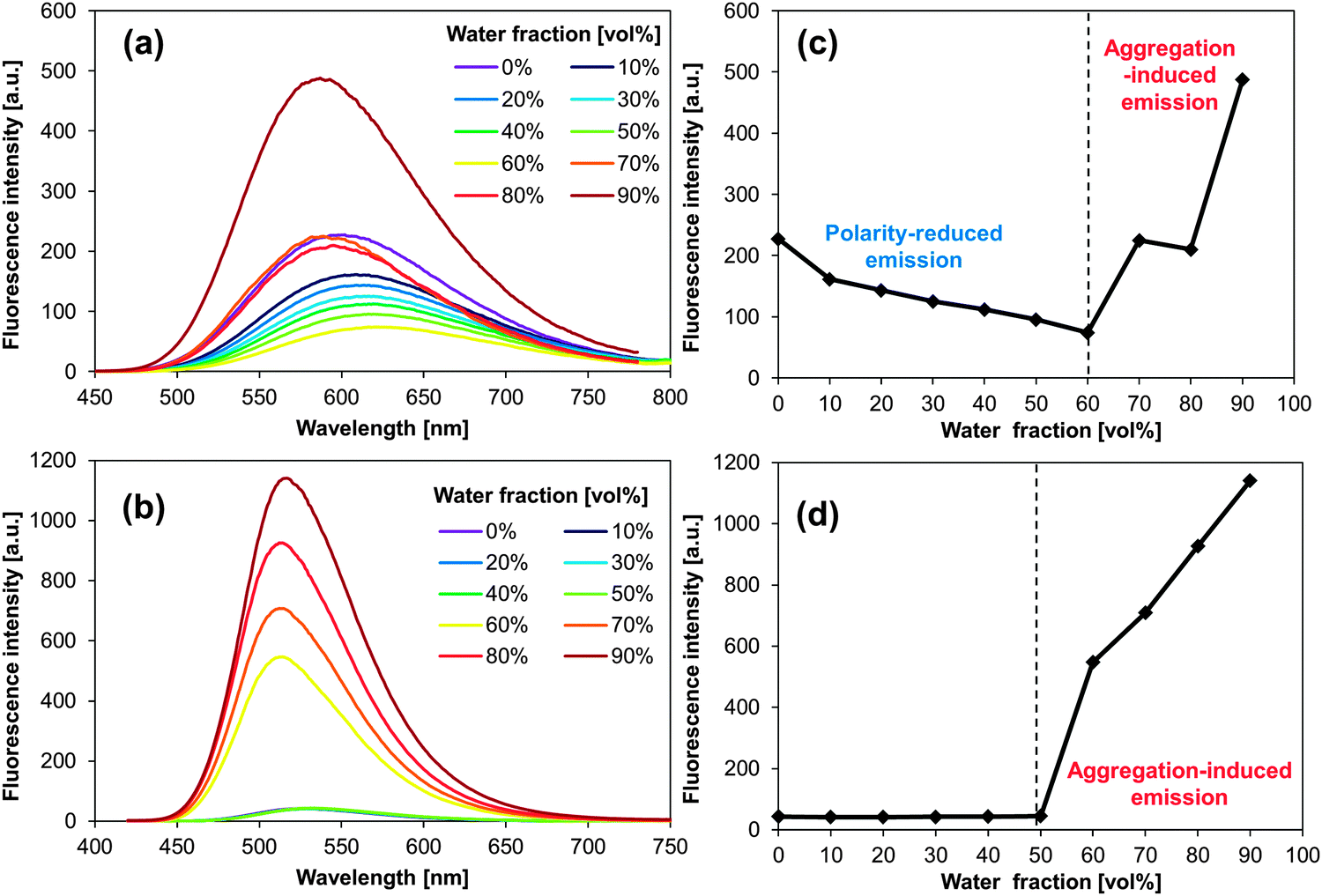 | ||
| Fig. 2 Fluorescence spectra of (a) 1,4-BPA and (b) 9,10-BPA in THF–water mixtures. Maximum fluorescence intensity of (c) 1,4-BPA and (d) 9,10-BPA as a function of water fraction [vol%]. | ||
Effects of aggregation on the radiative and non-radiative transition pathways of PAs and BPAs
In contrast to the other regioisomers, only para-substituted BPAs such as 1,4-BPA and 9,10-BPA exhibit AIE. In order to understand this unusual phenomenon, the correlation between aggregation (or crystallisation) and radiative or non-radiative transition pathways of PAs and BPAs should be addressed, as well as the reasons for the high fluorescence of 1,4-BPA and 9,10-BPA in the solid state.Firstly, it is important to understand how aggregation changes the fluorescent species in PAs and BPAs. The relationship between the aggregation state of the solids (i.e. colloidal suspension, or neat powder) and the fluorescence quantum yield should provide an insight into the fluorescent species in the solid state. If the aggregation-induced emission of 1,4-BPA and 9,10-BPA involves highly delocalised excitons, as reported for anthracenes and typical aromatic hydrocarbons, the fluorescence quantum yields should strongly depend on the conditions of the solids.5 However, the fluorescence quantum yields for almost all PAs and BPAs (except 9-PA) remain essentially unaffected by the condition of the solid (Table 1). Therefore, the fluorescent excitons of 1,4-BPA and 9,10-BPA are likely to be localised on a single, or very few chromophores.
Interestingly, 9-PA exhibits strong fluorescence (Φfl ≈ 0.71) only as a neat powder, but not in a suspension. The X-ray crystallographic analysis from a single crystal of 9-PA revealed a herringbone structure (Fig. S62, ESI†), wherein the anthracene moieties adopt a configuration that avoids π-stacking, which is disadvantageous with respect to the formation of strong intermolecular interactions and Frenkel excitons.38 However, given the lack of spectroscopic data for large and high-quality single crystals, which should be necessary to discuss the solid-state fluorescence properties of 9-PA accurately, we refrained from addressing this question herein.
Accordingly, the diffuse-reflectance spectra of the PA and BPA derivatives (Fig. S45–S52, ESI†) are comparable to their absorption spectra, and peaks associated with delocalised Frenkel excitons, i.e. J- or H-aggregates, were not observed.39 Thus, it seems likely that the aggregation does not significantly influence the fluorescent species in PAs and BPAs. Given that aggregation does not strongly affect radiative pathways, the aggregation effect on the non-radiative transition pathways of PAs and BPAs should be more important. In order to analyse the effect of aggregation on radiative and non-radiative transition rates, fluorescence lifetime measurements were carried out. However, as shown in Table S1 (ESI†), the fluorescence lifetimes measured in colloidal suspensions consisted of two components. Therefore, the lifetimes of bi-exponential decays have to be approximated to a single component to derive the transition rate constants. Sillen et al. have reported that the amplitude average lifetime is an effective parameter to derive the transition rate constants of multi-exponential decays caused by the exposure of fluorescent molecules to different environments.40 As colloidal suspensions contain at least two separate environments (i.e. the aggregation and solution state), the amplitude average lifetime was used to derive the transition rate constants. The derived radiative transition rate constants (kf) remain constant at ∼3–8 × 107 s−1, independent of the sample conditions such as solvent polarity or aggregation. On the other hand, the non-radiative transition rate constants (knr) for the aggregation state of PA (4–9 × 107 s−1) and BPA (2–3 × 108 s−1) are an order of magnitude higher than the corresponding knr values in solution (PA: 3–9 × 106 s−1; BPA: 0.4–5 × 107 s−1). The exceptions here are 1,4-BPA and 9,10-BPA, which exhibit exclusively AIE; their knr values (∼2–3 × 107 s−1) suggest that their non-radiative transitions are significantly slower than those of other BPA derivatives (see Table S1, ESI†).
Thus, it is feasible to conclude that 1,4-BPA and 9,10-BPA operate a fluorescence mechanism that avoids the non-radiative transition pathway, which is intrinsic to solid-state PAs and BPAs. Due to this unique mechanism, aggregation only serves to restrict the intramolecular vibrations, which trigger AIE phenomena.1,2
Relaxation of the excited state in 1,4-BPA and 9,10-BPA
In order to extend this method and prepare a variety of simple but bright AIE luminogens based on 1,4-BPA and 9,10-BPA, a concrete design strategy must be established. However, prior to doing this, the intrinsic non-radiative transition pathway of BPAs and the mechanism by which 1,4-BPA and 9,10-BPA overcome deactivation should be determined.Usually, non-radiative transition pathways in the solid state arise from energy transfers involving Forster or Dexter mechanisms,6,41 excimer or exciplex formation,6 the presence of polaronic species,42 or self-absorption.5 However, the photophysical data obtained in this study were unable to provide consistent and/or direct evidence for a specific mechanism underlying the fluorescence quenching of PA and BPA derivatives in the solid state. For instance, if we assume that the non-radiative pathway occurs by self-absorption, a colloidal suspension should experience a stronger bathochromic shift of its fluorescence relative to those of diluted and non-polar solutions. Even though 1,5-BPA and 1,8-BPA follow this paradigm, suspensions of 2,6-BPA exhibit a more hypsochromically shifted fluorescence emission than those of the solutions (Fig. S35–S44, ESI†). Accordingly, it is hardly debatable that further studies, focusing on a detailed analysis of the photophysical processes in the solid state, are required.
Nevertheless, the efficiency of the non-radiative transitions can be elucidated from X-ray crystallographic analyses and from the photophysical properties of dilute solutions. Fig. 3 shows the molecular structures of 1,4-BPA, 1,5-BPA, and 9,10-BPA, obtained from single crystal X-ray diffraction studies. It is widely accepted that undesirable interchromophoric interactions can be suppressed by separating chromophores spatially. Hence, a commonly employed design principle for the enhancement of solid-state emission is based on the use of bulky substituents and multiple aryl–aryl bonds.6,7 However, BPAs lack such substituents and bonds, and the anthracene chromophores are packed in such close proximity that Dexter-type energy transfers should be possible (i.e. interchromophoric distance ≤10 Å).41 Even though molecules of 1,4-BPA are in close proximity in the crystal structure, their orthogonal packing may minimise interchromophoric interactions, while simultaneously enhancing the fluorescence brightness (Fig. 3a). As the unit cells of the 9,10-BPA crystals lack a molecule at the body-centred position, 9,10-BPA molecules are packed in a face-to-face manner and arranged more closely compared to 1,4-BPA and 1,5-BPA (Fig. 3b). Thus, when only the electronic interaction with neighbouring chromophores is considered, 9,10-BPA is more likely to undergo fluorescence quenching than other BPAs. However, close packing is, at the same time, favourable to supress internal conversion. Recent theoretical studies43 demonstrated that a close packing in the solid state effectively restricts a low-frequency vibrational mode, which is strongly coupled with the reorganization energy that plays a pivotal role in fast internal conversions. This hypothesis can be applied to the AIE behaviour of 9,10-BPA: provided that the internal conversion rate of 9,10-BPA is, as for 9,10-diphenylanthracene,44 governed by a low-frequency mode such as a C–N rotation, it is reasonable to assume that the close packing of 9,10-BPA leads to a slow non-radiative transition. To quantitatively analyse the effect of the close packing, we will conduct experimental and theoretical investigations using model compounds.
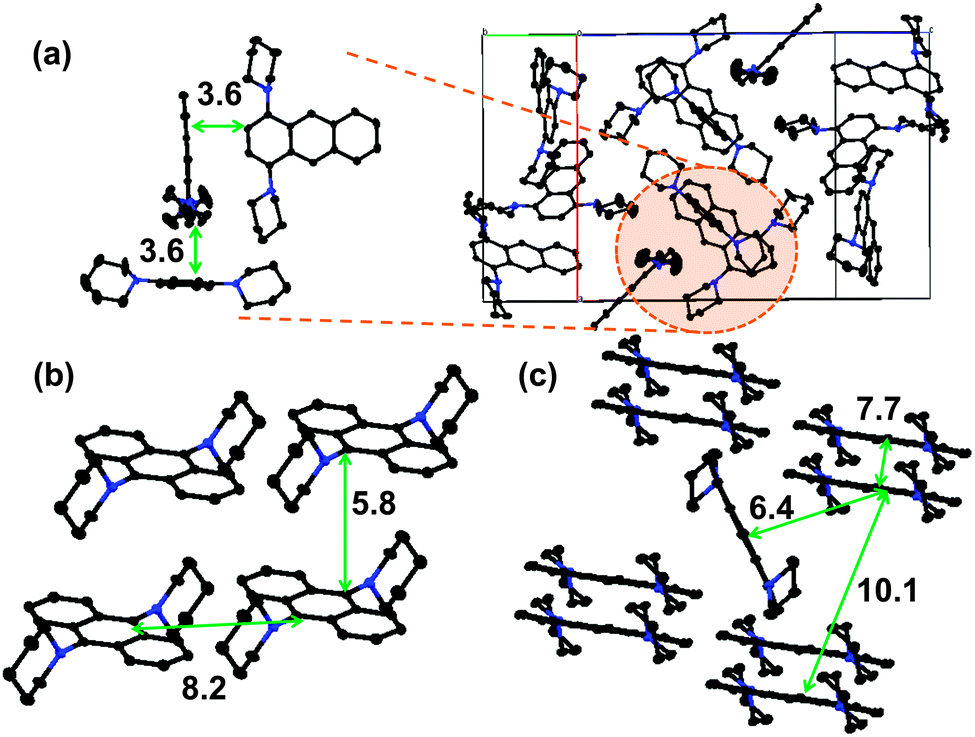 | ||
| Fig. 3 Molecular packing in the unit cell of (a) 1,4-BPA, (b) 9,10-BPA, and (c) 1,5-BPA. The number values refer to the closest distances (in Å) between neighbouring chromophores. | ||
It is noteworthy that the shape of the fluorescence spectra (Fig. S53–S60, ESI†) for several PAs and BPAs significantly depends on the aggregation condition of the solid (i.e. colloidal suspension, 1 mM in NaBr, or neat powder), which is indicative of their mechanofluorochromic properties. However, this spectral change is less pronounced for AIE-active 1,4-BPA and 9,10-BPA. To impart these AIE-active dyes with mechanofluorochromic properties, we designed 1,4-BPA and 9,10-BPA analogues with a bulkier dialkylamine group.45
The photophysical properties of dilute solutions, especially the Stokes shifts, are also relevant to the non-radiative transition rates in the solid state.47 Large Stokes shifts imply a marginal overlap between the absorption and fluorescence spectra, which prevents energy transfer41 and self-absorption. Fig. 4 shows a plot of the Stokes shifts of some PA and BPA derivatives in toluene, THF, and DMF as a function of solvent polarity parameter (fL) of each solvent.48 Since the Stokes shifts and other spectroscopic features of PA are similar to those of 1-, 2-, and 9-aminoanthracenes49 and their dimethylamine analogs,21,50 absorption and fluorescence spectra were assigned accordingly. While S0 → S1 absorptions (Fig. S27 and S33, ESI†) of 1-PA and 9-PA stem from symmetry-allowed mixing between the 1La state of anthracene51 and the 1CT (singlet charge transfer) state, a more red-shifted S0 → S1 absorption (Fig. S31, ESI†) is observed for 2-PA, which results from the mixing between the optically forbidden 1Lb state of anthracene and its 1CT state.51 Thus, the resultant Stokes shift of 2-PA is smaller than 1-PA despite small steric hindrance against C–N rotation. As the character of the S0–S1 absorption/fluorescence is very different for each PA, the effect of introducing a second piperidine group should be evaluated by comparison with the corresponding PA. As shown in Fig. 4, the Stokes shifts of 1,5-BPA, 1,8-BPA, and 2,6-BPA are smaller than those of the corresponding monosubstituted analogues, i.e.1-PA and 2-PA. This decreased Stokes shift can be ascribed to the symmetric excited-state dipole moment of 1,5-BPA, 1,8-BPA, and 2,6-BPA, as their Stokes shifts are less dependent on solvent polarity than those of the PAs. Conversely, 1,4-BPA and 9,10-BPA exhibited Stokes shifts, which are significantly larger than those of 1-PA and 9-PA, even though the Stokes shifts of these BPAs are equally less dependent on solvent polarity than corresponding PAs. In dilute solution, the Stokes shift of a fluorophore is determined by dielectric interactions with solvent molecules (outer-sphere reorganisation) and the deformation of solute molecules induced by excited-state energy relaxation (inner-sphere reorganisation). When considering small degrees of solvatochromism for BPAs into account, prominent Stokes shifts of 1,4-BPA and 9,10-BPA should arise from their large inner-sphere reorganization energy. It can therefore be concluded that only the second piperidyl groups at the para-position, in e.g.1,4-BPA and 9,10-BPA, contribute significantly to the excited-state energy relaxation and hence induce a larger Stokes shift.
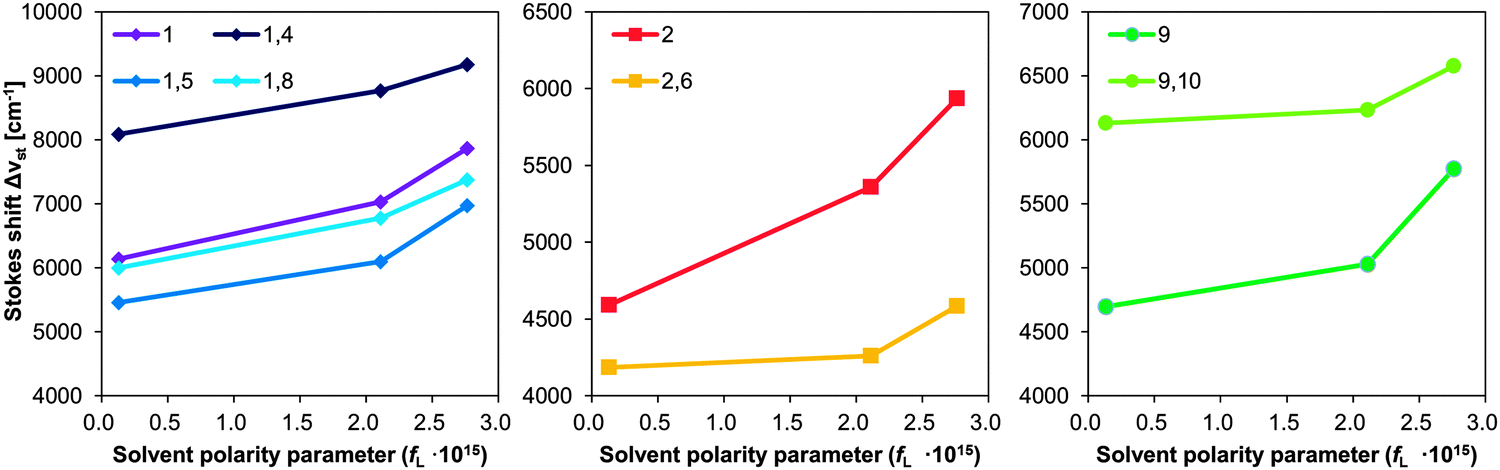 | ||
| Fig. 4 Stokes shifts of PA and BPA derivatives plotted against the solvent polarity parameter (fL).24 The solvent polarity parameter is obtained from the following equation: fL = 2Δf/hc, Δf = (ε − 1)/(2ε + 1) − (n2 − 1)/(2n2 + 1), where c refers to the speed of light [cm s−1], h to the Planck constant [erg s], n to the refractive index,46 and ε to the dielectric constant of each solvent.46 | ||
In order to gain a better understanding of the origin of the large Stokes shifts in 1,4-BPA, we performed time-dependent density functional theory (TD-DFT) calculations for 1,4-BPA and 1,5-BPA at the ωB97X-D/6-311G(d,p) level of theory (see ESI†) and the results are summarised in Fig. 5. The calculated HOMOs and LUMOs for both 1,4-BPA and 1,5-BPA are derived from the anthracene 1b2g and 1b3u orbital, respectively.52 Thus, the HOMO–LUMO transitions in these BPAs, which lead to the first singlet excited states S1, are based on the S0–1La transition (π–π* character) of anthracene53 (Tables S7 and S8, ESI†).
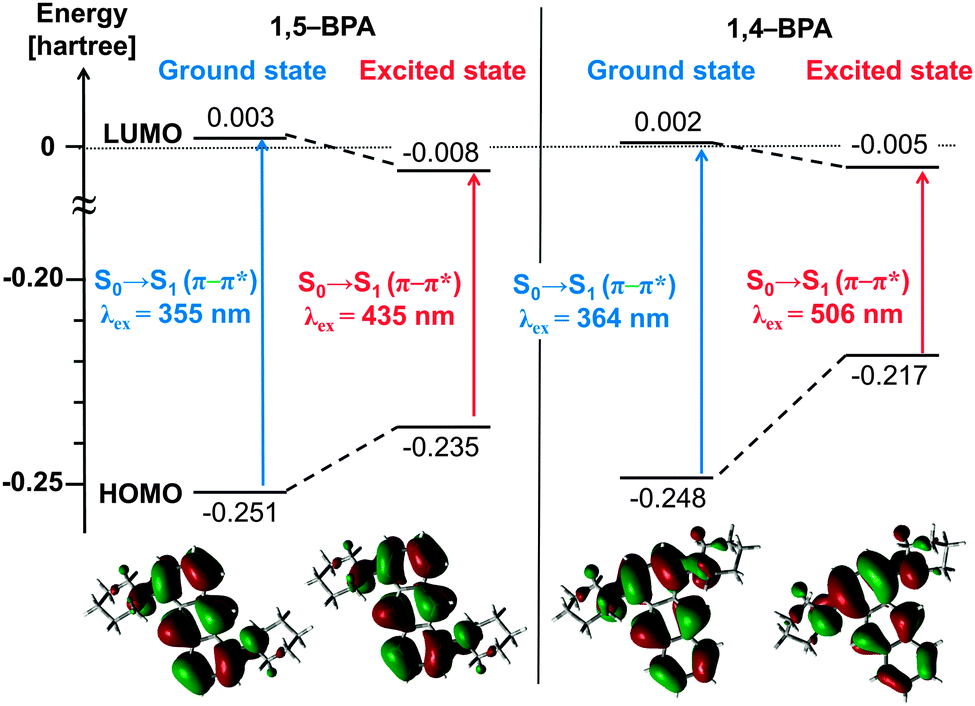 | ||
| Fig. 5 TD-DFT calculations for the energy levels of the HOMO and LUMO of the equilibrium structures for the ground (S0) and excited state (S1), as well as the corresponding wavelengths for the S0–S1 transitions (top); Kohn–Sham orbitals for the HOMO of each structure. All calculations were carried out at the ωB97X-D/6-311G(d,p) level of theory. | ||
The Stokes shifts for the S0–S1 transition in 1,4-BPA are significantly larger than for that in 1,5-BPA. The distribution of the HOMO does not show any significant change upon relaxation of 1,5-BPA, whereas the HOMO of 1,4-BPA clearly converges on the substituted benzene ring of the anthracene moiety after the structural relaxation. It can thus be concluded that the large Stokes shift of 1,4-BPA originates from the destabilisation of the HOMO, which is caused by the strong electronic repulsion between two piperidyl groups in close spatial proximity.
For the evaluation of the structural change induced by the inner-sphere reorganisation, we calculated the change of the dihedral angle between the anthracene plane (defined by atoms 4, 12, and 14; Tables S3–S6, ESI†) and the piperidyl group (defined by atoms 14, 18, and 20; Tables S3–S6, ESI†). For 1,5-BPA, dihedral angles of 51° (S0) and 44° (S1) were obtained, while angles of 52° (S0) and 39° (S1) were obtained for 1,4-BPA. Twisted intramolecular charge-transfer (TICT) states often display excited-state species with large Stokes shifts,54 and their inner-sphere reorganisation energy is comparable to that of 1,4-BPA. However, the inner-sphere reorganisation of TICT dyes demands a significantly larger change of the torsion angle between the donor and acceptor.55,56 Notably, the change of the torsion angle from S0 geometry to S1 geometry is only moderate for 1,4-BPA (13°), which suggests that 1,4-BPA can, in contrast to TICT dyes, complete the inner-sphere reorganisation even under restricted mobility in the solid state. In fact, the restricted environment in the solid state prohibits the formation of the TICT state, which frequently results in AIE,12,57 albeit concomitant with decreased Stokes shifts due to the limited inner-sphere reorganisation in the excited state. This trade-off relationship in TICT dyes requires elaborate and prudent design strategies.34 Given that electronic repulsion is able to induce large Stokes shifts with a minimal structural relaxation, twisted donors in spatial proximity such as in 1,4-BPA represent a simple, yet viable option to simultaneously obtain effective AIE and large Stokes shifts.
Although we performed TD-DFT calculations for 9,10-BPA as well, the results were so complicated that we were unable to interpret the underlying photophysical processes comprehensively. Considering its crystal structure, the mechanism for the solid-state emission and AIE in 9,10-BPA may be different from that in 1,4-BPA. Future studies will investigate the photophysical properties of 9,10-BPA by spectroscopic measurements, the synthesis of model compounds, and computational calculations.
Conclusion
In this study, we synthesised various regioisomers of piperidylanthracene (PA) and bis(piperidyl)anthracene (BPA), and evaluated their photophysical properties. The result revealed that only 1,4-BPA and 9,10-BPA exhibit AIE, and that their fluorescence colour can be tuned by changing the relative position of the piperidyl groups.Results from diffuse-reflectance spectroscopy and fluorescence lifetime measurements demonstrated that PA and BPA intrinsically possess an efficient non-radiative transition pathway in the solid state, and that 1,4-BPA and 9,10-BPA may overcome this pathway. While the molecular packing of 1,4-BPA in the crystalline state exhibited weak interchromophoric interactions, that of 9,10-BPA is indicative of the importance of suppression of intramolecular vibrations. Spectroscopic measurements in dilute solutions indicated that the Stokes shifts of 1,4-BPA and 9,10-BPA are considerably larger than those of their corresponding PA analogues. Accordingly, the spectroscopic characteristics of 1,4-BPA and 9,10-BPA are suitable for the suppression of self-absorption and energy transfer in the solid state. TD-DFT calculations corroborated that the large Stokes shift of 1,4-BPA originates from a drastic destabilisation of the HOMO, induced by a minimal structural relaxation in the excited state.
Nevertheless, several important points, such as e.g. the intrinsic quenching process of PAs and BPAs in the solid state, the mechanism, by which 9,10-BPA overcomes its quenching path with a close face-to-face packing, as well as the relaxation process of the excited state of 9,10-BPA still remain to be revealed. Investigations in these directions are currently in progress in our laboratory, and we also aim to apply the excellent photophysical properties of 1,4-BPA and 9,10-BPA in various functional molecules modifying dialkylamine sunstituents.
Experimental section
Instruments
1H NMR and 13C NMR spectra were recorded on a 400 MHz JEOL LMN-EX400 or a 300 MHz BRUKER DPX300 spectrometer using tetramethylsilane (TMS) as the internal standard. FT-IR spectra were recorded on a JASCO FT-IR 469 plus spectrometer. Melting points were recorded on a Yanaco micro melting point apparatus MP-500P. Thermogravimetric analyses were carried out on a TG/DTA 6200 SII Seiko Instruments Inc. EXSTAR 6000 thermal analyzer at a heating rate of 5 °C min−1 under a nitrogen atmosphere. MS spectra (FAB+) were obtained using a JEOL JMS700 mass spectrometer.All photophysical measurements performed in solution were carried out using dilute solutions with optical densities (ODs) around 0.1 at the maximum absorption wavelength in 1 cm path length quartz cells at room temperature (298 K). In addition, toluene, THF, and DMF solutions of all samples were deaerated by bubbling with argon gas for 15 min prior to the measurements. For photophysical measurements of the aggregate state, colloidal suspensions (THF:H2O = 1:9; v/v) with a concentration of 1.0 × 10−4 M were prepared, and aggregate formation was confirmed by the severe broadening of the UV-vis spectra. All photophysical measurements of these colloidal suspensions were conducted immediately after the aggregate formation in order to avoid experimental errors due to surface oxidation. UV-vis spectra were recorded on a JASCO V-670 UV-vis spectrophotometer. Fluorescence spectra were recorded on a JASCO FP-6500 spectrofluorometer. The wavelengths obtained by the fluorescence spectrometer were converted to the wavenumber using the equation I(![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ) = λ2I(λ).58 Absolute quantum yields were measured using a Hamamatsu Photonics Quantaurus QY apparatus. Fluorescence lifetime values were obtained from the most intense peaks using a Hamamatsu Photonics OB 920 fluorescence lifetime spectrometer equipped with an LED lamp (343 nm).
) = λ2I(λ).58 Absolute quantum yields were measured using a Hamamatsu Photonics Quantaurus QY apparatus. Fluorescence lifetime values were obtained from the most intense peaks using a Hamamatsu Photonics OB 920 fluorescence lifetime spectrometer equipped with an LED lamp (343 nm).
Diffuse-reflectance spectra
Diffuse-reflectance spectra were recorded on a JASCO FP-6500 spectrofluorometer equipped with an integration sphere detector. Thus, the experimental error arising from the fluorescence, typically encountered with diffuse-reflectance spectrometers producing polychromatic outgoing light, was avoided. Samples and references were charged in a JASCO powder sample cell to obtain a sufficiently thick powder layer. Just before each measurement, the synchronous spectrum of the NaBr powder was measured as a reflectance spectrum of a standard reference rstandard(λ). Then, a reflectance spectrum of each sample rsample(λ) was obtained following the same procedure. In addition to neat powder samples, the spectra of samples adsorbed on the NaBr powder were measured at a concentration of 1.0 × 10−3 M. The obtained reflectance spectra rsample(λ) and rstandard(λ) were converted to Kubelka–Munk functions f(r∞) using the following equation:59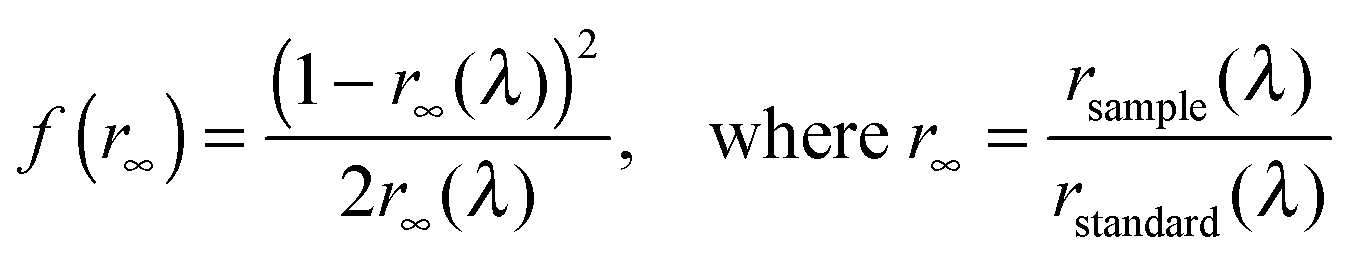
All diffuse-reflectance spectra are displayed as plots of the Kubelka–Munk functions, i.e. f(r∞) as a function of the wavelength, λ.
DLS measurement
All of DLS measurements were performed on a Wyatt DynaPro NanoStar. Autocorrelation functions attained from DLS measurement (Fig. S18, ESI†) clearly indicated the formation of aggregates in THF:H2O = 1:9 (v/v), and the autocorrelation functions of all sample suspension are smooth and continuous, exhibiting exponential decay from their maxima to a value of 1. Blank solutions (THF:H2O = 1:9; v/v) did not show any significant autocorrelation.
The particle size distribution (Fig. S26–S29, ESI†) was calculated by a regularisation analysis60 of the autocorrelation functions. Bimodal distributions were observed for 2-PA and 1,8-BPA, while other PA and BPA regioisomers exhibited unimodal distributions. Percentage polydispersities (i.e. the width of each peak normalised to the mean size of the peak) of all peaks were smaller than 20%, and hydrodynamic radii Rh are included in the particle size distributions.
Materials
Unless otherwise noted, all reagents and chemicals were used as received without further purification. 1-Bromoanthracene, 2-bromoanthracene, 9-bromoanthracene, 1,4-diaminoanthraquinone, 1,5-dibromoanthracene, 1,8-dibromoanthracene, 2,6-dibromoanthracene, 9,10-dibromoanthracene, sodium tert-butoxide and (±)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) were obtained from TCI (Tokyo, Japan). The PEPPSI™-IPr catalyst was purchased from Sigma-Aldrich Japan (Tokyo, Japan). Palladium(II)diacetate and dehydrated 1,4-dioxane were purchased from Wako Pure Chem (Tokyo, Japan). Dehydrated THF and copper(II)bromide were obtained from Kanto Chem (Tokyo, Japan). Spectrograde toluene, THF, DMF, magnesium sulphate and sodium carbonate were purchased from Nacalai Tasque (Kyoto, Japan). Pure water used for photophysical measurements was prepared using a Milli-Q® water purification system (Merck Millipore, United States).Synthesis and characterization of PAs and BPAs
1H NMR spectra and 13C NMR spectra are summarized in the ESI.†:hexane = 2:3 as the eluent to yield crude 1-PA. The crude product thus obtained was further purified by recrystallisation from hexane to afford 1-PA (0.30 g) as a pale yellow solid. Yield: 98%; mp: 83.4–84.1 °C; 1H NMR (400 MHz, CDCl3): δ 8.74 (s, 1H), 8.39 (s, 1H), 8.04 (dd, 4J = 2.9 Hz, 3J = 6.0 Hz, 1H), 7.97 (dd, 4J = 2.9 Hz, 3J = 6.0 Hz, 1H), 7.68 (d, 3J = 8.4 Hz, 1H), 7.44 (dd, 4J = 2.9 Hz, 3J = 6.4 Hz, 2H), 7.39–7.35 (m, 1H), 6.99 (d, 3J = 7.1 Hz, 1H), 3.13 (br, 4H), 1.92 (m, 4H), 1.72 (m, 2H) ppm (Fig. S1, ESI†); 13C NMR (100 MHz, CDCl3): δ 151.0 (Ar), 133.0 (Ar), 131.5 (ArN(CH2CH2CH2CH2CH2)), 131.2 (Ar), 128.8 (Ar), 127.9 (Ar), 127.8 (Ar), 126.5 (Ar), 125.4 (Ar), 125.3 (Ar), 125.0 (Ar), 123.1 (Ar), 122.6 (Ar), 113.0 (Ar), 54.6 (ArN(CH2CH2CH2CH2CH2)), 26.7 (ArN(CH2CH2CH2CH2CH2)), 24.7 (ArN(CH2CH2CH2CH2CH2)) ppm (Fig. S2, ESI†); FT-IR (KBr): 2933 (–CH2– stretch), 1615 (Ar ring stretch), 1537 (Ar ring stretch), 1401 (Ar–N stretch), 735 (Ar–H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C19H19N) requires m/z 261.1517, found m/z 261.1512.
:hexane = 1:4; (2) chloroform:hexane = 1:1), followed by recrystallisation of the crude product from hexane to afford 1,4-BPA (0.28 g) as a yellow solid. Yield: 83%; mp: 225.3–228.7 °C (decomposition onset at T = 200 °C); 1H NMR (300 MHz, CDCl3): δ 8.78 (s, 2H), 8.05 (dd, 4J = 3.2 Hz, 3J = 6.4 Hz, 2H), 7.47 (dd, 4J = 3.2 Hz, 3J = 6.4 Hz, 2H), 6.96 (s, 2H), 3.27 (br, 8H), 1.92 (m, 8H), 1.72 (br, 4H) ppm (Fig. S3, ESI†); 13C NMR (75 MHz, CDCl3): δ 146.8 (Ar), 131.2 (Ar), 129.2 (ArN(CH2CH2CH2CH2CH2)), 128.7 (Ar), 125.2 (Ar), 122.9 (Ar), 113.3 (Ar), 55.0 (ArN(CH2CH2CH2CH2CH2)), 26.9 (ArN(CH2CH2CH2CH2CH2)), 24.9 (ArN(CH2CH2CH2CH2CH2)) ppm (Fig. S4, ESI†); FT-IR (KBr): 2935 (–CH2– stretch), 1610 (Ar ring stretch), 1335 (Ar–N stretch), 745 (Ar–H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C24H28N2) requires m/z 344.2252, found m/z 344.2259.
:hexane = 1:1), followed by recrystallisation from hexane to afford 1,5-BPA (0.47 g) as a yellow solid. Yield: 91%; mp: 248.5–249.4 °C; 1H NMR (300 MHz, CDCl3): δ 8.68 (s, 2H), 7.71 (dd, 4J = 0.9 Hz, 3J = 8.4 Hz, 2H), 7.36 (dd, 3J = 7.2 Hz, 3J = 8.4 Hz, 2H), 6.97 (dd, 4J = 0.9 Hz, 3J = 7.2 Hz, 2H), 3.13 (br, 8H), 1.93–1.88 (m, 8H), 1.71 (br, 4H) ppm (Fig. S5, ESI†); 13C NMR (100 MHz, CDCl3): δ 150.9 (Ar), 133.7 (Ar), 127.9 (ArN(CH2CH2CH2CH2CH2)), 125.2 (Ar), 123.9 (Ar), 123.1 (Ar), 113.0 (Ar), 54.7 (ArN(CH2CH2CH2CH2CH2)), 26.8 (ArN(CH2CH2CH2CH2CH2)), 24.9 (ArN(CH2CH2CH2CH2CH2)) ppm (Fig. S6, ESI†); FT-IR (KBr): 2934 (–CH2– stretch), 1614 (Ar ring stretch), 1540 (Ar ring stretch), 1348 (Ar–N stretch), 733 (Ar–H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C24H28N2) requires m/z 344.2252, found m/z 344.2259.
:hexane = 2:3), followed by recrystallisation from hexane to yield 1,8-BPA (0.41 g) as a yellow solid. Yield: 80%; mp: 248.1–249.2 °C (the initially yellow solid turned into a white crystalline solid at 247.5 °C); 1H NMR (300 MHz, CDCl3): δ 9.17 (s, 1H), 8.34 (s, 1H), 7.65 (d, 3J = 8.4 Hz, 2H), 7.36 (dd, 3J = 7.1 Hz, 3J = 8.4 Hz, 2H), 6.97 (d, 3J = 7.1 Hz, 2H), 3.14 (br, 8H), 1.95–1.93 (m, 8H), 1.72 (br, 4H) ppm (Fig. S7, ESI†); 13C NMR (100 MHz, CDCl3): δ 151.6 (Ar), 133.0 (Ar), 127.5 (ArN(CH2CH2CH2CH2CH2)), 126.9 (Ar), 125.5 (Ar), 123.0 (Ar), 119.1 (Ar), 113.1 (Ar), 54.9 (ArN(CH2CH2CH2CH2CH2)), 27.1 (ArN(CH2CH2CH2CH2CH2)), 25.0 (ArN(CH2CH2CH2CH2CH2)) ppm (Fig. S8, ESI†); FT-IR (KBr): 2938 (–CH2– stretch), 1615 (Ar ring stretch), 1562, 1448, 1332 (Ar–N stretch), 1244, 896, 743 (Ar–H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C24H28N2) requires m/z 344.2252, found m/z 344.2259. These characterization data are consistent with those reported in the literature.28a
:hexane = 1:3), followed by recrystallisation from hexane to afford 2-PA (0.32 g) as a pale yellow solid. Yield: 63%; mp: 168.1–169.1 °C (lit. 162–164 °C (ref. 63)); 1H NMR (400 MHz, CDCl3): δ 8.27 (s, 1H), 8.19 (s, 1H), 7.92 (dd, 3J = 7.8 Hz, 3J = 7.8 Hz, 2H), 7.86 (d, 3J = 9.3 Hz, 1H), 7.42–7.32 (m, 3H), 7.17 (d, 4J = 2.0 Hz, 1H), 7.39–7.35 (m, 1H), 6.99 (d, 3J = 7.1 Hz, 1H), 3.30 (t, 3J = 5.4 Hz, 4H), 1.82–1.76 (m, 4H), 1.67–1.63 (m, 2H) ppm (Fig. S9, ESI†); 13C NMR (100 MHz, CDCl3): δ 149.5 (Ar), 133.3 (Ar), 132.4 (Ar), 130.3 (Ar), 129.0 (Ar), 128.3 (Ar), 128.1 (Ar), 127.7 (Ar), 125.9 (Ar), 125.3 (Ar), 124.1 (Ar), 123.9 (Ar), 121.8 (Ar), 108.4 (Ar), 51.0 (ArN(CH2CH2CH2CH2CH2)), 26.1 (ArN(CH2CH2CH2CH2CH2)), 24.5 (ArN(CH2CH2CH2CH2CH2)) ppm (Fig. S10, ESI†); FT-IR (KBr): 2932 (–CH2– stretch), 1625 (Ar ring stretch), 1483 (Ar ring stretch), 1344 (Ar–N stretch), 741 (Ar–H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C19H19N) requires m/z 261.1517, found m/z 261.1523.
:hexane = 1:3), followed by recrystallisation from hexane to afford 9-PA (0.088 g) as a pale yellow solid. Yield: 29%; mp: 145.2–146.0 °C; 1H NMR (300 MHz, CDCl3): δ 8.52–8.48 (m, 2H), 8.27 (s, 1H), 8.01–7.96 (m, 2H), 7.48–7.40 (m, 4H), 3.47 (t, 3J = 5.0 Hz, 4H), 1.92–1.77 (m, 6H) ppm (Fig. S13, ESI†); 13C NMR (100 MHz, CDCl3): δ 145.8 (Ar), 132.6 (Ar), 130.5 (Ar), 128.8 (Ar), 125.1 (Ar), 125.0 (Ar), 124.7 (Ar), 124.3 (Ar), 53.1 (ArN(CH2CH2CH2CH2CH2)), 27.5 (ArN(CH2CH2CH2CH2CH2)), 24.9 (ArN(CH2CH2CH2CH2CH2)) ppm (Fig. S14, ESI†); FT-IR (KBr): 2939 (–CH2– stretch), 1623 (Ar ring stretch), 1491 (Ar ring stretch), 735 (Ar–H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C19H19N) requires m/z 261.1517, found m/z 261.1524.
:ethyl acetate = 2:1) to afford 9,10-BPA (310 mg) as a yellow solid. Yield: 60%; 1H NMR (300 MHz, CDCl3): δ 8.50 (dd, 4J = 3.4 Hz, 3J = 6.8 Hz, 4H), 7.43 (dd, 4J = 3.3 Hz, 3J = 6.8 Hz, 4H), 3.54 (t, 3J = 4.5 Hz, 8H), 1.86–1.79 (m, 12H) ppm (Fig. S15, ESI†); 13C NMR (75 MHz, CDCl3): δ 143.8 (ArN(CH2CH2CH2)), 131.6 (Ar), 125.8 (Ar), 124.6 (Ar), 53.2 (ArN(CH2CH2CH2CH2CH2)), 27.8 (ArN(CH2CH2CH2CH2CH2)), 25.1 (ArN(CH2CH2CH2CH2CH2)) ppm (Fig. S16, ESI†); FT-IR (KBr): 2936 (–CH2– stretch), 2847 (–CH2– stretch), 1619 (Ar ring stretch), 1401 (Ar–N stretch), 771 (Ar–H) cm−1; HRMS (FAB+) exact mass calculated for [M]+ (C24H28N2) requires m/z 344.2252, found m/z 344.2262. The melting point could not be determined due to decomposition at 280 °C.
References
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- (a) L. Porres, A. Holland, L. O. Palsson, A. P. Monkman, C. Kemp and A. Beeby, J. Fluoresc., 2006, 16, 267–272 CrossRef CAS PubMed; (b) Y. Kawamura, H. Sasabe and C. Adachi, Jpn. J. Appl. Phys., 2004, 43, 7729–7730 CrossRef CAS.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961–2965 CAS.
- M. Shimizu and T. Hiyama, Chem. – Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed.
- S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS PubMed.
- (a) W. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed; (b) J. Huang, N. Sun, P. Chen, R. Tang, Q. Li, D. Ma and Z. Li, Chem. Commun., 2014, 50, 2136–2138 RSC.
- J. Mei, J. Wang, J. Z. Sun, H. Zhao, W. Yuan, C. Deng, S. Chen, H. H. Y. Sung, P. Lu, A. Qin, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, Chem. Sci., 2012, 3, 549–558 RSC.
- (a) H. Wang, Y. Huang, X. Zhao, W. Gong, Y. Wang and Y. Cheng, Chem. Commun., 2014, 50, 15075–15078 RSC; (b) S. Y. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed; (c) N. B. Shustova, T. C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dinca, J. Am. Chem. Soc., 2012, 134, 15061–15070 CrossRef CAS PubMed; (d) A. Battisti, F. Tantussi, F. Fuso, M. Allegrini, G. Ruggeri and A. Pucci, Macromol. Rapid Commun., 2014, 215, 499–506 CrossRef PubMed; (e) X. Yao, X. Ma and H. Yuan, J. Mater. Chem. C, 2014, 2, 5155–5160 RSC.
- (a) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed; (b) X. Zhang, X. Zhang, L. Tao, Z. Chi, J. Xu and Y. Wei, J. Mater. Chem. B, 2014, 2, 4398–4414 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Sun, J. Liu, Z. Li, S. Zhang, J. Sun, H. S. Kwok and B. Z. Tang, Appl. Phys. Lett., 2007, 91, 011111 CrossRef PubMed.
- (a) L. Zhu and Y. Zhao, J. Mater. Chem. C, 2013, 1, 1059–1065 RSC; (b) R. Wei, Y. He, X. Wang and P. Keller, Macromol. Rapid Commun., 2014, 35, 1571–1577 CrossRef CAS PubMed.
- (a) X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem., 2011, 21, 1788–1796 RSC; (b) X. Zhang, Z. Chi, J. Zhang, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. B, 2011, 115, 7606–7611 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- (a) G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC; (b) M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- M. Chen, L. Li, H. Nie, J. Tong, L. Yan, B. Xu, J. Z. Sun, W. Tian, Z. Zhao, A. Qin and B. Z. Tang, Chem. Sci., 2015, 6, 1932–1937 RSC.
- (a) Z. Zhao, S. Chen, X. Shen, F. Mahtab, Y. Yu, P. Lu, J. W. Y. Lam, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 686–688 RSC; (b) Z. Zhao, P. Lu, Z. Wang, C. Y. K. Chan, H. H. Y. Sung, I. D. Williams, Y. Ma and B. Z. Tang, Chem. Sci., 2011, 2, 672–675 RSC.
- S. Sasaki, Y. Niko, K. Igawa and G. Konishi, RSC Adv., 2014, 4, 33474–33477 RSC.
- E. Hasegawa, S. Takizawa, T. Seida, A. Yamaguchi, N. Yamaguchi, N. Chiba, T. Takahashi, H. Ikeda and K. Akiyama, Tetrahedron, 2006, 62, 6581–6588 CrossRef CAS PubMed.
- J. Dey and I. M. Warner, J. Phys. Chem. A, 1997, 101, 4872–4878 CrossRef CAS.
- (a) D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC; (b) J. P. Wolfe, S. Wagaw and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 7215–7216 CrossRef CAS.
- (a) M. G. Organ, M. Abdel-Hadi, S. Avola, I. Dubivyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien, M. Sayah and C. Valente, Chem. – Eur. J., 2008, 14, 2443–2452 CrossRef CAS PubMed; (b) Y. Suzuki, N. Fukui, K. Murakami, H. Yorimitsu and A. Osuka, Asian J. Org. Chem., 2013, 2, 1066–1071 CrossRef CAS PubMed.
- Y. Chung, B. F. Duerr, T. A. Mckelvey, P. Nanjappan and A. W. Czarnik, J. Org. Chem., 1989, 54, 1018–1032 CrossRef CAS.
- T. W. Campbell, V. E. Mccoy, J. C. Kauer and V. S. Foldi, J. Org. Chem., 1961, 26, 1422–1426 CrossRef.
- The only report relevant to 1,4-BPA describes 1,4-diaminoanthracene and its UV-vis spectrum; see: E. A. Venediktov, V. A. Perfil'ev and B. D. Berezin, Russ. J. Phys. Chem., 1985, 59, 1824–1826 Search PubMed.
- The synthesis of a dimethylamine analogue of 1,5-BPA was reported, but unfortunately photophysical properties were not described; see: T. Harada, T. Hiramatsu and T. Yamaji, Synth. Commun., 1981, 11, 379–384 CrossRef CAS PubMed.
- The synthesis of 1,8-BPA was previously reported, albeit in the absence of any photophysical measurements. UV-vis spectra are only available for 1,8-diaminoanthracene; see: (a) I. P. Beletskaya, A. G. Bessemertnykh, A. D. Averin, F. Denat and R. Guilard, Eur. J. Org. Chem., 2005, 281–305 CrossRef CAS PubMed; (b) A. Dahan, T. Ashkenazi, V. Kuznetsov, S. Makievski, E. Drug, L. Fadeev, M. Bramson, S. Schokory, E. R. Kemelmakher and M. Gozin, J. Org. Chem., 2007, 72, 2289–2296 CrossRef CAS PubMed.
- (a) The synthesis of a dimethylamine analogue of 2,6-BPA was reported, albeit in the absence of any photophysical measurements. UV-vis spectra are only available for 2,6-diaminoanthracene; see: F. Keller and C. Ruchardt, J. Prakt. Chem., 1998, 340, 642–648 CrossRef CAS PubMed; (b) W. S. Trahanovsky, J. L. Tunkel, J. C. Thoen and Y. Wang, J. Org. Chem., 1995, 60, 8407–8409 CrossRef CAS.
- The synthesis of a dimethylamine analogue of 9,10-BPA was reported, and the photophysical properties were described as “non-fluorescent”; see: ref. 20.
- (a) M. P. Aldred, C. Li and M. Q. Zhu, Chem. – Eur. J., 2012, 18, 16037–16045 CrossRef CAS PubMed; (b) A. J. Qin, L. Tang, J. W. Y. Lam, C. K. W. Jim, Y. Yu, H. Zhao, J. Z. Sun and B. Z. Tang, Adv. Funct. Mater., 2009, 19, 1891–1900 CrossRef CAS PubMed.
- K. T. Wong, Y. Y. Chien, R. T. Chen, C. F. Wang, Y. T. Lin, H. H. Chiang, P. Y. Hsieh, C. C. Wu, C. H. Chou, Y. O. Su, G. H. Lee and S. M. Peng, J. Am. Chem. Soc., 2002, 124, 11576–11577 CrossRef CAS PubMed.
- (a) A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273–4276 CrossRef CAS PubMed; (b) M. Shimizu, Y. Takeda, M. Higashi and T. Hiyama, Angew. Chem., Int. Ed., 2009, 355, 3707–3710 CrossRef PubMed; (c) R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed.
- R. Hu, E. Lager, A. A. Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. P. Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CAS.
- (a) H. Deng, B. Liu, C. Yang, G. Li, Y. Zhuang, B. Li and X. Zhu, RSC Adv., 2014, 4, 62021–62029 RSC; (b) C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeil, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS PubMed.
- (a) L. Schermelleh, R. Heintzmann and H. Leonhardt, J. Cell Biol., 2010, 190, 165–175 CrossRef CAS PubMed; (b) B. Huang, Curr. Opin. Chem. Biol., 2010, 14, 10–14 CrossRef CAS PubMed; (c) E. A. J. Erijiman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387–1395 CrossRef PubMed.
- (a) H. Schill, S. Nizamov, F. Bottanelli, J. Bierwagen, V. N. Belov and S. W. Hell, Chem. – Eur. J., 2013, 19, 16556–16565 CrossRef CAS PubMed; (b) Y. Yang, M. Lowry, X. Xu, J. O. Escobedo, M. S. Vazquez, L. Wong, C. M. Schowalter, T. M. Jensen, F. R. Fronczek, I. M. Warner and R. M. Strongin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8829–8834 CrossRef CAS PubMed; (c) P. Horvath, P. Sebej, T. Solomek and P. Klan, J. Org. Chem., 2015, 80, 1299–1311 CrossRef CAS PubMed; (d) J. F. Araneda, W. E. Piers, B. Heyne, M. Parvez and R. McDonald, Angew. Chem., Int. Ed., 2011, 50, 12214–12217 CrossRef CAS PubMed; (e) Y. Y. Wu, Y. Chen, G. Z. Gou, W. H. Mu, X. J. Lv, M. L. Du and W. F. Fu, Org. Lett., 2012, 14, 5226–5229 CrossRef CAS PubMed.
- (a) D. W. Schlosser and M. R. Philpott, Chem. Phys., 1980, 49, 181 CrossRef CAS; (b) H. Yamagata, J. Norton, E. Hontz, Y. Oliver, D. Beljonne, J. L. Bredas, R. J. Silbey and F. C. Spano, J. Chem. Phys., 2011, 134, 204703 CrossRef CAS PubMed.
- (a) R. Steiger, R. Pugin and J. Heier, Colloids Surf., B, 2009, 74, 484–491 CrossRef CAS PubMed; (b) H. B. Fu, Y. Q. Wang and J. N. Yao, Chem. Phys. Lett., 2000, 322, 327–332 CrossRef CAS.
- A. Sillen and Y. Engelborghs, Photochem. Photobiol., 1998, 67, 475–486 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, 2010, pp. 388–413 Search PubMed.
- (a) O. G. Reid, R. D. Pensack, Y. Song, G. D. Scholes and G. Rumbles, Chem. Mater., 2014, 26, 561–571 CrossRef CAS; (b) A. K. Thomas, J. A. Garcia, J. U. Sanchez, J. Gao and J. K. Grey, ACS Nano, 2014, 8, 10559–10568 CrossRef CAS PubMed.
- (a) C. Deng, Q. Peng, Y. Niu and Z. Shuai, J. Comput. Chem., 2012, 33, 1862–1869 CrossRef PubMed; (b) Q. Wu, Q. Peng, Y. Niu, X. Gao and Z. Shuai, J. Phys. Chem. A, 2012, 116, 3881–3888 CrossRef CAS PubMed; (c) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- A. P. Darmanyan, Chem. Phys. Lett., 1982, 91, 396–400 CrossRef CAS.
- X. Zhang, Z. Chi, B. Xu, C. Chen, X. Zhou, Y. Zhang and S. Liu, J. Mater. Chem., 2012, 22, 18505–18513 RSC.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, Taylor & Francis, Boca Raton, 2006, pp. 536–540 Search PubMed.
- A. Iida and S. Yamaguchi, Chem. Commun., 2009, 3002–3004 RSC.
- (a) E. L. Lippert, Organic Molecular Photophysics, Wiley, New York, 1975 Search PubMed; (b) N. Mataga and T. Kubota, Molecular Interaction and Electronic Spectra, Dekker, New York, 1970 Search PubMed; (c) A. Fin, A. V. Jentzsch, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2012, 51, 12736–12739 CrossRef CAS PubMed.
- (a) K. Rotkiewicz and Z. R. Grabowski, Trans. Faraday Soc., 1969, 65, 3263–3278 RSC; (b) M. Tichy and R. Zahradnik, J. Phys. Chem., 1969, 73, 534–544 CrossRef CAS.
- S. Watanabe, K. Kumagai, M. Hasegawa, M. Kobayashi, J. Okubo, T. Yoshinaga, H. Hiratsuka and T. Hoshi, Bull. Chem. Soc. Jpn., 2000, 73, 1783–1789 CrossRef CAS.
- R. P. Steiner and J. Michl, J. Am. Chem. Soc., 1978, 100, 6861–6867 CrossRef CAS.
- S. Zilberg, Y. Haas and S. Shaik, J. Chem. Phys., 1995, 99, 16558–16565 CrossRef CAS.
- N. Nijegorodov and R. Mabbs, Spectrochim. Acta, Part A, 2000, 56, 2157–2166 CrossRef.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed.
- T. Tahara and H. Hamaguchi, Chem. Phys. Lett., 1994, 217, 369 CrossRef CAS.
- (a) A.-D. Gorse and M. Pesquer, J. Phys. Chem., 1995, 99, 4039–4049 CrossRef CAS; (b) C. J. Jodicke and H. P. Luthi, J. Am. Chem. Soc., 2003, 125, 252–264 CrossRef PubMed; (c) T. Taniguchi, J. Wang, S. Irie and S. Yamaguchi, Dalton Trans., 2013, 42, 620–624 RSC.
- (a) J. Li, Y. Qian, L. Xie, Y. Yi, W. Li and W. Huang, J. Phys. Chem. C, 2015, 119, 2133–2141 CrossRef CAS; (b) Y. Qian, M.-M. Cai, L. H. Xie, G.-Q. Yang, S.-K. Wu and W. Huang, ChemPhysChem, 2011, 12, 397–404 CrossRef CAS PubMed.
- L. R. Lakowicz, Principles of Fluorescence Spectroscopy Third Edition, Springer Science+Business Media, LLC, New York, 2006, p. 54 Search PubMed.
- G. Kortüm, W. Braun and G. Herzog, Angew. Chem., Int. Ed., 1963, 2, 333–341 CrossRef PubMed.
- B. Chu, Laser Light Scattering Basic Principles and Practice Second Edition, Dover Publications, Mineola, 2007 Search PubMed.
- S. K. Lee, W. J. Yang, J. J. Choi, C. H. Kim, S.-J. Jeon and B. R. Cho, Org. Lett., 2005, 7, 323–326 CrossRef CAS PubMed.
- S. E. Wheeler, A. J. McNeil, P. Muller, T. M. Swager and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 3304–3311 CrossRef CAS PubMed.
- A. Richardson, K. R. Brower and E. D. Amstutz, J. Org. Chem., 1956, 21, 890–891 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data, quantum chemical calculations, and X-ray crystal structure analyses. CCDC 1055226–1055229. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc00946d |
| This journal is © The Royal Society of Chemistry 2015 |