
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and electrical properties of layered perovskite type Pr2Ti2O7: experimental and theoretical investigations†
Sadequa J.
Patwe
a,
Vasundhara
Katari
a,
Nilesh P.
Salke
b,
Sudhanshu K.
Deshpande
c,
Rekha
Rao
b,
Mayanak K.
Gupta
b,
Ranjan
Mittal
b,
S. Nagabhusan
Achary
*a and
Avesh K.
Tyagi
*a
aChemistry Division, Bhabha Atomic Research Centre, Mumbai 400085, India. E-mail: sachary@barc.gov.in; acharysn@rediffmail.com; aktyagi@barc.gov.in; Fax: +91-22-25505151; Tel: +92-22-25592328
bSolid State Physics Division, Bhabha Atomic Research Centre, Mumbai 400085, India
cUGC-DAE Consortium for Scientific Research, BARC, Mumbai 400085, India
First published on 31st March 2015
Abstract
In this communication we report the details of the structural and thermal properties of monoclinic layered perovskite type Pr2Ti2O7 (PTO) using ambient to higher temperature XRD and Raman spectroscopic studies. The monoclinic (P21) structure is found to be the stable structure of PTO compared to the orthorhombic Pna21, Cmc21 or Cmcm and monoclinic P21/m structures. The crystal structure is further supported by the ab initio total energy calculations using density functional theory (DFT) formalism. The total energy calculation and structural relationship favour the ferroelectric (P21) to paraelectric (P21/m) displacive transition. The calculated electric polarization as observed from the displacement of ions is ∼8.3 μC cm−2. The calculated electron density of states indicated a band gap of about 2.7 eV, which closely agrees with that measured by UV-Vis diffuse reflectance spectroscopy. Variable temperature XRD and differential thermal analysis studies revealed no structural transition to Cmc21 in the temperature range from ambient to 1473 K as reported for analogous rare-earth titanates, like La2Ti2O7 and Nd2Ti2O7. A partial decomposition of PTO to cubic perovskite type structure is observed at around 1673 K. The measurement of field dependent electric polarization indicates the ferroelectric nature of PTO. The electrical properties of PTO have also been investigated by ac impedance spectroscopic studies from 173 to 1073 K. The low temperature dielectric data indicate two different types of relaxations, one at a lower frequency region and strongly temperature dependent while the other at a higher frequency region (>1 kHz) and nearly temperature independent. The low and high frequency relaxations have been attributed to the thermally activated polarization process arising from the grain boundaries and dipolar orientations, respectively. The activation energy for a thermally activated low frequency relaxation process is 0.38 eV, which is similar to the interfacial polarizations due to ionic movements. An appreciable contribution of ionic conductivity in PTO is observed at still higher temperature (∼700 K). The activation energy for ionic conductivity is about 0.60 eV.
1. Introduction
A2B2O7 type compositions with A = trivalent rare-earth ions and B = tetravalent ions, like Ti4+, Zr4+, Hf4+ and Sn4+, have been of interest due to their interesting crystal chemistry and several technological applications based on their dielectric, ferroelectric and piezoelectric as well as ionic conductivity. Depending on the radius of the cations, the A2B2O7 type compositions of rare-earth ions can be grouped into three categories, namely, (a) fluorite (rA/rB < 1.48), (b) pyrochlore (1.48 < rA/rB < 1.78) and (c) perovskite (rA/rB > 1.78) structure types.1,2 For Ti4+ as the B site cation, the A2B2O7 type rare-earth (Ln) titanates form monoclinic perovskite type structure for A = La, Nd and Pr while all others form cubic pyrochlore type structures.3 The monoclinic perovskite type titanates have attracted attention due to their high Curie temperature, ferroelectric and piezoelectric properties, high dielectric constants, nonlinear optical and photocatalytic properties.4–12 Early studies on phase diagrams in Ln2O3–TiO2 systems by Shcherbakova et al.3 had revealed that both monoclinic or cubic Ln2Ti2O7 phases have higher melting temperature (>1450 °C). Nanamatsu et al.6 have reported ferroelectric and nonlinear optical activity in monoclinic La2Ti2O7. From the high temperature dielectric studies, Yan et al.4 have suggested that the ferroelectric to paraelectric transition temperature of La2Ti2O7 and Nd2Ti2O7 is above 1073 K. It has been reported that the octahedral network and interstitial cations of Ln2Ti2O7 play a significant role in their electronic as well as dielectric properties.11,13–15Despite the structural similarity in all the three compositions, viz. A2Ti2O7 (A = La, Nd and Pr), much research attention has been devoted to La2Ti2O7 or Nd2Ti2O7 while Pr2Ti2O7 is still under explored. Among the monoclinic rare-earth titanates Pr2Ti2O2 and Ce2Ti2O7 are likely to have exceptional behaviour due to intrinsic defects arising from the variable valence states of Pr and Ce. Recently, Atuchin et al.16 have reported spectroscopic and electronic properties of Pr2Ti2O7. The investigation of the optical and dielectric properties of Pr2Ti2O7 indicates higher non-linear optical activity and a dielectric constant of about 27–32.13 Literature also shows that the ferroelectric transition of Pr2Ti2O7 depends on the nature of sample preparation. The ferroelectric transition at around 740 °C has been reported in the spark plasma sintered sample of Pr2Ti2O7.7 Sun et al. have reported coupled magnetism and ferroelectric properties in nanocrystalline Pr2Ti2O7, where the magnetism is associated with the oxygen vacancies while the ferroelectricity has been assigned to the structural features.17 The frequency and temperature dependent dielectric studies of Sun et al.17 indicated an anomaly near the suggested ferroelectric transition temperature (570 K) and the lowering of Tc has been claimed for the crystallite size effects. This value of reported transition temperature for Pr2Ti2O7 is the lowest temperature reported for any of the monoclinic Ln2Ti2O7 phases.3,4,7,17
Though such titanates have been of technological importance, their structure and phase transitions are still obscure in literature. The structure of Ln2Ti2O7 (Ln = La and Nd) has been explained by a monoclinic (P21) lattice.3,18–20 However, a closely related orthorhombic (Pna21) has also been assigned for the structure of La2Ti2O7.21,22 In addition Scheunemann & Müller-Buschbaum22 and Harvey et al.23 have reported a monoclinic super-structure with P21 symmetry for the exact crystal structure of Nd2Ti2O7. High temperature structural studies on La2Ti2O7 indicate a structural transition from P21 to Cma21 above 1053 °C and both low and high temperature phases differ by the shift of the rare-earth ions as well as distortion and tilt of the TiO6 octahedral units.24 From the structural analyses the authors have indicated that the polarization in these structures arises mainly from shift of the rare-earth ions and partly from the rotation of the deformed TiO6 octahedra. Recently, Ishizawa et al.25 have investigated the crystal structure of Nd2Ti2O7 and indicated that the sub-cell represents the actual structure of Nd2Ti2O7 which is related to the earlier reported super-structure by the differences in the shift of cations along one of the axes, i.e. along the polarization direction. From density functional calculations, Pruneda et al.,26 Zhang et al.27 and Xiao et al.28 have reported the structure and elastic properties of the cubic phase of La2Ti2O7 and Nd2Ti2O7, while such cubic phases have never been observed experimentally. Theoretical analysis of Lopez-Perez and Iniguez15 indicated that the transformation from Cmc21, the real high temperature structure of La2Ti2O7, to a paraelectric (Cmcm) structure is related to the shift of cations and distortion of octahedra and is a topological ferroelectric transition.15 Similar studies on La2Ti2O7 and Nd2Ti2O7 by Bruyer and Sayede14 indicated that the monoclinic (P21) structure is the stable phase under ambient conditions. This study also indicates that the transformation from the ferroelectric to paraelectric phase may occur as P21 to P21/m structural transition. However, the prototype P21/m or Cmcm phases have never been observed experimentally for any of these titanates up to melting temperature. Besides these studies there are no other reports available in literature to confirm the dielectric properties as well as high temperature structural properties of Pr2Ti2O7. In view of this we have prepared Pr2Ti2O7 and investigated the structure and electrical properties in a wider range of temperature and frequency.
2. Experimental
The polycrystalline sample of Pr2Ti2O7 was prepared by solid state reaction of appropriate amounts of Pr6O11 and TiO2. Pr6O11 was preheated at 1073 K to remove any adsorbed moisture. TiO2 was heated at 1273 K to remove any adsorbed moisture or hydroxyl groups. About 7 g of the homogenous mixture was prepared by mixing stoichiometric amounts of the reactants in acetone media. The mixed powder was pressed into pellets (about 1 inch diameter and 5 mm height) and slowly heated to 1173 K and held for 24 h. The pellet obtained after cooling to room temperature was rehomogenized and heated at 1373 K for another 24 h. The completion of the reaction is ensured after this heating step. The pellet is then rehomogenized and then pressed into pellets of about 1 cm diameter and 2 mm height. These pellets were sintered at 1573 K for 24 h and cooled at the rate of 2 K min−1 to ambient temperature. Well sintered pellets obtained after this heat treatment were used for all the studies.The product obtained after the final sintering step was characterized by powder XRD data recorded on a rotating anode based X-ray diffractometer (Ragaku, Japan). The powder sample of Pr2Ti2O7 was pressed into a groove of about 1 mm of a glass sample holder and XRD data were collected from 5 to 100° with a step width of 0.02° and time per step is 5 s. The XRD patterns at several temperatures in between ambient to 1000 °C were recorded on a powder X-ray diffractometer (X-pert Pro Panalytical) equipped with Anton Parr high temperature attachment. The well ground sample was smeared on a platinum strip sample holder-cum-heater. The sample is heated to a desired temperature at a rate of 20 K min−1 and held for 5 m for equilibration. The HT-XRD patterns were recorded in the two-theta range of 10–80° with a step width and a step time of 0.02° and 1.5 s, respectively. Monochromatized Cu-Kα radiation was used for recording the XRD data at ambient and higher temperature. The powder XRD data were analyzed by the Rietveld refinement method using GSAS and Fullprof 2000 software packages.29,30
Raman spectroscopic measurements in the temperature range of 298–1073 K were carried out by using the Linkam variable temperature stage (Model-TS 1500) in back-scattering geometry. The spectrum of the polycrystalline sample of Pr2Ti2O7 was excited using a 532 nm laser line with a power of 15 mW. Scattered light was analyzed by using a home built 0.9 m single monochromator, coupled with an edge filter and detected by a cooled CCD.31 The entrance slit was kept at 50 μm, which gives a spectral band pass of 3 cm−1.
Thermal stability and oxidation behavior of the sample in dry oxygen and argon were studied by a simultaneous thermogravimetric (TG) and differential thermal analyses (DTA) method. The TG and DTA traces were recorded while heating about 100 mg of sample to 1673 K on a thermobalance (SETARAM Instrumentation, France). Typical heating rate 10 K min−1 was used for recording the TG-DTA data. The residue obtained after the TG-DTA runs was also characterized by the powder XRD study.
The microstructure of sintered samples was studied by using a scanning electron microscope (Zeiss, Germany). The micrographs at different magnifications were recorded on both top and fractured surfaces of sintered pellets.
The electric field dependent electric polarizations (PE-loops) were recorded at ambient temperature by using an Aixacct TF2000 (Aixacct GmbH, Germany) ferroelectric analyzer. Polarization data were recorded by using platinum paste coated pellets of the sample (2 mm thickness).
The UV-visible (UV-Vis) diffuse reflectance spectrum was recorded on a two-beam spectrometer (V-670, JASCO) using a fine powder sample.
For impedance measurements, platinum paste coated on two parallel faces of cylindrical pellets were used. The dielectric properties of the pellets were measured in a parallel-plate capacitor configuration using flat gold-plated electrodes. Low temperature (173 to 473 K) impedance measurements were carried out by using a Novocontrol Alpha AN impedance analyzer (Novocontrol Technologies, Germany) and a Quatro nitrogen cryosystem. The complex dielectric data were recorded at a series frequency range of 100 Hz to 5 MHz at several temperatures while heating from 173 K to 473 K. The electrical properties between 300 and 1073 K were investigated by using an impedance analyzer (Solartron impedance analyzer, Model 1290). In both cases, the rms bias voltage of 0.5 V was used for data collection. The impedance data were measured in the frequency range of 1 MHz to 1 Hz at several temperatures while heating the sample. The dielectric and impedance data were analyzed by using Winfit and Z-view software packages.
3. Density functional calculations
The total energy calculations for various structural models of Pr2Ti2O7 have been carried out by using ab initio density functional theory (DFT). The calculations were performed by using the projector-augmented wave formalism32 of the Kohn–Sham formulation of the density-functional theory (DFT)33,34 at the generalized gradient approximation (GGA) level, implemented in the Vienna ab initio simulation package (VASP).35,36 The GGA was formulated by the Perdew–Burke–Ernzerhof (PBE) density functional approximations.37,38 All results are well converged with respect to k mesh and energy cutoff for the plane-wave expansion. The total energy calculations have been carried out by using a cutoff of 820 eV of plane wave kinetic energy. k-points mesh was generated according to the Monkhorst–Pack (MP) scheme.37–39 The break conditions for the self-consistent field and for the ionic relaxation loops were set to 10−8 eV Å-1 and 10−4 eV Å−1, respectively. The Hellmann–Feynman forces following the geometry optimization were less than 10−4 eV Å−1. The numbers of valence electrons used in oxygen, titanium and praseodymium atomic pseudo-potentials are 6, 4 and 11, respectively. Full geometry optimizations along with unit cell parameters were carried out on various structural models of Pr2Ti2O7. The variation of energy with volume at 0 K and electron density of states (dos) was calculated for all the five relaxed structures.4. Results and discussion
The observed reflections of the powder XRD data of the Pr2Ti2O7 (PTO) sample obtained after final sintering are quite similar to the reported monoclinic (JC-PDS-PDF: 35-0267) as well as orthorhombic (JC-PDS-PDF: 35-0224) phases. As mentioned in the introduction section the structure of PTO and analogous rare-earth titanates has been explained by diversified symmetries, viz. monoclinic (P21 and P21/m) and orthorhombic (Pna21, Cmc21 and Cmcm). The ferroelectric phase of PTO is likely to have P21, Pna21 or Cmc21 symmetry, while the paraelectric phase is likely to have monoclinic P21/m or orthorhombic Cmcm symmetry.20,21,24,32 It can be mentioned here that all the structures are closely related and hence their XRD patterns show only marginal differences. In order to elucidate the structure of PTO, we have considered the reported structural details of monoclinic (P21) of PTO and orthorhombic (Pna21 and Cmc21) symmetries of La2Ti2O7. Initial models for other structures were obtained by structural transformations as shown in Fig. 1. Thus the Rietveld refinements of the powder X-ray diffraction (XRD) were carried by considering all the possible models based on Ln2Ti2O7 and related structure types, viz. P21, P21/m. Pna21, Cmc21 and Cmcm symmetries. The peaks and background of the diffraction pattern were modeled by using a pseudo-Voigt profile function and a shifted Chebyschev polynomial function, respectively. Initially background parameters along with the scale were refined and unit cell parameters, half-width (U, V and W) and mixing (η) parameters of pseudo-Voigt function and peak asymmetry corrections were added in the subsequent refinement cycles. All the intense peaks could be explained by any of the considered models. However the intensities of some weak peaks, in particular peaks at two theta ∼35.0, 37.5°, favour orthorhombic (Pna21)21 and monoclinic (P21)23 structures compared to the orthorhombic Cmc21 or Cmcm structures.20 The refinement of the position coordinates of all the possible models was subsequently carried out to further ascertain the structure of PTO. The goodness of fit was monitored from the residual (R-values) of refinements and differences between the observed and calculated diffraction patterns. It can be mentioned here that the monoclinic unit cell reported by Scheunemann and Müller-Buschbaum22 and Harvey et al.23 was also considered for refinements. The refinements with this monoclinic super-structure model indicate closely similar residuals while the unit cell parameter is doubled along the c-axis. Despite the structure consisting of twice the number of atoms in the asymmetric unit and having a larger unit cell, no better residuals as well as profile could be obtained in the super-structure model of monoclinic P21 structure. Hence, this model was not considered for a further study. Also, the Pna21 symmetry has larger unit cell parameters and should exhibit extra reflections as well as detectable intensity at 2θ – 12.4 and 23.5°. The present observed powder XRD pattern is not in agreement with these aspects. Additionally, a weak peak observed at 2θ – 14.4° is attributable only to P21 symmetry. Thus we support the monoclinic P21 structure as the actual structure of PTO.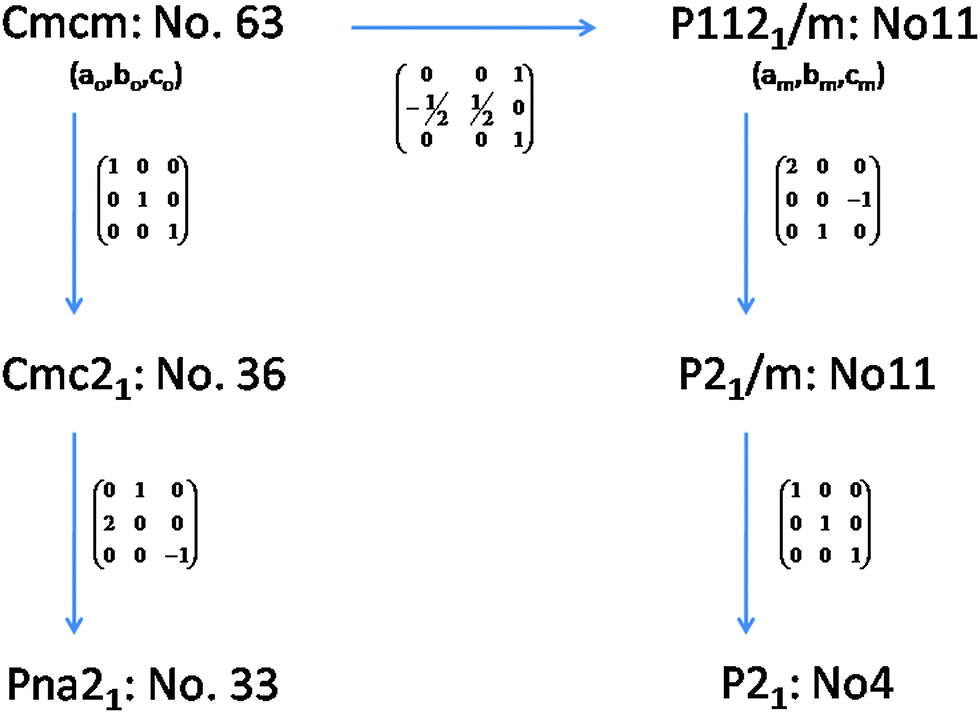 | ||
| Fig. 1 Relations between different model structures of Pr2Ti2O7. | ||
The refined unit cell parameters and the residuals of the refinements with different structural models are summarized in Table 1. A comparison of the residuals obtained in different models indicates that the monoclinic (P21) structure with unit cell parameters: a = 7.7152(1), b = 5.4878(1), c = 13.0042(2) Å and β = 98.552(4)° is the best model for the observed diffraction data. The refined position coordinates and typical inter-atomic distances for the monoclinic (P21) structure are given in Tables 2 and 3, respectively. The typical Rietveld refinement plot of the final cycle for P21 structure is shown in Fig. 2. The refined structural parameters and Rietveld refinement plots for other considered models are provided as ESI† (S-I and S-II) to this manuscript.
| P21 (No. 4) | Pna21 (No. 33) | Cmc21 (No. 36) | P21/m (No. 11) | Cmcm (No. 63) | |
|---|---|---|---|---|---|
| a (Å) | 7.7152(1) | 25.7197(4) | 3.8575(1) | 7.7146(2) | 3.8573(1) |
| b (Å) | 5.4878(1) | 7.7151(1) | 25.7194(5) | 5.4874(1) | 25.7197(6) |
| c (Å) | 13.0042(2) | 5.4877(1) | 5.4877(1) | 13.0036(4) | 5.4872(1) |
| β (°) | 98.552(4) | 98.531(7) | |||
| V | 544.47(2) | 1088.92(3) | 544.46(2) | 544.40(3) | 544.38(2) |
| Z | 4 | 8 | 4 | 4 | 4 |
| ρ | 5.973 g cc−1 | 5.973 g cc−1 | 5.973 g cc−1 | 5.974 g cc−1 | 5.974 g cc−1 |
| R p | 0.0616 | 0.0804 | 0.0834 | 0.1192 | 0.1066 |
| R wp | 0.0775 | 0.1001 | 0.1084 | 0.1496 | 0.1433 |
| χ 2 | 3.758 | 5.899 | 6.791 | 10.64 | 10.72 |
| R F 2 | 0.0630 | 0.0892 | 0.1008 | 0.1856 | 0.1603 |
| a | 7.7540 | 26.3100 | 3.8502 | 7.8665 | 3.92706 |
| b | 5.5610 | 7.7550 | 25.6992 | 5.5450 | 26.39951 |
| c | 13.3010 | 5.5600 | 5.7720 | 13.3455 | 5.54500 |
| β (°) | 98.436 | 98.529 | |||
| E/atom (eV) | −8.6630 | −8.6630 | −8.6526 | −8.6166 | −8.6009 |
| Atoms | Wyc. | x | y | z | 100 × Uiso (Å2) |
|---|---|---|---|---|---|
| Space group: P21 (No. 4), Z = 4 (Rp: 6.16, Rwp: 7.75, χ2 = 3.76; RF2 = 6.29). a = 7.7152(1), b = 5.4878(1), c = 13.0042(2) Å, β = 98.552(4), V = 544.47(1) Å3. a = 7.7540, b = 5.5610, c = 13.3010 Å, β = 98.47° (DFT results). | |||||
| Pr1 | 2a | 0.2841(8) | 0.2539(24) | 0.11325(20) | 2.21(7) |
| 0.2815 | 0.2536 | 0.1218 | |||
| Pr2 | 2a | 0.7763(9) | 0.2559(6) | 0.09940(21) | 2.49(7) |
| 0.7706 | 0.2651 | 0.0887 | |||
| Pr3 | 2a | 0.3557(9) | 0.7988(5) | 0.39211(21) | 3.22(8) |
| 0.3506 | 0.7958 | 0.3813 | |||
| Pr4 | 2a | 0.8526(9) | 0.8421(5) | 0.41616(22) | 3.46(9) |
| 0.8543 | 0.8666 | 0.4288 | |||
| Ti1 | 2a | 0.0370(10) | 0.7619(7) | 0.1215(4) | 0.71(19) |
| 0.0315 | 0.7699 | 0.1198 | |||
| Ti2 | 2a | 0.5455(9) | 0.7682(7) | 0.1200(4) | 0.37(18) |
| 0.5282 | 0.7680 | 0.1222 | |||
| Ti3 | 2a | 0.0724(10) | 0.2972(8) | 0.3247(4) | 1.63(25) |
| 0.0719 | 0.2970 | 0.3199 | |||
| Ti4 | 2a | 0.5668(9) | 0.2934(7) | 0.3226(4) | 0.33(20) |
| 0.5847 | 0.3043 | 0.3253 | |||
| O1 | 2a | 0.7859(10) | 0.8481(10) | 0.1082(16) | 2.58(17) |
| 0.7774 | 0.8209 | 0.1097 | |||
| O2 | 2a | 0.2886(10) | 0.7248(10) | 0.0969(15) | 2.58(17) |
| 0.2729 | 0.6774 | 0.0921 | |||
| O3 | 2a | 0.0279(50) | 0.0213(9) | 0.0160(8) | 2.58(17) |
| 0.0439 | 0.0251 | 0.0199 | |||
| O4 | 2a | 0.4802(46) | 0.0578(9) | 0.0302(7) | 2.58(17) |
| 0.4667 | 0.0243 | 0.0217 | |||
| O5 | 2a | 0.0151(35) | 0.9512(9) | 0.2357(8) | 2.58(17) |
| 0.1171 | 0.9647 | 0.2280 | |||
| O6 | 2a | 0.4881(26) | 0.9621(9) | 0.2235(8) | 2.58(17) |
| 0.4967 | 0.9610 | 0.2307 | |||
| O7 | 2a | 0.0447(65) | 0.4576(9) | 0.1842(8) | 2.58(17) |
| 0.0223 | 0.4512 | 0.1828 | |||
| O8 | 2a | 0.5776(46) | 0.4591(9) | 0.1822(8) | 2.58(17) |
| 0.5674 | 0.4476 | 0.1810 | |||
| O9 | 2a | 0.0734(50) | 0.5244(10) | 0.4222(10) | 2.58(17) |
| 0.0781 | 0.5646 | 0.4057 | |||
| O10 | 2a | 0.6232(51) | 0.5605(9) | 0.4002(13) | 2.58(17) |
| 0.6266 | 0.5894 | 0.3878 | |||
| O11 | 2a | 0.1533(40) | 0.0521(9) | 0.4200(12) | 2.58(17) |
| 0.1304 | 0.0780 | 0.4284 | |||
| O12 | 2a | 0.5954(59) | 0.0635(9) | 0.4263(10) | 2.58(17) |
| 0.5901 | 0.0970 | 0.4407 | |||
| O13 | 2a | 0.3230(11) | 0.3908(10) | 0.3168(16) | 2.58(17) |
| 0.3273 | 0.3607 | 0.3060 | |||
| O14 | 2a | 0.8181(10) | 0.2440(10) | 0.3124(15) | 2.58(17) |
| 0.8261 | 0.2042 | 0.3055 | |||
| Bonds | Dist. (Å) | Bonds | Dist. (Å) | Bonds | Dist. (Å) | Bonds | Dist. (Å) |
|---|---|---|---|---|---|---|---|
| Pr1_O1 | 2.896(20) | Pr3_O6 | 2.705(13) | Ti1_O1 | 1.976(9) | Ti3_O5 | 2.233(8) |
| Pr1_O2 | 2.912(15) | Pr3_O9 | 2.723(32) | Ti1_O2 | 2.024(10) | Ti3_O7 | 2.010(9) |
| Pr1_O2 | 2.594(15) | Pr3_O10 | 2.433(34) | Ti1_O3 | 1.971(8) | Ti3_O9 | 1.777(10) |
| Pr1_O3 | 2.528(29) | Pr3_O10 | 3.041(16) | Ti1_O3 | 2.220(9) | Ti3_O11 | 1.872(11) |
| Pr1_O3 | 3.093(28) | Pr3_O11 | 2.161(25) | Ti1_O5 | 1.840(10) | Ti3_O13 | 2.018(10) |
| Pr1_O4 | 2.258(28) | Pr3_O12 | 2.341(35) | Ti1_O7 | 1.856(7) | Ti3_O14 | 1.966(9) |
| Pr1_O6 | 2.535(16) | Pr3_O12 | 2.668(12) | ||||
| Pr1_O7 | 2.45(4) | Pr3_O13 | 2.442(10) | ||||
| Pr1_O8 | 2.568(31) | ||||||
| Pr1_O13 | 2.724(20) | Pr3_O10 | 3.187(24) | ||||
| 8 | |||||||
| CN | 10 | CN | 8 | CN | 6 | CN | 6 |
| BVS | 2.98(9) | BVS | 3.00(1) | BVS | 4.04(5) | BVS | 4.12(5) |
| Distt | 79.80 × 10−4 | Distt | 100.62 × 10−4 | Distt | 40.39 × 10−4 | Distt | 47.50 × 10−4 |
| Pr2_O1 | 2.241(6) | Pr4_O5 | 2.883(17) | Ti2_O1 | 1.934(10) | Ti4_O6 | 2.259(8) |
| Pr2_O2 | 2.533(20) | Pr4_O9 | 2.431(27) | Ti2_O2 | 1.975(9) | Ti4_O8 | 2.053(10) |
| Pr2_O3 | 2.688(32) | Pr4_O9 | 2.322(13) | Ti2_O4 | 1.992(9) | Ti4_O10 | 1.796(10) |
| Pr2_O3 | 2.707(27) | Pr4_O10 | 2.336(30) | Ti2_O4 | 2.252(9) | Ti4_O12 | 1.836(10) |
| Pr2_O4 | 2.569(30) | Pr4_O11 | 2.584(29) | Ti2_O6 | 1.822(10) | Ti4_O13 | 1.946(9) |
| Pr2_O4 | 2.917(22) | Pr4_O11 | 2.666(15) | Ti2_O8 | 1.880(7) | Ti4_O14 | 1.982(10) |
| Pr2_O5 | 2.891(17) | Pr4_O12 | 2.35(4) | ||||
| Pr2_O7 | 2.46(4) | Pr4_O14 | 2.579(11) | ||||
| Pr2_O8 | 2.290(27) | ||||||
| Pr2_O14 | 2.741(19) | ||||||
| CN | 10 | CN | 8 | CN | 6 | CN | 6 |
| BVS | 3.38 | BVS | 2.98(9) | BVS | 4.12(6) | BVS | 4.17(6) |
| Distt | 69.99 × 10−4 | Distt | 79.80 × 10−4 | Distt | 50.82 × 10−4 | Distt | 59.18 × 10−4 |
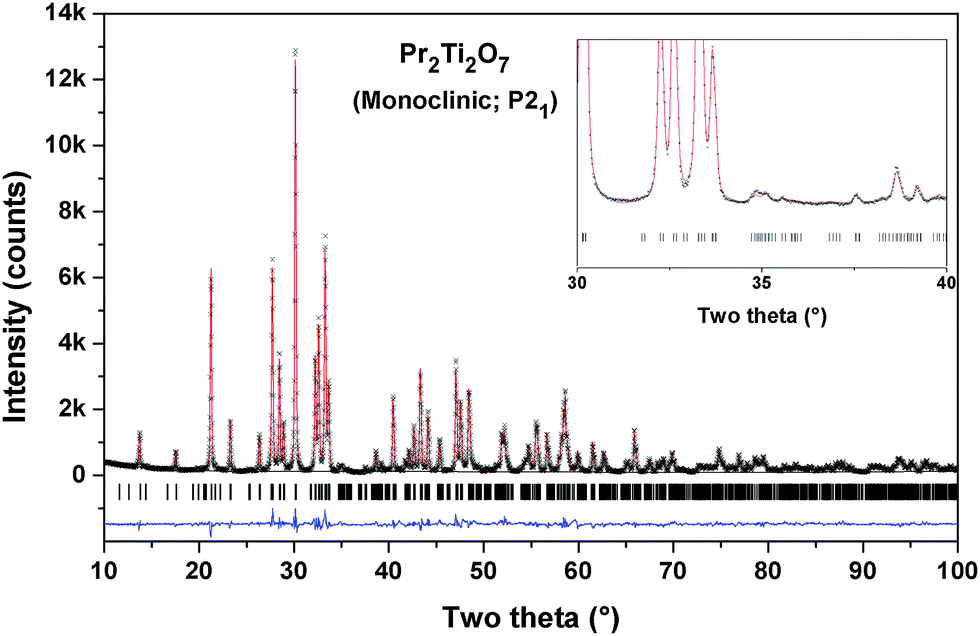 | ||
| Fig. 2 Rietveld refinement plot of Pr2Ti2O7 in the P21 model. | ||
The analyses of the refined structural parameters of PTO at ambient temperature in P21 symmetry indicate that the structure has crystallographically distinguishable four Pr (Pr1 to Pr4) atoms, four Ti (Ti1 to Ti4) atoms and fourteen oxygen atoms. The typical crystal structure of PTO and its projection along the a- and c-axes are shown in Fig. 3(a and b). All the TiO6 octahedra of the PTO are distorted and connected by sharing the corners to form a perovskite type octahedral net. However, the periodicity disrupts along the c-axis, which render it a layered structure. The interstices in the perovskite slabs are filled by the Pr1 and Pr2 atoms. The Pr3 and Pr4 are located closer to interlayer interstices. The shift of these atoms from the ab-plane causes a variation in the stacking of the perovskite slabs in the PTO structure. The transformation of perovskite to layered perovskite systems has been reported earlier in the literature.40 The excess oxygen contents compared to the stoichiometry of the perovskite composition cleave the octahedral connections and in turn lead to layered structures with AnBnO3n+1 homologues. In the PTO structure, all the Ti atoms (Ti1, Ti2, Ti3 and Ti4) have octahedral coordination with the anions with Ti–O bond lengths in between 1.83 to 2.25 Å, while the Pr3+ ions are 10 (Pr1, Pr2) or 8 (Pr3 and Pr4) coordinated with the oxygen atoms. The Pr3+ ions inside the perovskite layers show larger distortion and dispersion in bond lengths compared to those closer to the outside of layers (Table 3). The local distortions around the cations and shift of the interstitial cations in such layered perovskites show lower symmetric structure and often a ferroelectric structure, which is explained later in this manuscript.
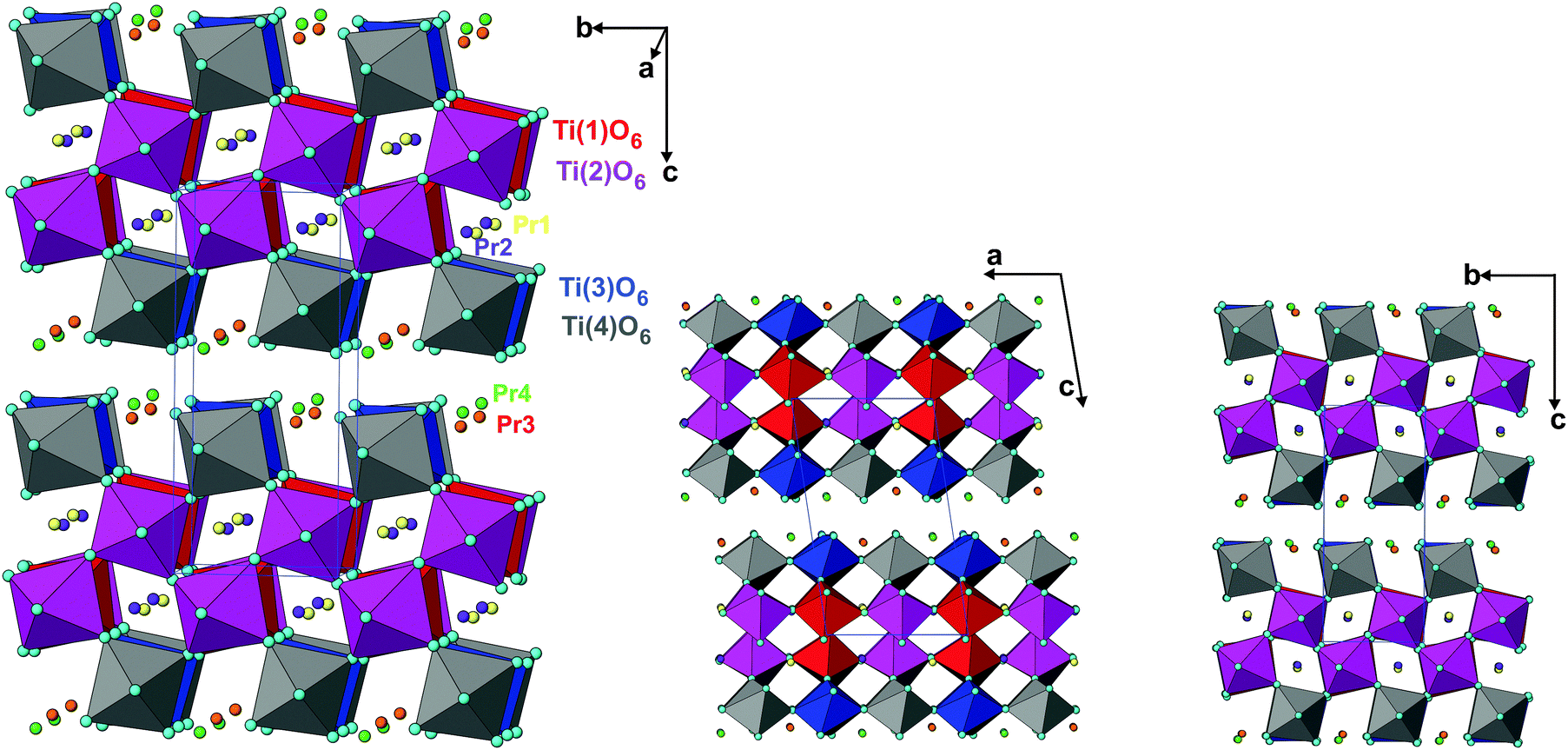 | ||
| Fig. 3 Crystal structure of Pr2Ti2O7 in different projections (TiO6 octahedra are shown, isolated spheres are Pr atoms). | ||
In order to further ascertain the structure and phase transformation in PTO, total energy calculations in DFT formalism were carried out. The calculated total energy and unit cell parameters of PTO in all the possible symmetry models are included in Table 1. The details of calculated structural parameters for P21 symmetry are included in Table 2 and those for other symmetry models are provided as ESI† (S-II) to this manuscript. It is observed that the PTO has almost equivalent energy in the P21 and Pna21 symmetry, while those for the ferroelectric Cmc21 and other paraelectric (P21/m and Cmcm) model structures have higher values (about 0.05 eV per atom). Thus the later models are not energetically favorable for the structure of PTO. From the comparison of energies, it can be suggested that the P21 or Pna21 ferroelectric structures can be retained at ambient temperature. It can be mentioned here that the structure in Pna21 and P21 symmetries are closely related. Typical unit cell relations between the orthorhombic Pna21 and monoclinic P21 structures are depicted in Fig. 4. The transformation from Pna21 to P21 symmetry resulted from the shift of the Pr atoms and octahedral slabs along the c-axis, which destroy two of the glide planes of the former. As mentioned earlier the Pna21 symmetry cannot account for the weak reflections at 2θ ∼ 14.5°. Also, the absence of measureable intensity at 2θ ∼ 12.4 and 23.5° in the observed XRD data support for monoclinic P21 structure over the orthorhombic Pna21 for PTO. Further the variations of energies with volume for all the five relaxed structures were calculated and they are given as ESI† (SI–SIII). In a wider range of volume, the variation of energy shows an almost similar trend for all except the Cmc21 structure. Similar to the fully relaxed states, the Pna21 and P21 structures do not show any difference with the change in volume. Almost identical energies for monoclinic (P21) and orthorhombic (Pna21) structures can be due to the fact that calculations are made by DFT at 0 K, without including the entropy contribution. The differences in the contributions of entropy at finite temperature may favor the monoclinic (P21) structure compared to the orthorhombic (Pna21) structure. At a larger volume (above the molar volume (Vm) 152 Å3), the orthorhombic Cmc21 structure is favored over orthorhombic Pna21 and monoclinic P21 structures.
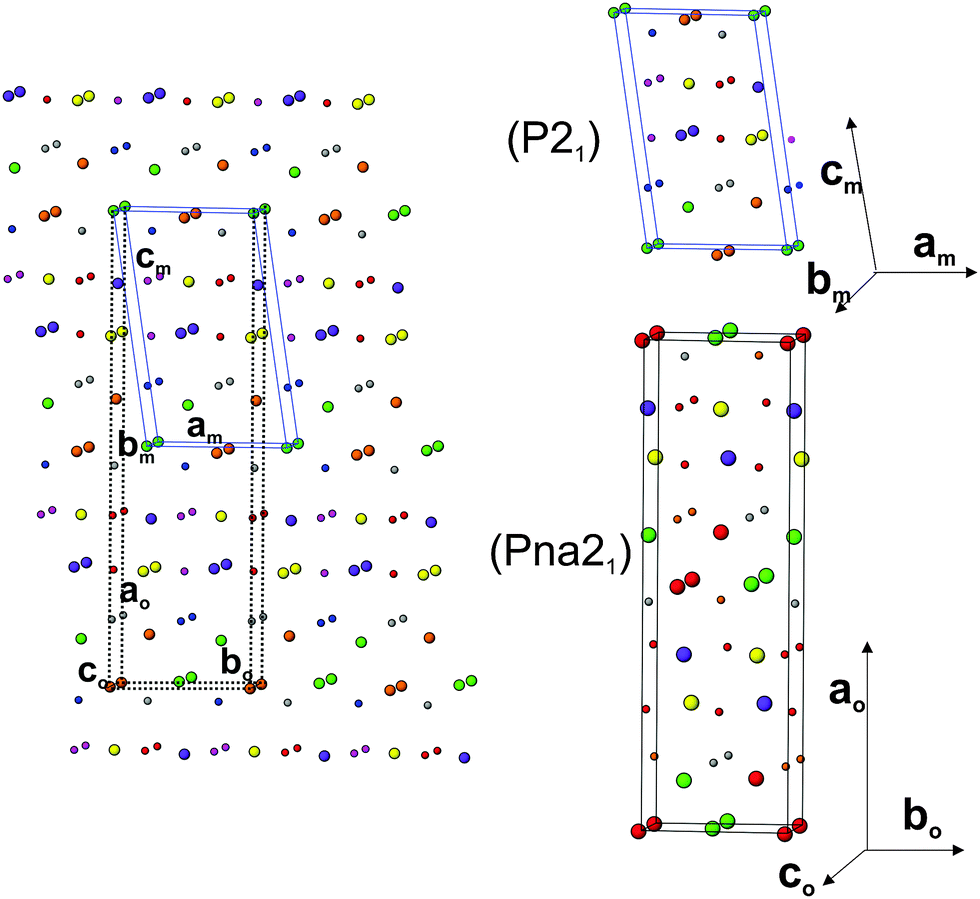 | ||
| Fig. 4 Comparison of monoclinic P21 and orthorhombic Pna21 structures of Pr2Ti2O7. Only cations are shown (larger sphere: Pr3+ and small sphere: Ti4+). The unit cell are related as: ao = 2cmcos(β − 90), bo= am, co= bm. | ||
In order to understand the electronic properties of PTO, the electron density of states (DOS) for all the relaxed structures were calculated. The total electronic density of states and the Pr, Ti, and O contributions (projected within their atomic spheres) are shown in Fig. 5. From the calculated DOS, it is inferred that the lower lying states below −10 eV originate mainly from O 2s and Pr np orbitals. The states in the valence band region from −5 to 0 eV are mainly composed of O 2p orbitals which is hybridized with Ti 3d orbitals. This suggests that the nature of the Ti–O bonding is largely covalent. Further, the states in the conduction band region from 2 to 6 eV have a dominant contribution from Ti 3d orbitals. The O 2p orbitals also contribute a little to the conduction band and hybridized to a small extent with Ti 3d orbitals. The band gaps for the ferroelectric structures, like P21, Pna21 and Cmc21 structures are almost similar and are close to 2.7 eV, while those for the paraelectric phases, like P21/m and Cmcm structures are low (∼2.0 eV). This suggests that ferroelectric to paraelectric phase transition would decrease the band gap. The experimental optical band gap of PTO as observed from the absorption spectra (SI–SIV, ESI†) is 3.10 eV, which is in agreement with the earlier reported value.11 As commonly observed the band gap calculated by DFT is often underestimated compared to the true experimental gap.41 This difference can be attributed to the limitation of the Kohn–Sham formalism to account the discontinuity of the exchange–correlation function.
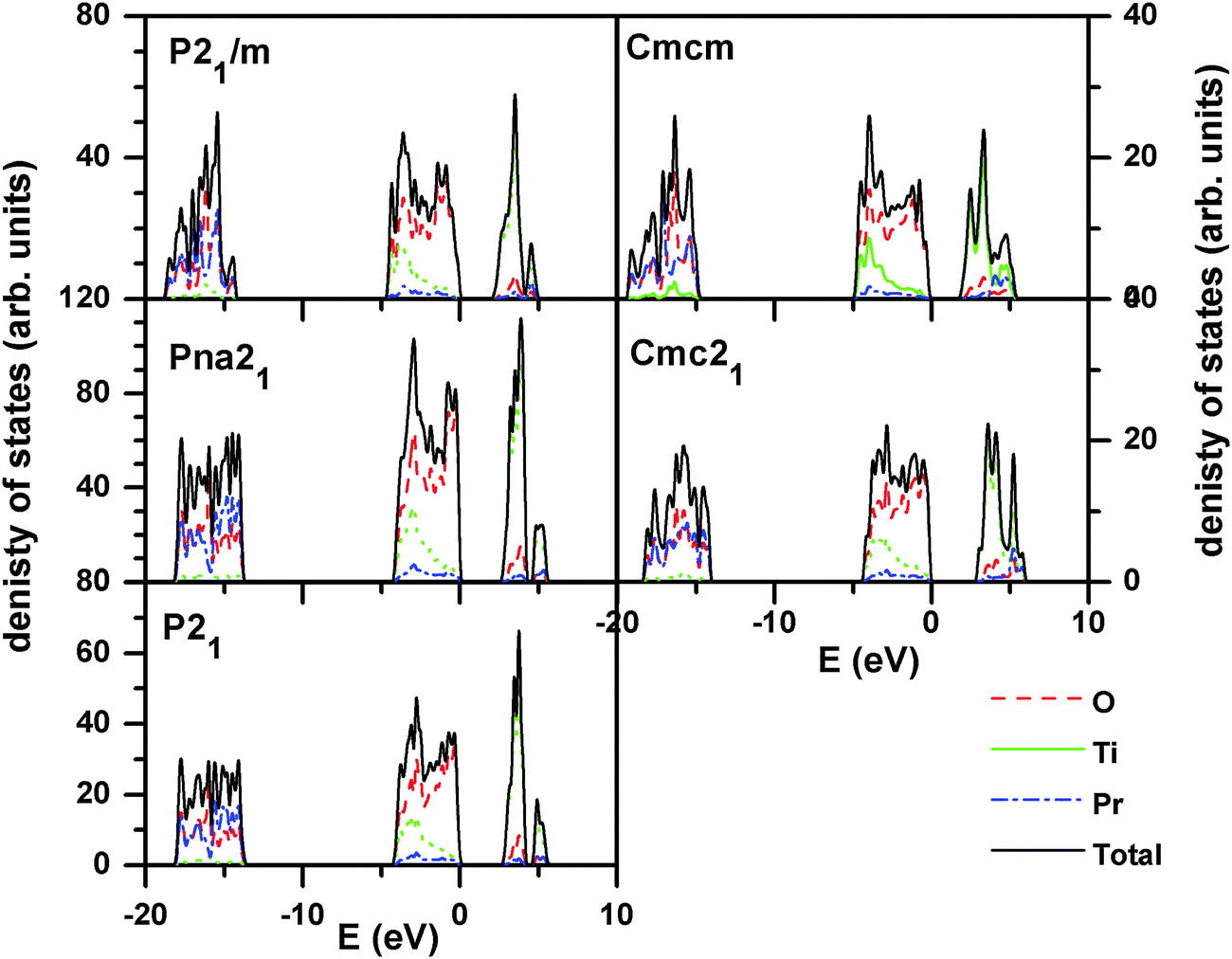 | ||
| Fig. 5 Calculated density of states of Pr2Ti2O7 for different structures. (For P21, Pna21 and Cmc21 models, the band gaps are similar (∼2.7 eV) while for the paraelectric Cmcm and P21/m, the band gaps have lower values (∼2 eV)). | ||
The thermal properties like phase transition and stability of Pr2Ti2O7 were investigated by thermogravimetric and differential thermal analyses (TG-DTA) in oxygen as well as in an inert atmosphere. Typical TG-DTA traces recorded while heating the sample from ambient to 1673 K in an oxygen or argon atmosphere are shown in Fig. 6. No weight gain except the apparent baseline shift as seen from the TG traces recorded under argon or oxygen atmosphere suggests the retention of the 3+ state of praseodymium until the highest temperature. The DTA traces in both argon and oxygen atmospheres show deviations around 900 °C and then a sharp deviation above around 1300 °C. The TG residues obtained indicate no sign of melting of the sample. Shcherbakova et al.3 have compiled the melting temperature for various rare-earth titanates and mentioned the melting temperature of Pr2Ti2O7 as 1560 °C. Thus, this deviation cannot be attributed to the melting point. The sharp endothermic peak above 1300 °C coincides with the reported ferroelectric to paraelectric transition of PTO.7 Yan et al.4 have investigated analogous titanates, namely, La2Ti2O7 and Nd2Ti2O7 and observed that the ferroelectric to paraelectric transitions are at 1482 °C and 1461 °C, respectively. Thus, the ferroelectric to paraelectric transition of Pr2Ti2O7 though cannot be ruled out at 1300 °C but more expected above 1400 °C only. It can also be mentioned that at higher temperature, Ti4+ has a tendency to reduce to Ti3+ leaving oxygen vacancy. A feeble deviation in the TG traces at the corresponding temperatures of deviations in the DTA traces may be a support for this.
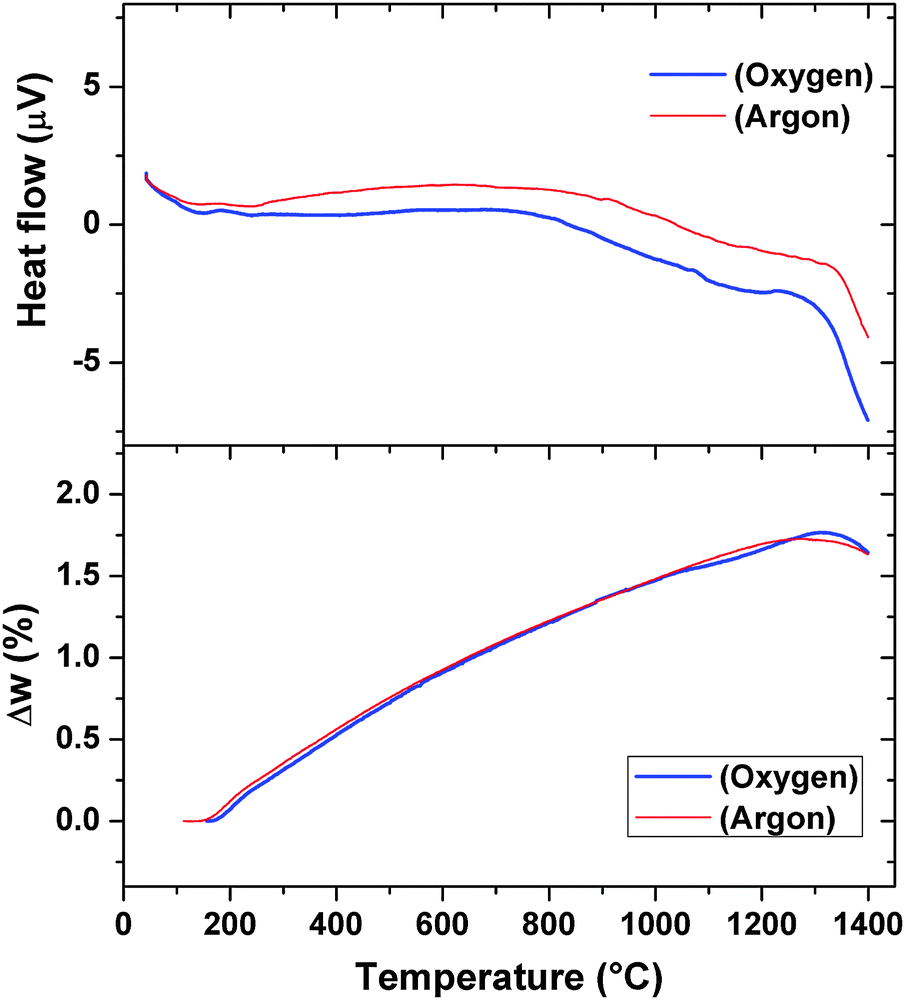 | ||
| Fig. 6 TG/DTA curves of Pr2Ti2O7 while heating from ambient to 1673 K. (heating rate = 10 °C min−1). | ||
The XRD patterns of the residue obtained after TG-DTA runs indicate the presence of the original monoclinic P21 structure (Fig. 7). However, several extra reflections attributable to the Ti rich perovskite type phase, (Pr2Ti3O9), are observed in both the XRD patterns. This suggests a partial decomposition of the Pr2Ti2O7 composition at higher temperature. The Rietveld refinements of the powder XRD patterns of the samples obtained after TG runs suggest no significant change in the unit cell or structural parameters of the PTO lattice compared to the original ambient temperature phases. Gao et al.7 and Sun et al.17 have observed a dielectric anomaly at lower temperature, viz. below 800 K, which the authors have attributed to the ferroelectric to paraelectric phase transition. The lowering of the ferroelectric Tc has been attributed to the preparation procedure and/or the nanocrystalline nature of the sample.4,7,17 Such anomaly is also seen in the present study on the dielectric and electrical properties. However, differential scanning calorimetric (DSC) investigation of PTO also did not show any anomaly attributable to phase transition up to 873 K (S-IV, ESI†). The deviation in the DTA pattern above 1073 K observed in DTA traces recorded both in oxygen or argon atmosphere plausibly can be attributed to this affect. However no structural changes are noticed in both in situ high temperature XRD and Raman spectroscopic investigations of the present study. This indicates either the absence of any structural transition or a transition accompanied by a very low energy change. Considering, the high temperature structural studies, it is concluded that the deviations in DTA traces are not related to the structural transition accompanied with ferroelectric to paraelectric phase transition. The details of the high temperature behavior of the PTO, as observed by in situ variable temperature powder XRD studies, are explained below.
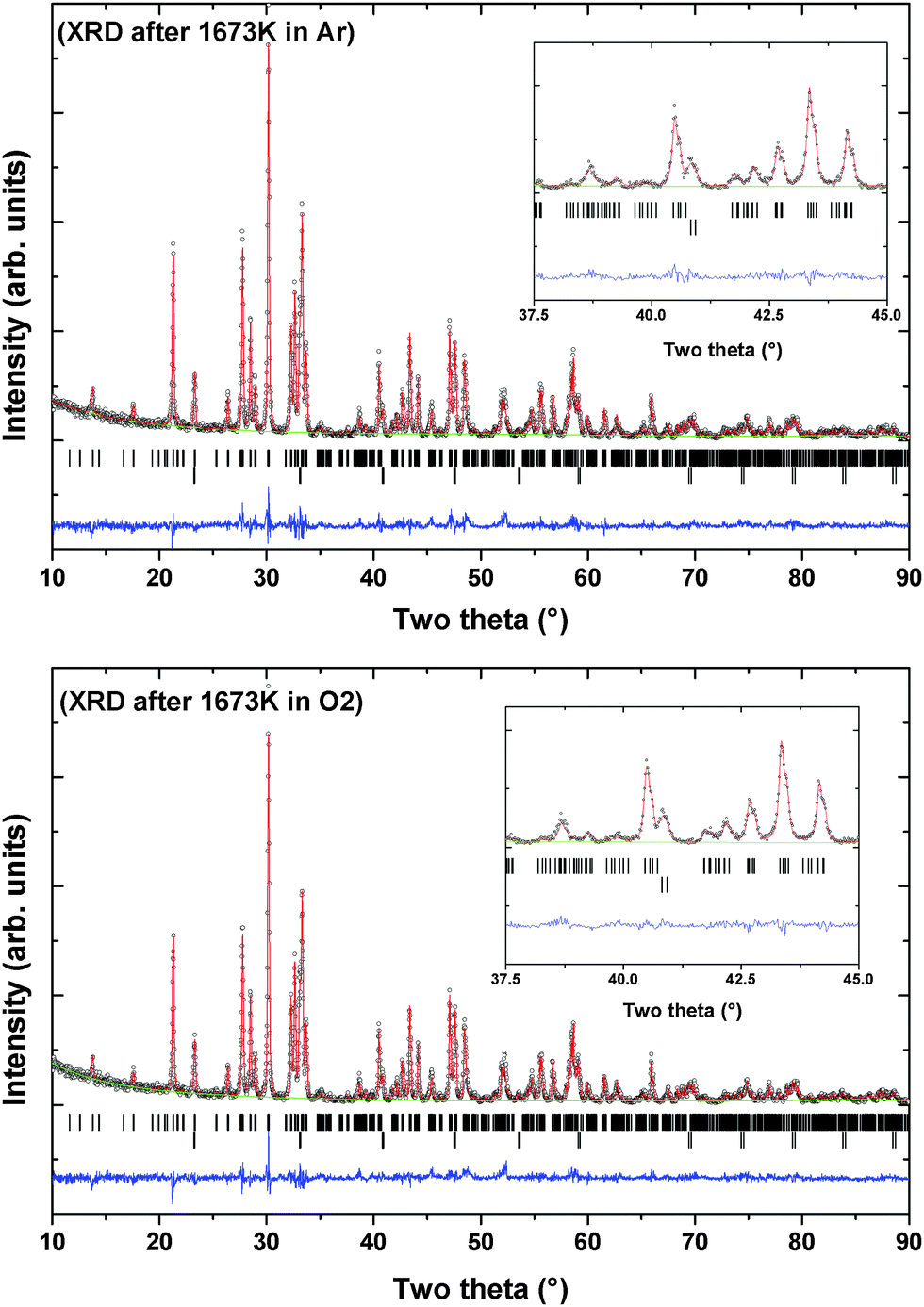 | ||
| Fig. 7 XRD patterns of Pr2Ti2O7 recorded after heating at 1673 K in argon and oxygen atmospheres. (Argon): a = 7.7158(2) Å, b = 5.5878(2) Å, c = 13.0031(4) Å, β = 98.572(6)°, V = 544.43(2) Å3. Rp = 9.78%, Rwp = 12.9% and χ2 = 1.749 (Oxygen): a = 7.7159(2) Å, b = 5.5878(2) Å, c = 13.0021(3) Å, β = 98.582(4)°, V = 544.39(2) Å3. Rp = 9.63%, Rwp = 12.7% and χ2 = 1.884. | ||
Powder XRD patterns of PTO recorded at successive temperatures between ambient to 1273 K are shown in Fig. 8. The XRD patterns recorded at higher temperature are almost similar to that observed at ambient temperature except the shift in positions due to the thermal effect. Considering the refined structural parameters observed at ambient temperature, the XRD data recorded at different temperatures were refined using the Rietveld refinement method. The refined unit cell parameters observed for ambient temperature phases are comparable to those observed in the ambient temperature data recorded on a high temperature stage. The observed unit cell parameters at different temperatures are given in Table 4. A comparison of the variations of unit cell parameters observed at different temperatures indicates a smooth variation with temperature. In order to see the possible phase transition, the XRD data recorded at and beyond 873 K were analyzed by considering the possible symmetries of paraelectric (P21/m; Cmcm) and ferroelectric (Cmc21) phases. However, the observed peaks, in particular the differentiating peak at two-theta ∼37.5°, could not be accounted by these models. A typical Rietveld refinement plot of the powder XRD data recorded at 1273 K is shown in Fig. 9. Similar features are also observed in the XRD pattern of PTO recorded at 1473 K (shown as SI–SVI, ESI†), which suggests that PTO does not have any structural transition up to 1473 K. However, as observed from the variation of energy versus volume (SI–SIII, ESI†) the orthorhombic (Cmc21) structure has higher stability over the monoclinic (P21) or orthorhombic (Pna21) structures above the molar volume (Vm) 152 Å3. From the high temperature XRD studies, it is observed that the unit cell volume at 1473 K is 565.69(3) Å3, i.e. Vm = 141.4 Å3. Considering the average volume thermal expansion coefficient (between RT–1473 K) as 32.3 × 10−6 K−1, the expected unit cell volume of monoclinic (P21) at 1673 K is only about 569.2 Å3 (Vm = 142.3 Å3). Thus a phase transition from monoclinic ferroelectric (P21) to orthorhombic (Cmc21) can only be expected at still higher temperature. Hence it can be mentioned here that the ferroelectric to paraelectric phase transition takes place above 1473 K, as observed in other analogous titanates.3,4,14
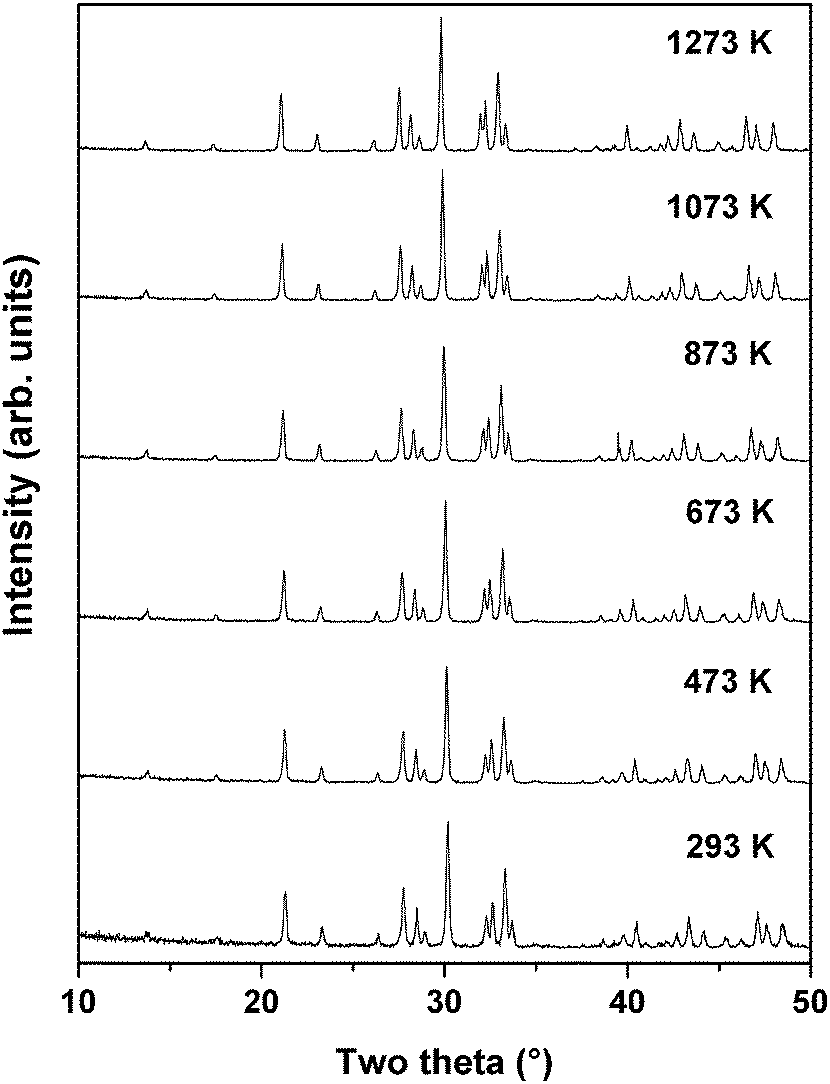 | ||
| Fig. 8 Typical powder XRD patterns of PTO recorded at different temperatures. | ||
| Temp. (K) | a (Å) | b (Å) | c (Å) | β (°) | V (Å)3 |
|---|---|---|---|---|---|
| 300 | 7.7184(4) | 5.4898(3) | 13.0076(9) | 98.59(2) | 544.98(6) |
| 473 | 7.7336(3) | 5.5003(2) | 13.0224(7) | 98.57(2) | 547.75(4) |
| 673 | 7.7518(3) | 5.5135(2) | 13.0423(7) | 98.56(2) | 551.21(4) |
| 873 | 7.7683(3) | 5.5246(2) | 13.0613(7) | 98.59(1) | 554.26(4) |
| 1073 | 7.7882(3) | 5.5387(2) | 13.0833(7) | 98.58(1) | 558.05(4) |
| 1273 | 7.8107(3) | 5.5543(2) | 13.1080(6) | 98.59(1) | 562.28(4) |
| 1473 | 7.8264(2) | 5.5658(1) | 13.1338(4) | 98.60(1) | 565.69(3) |
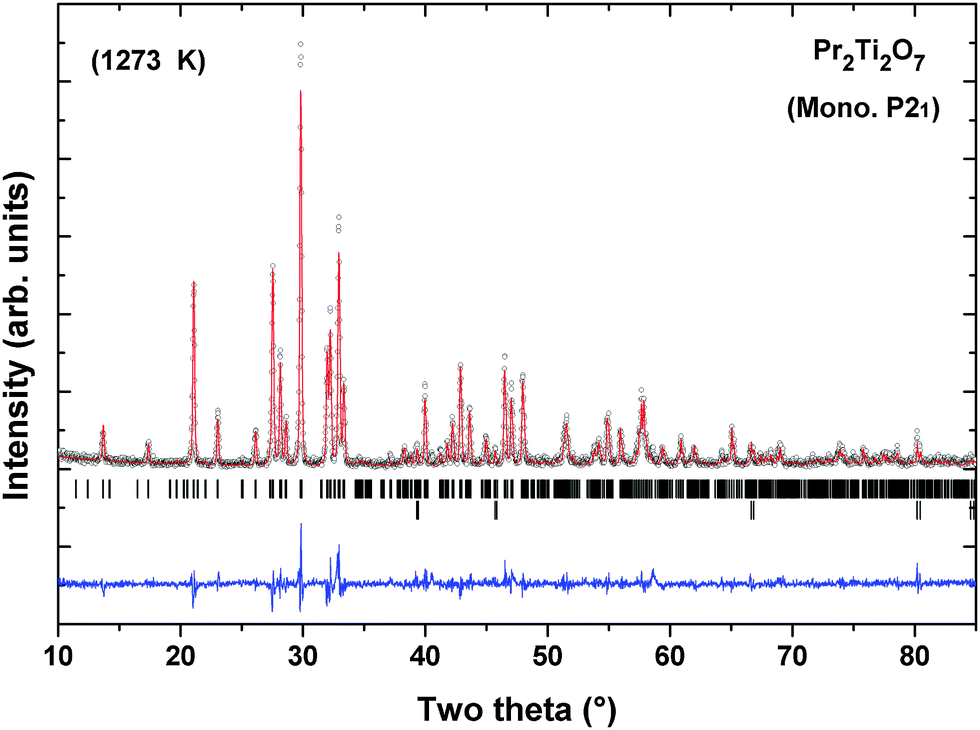 | ||
| Fig. 9 Typical Rietveld refinement plot of XRD data of PTO recorded at 1273 K. Lower vertical ticks indicate the Bragg position (upper panel: Pr2Ti2O7 and lower panel: sample holder) (Rp = 15.7%, Rwp = 20.8%, χ2 = 1.67). | ||
Earlier studies on isostructural compound La2Ti2O7 indicated a structural transition from ferroelectric (P21) to another ferroelectric (Cmc21) transition in between 993 to 1053 K,24 which is similar to that reported for Ca2Nb2O7.42 In a recent study, Herrera et al. have reported both P21 and Cmc21 structures of La2Ti2O7 at ambient temperature, in the sample prepared by the xerogel method by heating at 1073 and 1273 K, respectively.43 However, the reported XRD pattern indicated the poorly crystalline nature of the phase where the differentiation of these two symmetries is not easy. In the present study and earlier studies on La2Ti2O7 and related materials prepared by high temperature reactions show only P21 structure at ambient temperature.18,20,24,25 Since the differentiation of these probable symmetries is difficult as they are mainly based on the presence or absence of weak reflections, the claim for the stabilization of Cmc21, which is also energetically unfavorable compared to Pna21 or P21 structures, cannot be relied. Thus we suggest no structural transition in PTO up to 1473 K.
Further to confirm the high temperature behavior of PTO, in situ high temperature Raman spectroscopic investigations were carried out. Raman modes being sensitive to the variation of local coordination and distortion as well as to symmetry, the analyses of temperature dependent Raman mode can provide clear information on these features. Under ambient conditions, Pr2Ti2O7 has monoclinic structure with space group P21 (point group: C2) and four formula units per unit cell which gives 132 normal modes of vibration. Factor group analysis gives irreducible mechanical representations for optic and acoustic modes as Γoptic = 65A + 64B and Γacoustic = A + 2B, respectively. From group theoretical calculations, 129 Raman active modes are expected in Pr2Ti2O7. The earlier report suggested the presence of photoluminescence lines in Pr2Ti2O7.44 Hence to verify this, Raman spectra of Pr2Ti2O7 at different laser excitation wavelengths under ambient conditions were recorded and they are given as supplementary data to this manuscript (SI–SVII, ESI†). It is observed that the excitation wavelengths, 532, 514 and 785 nm give identical spectra, while the excitation wavelengths lower than 488 nm show dominating luminescence in the Raman spectra. This indicates that all the peaks in the spectra with 532 nm are Raman modes. All the spectra were recorded at an excitation wavelength of 532 nm and they are presented in this manuscript. Fig. 10 shows the Raman spectra of PTO at different temperatures in between 298–1073 K. At ambient temperature 32 distinct Raman modes could be clearly identified. The less number of observed Raman modes than expected may be due to the degeneracy or the weak nature of some modes. Low frequency modes in the range 50–490 cm−1 are attributed to the Pr–O vibrations.16 Modes in the range 490–575 cm−1 are assigned to the vibrations of the distorted TiO6 octahedron.16 The modes above 600 cm−1, particularly the high frequency modes at 785 and 814 cm−1, are assigned to stretching modes of Ti–O vibrations. These stretching modes are separated from the rest of the modes with a gap. This feature is similar to the gap in between the stretching and bending vibrations of VO4 tetrahedra in ABO4 type orthovanadates.45 Since the TiO6 octahedra are distorted and the bond strength of Ti–O is weaker compared to V–O bonds of VO4 tetrahedra of orthovanadates, the gap between the bending and stretching modes is less in Pr2Ti2O7 compared to that observed in orthovanadates.
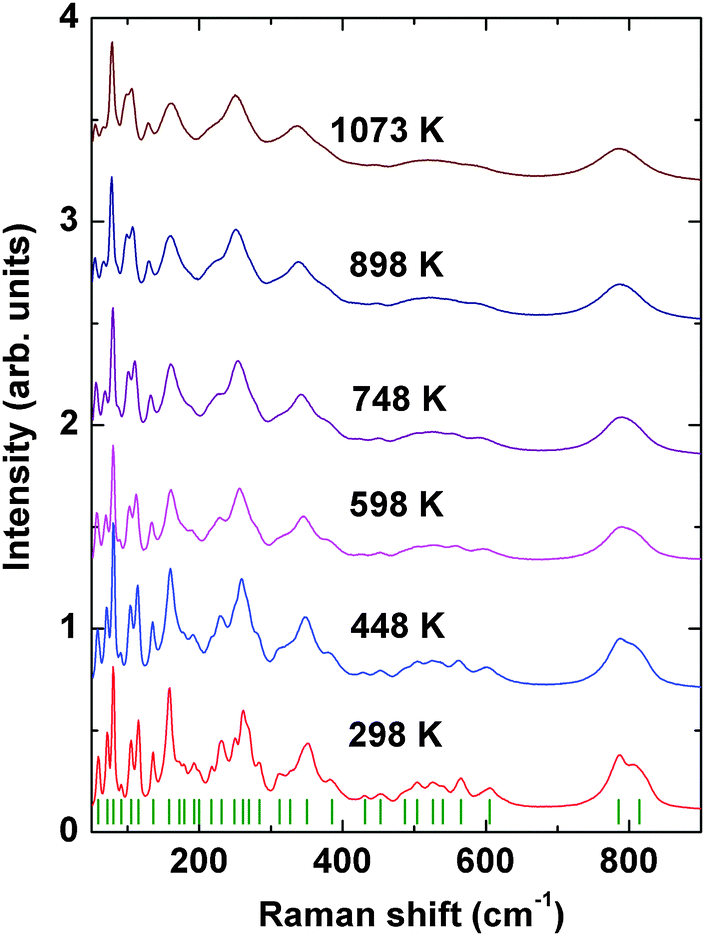 | ||
| Fig. 10 Raman spectra of Pr2Ti2O7 at different temperatures. | ||
The evolution of Raman modes with temperature were was followed by comparing with the modes observed at ambient temperature. The temperature dependencies of Raman mode frequencies of PTO are shown in Fig. 11. It is also observed that several weak modes could not be followed at higher temperatures due to overlapping caused by temperature induced broadening. From the variations of mode frequency with temperature, it can be seen that almost all the modes soften with increasing temperature. Table 5 shows the temperature coefficients of Raman mode frequencies of PTO. Most of the Raman mode frequencies show a linearly decreasing trend in the range 298–1073 K, which indicates that the anharmonicity in PTO is predominantly due to three phonon decay processes. However, typical trend for some of the modes show anomalous behaviour. The modes at 217 and 814 cm−1 show a negligible change in frequency in the temperature range. The lattice mode at 158 cm−1, the mode at 312 cm−1, which has been attributed to the Pr–O vibration, and the mode at 785 cm−1 due to Ti–O stretching vibrations show an increase in frequency with temperature. Generally, higher order terms of phonon–phonon interactions are responsible for such a kind of anomaly. This anomalous behaviour could also be associated with symmetrization of the distorted PrOn and TiO6 polyhedra. At higher temperature, it is possible that phonon anomaly could bring instability in the structure, which could lead to ferroelectric to paraelectric phase transition. The smooth variation of Raman modes within the temperature range 298–1073 K suggests the absence of any structural transition. Thus the Raman spectroscopic studies do not show any signatures of phase transition in the temperature range studied; but the possibility of ferroelectric to paraelectric phase transition at higher temperature cannot be ruled out.
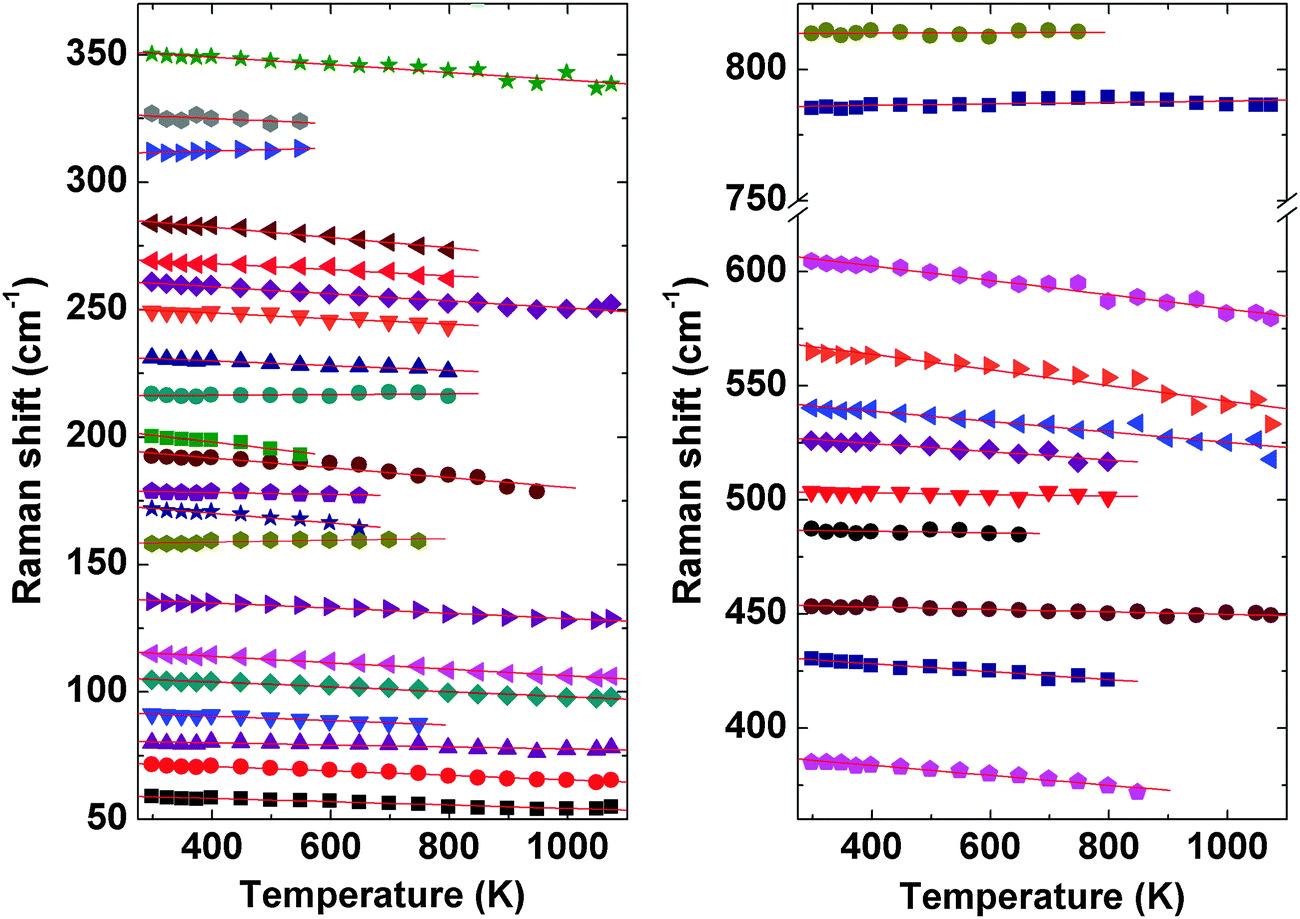 | ||
| Fig. 11 Temperature dependence of Raman mode frequencies of Pr2Ti2O7. | ||
| Mode ω (cm−1) | dω/dT (cm−1 K−) | Mode ω (cm−1) | dω/dT (cm−1 K−) |
|---|---|---|---|
| 59 | −0.007 | 269 | −0.012(1) |
| 72 | −0.009 | 284 | −0.020(1) |
| 80 | −0.004(1) | 312 | 0.006(2) |
| 91 | −0.009(1) | 327 | −0.010(4) |
| 105 | −0.010 | 350 | −0.015(1) |
| 115 | −0.013 | 385 | −0.022(1) |
| 136 | −0.010(1) | 431 | −0.017(1) |
| 158 | 0.003 | 453 | −0.005(1) |
| 172 | −0.019(2) | 487 | −0.003(2) |
| 179 | −0.004(1) | 504 | −0.003(1) |
| 193 | −0.019(2) | 526 | −0.018(2) |
| 200 | −0.027(4) | 540 | −0.023(2) |
| 217 | 0 | 565 | −0.034(3) |
| 231 | −0.010(1) | 605 | −0.031(1) |
| 249 | −0.012(1) | 785 | 0.003(1) |
| 261 | −0.014(1) | 814 | 0 |
Microstructural features of sintered PTO samples were investigated by scanning electron microscopy. The SEM pictures of the top and fractured surfaces of the prepared pellets are given as ESI† (S-VII) to this manuscript. Irregular micron sized well connected grains are observed under the preparation conditions. However, the observed grains are quite different in shape and connections from the samples prepared from the low temperature solution processing and spark plasma sintered samples.4,7 However, in the present case no such preferred orientation or well developed facets are observed under the employed sintering condition. At higher sintering temperature, such layered materials can exhibit highly oriented grain growth as seen by Gao et al.7 The differences might be attributed to the differences in the sintering temperature. Since a partial decomposition at higher temperature is observed in this study, further high temperature processing was not been carried out. Besides, at higher temperature differences in the composition and oxidation state of cations may also occur in PTO.
The measurement of electric polarization (P) with applied potential (E) at ambient temperature show typical ferroelectric like loops and is in agreement with the ferroelectric nature of PTO. A typical PE loop of PTO is shown in Fig. 12. No saturation of polarization is observed up to the maximum applied voltage of this study. This might be due to higher loss arising from the low density of the studied pellets. The observed maximum polarization (Ps), remanent polarization (Pr) and coercive field (Ec) are 0.017 μC cm−2, 0.005 μC cm−2 and 235 V, respectively. A typical loss current of about 0.1 μA is observed at 1200 V.
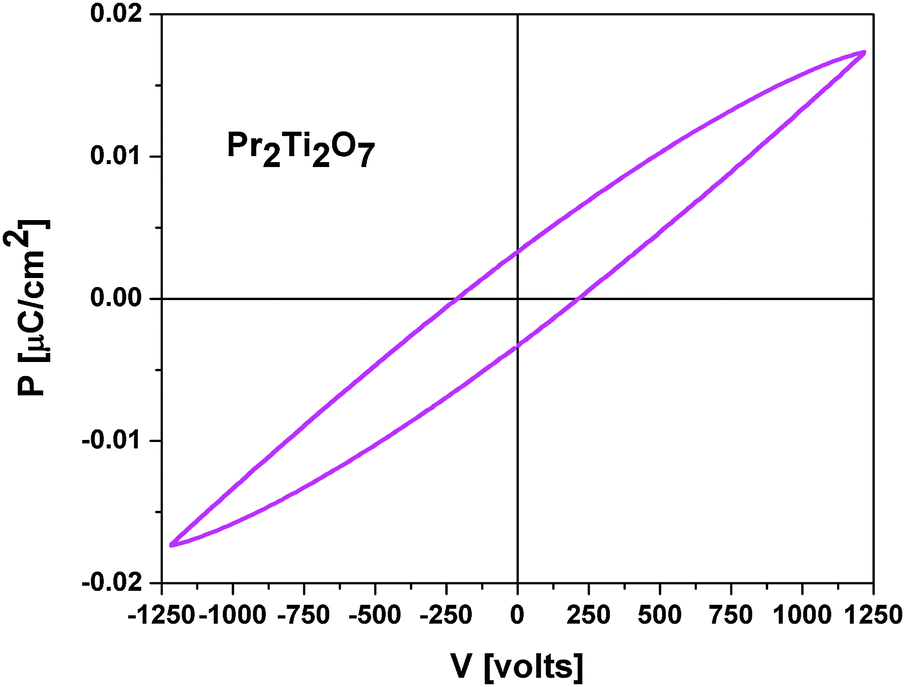 | ||
| Fig. 12 Ambient temperature PE loop of Pr2Ti2O7. | ||
Further to understand the ferroelectric to paraelectric phase transition, the refined structural models are compared. All the structures can be compared by using unit cell and symmetry relations are as shown in Fig. 1. It can be mentioned here that the paraelectric P21/m or Cmcm structures differ from the parent ferroelectric P21 structure in both octahedral distortion as well as displacement of the metal ion positions. Recently, the density functional calculations on a similar layered perovskite La2Ti2O7 and Nd2Ti2O7 indicated a similar energy variation between different symmetries and the authors concluded that the transformation from monoclinic P21 to Cmcm may occur through an intermediate P21/m phase.14 The total energy calculations of this study also support for a similar transformation sequence. Similar studies on the ferroelectric transition by Lopez-Perez et al.15 suggested the shift of the position of rare-earth and the rotation of TiO6 octahedral units are linked to the transition. From the energy variations, it can be suggested that the structure may transform into another ferroelectric Cmc21 phase before transformation to paraelectric phases. But the present in situ high temperature XRD and Raman spectroscopic investigations indicate no distinct phase transition in PTO.
In order to compare the ferroelectric and paraelectric phase of PTO, it is ideal to compare the structure with P21/m and P21 symmetries due to the simple symmetrically related displacive and strain free nature of lattice transformation. The two structures in the unit cell of P21 and the polarization arising from the ionic displacements along the polar axis, i.e. b-axis, are shown in Fig. 13. The spontaneous polarization as calculated from the shift of the ions in the b-axis is 8.3 μC cm−2. The calculated polarization of PTO is comparable to that reported for La2Ti2O7 (7.4–7.7 μC cm−2).14,15 It can be mentioned here that the electric polarization observed for La2Ti2O7 and Nd2Ti2O7 is in the range of 5 to 9 μC cm−24,46 while that of Pr2Ti2O7 thin films is about 1–4 μC cm−2.17,47 The lower observed experimental values might be due to relatively higher conducting contribution as well as the polycrystalline nature of the sample in the present study.
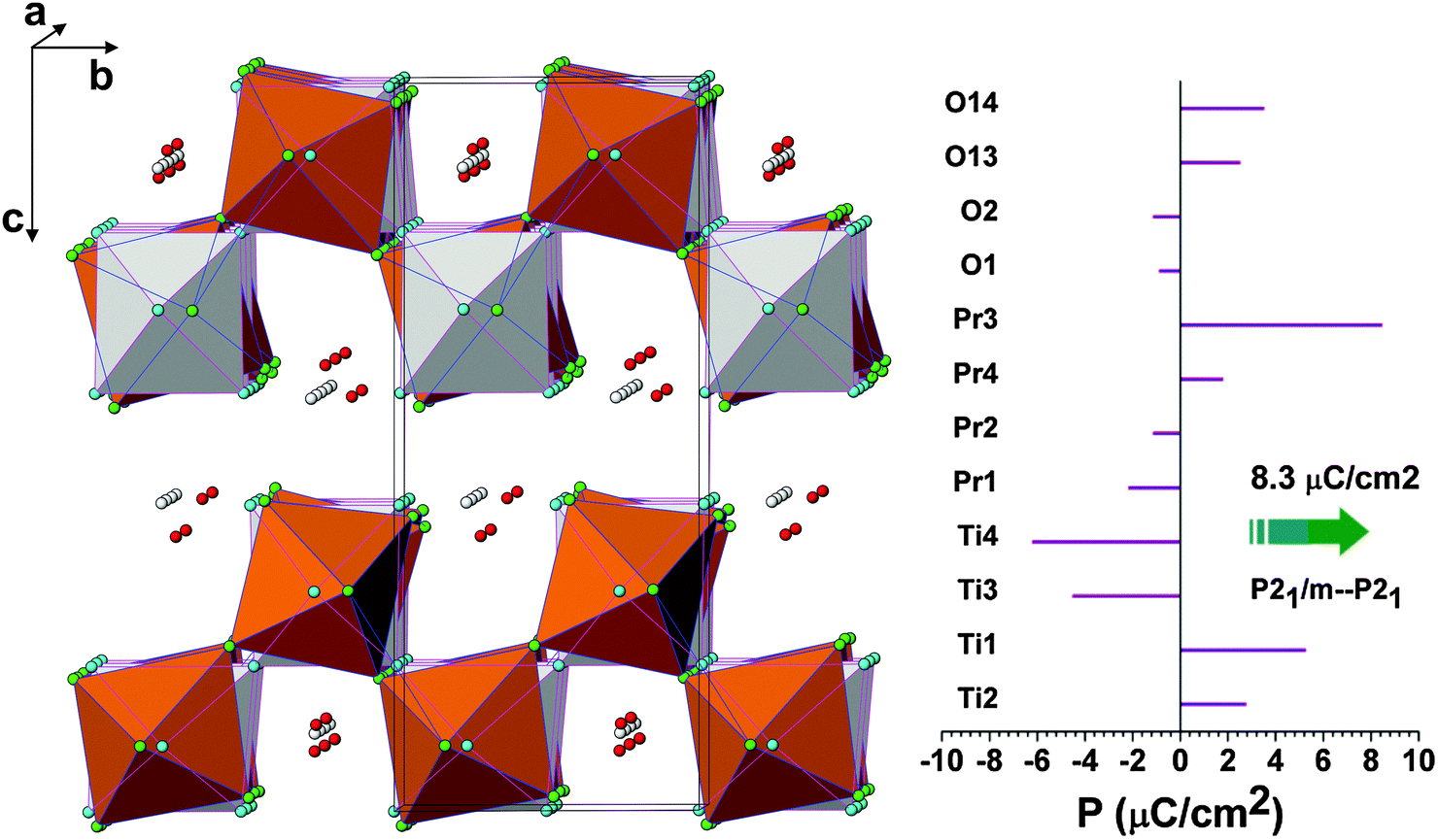 | ||
| Fig. 13 Comparison of monoclinic ferroelectric P21 and paraelectric P21/m structures of Pr2Ti2O7. Light shades: P21/m structure and colour: P21 structure. The spontaneous polarizations along the b-axis due to the shift of various ions are shown in the right hand side figure. | ||
Additional electrical properties of PTO were studied by the impedance spectroscopic investigation. The temperature dependent permittivity and loss spectra in between 173 to 473 K are shown in Fig. 14. In the studied frequency range, both the spectra indicate two different types of dielectric responses. The low frequency relaxations show significant temperature dependence, with the loss peak moving towards higher frequencies as the temperature rises, whereas the high frequency relaxation is almost invariant with temperature. The permittivity spectra recorded up to 473 K show almost similar behavior. However, at still higher temperature the permittivity shows sharp rise at lower frequency compared to that at higher frequency. A comparison of the temperature dependent permittivity and loss features suggests that at higher temperature an appreciable ion migration causes interfacial polarization leading to sharp rise in permittivity due to the Maxwell Wagner (MW) effect.
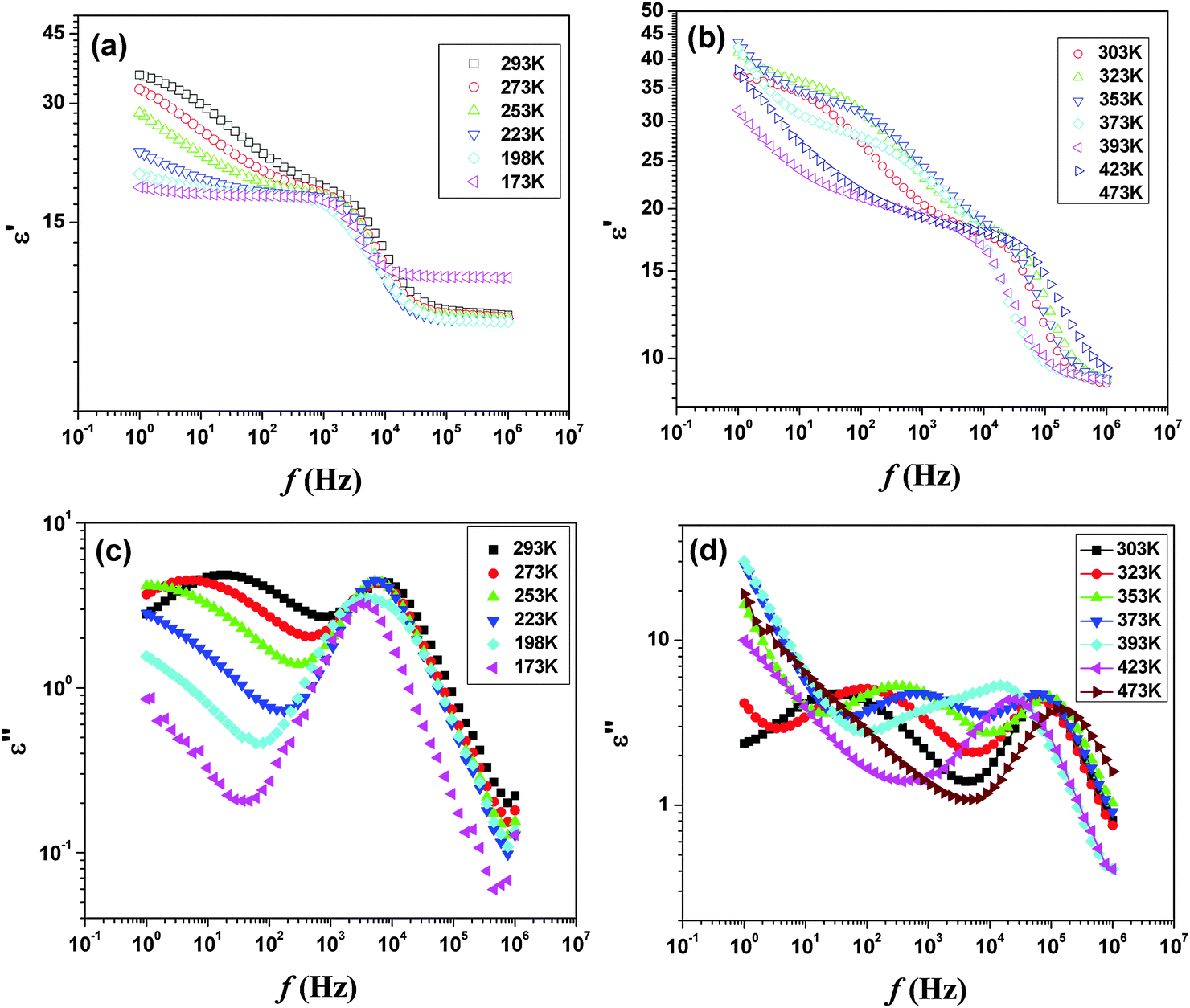 | ||
| Fig. 14 Temperature dependent ε′ (a and b) and ε′′ (c and d) of Pr2Ti2O7in the low temperature region. | ||
The frequency-dependent complex permittivity ε*(ω) can be most generally represented by the Havriliak–Negami (H–N) model as given below48
| ε*(ω) = ε∞ + (εs − ε∞)/[1 + (iωτ)α]β | (1) |
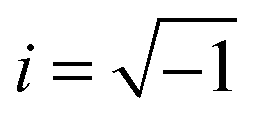 , and α and β are exponents such that α > 0, αβ ≤ 1. The contribution due to electrical conductivity can be incorporated by adding the term (−i[σdc/(ε0ω)]n) to eqn (1), where σdc is the dc-conductivity, ε0 is the permittivity of free space, and n ≈ 1.
, and α and β are exponents such that α > 0, αβ ≤ 1. The contribution due to electrical conductivity can be incorporated by adding the term (−i[σdc/(ε0ω)]n) to eqn (1), where σdc is the dc-conductivity, ε0 is the permittivity of free space, and n ≈ 1.
We have further analyzed these two dielectric responses by fitting two H–N relaxation models and a conductivity contribution to the measured dielectric loss spectra as below
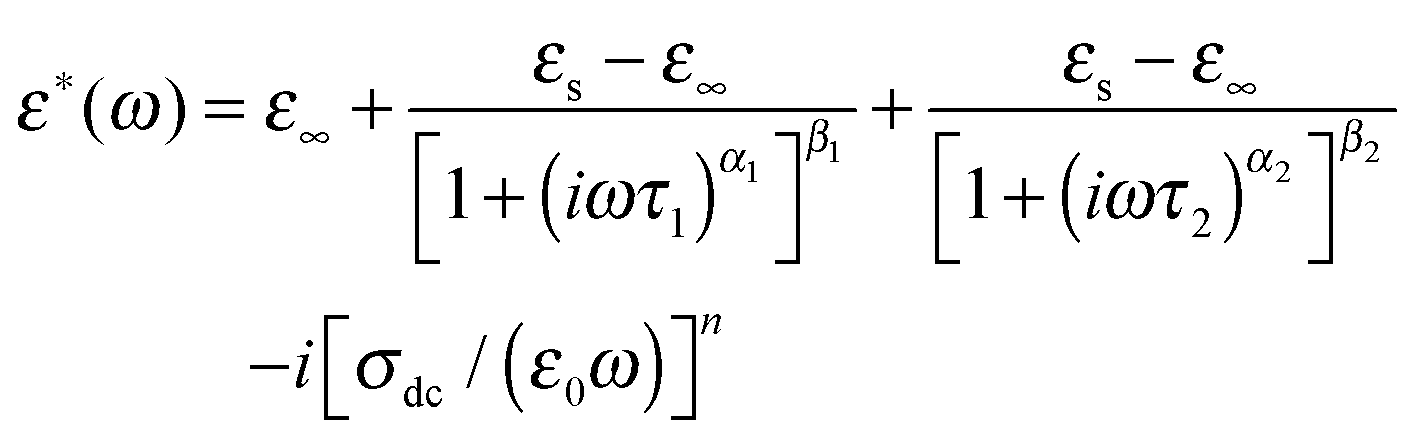 | (2) |
The relaxation time τ1 extracted from this fitting was found to follow the Arrhenius relation eqn (3), which suggests that the relaxation is a thermally activated process
τ1 = τ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(E1/kBT) exp(E1/kBT) | (3) |
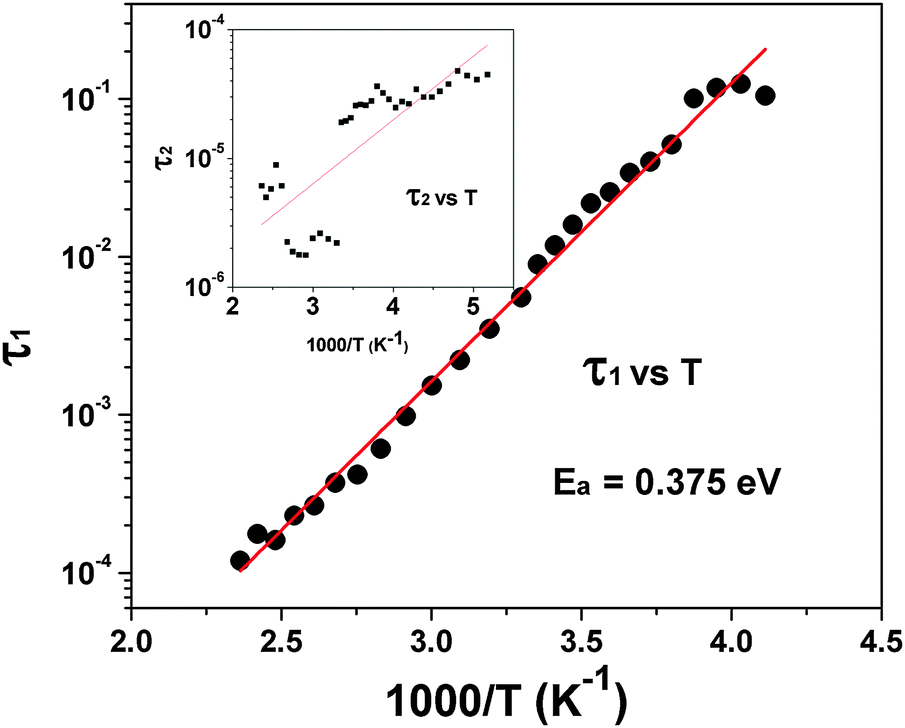 | ||
| Fig. 15 Typical Arrhenius fit for low temperature relaxation (τ1) of Pr2Ti2O7. The temperature dependency of τ2 is shown as the inset. | ||
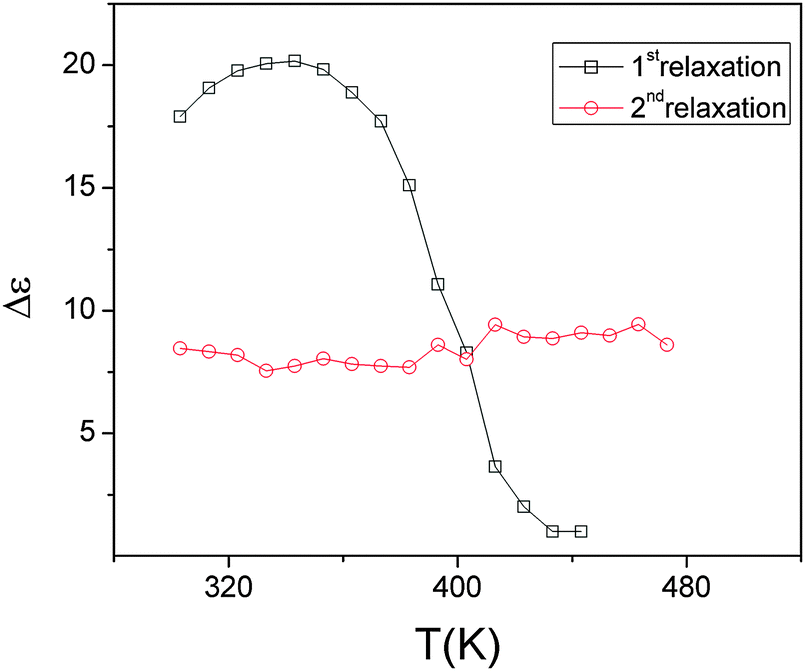 | ||
| Fig. 16 Variation of the dielectric strength of the two low temperature relaxations of Pr2Ti2O7 with temperature. | ||
To follow the dielectric properties at still higher temperature, the impedance spectroscopic studies at higher temperature were carried out. The variations of permittivity and tanδ with temperature are shown in Fig. 17. The temperature dependent permittivity shows a peak like feature around 673 K, which is similar to the ferroelectric like transition observed by Sun et al.17 Despite the crystalline nature of the present studied sample, this anomaly coincides with the results of Sun et al.17 A gradually decreasing trend of permittivity and shifting of peak temperature towards higher temperature are observed with increasing frequency. The variation of peak temperature with frequency follows Arrhenius relation and thus again support for a thermal activated process in PTO. At lower frequencies, a sharp rise in permittivity is observed. Such behaviours are expected for an ionic conductor due to the polarization of electrodes. This indicates that the low frequency dielectric anomaly is not related to the polar orientation. A sharp increase in loss tangent at higher temperature is observed in the variation of tanδ with temperature. Though the values of loss tangent are small at lower temperature, they show considerably larger values at higher temperature. Thus at higher temperature, PTO shows appreciable ionic conduction which can be either due to the formation of intrinsic defects or annihilation of defect clusters. Thus the dielectric anomaly is related to ionic movement in the system. In order to understand the electrical conductivity the total conductivity was extracted from the impedance data and the temperature dependent conductivities are shown in Fig. 18. From Fig. 18, it is observed that the conductivity decreases appreciably below 873 K. The activation energy for conductivity as obtained from Arrhenius relation is 0.60 eV. The observed activation energy is more similar for a mixed ionic conductor as observed earlier in Pr2Zr2O7 and other related systems.50,51 It can be mentioned here even though Pr2Ti2O7 and Pr2Zr2O7 have a different crystal structure, the activation energies for electrical conductivities are not significantly different, viz. Pr2Ti2O7 (0.60 eV) and Pr2Zr2O7 (0.42 eV).50 In both cases the mixed electronic conductivities are expected from the variation of the oxidation state of Pr, due to partial oxidation of Pr3+ to Pr4+. However, Pr2Zr2O7 show a significant oxidation of Pr3+ to Pr4+ at higher temperature in air due to the more open nature of pyrochlore structure compared to the perovskite structure of Pr2Ti2O7. Also the inter-cation (Pr3+–Pr3+, electron hopping sites) distances are larger in Pr2Ti2O7 compared to those in Pr2Zr2O7. In addition appreciable amounts of interstitial oxygen ions in Pr2Zr2O7+x favor oxide ion conduction in the lattice. Thus the observed activation energy of Pr2Ti2O7 is little higher compared to Pr2Zr2O7, but not appreciably different.
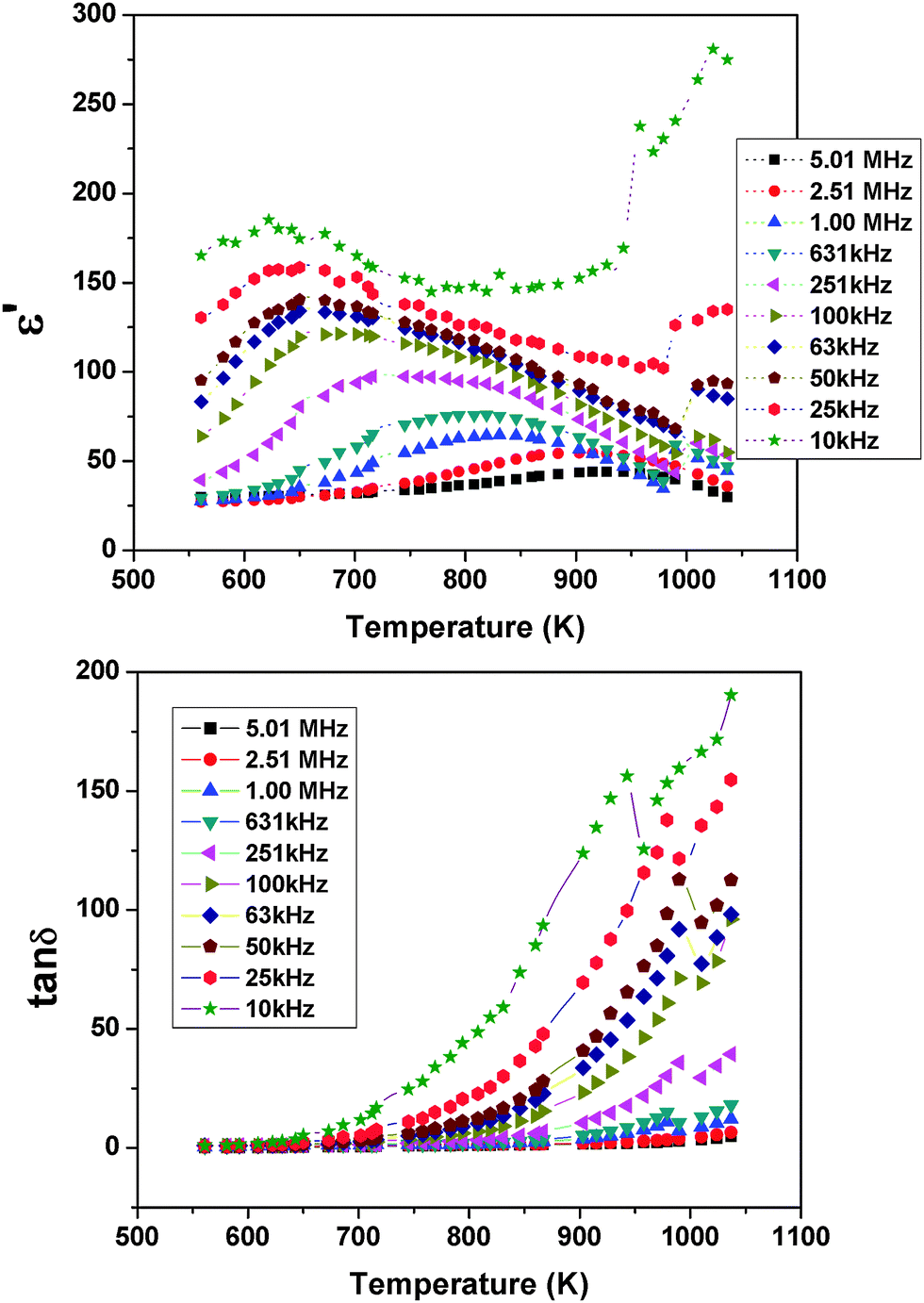 | ||
| Fig. 17 Temperature dependent ε′ and tanδ of Pr2Ti2O7 at selected frequencies in the high temperature region. | ||
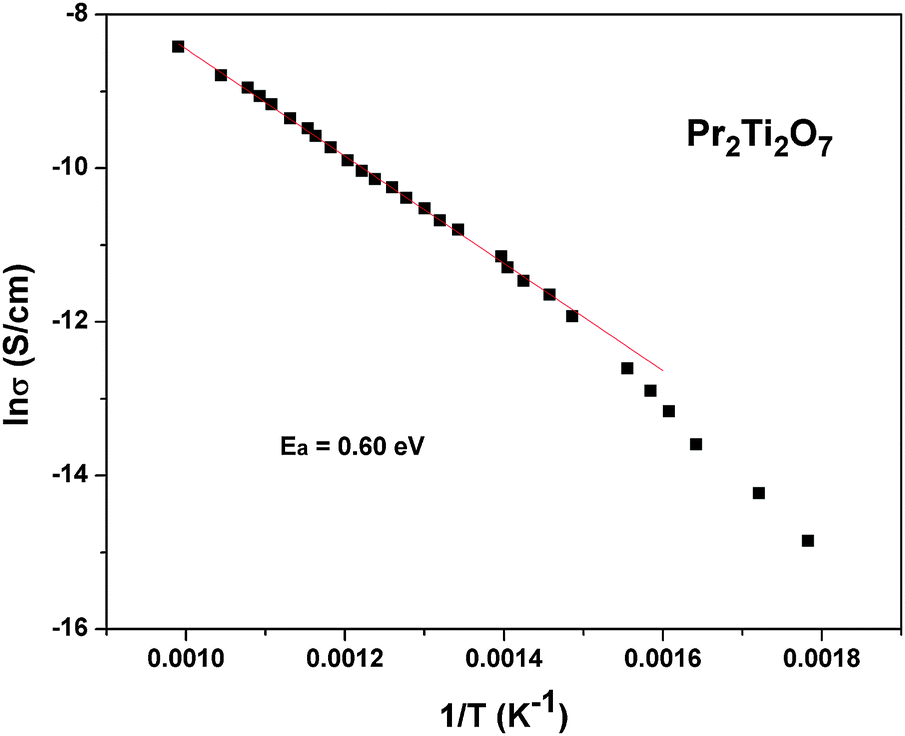 | ||
| Fig. 18 Variation of total conductivity of Pr2Ti2O7 with temperature at high temperature (Arrhenius fits are shown as a solid line). | ||
From the dielectric and impedance data it is observed that the electrical properties of Pr2Ti2O7 are cumulative effects of the grain boundary and ionic and/or electronic defects. The dielectric behaviour is similar to any of the ferroelectric materials while at higher temperature the migration of ions, in particular anions and electrons due to the feeble valence fluctuation of Pr3+ may occur. The anionic defects can be easily formed in the perovskite sub-lattice, either during the preparation or due to a slight deviation in the composition. Since the ferroelectric properties are related to the metal ion shifts and distortion of the TiO6 octahedra, a feeble deviation in oxygen may not alter the polar character drastically, but at higher temperature they appear as a loss contribution due to ion or electron movement.
5. Conclusions
From the ambient temperature XRD and Raman studies, a monoclinic P21 structure is established as the stable structure of Pr2Ti2O7. The crystal structure is further supported from the ab initio total energy calculations in DFT formalism. From the density functional calculations, it is observed that Pna21 and P21 have almost similar stability while the paraelectric P21/m or Cmcm structures have least stability at ambient temperature. A partial decomposition at 1673 K is noticed in Pr2Ti2O7. From the analyses of symmetry and structure, it was inferred that the ferroelectric (P21) to paraelectric (P21/m) transition is due to the displacement of cations along the b-axis and the rotation of TiO6 octahedral units around the a-axis. No structural transition to Cmc21 from ambient to 1473 K was observed. From the volume dependent energy calculations and high temperature XRD studies, it is revealed that PTO may transform to another ferroelectric orthorhombic (Cmc21) structure at a still higher temperature. The electron density of state calculations revealed that the ferroelectric Pr2Ti2O7 has a band gap of ∼2.7 eV which is close to the experimentally observed band gap of 3.10 eV. The measurement of field dependent electric polarization indicates the ferroelectric nature of PTO. The low temperature dielectric data indicate two different types of relaxations, one at the lower frequency side due to the thermally activated polarization process at the grain boundaries and other at higher frequency due to dipolar reorientation. The activation energy for thermally activated relaxation process is 0.38 eV and can be attributed to interfacial polarization due to ionic movements. At still higher temperature the dielectric properties are dominated by ionic conduction.Acknowledgements
The Department of Atomic Energy's Science Research Council (DAE-SRC) is acknowledged for supporting this work, vide sanction number no. 2010/21/9-BRNS/2025 dated 7-12-2010.References
- M. A. Subramanian, G. Aravamudan and G. V. Subba Rao, Prog. Solid State Chem., 1983, 15, 55 CrossRef CAS.
- A. W. Sleight, Inorg. Chem., 1968, 7, 1704 CrossRef CAS.
- L. G. Shcherbakova, L. G. Mamsurova and G. E. Sukhanova, Russ. Chem. Rev., 1979, 48, 228 CrossRef PubMed.
- H. Yan, H. Ning, Y. Kan, P. Wang and M. Reece, J. Am. Ceram. Soc., 2009, 92, 2270 CrossRef CAS PubMed.
- T.-M. Pan, M.-D. Huang, C.-W. Lin and M.-H. Wu, Sens. Actuators, B, 2010, 144, 139 CrossRef CAS PubMed.
- S. Nanamatsu, M. Kimura, K. Doi, S. Matsushita and N. Yamada, Ferroelectrics, 1974, 8, 511 CrossRef CAS.
- Z. P. Gao, H. X. Yan, H. P. Ning and M. J. Reece, Adv. Appl. Ceram., 2013, 112, 69 CrossRef CAS PubMed.
- M. Kimura, S. Nakamatsu, T. Kawamura and S. Matsushita, Jpn. J. Appl. Phys., 1974, 13, 1473 CrossRef CAS.
- N. A. Zakharov, V. S. Krikorov, E. F. Kustov and S. Y. Stefanovich, Phys. Status Solidi A, 1978, 50, K13 CrossRef CAS PubMed.
- N. A. Zakharov, S. Y. Stefanovich, E. F. Kustov and Y. N. Venevtsev, Krist. Tech., 1980, 15, 29 CrossRef CAS PubMed.
- D. W. Hwang, J. S. Lee, W. Li and S. H. Oh, J. Phys. Chem. B, 2003, 107, 4963 CrossRef CAS.
- Z. Shao, S. Saitzek, P. Roussel, O. Mentre, F. P. Gheorghiu, L. Mitoseriu and R. Desfeux, J. Solid State Chem., 2010, 183, 1652 CrossRef CAS PubMed.
- C. Wenger, G. Lupina, M. Lukosius, O. Seifarth, H.-J. Mussig, S. Pasko and C. Lohe, J. Appl. Phys., 2008, 103, 104103 CrossRef PubMed.
- E. Bruyer and A. Sayede, J. Appl. Phys., 2010, 108, 053705 CrossRef PubMed.
- J. Lopez-Perez and J. Iniguez, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 075121 CrossRef.
- V. V. Atuchin, T. A. Gavrilova, J.-C. Grivel, V. G. Kesler and I. B. Troitskaia, J. Solid State Chem., 2012, 195, 125 CrossRef CAS PubMed.
- L. Sun, L. Ju, H. Qin, M. Zhao, W. Su and J. Hu, Physica B, 2013, 431, 49 CrossRef CAS PubMed.
- P. M. Gasperin, Acta. Cryst. B, 1975, 31, 2129 CrossRef.
- P. A. Kozmin, N. A. Zakharov and M. D. Surazhskaya, Inorg. Mater., 1997, 33, 850 CAS.
- N. Ishizawa, F. Maruo, S. Iwai, M. Kimura and T. Kawamura, Acta Crystallogr., Sect. B: Struct. Sci., 1980, 36, 763 CrossRef.
- K. Scheunemann and H. Müller-Buschbaum, J. Inorg. Nucl. Chem., 1974, 36, 1965 CrossRef CAS.
- K. Scheunemann and H. Müller-Buschbaum, J. Inorg. Nucl. Chem., 1975, 37, 1879 CrossRef CAS.
- E. J. Harvey, S. E. Ashbrook, G. R. Lumpkin and S. A. T. Redfern, J. Mater. Chem., 2006, 16, 4665 RSC.
- N. Ishizawa, F. Marumo and S. I. Iwai, Acta Crystallogr., Sect. B: Struct. Sci., 1982, 38, 368 CrossRef.
- N. Ishizawa, K. Ninomiya, T. Sakakura and J. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, i19 CAS.
- J. M. Pruneda and E. Artacho, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085107 CrossRef.
- Z. L. Zhang, H. Y. Xiao, X. T. Zu, F. Gao and W. J. Weber, J. Mater. Res., 2009, 24, 1335 CrossRef CAS.
- H. Y. Xiao, L. M. Wang, X. T. Zu, J. Lian and R. C. Ewing, J. Phys.: Condens. Matter, 2007, 19, 346203 CrossRef.
- A. C. Larson and R. B. van Dreele GSAS, General Structure Analysis System. Los Alamos National Laboratory, Report LA-UR 86-748, 2000.
- Rodriguez-Carvajal, J. Fullprof 2000: A Program for Rietveld, Profile Matching and Integrated Intensity Refinements for X-ray and Neutron Data. Version 1.6, Laboratoire Leon Brillouin, Gif sur Yvette, France, 2000 Search PubMed.
- A. P. Roy, S. K. Deb, M. A. Rekha and A. K. Sinha, J. Pure Appl. Phys., 1992, 30, 724 CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev. B: Condens. Matter Mater. Phys., 1964, 136, 864 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev. A: At., Mol., Opt. Phys., 1965, 140, 1133 Search PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- R. L. C. Park, J. Am. Ceram. Soc., 1995, 78, 3171 CrossRef PubMed.
- V. Panchal, D. Errandonea, A. Segura, P. Rodriguez-Hernandez, A. Muñoz A, S. Lopez-Moreno and M. Bettinelli, J. Appl. Phys., 2011, 110, 043723 CrossRef PubMed.
- J. K. Brandon and H. D. Megaw, Philos. Mag., 1970, 21, 189 CrossRef CAS.
- G. Herrera, J. Jiménez-Mierc and E. Chavira, Mater. Charact., 2014, 89, 13 CrossRef CAS PubMed.
- S. Saha, S. Prusty, S. Singh, R. Suryanarayanan, A. Revcolevschi and A. K. Sood, J. Solid State Chem., 2011, 184, 2204 CrossRef CAS PubMed.
- D. Errandonea, S. N. Achary, J. Pellicer-Porres and A. K. Tyagi, Inorg. Chem., 2013, 52, 5464 CrossRef CAS PubMed.
- W. S. Kim, S.-M. Ha, S. Yun and H.-H. Park, Thin Solid Films, 2002, 420–421, 575 Search PubMed.
- A. Bayart, S. Saitzek, A. Ferri, R. Pouhet, M.-H. Chambrier, P. Roussel and R. Desfeuxa, Thin Solid Films, 2014, 553, 71 CrossRef CAS PubMed.
- A. Schönhals and F. Kremer, in Broadband Dielectric Spectroscopy, ed. F. Kremer and A. Schönhals, Springer Verlag, Berlin, 2003 Search PubMed.
- C. C. Wang, H. B. Lu, K. J. Jin and G. Z. Yang, Phys. Lett. B, 2008, 22, 1297 CAS.
- K. Vasundhara, S. N. Achary and A. K. Tyagi, Int. J. Hydrogen Energy, 2015, 40, 4252 CrossRef CAS PubMed.
- J. Zhang, W. Peng, Z. Chen, H. Chen and L. Han, J. Phys. Chem. C, 2012, 116, 19182 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Rietveld refinement plots and structural parameters of Pr2Ti2O7 in different model symmetries, differential scanning calorimetric traces and scanning electron microscopy images of Pr2Ti2O7. See DOI: 10.1039/c5tc00242g |
| This journal is © The Royal Society of Chemistry 2015 |