
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antibacterial 45S5 Bioglass®-based scaffolds reinforced with genipin cross-linked gelatin for bone tissue engineering†
Wei
Li‡
a,
Hui
Wang‡
b,
Yaping
Ding
c,
Ellen C.
Scheithauer
a,
Ourania-Menti
Goudouri
a,
Alina
Grünewald
a,
Rainer
Detsch
a,
Seema
Agarwal
b and
Aldo R.
Boccaccini
*a
aInstitute of Biomaterials, Department of Materials Science and Engineering, University of Erlangen-Nuremberg, Cauerstrasse 6, 91058 Erlangen, Germany. E-mail: aldo.boccaccini@ww.uni-erlangen.de; Fax: +49 9131 85 28602; Tel: +49 9131 85 28601
bUniversity of Bayreuth, Macromolecular Chemistry II and Bayreuth Center for Colloids and Interfaces, Universitaetsstrasse 30, 95440 Bayreuth, Germany
cInstitute of Polymer Materials, Department of Materials Science and Engineering, University of Erlangen-Nuremberg, Martensstrasse 7, 91058 Erlangen, Germany
First published on 9th March 2015
Abstract
45S5 Bioglass® (BG) scaffolds with high porosity (>90%) were coated with genipin cross-linked gelatin (GCG) and further incorporated with poly(p-xylyleneguanidine) hydrochloride (PPXG). The obtained GCG coated scaffolds maintained the high porosity and well interconnected pore structure. A 26-fold higher compressive strength was provided to 45S5 BG scaffolds by GCG coating, which slightly retarded but did not inhibit the in vitro bioactivity of 45S5 BG scaffolds in SBF. Moreover, the scaffolds were made antibacterial against both Gram-positive and Gram-negative bacteria by using polyguanidine, i.e. PPXG, in this study. Osteoblast-like cells (MG-63) were seeded onto PPXG and GCG coated scaffolds. PPXG was biocompatible with MG-63 cells at a low concentration (10 μg mL−1). MG-63 cells were shown to attach and spread on both uncoated and GCG coated scaffolds, and the mitochondrial activity measurement indicated that GCG coating had no negative influence on the cell proliferation behavior of MG-63 cells. The developed novel antibacterial bioactive 45S5 BG-based composite scaffolds with improved mechanical properties are promising candidates for bone tissue engineering.
1. Introduction
Tissue and organ failure is a major health problem. Among them, bone is one of the most common tissues necessitating replacement or repair as bone failure can widely result from trauma, tumor, bone related diseases or aging.1 Using scaffolds made from engineered biomaterials is an effective approach to restore function of damaged bone or to regenerate bone tissues.2 An ideal scaffold should act as a temporary template to support cell activity and to induce extracellular matrix deposition until new bone forms in the defect sites.3,4 The essential properties that an ideal scaffold should possess for bone tissue engineering applications have been comprehensively discussed in detail in the literature,2,5–7 and include suitable mechanical properties, bioactivity and 3D pore architecture.45S5 Bioglass® (BG)-based scaffolds fabricated by the foam replication method meet several important properties of an ideal bone tissue engineering scaffold, due to the intrinsic bioactivity, biocompatibility, osteogenic and angiogenic effects of 45S5 BG,8–10 and the high porosity and interconnected large pore structure derived from the foam replication method.11,12 The high porosity and large pore size of such scaffolds are favorable for osteogenesis and vascularization throughout the entire 3D structure.12,13 However, the high porosity also limits the mechanical properties of the scaffolds.11 Besides the concern of mechanical properties, antibacterial action should also be taken into consideration since the risk of infection exists during scaffold implantation which may eventually lead to implantation failure. To this end, in previous efforts 45S5 BG scaffolds have been coated with polymers (e.g. poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV)14 or polycaprolactone (PCL)/chitosan15), and these polymers were shown to not only enhance the mechanical properties of the scaffolds without significantly sacrificing the porosity and pore size but also impart an antibiotic release function to the scaffolds. It is worth noting that the compressive strength of these coated scaffolds (0.1–0.2 MPa), although improved, still falls close to the lower bound of the values for cancellous bone.16 Therefore, the relatively low mechanical properties still limit the potential application of these coated scaffolds.
The present research aims at developing 45S5 BG-based scaffolds with a novel coating based on gelatin for increased mechanical properties (compressive strength and the work of fracture) and antibacterial effect. Gelatin, a water soluble natural polymer, has been shown to be able to considerably improve the mechanical properties of bioactive glass/ceramic scaffolds due to its strengthening and toughening effects which can be linked to a micron-scale crack-bridging mechanism.17,18 However, gelatin, in its original state, dissolves/degrades rapidly in aqueous solution,19,20 which may lead to the quick loss of its reinforcing effects on scaffolds. In order to decrease the dissolution/degradation rate, gelatin has been chemically cross-linked with crosslinking reagents, such as glutaraldehyde or genipin.19–21 Genipin was reported to be much less cytotoxic than glutaraldehyde.22 Therefore, in the present study, genipin is selected as the crosslinking agent for gelatin which will be used for coating the 45S5 BG scaffolds prepared by the foam replication method.
As mentioned above, an antibiotic release function can be incorporated into the scaffolds in order to reduce and combat bacterial infection, which is one of the major complications associated with implants.23,24 However, the emergence of resistance of bacteria to antibiotics becomes a common phenomenon, because inappropriate antibacterial treatment and overuse of antibiotics accelerate the evolution of resistant strains.25 Therefore, there is a particular interest in the development of new biocides in order to fight infections. Biocidal cationic polymers, such as polyguanidines, have attracted considerable attention for their high antibacterial activity and low toxicity to humans, and they have been widely investigated or used as disinfectants or biocides in ophthalmology, water systems, topical wounds and environments.26–29 The antibacterial action of the polyguanidines starts with the interaction of positively charged polymer molecules with the bacteria which carry a net negative charge on their surface due to negatively charged lipids in the cell membrane, and followed by the hole-formation i.e., perturbations of the polar headgroups and the hydrophobic core region of the lipids membranes killing the bacteria.30,31
Based on the facts discussed above, in order to incorporate an effective antibacterial function to the gelatin coated 45S5 BG-based scaffolds, poly(p-xylyleneguanidine) hydrochloride (PPXG), which belongs to the polyguanidines, was used as the antibacterial agent. To the best of our knowledge, polyguanidine has not been used as an antibacterial agent in bone tissue engineering scaffolds; this work was thus dedicated to fabricate and characterize 45S5 BG scaffolds which were coated with genipin cross-linked gelatin (GCG) and then incorporated with PPXG. This study is focused on the investigation of the antibacterial effect and biocompatibility that PPXG can confer to the 45S5 BG scaffolds. In addition, the influence of GCG coating on the mechanical properties and bioactivity of the 45S5 BG scaffolds was also studied.
2. Materials and methods
2.1. Preparation of 45S5 BG scaffolds
Commercially available melt-derived 45S5 BG powder (mean particle size ∼5 μm) and polyurethane (PU) foams (45 pores per inch, Eurofoam, Troisdorf, Germany) were used for preparing the scaffolds by the foam replication method.11,32 In brief, the slurry was prepared by dissolving 6% w/v polyvinyl alcohol (PVA) (Mw ∼ 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Merck, Darmstadt, Germany) in water, and followed by adding 45S5 BG powder to the PVA solution up to a concentration of 50 wt%. PU foams were immersed and rotated in the slurry, and then taken out from the slurry. The extra slurry was completely squeezed out from the foams. The foams were left to dry at room temperature followed by repeating the procedure described above one more time. The obtained green bodies were heated at 400 °C for 1 h to decompose the PU foams, and then at 1100 °C for 2 h to densify the glass network. The heating and cooling rates used were 2 °C min−1 and 5 °C min−1, respectively.
000, Merck, Darmstadt, Germany) in water, and followed by adding 45S5 BG powder to the PVA solution up to a concentration of 50 wt%. PU foams were immersed and rotated in the slurry, and then taken out from the slurry. The extra slurry was completely squeezed out from the foams. The foams were left to dry at room temperature followed by repeating the procedure described above one more time. The obtained green bodies were heated at 400 °C for 1 h to decompose the PU foams, and then at 1100 °C for 2 h to densify the glass network. The heating and cooling rates used were 2 °C min−1 and 5 °C min−1, respectively.
2.2. Synthesis and antibacterial activity of PPXG
PPXG was synthesized by condensation polymerization of p-xylylenediamine and guanidine hydrochloride in melt according to the literature.29,33 A dry 100 mL three necked round bottom flask equipped with a thermometer and a reflux condenser was charged with guanidine hydrochloride (6.18 g, 50.00 mmol) and p-xylylenediamine (4.78 g, 50.00 mmol). The reagents were heated up to 150 °C. The polycondensation reaction was stopped after 5 hours by cooling the reaction flask in an ice bath and the polymer was obtained as a colorless transparent solid. The polymer was structurally characterized using 1D (1H and 13C), 2D heteronuclear single quantum coherence (HSQC) NMR and atmospheric pressure chemical ionization (APCI) spectroscopic techniques (Fig. S1 and S2; see ESI†). The molecular weight of the polymer (Mn:2200, Mw:2500, PDI: 1.12) was determined by MALDI-TOF MS (Fig. S3, ESI†). In addition, the thermal behavior of the polymer was analyzed using differential scanning calorimetry (DSC) and thermogravimetry (TG) (Fig. S4 and S5, ESI†).
Minimal inhibitory concentration (MIC) and minimal bactericidal concentration (MBC) were evaluated to determine the antibacterial activity of PPXG. For a dilution series of the PPXG solution starting from 1000 μg mL−1, each 500 μL was prepared in a sterile 24 well plate (Greiner bio-online). Equal volumes of bacteria (Bacillus subtilis (B. subtilis) or Escherichia coli (E. coli); 106 cfu mL−1) were added and incubated for 24 h at 37 °C. After this the wells were visually evaluated for bacterial growth. The lowest concentration which remained transparent was taken as the MIC. To determine the MBC, 100 μL of solution was removed from each clear well and spread on nutrient agar plates and incubated for a further 24 h at 37 °C. The lowest concentration of biocide at which no colony formation was observed was taken as the MBC. Each test was done in quadruplicate.
2.3. Incorporation of GCG coating and PPXG
Gelatin–genipin solution at a concentration of 5% w/v was prepared by dissolving gelatin (type A, Sigma-Aldrich, St. Louis, MO, USA) and genipin (Wako, Osaka, Japan) together in a distilled water–ethanol mixture (5 vol% ethanol) under magnetic stirring at 50 °C. The amount of genipin in the gelatin–genipin mixture was 1 wt%. The 45S5 BG scaffolds were then completely immersed in the gelatin–genipin mixture solution for 1 min under vacuum, and then dried at room temperature for 1 day. Subsequently the above coating procedure was repeated one more time.In order to load different amounts of PPXG directly into the GCG coated scaffolds, PPXG was dissolved in methanol at concentrations of 40, 120 and 200 μg mL−1, respectively. Then 0.5 mL PPXG solution of each concentration was dripped onto the GCG coated scaffolds from different sides, followed by drying at room temperature for 1 day.
2.4. Characterization of scaffolds
The porosity of scaffolds before (p1) and after (p2) coating with GCG was calculated by eqn (1) and (2):
| p1 = 1 − M1/(ρBGV1) | (1) |
| p2 = 1 − (M1/ρBG + (M2 − M1)/ρGCG)/V2 | (2) |
2.5. In vitro bioactivity and degradation tests
The in vitro bioactivity test was performed using the standard procedure described by Kokubo et al.35 The scaffolds with dimensions of 10 mm × 8 mm × 8 mm were immersed in 50 mL of SBF and maintained at 37 °C in a shaking incubator (90 rpm) for 1, 3, 7, 14 and 28 days. The SBF was replaced twice a week during the test. Once removed from the incubator, the scaffolds were rinsed with deionized water and left to dry at room temperature. Afterwards, SEM, XRD and FTIR were used to assess hydroxyapatite (HA) formation on the scaffolds. Weight loss of the samples was calculated using eqn (3):| Weight loss (%) = (M1 − M2)/M1 × 100% | (3) |
In addition to uncoated 45S5 BG scaffolds, the degradation behavior of pure GCG films was also studied in SBF following the procedure as described above for comparison with the degradation behavior of GCG coated 45S5 BG scaffolds. GCG films were prepared by drying the GCG solution used for coating scaffolds in a petri dish.
2.6. Antibacterial test
Antibacterial activity was characterized by Kirby–Bauer test and time-dependent shaking flask test. E. coli (DSM No. 1077, K12 strain 343/113, DSMZ) as the gram-negative and B. subtilis (DSM No. 2109, ATCC 11774, ICI 2/4 strain, DSMZ) as the gram-positive test organism were used.29 Tryptic soy broth (TSB) (Sigma-Aldrich, Germany) was used as a nutrient for E. coli (30 g L−1 in distilled water for liquid nutrient; 15 g L−1 agar–agar in addition for nutrient agar plates) and peptone/meat extract medium for B. subtilis (5 g L−1 peptone and 3 g L−1 meat extract in distilled water for the liquid nutrient; 15 g L−1 agar–agar in addition for nutrient agar plates). Both strains were preserved on nutrient agar plates and liquid cultures were grown by inoculation of the liquid nutrient with a single bacteria colony using an inoculation loop. The inoculated broth was incubated under shaking at 37 °C until the optical density at 578 nm had increased by 0.125 indicating a cell density of 107–108 cfu mL−1. To obtain the final bacterial suspensions the inoculated broth was diluted with the liquid nutrient to an approximate cell density of 106 cfu mL−1.2.7. In vitro biocompatibility test
In vitro biocompatibility tests were carried out using the human osteosarcoma cell line MG-63 (Sigma-Aldrich, Germany). Cells were cultured at 37 °C in a humidified atmosphere of 95% air and 5% CO2 in DMEM (Dulbecco's modified Eagle's medium, Gibco, Germany) containing 10 vol% fetal bovine serum (FBS, Sigma-Aldrich, Germany) and 1 vol% penicillin/streptomycin (Gibco, Germany). Cells were grown to confluence in 75 cm2 culture flasks (Nunc, Denmark), and afterwards harvested using Trypsin/EDTA (Gibco, Germany) and counted using a hemocytometer (Roth, Germany).PPXG is water soluble and dissolves in aqueous medium rather quickly. Since the pH of BG scaffolds needs to be regulated in aqueous medium before seeding the cells, PPXG preloaded on GCG coated 45S5 BG scaffolds will not be present on the scaffold anymore after the pH regulation step. Therefore, in this study, the in vitro biocompatibility tests were carried out in two steps rather than directly on GCG coated 45S5 BG scaffolds loaded with PPXG. Firstly, a preliminary test was performed on PPXG, genipin and GCG in order to understand the behavior of MG-63 cells in the presence of these individual components of the GCG coated 45S5 BG scaffolds. This test was carried out in a short term (2 days), because these components will rather quickly dissolve in the cell culture medium which thus makes long term testing impossible as the cell culture medium needs to be changed every few days. Cell cultivation in the well plate without any material was used as a control. Secondly, MG-63 cells were directly cultured onto the uncoated and GCG coated 45S5 BG scaffolds. Uncoated 45S5 BG scaffolds were used as a control.
For preparing the samples, PPXG, genipin and GCG were sterilized by filtering their respective solution through a 0.22 μm syringe filter. PPXG was dissolved in distilled water, while genipin or gelatin–genipin mixture was dissolved in a distilled water–ethanol mixture solution (5 vol% ethanol). Uncoated 45S5 BG scaffolds were sterilized at 160 °C for 2 h in a furnace (Nabertherm, Germany). GCG coated 45S5 BG scaffolds were prepared by using sterilized GCG solution and sterilized uncoated 45S5 BG scaffolds.
000 MG-63 cells in 0.6 mL cell culture medium were seeded into each well, and cells were cultivated for 2 days without changing the culture medium. Therefore, the tested concentration of PPXG was 10 μg mL−1, 30 μg mL−1 and 50 μg mL−1, genipin was 50 μg mL−1, and GCG was 1 mg mL−1 and 5 mg mL−1. The water soluble tetrasodium (WST) test, a colorimetric assay, was used to assess the cell viability. After cell cultivation, cell culture medium was removed and samples were washed with 0.5 mL phosphate buffered saline (PBS). Afterwards, 0.25 mL WST medium (containing 1 vol% of WST reagent (Cell Counting Kit-8, Sigma) and 99 vol% of DMEM medium) was added and incubated for 2 h. After incubation, 0.1 mL of the supernatant was transferred to a 96-well culture plate and spectrometrically measured using a microplate reader (PHOmo, anthos Mikrosysteme GmbH, Germany) at 450 nm. To analyze the adherent growth of cells on the samples, green Calcein AM (Molecular Probes, The Netherlands) cell-labelling solution were used for staining the cytoplasm of the cells. After removing the cell culture medium, 0.25 mL staining solution (0.5 vol% of dye labelling solution and 99.5 vol% of PBS) was added and incubated for 30 min. Afterwards, the solution was removed and the samples were washed with 0.5 mL PBS. Cells on the surfaces were fixed by 3.7 vol% paraformaldehyde. Samples were washed again and blue fluorescent DAPI (4′,6-diamidino-2-phenylindole dihydrochloride, Roche, Basel, Switzerland) was added to label the nucleus. After 5 minutes of incubation, the solution was removed and the samples were left in PBS for microscopic viewing using a fluorescence microscope (Axio Scope, ZEISS, Germany).
2.8. Statistical analysis
All quantitative data were expressed as the mean ± standard deviation. Statistical analysis was performed with one-way analysis of variance (ANOVA) using Microsoft Excel 2010 (Microsoft, Redmond, WA, USA). A value of P < 0.05 was considered statistically significant.3. Results and discussion
3.1. Morphology of scaffolds
Typical morphologies of the uncoated (Fig. 1(a) and (b)) and GCG coated (Fig. 1(c) and (d)) 45S5 BG scaffolds were observed by SEM. The uncoated scaffolds (Fig. 1(a)) exhibited a highly interconnected pore structure. The porosity and pore size were determined to be 95% and 200–550 μm, respectively. After coating with GCG (Fig. 1(c)), the interconnected pore structure of the scaffolds was maintained since only very few pores were clogged by the coating, and the porosity slightly decreased to 93%. The amount of GCG in the coated scaffolds was determined to be 15 ± 2 wt%. As shown in the cross section image at a high magnification, the strut of the scaffold is homogeneously covered by the GCG coating (Fig. 1(d)), and the GCG coating firmly adheres to the strut (Fig. 1(d) and (e)), which is qualitatively confirmed by the fact that the GCG coating did not peel off during cutting of the scaffolds. Moreover, it is worth noting that the voids of the hollow struts, which result from the burning out of polyurethane during the foam replication method (Fig. 1(b)),11 were mostly filled with the GCG (Fig. 1(d)). This filling effect could be attributed to the infiltration of the polymer solution into the hollow struts under the applied vacuum condition for coating the scaffolds, and it means many defects and cracks on the struts can be “repaired” by the GCG coating, as evidenced by the quite smooth surface of the GCG coated strut (Fig. 1(e)), thus expecting a positive contribution to the mechanical behavior of the scaffolds. | ||
| Fig. 1 SEM images of 45S5 BG scaffolds (a) and (b) before and (c)–(e) after coating with GCG. | ||
3.2. Degradation behavior
Gelatin films without any crosslinking completely dissolved at 37 °C in SBF within a few minutes, which would lead to the loss of their potential strengthening and toughening effects as a coating on scaffolds. Therefore, the main aim to crosslink gelatin is to decrease its dissolution/degradation rate. GCG films only exhibited 24% weight loss after immersion in SBF for 1 day, and their weight loss increased to 62% after 7 days (Fig. 2). GCG films were still present in SBF after 14 days, but they already broke up into small gelatinous blue pieces which therefore made the weight loss measurement impossible. Also, only small gelatinous blue pieces were visible in the SBF solution after 28 days. The decrease of the dissolution/degradation rate of gelatin after crosslinking with genipin has also been reported in other studies.21 It should be pointed out that although the dissolution/degradation behavior of the GCG film cannot be considered as GCG coating existed on the 45S5 BG scaffolds equally, it still could represent the gradual dissolution/degradation trend of GCG coating. Actually, this gradually dissolution/degradation trend of GCG coating on 45S5 BG scaffolds can be proved by the FTIR results of GCG coated scaffolds before and after immersion in SBF for different times. As shown in Fig. 3, compared to the spectra of uncoated 45S5 BG scaffolds, two new bands can be observed at 1660 cm−1 and 1540 cm−1. These bands are identified as amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (amide I) and amide N–H bending vibration (amide II),21,36,37 which indicate the presence of gelatin, in this particular case genipin cross-linked gelatin (GCG). The intensity of the amide I band (1660–1650 cm−1) and amide II band (1540 cm−1) decreased and almost disappeared as immersion time in SBF increased, suggesting the gradual dissolution/degradation of the GCG coating.
O stretching vibration (amide I) and amide N–H bending vibration (amide II),21,36,37 which indicate the presence of gelatin, in this particular case genipin cross-linked gelatin (GCG). The intensity of the amide I band (1660–1650 cm−1) and amide II band (1540 cm−1) decreased and almost disappeared as immersion time in SBF increased, suggesting the gradual dissolution/degradation of the GCG coating.
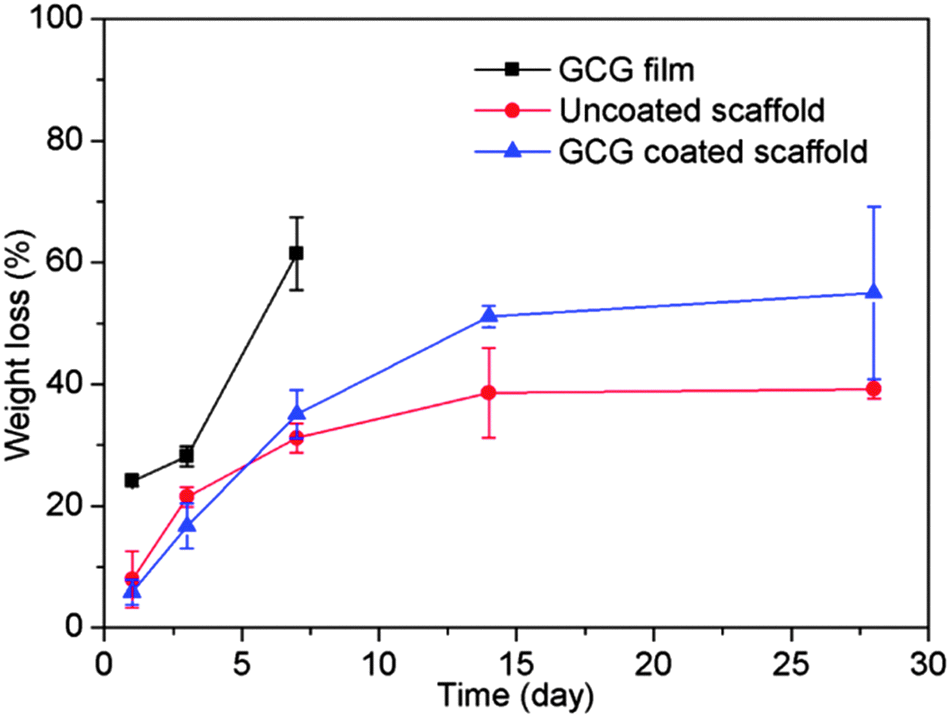 | ||
| Fig. 2 Degradation behaviors in SBF of GCG films, uncoated and GCG coated 45S5 BG scaffolds. | ||
 | ||
| Fig. 3 FTIR spectra of uncoated 45S5 BG scaffolds (labelled as uncoated), and GCG coated 45S5 BG scaffolds before (0 d) and after immersion in SBF for 3, 7, 14 and 28 days. | ||
As shown in Fig. 2, the weight loss of uncoated 45S5 BG scaffolds increases with immersion time; however the degradation rate is reduced with immersion time. The degradation of bioactive glass/ceramic-based scaffolds consists of the partial dissolution of the glass and crystalline phases and the formation of HA on the scaffold surface.38 The rapid weight loss at initial immersion times is due to the fast dissolution of the 45S5 BG surface upon immersion in SBF. As the immersion time increases, HA begins to form on the 45S5 BG scaffolds,14 which compensates the weight loss caused by dissolution and therefore reduces the overall degradation rate of the 45S5 BG scaffolds. The weight loss of GCG coated 45S5 BG scaffolds was similar to that of uncoated 45S5 BG scaffolds for up to 7 days, and then became faster after 7 days. The weight loss caused by the dissolution of the 45S5 BG surface should be slower in the presence of GCG coating at the initial immersion stage; however the GCG coating begins to gradually dissolve upon immersion in SBF which therefore results in the overall weight loss of the GCG coated 45S5 BG scaffolds increasing and eventually it becomes similar to that of the uncoated 45S5 BG scaffolds. As suggested by the dissolution/degradation behavior of the GCG film, the GCG coating on the 45S5 BG scaffolds is also likely to largely dissolve/degrade in SBF after 7 days. Moreover, as HA forms on both uncoated and GCG coated 45S5 BG scaffolds after 7 days, the higher weight loss of GCG coated scaffolds over uncoated scaffolds is assumed to be mainly attributed to the loss of the GCG coating. To a certain extent, this assumption is confirmed by the fact that the 12 wt% difference of the weight loss of uncoated and GCG coated scaffolds after 14 days of immersion in SBF is close to the amount (15 wt%) of GCG in the coated scaffolds.
3.3. In vitro bioactivity of GCG coated 45S5 BG scaffolds
As an assessment of bioactivity, HA formation on the surface of scaffolds upon immersion in SBF was characterized by FTIR, XRD and SEM. Fig. 3 shows FTIR spectra of GCG coated 45S5 BG scaffolds before and after immersion in SBF. The FTIR spectra of GCG coated scaffolds after 3, 7, 14 and 28 days of immersion in SBF present dual bands at 564 cm−1 and 602 cm−1 corresponding to the bending vibration of the P–O bond, which is characteristic of a crystalline phosphate phase.14,39–41 Furthermore, the band at 876 cm−1 and the dual broad bands at 1423–1455 cm−1 can be assigned to the stretching vibration of the C–O bond, suggesting that the formed HA is carbonated hydroxyapatite (cHA) rather than stoichiometric hydroxyapatite.14,39,40,42,43 It should be noted that, for GCG coated 45S5 BG scaffolds, the characteristic bands of cHA after 3 days of immersion in SBF were relatively weaker in comparison to that after 7 days. As shown in our previous study,14 for uncoated 45S5 BG scaffolds, the characteristic bands of cHA did not appear after 1 day of immersion in SBF, while these bands occurred after 3 days and their relative intensities were quite close to those that appeared after 7 days. This comparison between the FTIR spectra of uncoated and GCG coated 45S5 BG scaffolds after immersion in SBF suggests that the bioactivity of 45S5 BG scaffolds was maintained after coating with GCG, although the GCG coating may slightly retard the formation rate of cHA at the initial stage of immersion in SBF.Fig. 4 shows the XRD spectra of GCG coated scaffolds before and after immersion in SBF. The peaks in scaffolds before immersion in SBF correspond to the Na4Ca4(Si6O18) and Na2Ca4(PO4)2SiO4 phases, which have also been found in previous studies.14,44 Growing HA peaks (e.g. at 2θ = 25.8° and 31.7°) were observed on coated scaffolds after immersion in SBF for 7, 14 and 28 days. In addition, the crystallinity of the sintered scaffolds decreased with increasing immersion time in SBF as indicated by the gradual disappearance of the sharp peaks of the Na4Ca4(Si6O18) phase.
 | ||
| Fig. 4 XRD spectra of GCG coated 45S5 BG scaffolds before (0 d) and after immersion in SBF for 3, 7, 14 and 28 days. | ||
SEM images of GCG coated scaffolds after immersion in SBF for different times are shown in Fig. 5. After 3 days immersion in SBF, there were some apatite-like precipitates on the surface of struts. As the immersion time increased to 7 days, the struts were almost fully covered by HA crystals which can be clearly recognized by their well-known globular and cauliflower-like shape.
 | ||
| Fig. 5 SEM images showing HA formation on the surfaces of GCG coated 45S5 BG scaffolds after immersion in SBF for (a) and (b) 3 days and (c) and (d) 7 days. | ||
Based on the XRD, FTIR and SEM results described above, the bioactivity of the 45S5 BG scaffolds is confirmed to be maintained after coating with GCG. The explanation for HA formation on polymer coated bioactive glass/ceramic scaffolds has been given in our previous studies.14,17 Briefly, some areas of the struts are not fully covered by the polymer as a result of the surface roughness of the original struts. Thus, uncoated areas of the struts provide paths for SBF to penetrate the area underneath the coating. Besides, in the present study, GCG will gradually dissolve/degrade in the SBF which enables coated areas of the struts to be increasingly exposed to SBF. Thus, the established direct contact between SBF and the surface of bioactive glass/ceramic struts is essential to retain the intrinsic bioactivity of the scaffolds.
3.4. Mechanical properties
The mechanical properties of uncoated and GCG coated 45S5 BG scaffolds were investigated by the uniaxial compressive strength test. As indicated by the typical compressive stress–strain curves of these scaffolds (Fig. 6), the compressive strength of GCG coated scaffolds (1.04 ± 0.11 MPa) was significantly higher than that of uncoated scaffolds (0.04 ± 0.01 MPa). The area under the load–displacement curve (related to the work of fracture) of GCG coated scaffolds was calculated to be 285.6 ± 23.3 N mm, whereas it was only 5.0 ± 1.1 N mm for the uncoated scaffolds. It is worth pointing out that the uncoated scaffolds were completely broken into little pieces during compressive strength test, while the GCG coated scaffolds were able to partly maintain their cuboid shape despite being compressed (Fig. 7). Taking into consideration the high porosity (93%) of the fabricated GCG coated scaffolds, the achieved compressive strength (1.04 MPa) is obviously higher than the lower bound of the values for human cancellous bone (>0.15 MPa, porosity ∼90%).16 | ||
| Fig. 6 Typical compressive stress–strain curves of uncoated and GCG coated 45S5 BG scaffolds, showing a remarkable improvement of mechanical properties by the presence of the GCG coating. | ||
 | ||
| Fig. 7 Digital photographs of (a) and (b) uncoated and (c) and (d) GCG coated 45S5 BG scaffolds before ((a) and (c)) and after ((b) and (d)) compressive strength test. | ||
It is well-known that polymer coatings can not only fill microcracks on the strut surfaces but also fill the void of hollow struts.14,18,45 In other words, the polymer coatings turn the original weak and brittle struts into strong and tough composite struts, thus significantly improving the mechanical stability of the flaw sensitive glass/ceramic struts. As a consequence, the compressive strength and toughness of the uncoated scaffolds in the present study are considerably improved after coating with GCG. The strengthening and toughening effects in the present study are in broad agreement with other studies about polymer coated scaffolds,14,17,45,46 and they can be explained by the micron-scale crack-bridging mechanism.46–48
It is worth mentioning that the GCG coating provides much more significant strengthening and toughening effects than PHBV or PCL/chitosan coating on 45S5 BG scaffolds.14,49 Similarly, significant strengthening and toughening effects were also observed on non-cross-linked gelatin coated Biosilicate® scaffolds.17 The different degrees of strengthening and toughening effects obtained from different polymer coatings are likely to be determined by the wettability of polymer solution on the scaffold struts and the adhesion ability of the obtained polymer coating on the scaffold struts. Obviously, low viscosity gelatin aqueous solution is much easier to spread on and also infiltrate into the hydrophilic glass/ceramic struts than other polymer solutions in which synthetic polymers (e.g. PHBV or PCL) are dissolved in organic solvent (e.g. dichloromethane or chloroform). Also, the interface between the hydrophilic polymer (i.e. gelatin) and hydrophilic glass/ceramic strut is likely to be stronger than that between the hydrophobic polymer (e.g. PHBV or PCL) and hydrophilic struts, given the evidence that GCG adheres well to the surface of scaffold struts (Fig. 1(d)).
3.5. Antibacterial properties
PPXG exhibited high antibacterial activity as determined by MIC and MBC values. It showed MIC values of 7.81 μg mL−1 and 32.25 μg mL−1, and MBC values of 31.25 μg mL−1 and 62.50 μg mL−1 for B. subtilis and E. coli, respectively (Fig. S6 and S7, ESI†). PPXG was used for providing antibacterial property to GCG coated 45S5 BG scaffolds. The antibacterial property was tested using the Kirby–Bauer test and the samples were qualitatively checked for the zone of inhibition after incubation (Fig. 8(a) and (b)). Both the uncoated and GCG coated 45S5 BG scaffolds (labelled 1 and 2) without PPXG did not show any zone of inhibition to the B. subtilis and E. coli (Fig. 8(c) and (d)). GCG coated scaffolds loaded with PPXG showed an increasing zone of inhibition to the B. subtilis as the PPXG concentration increased (Fig. 8(c)). GCG coated scaffold loaded with 10 μg mL−1 PPXG did not clearly exhibit a zone of inhibition to E. coli (Fig. 8(d)). However, the zone of inhibition occurred and was further increased as the PPXG concentration increased. After checking the zone of inhibition, a swab from the area under the samples (Fig. 8(c) and (d)) was further transferred to a new agar plate using a sterile inoculation loop. After incubation, the colony formation was visually inspected. As shown in Fig. 8(e) and (f), for the PPXG loaded samples, the only bacteria which obviously existed under the scaffolds were E. coli at the PPXG concentration of 10 μg mL−1. In order to quantify the antibacterial properties, a time-dependent shaking flask test was further performed for up to 6 hours. A 6 hour post-implantation period has been identified during which prevention of bacterial adhesion is critical to the long-term success of an implant.23 Since both of the uncoated and GCG coated scaffolds without PPXG did not clearly show antibacterial properties to both B. subtilis and E. coli, they were not further included in the time-dependent test. As shown in Fig. 9, more than 95% of B. subtilis and E. coli were killed until 2 hours in the presence of GCG coated scaffolds loaded with 10−50 μg mL−1 PPXG, and these antibacterial effects were maintained until 6 hours with the only exception that the E. coli began to grow after 2 hours in the presence of GCG coated scaffolds only incorporated with 10 μg mL−1 PPXG. In other words, the difference of sensitiveness of B. subtilis and E. coli to PPXG becomes evident at 10 μg mL−1 after 2 hours. This would be explained by the different features of the bacterial cell wall. Although all bacteria have an inner membrane in their walls, gram-negative bacteria have a unique outer membrane which envelops a barrier function, i.e., prevents drugs from penetrating the cell wall. Therefore, E. coli, as one species of gram-negative bacteria, is likely to be more resistant to PPXG than B. subtilis which belongs to gram-positive bacteria. | ||
| Fig. 8 Kirby–Bauer test using B. subtilis and E. coli for samples 1: uncoated scaffold without PPXG, 2: GCG coated scaffold without PPXG, 3: GCG coated scaffold loaded with 10 μg mL−1 PPXG, 4: GCG coated scaffold loaded with 30 μg mL−1 PPXG, and 5: GCG coated scaffold loaded with 50 μg mL−1 PPXG. (a) and (b) after incubation for 24 h, (c) and (d) area under the incubated samples, (e) and (f) smears on the agar plate (bacterial growth after transferring swab from area under the samples to a new agar plate). | ||
 | ||
| Fig. 9 Time-dependent shaking flask test results of samples 3: GCG coated scaffold loaded with 10 μg mL−1 PPXG, 4: GCG coated scaffold loaded with 30 μg mL−1 PPXG, and 5: GCG coated scaffold loaded with 50 μg mL−1 PPXG. | ||
Incorporating an antibacterial agent in scaffolds can allow the scaffolds themselves to fight bacterial infection. GCG coated 45S5 BG scaffolds incorporated with PPXG show effective antibacterial effects on both gram-positive and gram-negative bacteria, and the antibacterial effects increase with PPXG concentration, suggesting that PPXG and also other biocidal cationic polymers belonging to polyguanidines are promising for the antibacterial purpose in bone tissue engineering scaffolds.
3.6. Biocompatibility of PPXG, genipin and GCG
The in vitro biocompatibility of PPXG, genipin and GCG was characterized by evaluating the cell proliferation and cell morphology. Cell proliferation was measured in terms of mitochondrial activity, and the cell morphology was observed using calcein AM that stains the cytoplasm of living cells. Apart from the calcein staining, cells were also stained with DAPI which gives information about the integrity of the nucleus. The concentration of the materials was calculated based on the volume of the used cell culture medium. The cell culture plate without any addition of material was used as a control. As shown in Fig. 10, the mitochondrial activity of MG-63 cells grown in the presence of 10 μg mL−1 PPXG is 79%, while it significantly decreases when the PPXG concentration increases. This result is in accordance with the fluorescence staining results of MG-63 cells as presented in Fig. 11(a)–(d), which also indicates a reduction in viable cell numbers as the PPXG concentration increases. As shown in Fig. 11(a) and (b), the cell shape, cell membrane integrity and nucleus integrity of MG-63 cells cultured in 10 μg mL−1 PPXG solution are quite similar to those of the control group. Taking into consideration the antibacterial test results in Section 3.5, PPXG concentration between 10–30 μg mL−1 would be a balanced concentration for both antibacterial properties and biocompatibility. As a natural crosslinking reagent, 50 μg mL−1 genipin enabled MG-63 cells to show 51% mitochondrial activity (Fig. 10), and the viable cells possessed intact nuclei and cell membranes (Fig. 11(e)). In addition, compared to the control group, the cell shape was not obviously affected by the genipin. MG-63 cells exhibited 59% mitochondrial activity at a GCG amount of 1 mg mL−1, and the mitochondrial activity decreased when the GCG amount increased to 5 mg mL−1. The relatively low mitochondrial activity of MG-63 cells in the present study on one hand may be due to the existence of genipin, while on the other hand it may mainly be due to the inhibition of MG-63 cell growth under overdose of gelatin.50,51 As shown in Fig. 11(f) and (g), compared to the control group, although an obvious reduction in viable cell numbers is observed, the cell shape is still similar to that of the control group. Interestingly, many of the MG-63 cells formed clusters on the 1 mg mL−1 GCG films (Fig. 11(f)) and were found to be considerably agglomerated on the 5 mg mL−1 GCG films as indicated by the large blue dot in Fig. 11(g). This result indicates that at such concentration of GCG, cell–material interactions are weaker than cell–cell interactions, which becomes even more obvious when the GCG concentration increases. | ||
| Fig. 10 Mitochondrial activity measurement of MG-63 cells in the presence of PPXG, genipin and GCG at different concentrations after 2 days of cultivation. The values are mean ± standard deviation. The asterisks indicate significant difference. ***P < 0.001. | ||
 | ||
| Fig. 11 Fluorescence images of MG-63 cells after 2 days of cultivation in the presence of PPXG, genipin and GCG at different concentrations. (a) Control group (cell culture plate), (b) PPXG 10 μg mL−1, (c) PPXG 30 μg mL−1, (d) PPXG 50 μg mL−1, (e) genipin 50 μg mL−1, (f) GCG 1 mg mL−1 and (g) GCG 5 mg mL−1. Calcein/DAPI staining: living cells (green)/nuclei (blue). | ||
3.7. Biocompatibility of scaffolds
Fig. 12 shows that the mitochondrial activity of MG-63 cells on GCG coated 45S5 BG scaffolds is slightly higher than on uncoated 45S5 BG scaffolds after 2 weeks of cultivation. However, the difference between the mitochondrial activity of these two groups does not reach statistical significance (P > 0.05). | ||
| Fig. 12 Mitochondrial activity measurement of MG-63 cells on GCG coated 45S5 BG scaffolds after 2 weeks of incubation, using uncoated 45S5 BG scaffolds as a control. The values are mean ± standard deviation. | ||
To visualize cell adhesion and cell distribution on the scaffolds, MG-63 cells were labelled with Vybrant™ cell-labelling solution. CLSM-images of uncoated and GCG coated scaffolds after 2 weeks of cell cultivation are shown in Fig. 13. MG-63 cells were seen to have grown on the strut surfaces of both uncoated and GCG coated scaffolds. As judged by visual inspection of the images, the amount of cells on GCG coated scaffolds seems to be higher than on uncoated scaffolds, which is in agreement with the results of the cell proliferation assay (Fig. 12). After cell cultivation for 2 weeks, the pores of uncoated scaffolds as well as GCG coated scaffolds were still open. This can be attributed to the highly porous and interconnected large pore structure of the scaffolds which facilitate oxygen and nutrient supply for the cells.
 | ||
| Fig. 13 CLSM images of MG-63 osteoblast-like cells on the surfaces of (a) uncoated and (b) GCG coated 45S5 BG scaffolds after 2 weeks of cultivation. The cells were stained red and the 45S5 BG surface can be seen in green. | ||
Furthermore, in order to reveal the cell–cell and cell–material interactions, the cell morphology, especially considering how cells attach and spread on both uncoated and GCG coated scaffolds, was observed by SEM. Representative images are presented in Fig. 14. Fig. 14(a) and (d) show that the strut surfaces of both uncoated and GCG coated scaffolds are well covered by cells, and the well flattened cells covering the scaffold struts tend to form a monolayer in both scaffold types. A closer observation of the gap among the cells showed that the strut surface of the GCG coated scaffold was smooth (Fig. 14(e)), while that of the uncoated scaffold was rougher (Fig. 14(b)). The smooth strut surface of the GCG coated scaffold is likely due to the remaining GCG coating on GCG coated scaffolds after 2 weeks of cell cultivation. Indeed, as shown in the FTIR results (Fig. 3), GCG does exist on GCG coated scaffolds after immersion in SBF for 14 days. At higher magnifications (Fig. 14(c) and (f)), the cells on both scaffold types displayed a typical osteoblastic phenotype with mainly elongated polygonal and flat structures as well as expressed filopodias in contact with the scaffold surface.32,52 Moreover, well developed microvilli were observed on the spread cells on both scaffold types, which indicates that the cells are highly active.
 | ||
| Fig. 14 SEM images of MG-63 cells on the strut surfaces of (a)–(c) uncoated and (d)–(f) GCG coated 45S5 BG scaffolds after 2 weeks of cultivation. The inset in (f) indicates the typical morphology of the microvilli. | ||
The quantitative result of WST assay indicates that GCG coating may have a slightly positive effect on the cell proliferation of MG-63 cells on 45S5 BG scaffolds. Indeed, GCG coatings have been shown to be able to significantly increase the mitochondrial activity of human mesenchymal stem cells on porous PCL scaffolds, however, the realization of this significant improvement of cell response is due to the fact that pure PCL scaffolds were less satisfactory in supporting cell adhesion and growth because of their hydrophobic nature.53 In contrast, uncoated 45S5 BG scaffolds (with their hydrophilic nature14) in the present study already could support suitable cell attachment and growth, as described above. The qualitative studies, i.e., CLSM and SEM images, confirmed that MG-63 cells could attach well and spread on uncoated 45S5 BG scaffolds, and the cell attachment, cell spreading and cell morphology were not significantly changed in the presence of GCG coating. All these results indicate that the GCG coating on the scaffolds seems to have no negative effects on cell activity, which is different from the biocompatibility results of GCG films, as shown in Section 3.6. The better biocompatibility of the GCG coating is due to the fact that part of the GCG is lost during the pH regulation of GCG coated scaffolds (pretreatment in DMEM) before starting the cell cultivation. The remaining GCG on the scaffolds is in a reduced amount. As discussed in Section 3.6, a relatively lower concentration of gelatin is able to favor the growth of MG-63 cells.50,51 Therefore, GCG coated 45S5 BG scaffolds, as well as GCG coating itself at a relatively low concentration are biocompatibility to MG-63 cells. The biocompatibility of GCG was also demonstrated in other studies.37,54–56 Especially, MG-63 cells were shown to attach on genipin cross-linked gelatin porous scaffolds, and the cells exhibited a fibroblastic and a polygonal like morphology after 2 weeks of cell culture.54
4. Conclusions
Significantly improved mechanical properties were conferred to 45S5 BG scaffolds by GCG coating. The GCG coating slightly retarded but did not inhibit the cHA formation on 45S5 BG scaffolds upon immersion in SBF, confirming the bioactive character of the coated scaffolds. Additionally, GCG coated 45S5 BG scaffolds were antibacterial against both Gram-positive and Gram-negative bacteria after the incorporation of polyguanidine, i.e. PPXG. In vitro tests indicated that PPXG was biocompatible with MG-63 cells at a low concentration, and the MG-63 cells could attach, spread and proliferate on the GCG coated scaffolds as on uncoated scaffolds. The obtained bioactive, antibacterial and biocompatible composite scaffolds with improved mechanical properties represent promising candidates for bone tissue engineering applications. They belong to a growing family of functionalized, polymer coated BG-based scaffolds with expected superior in vivo performance which, however, remains to be investigated in further studies.Acknowledgements
Wei Li and Yaping Ding would like to acknowledge the China Scholarship Council (CSC) for financial support. Financial support provided by the German Science Foundation (DFG) to Hui Wang is acknowledged.References
- F. Baino and C. Vitale-Brovarone, J. Biomed. Mater. Res., Part A, 2011, 97, 514–535 CrossRef PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- D. W. Hutmacher, Biomaterials, 2000, 21, 2529–2543 CrossRef CAS.
- A. J. Salgado, O. P. Coutinho and R. L. Reis, Macromol. Biosci., 2004, 4, 743–765 CrossRef CAS PubMed.
- S. Bose, M. Roy and A. Bandyopadhyay, Trends Biotechnol., 2012, 30, 546–554 CrossRef CAS PubMed.
- R. Detsch and A. R. Boccaccini, J. Tissue Eng. Regener. Med., 2014 DOI:10.1002/term.1851.
- J. Yang, T. Long, N.-F. He, Y.-P. Guo, Z.-A. Zhu and Q.-F. Ke, J. Mater. Chem. B, 2014, 2, 6611–6618 RSC.
- L. L. Hench, J. Mater. Sci.: Mater. Med., 2006, 17, 967–978 CrossRef CAS PubMed.
- J. R. Jones, Acta Biomater., 2013, 9, 4457–4486 CrossRef CAS PubMed.
- A. A. Gorustovich, J. A. Roether and A. R. Boccaccini, Tissue Eng., Part B, 2010, 16, 199–207 CrossRef CAS PubMed.
- Q. Z. Chen, I. D. Thompson and A. R. Boccaccini, Biomaterials, 2006, 27, 2414–2425 CrossRef CAS PubMed.
- R. Detsch, S. Alles, J. Hum, P. Westenberger, F. Sieker, D. Heusinger, C. Kasper and A. R. Boccaccini, J. Biomed. Mater. Res., Part A, 2015, 103, 1029–1037 CrossRef PubMed.
- V. Karageorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474–5491 CrossRef CAS PubMed.
- W. Li, P. Nooeaid, J. A. Roether, D. W. Schubert and A. R. Boccaccini, J. Eur. Ceram. Soc., 2014, 34, 505–514 CrossRef CAS PubMed.
- Q. Yao, P. Nooeaid, J. A. Roether, Y. Dong, Q. Zhang and A. R. Boccaccini, Ceram. Int., 2013, 39, 7517–7522 CrossRef CAS PubMed.
- K. A. Athanasiou, C. F. Zhu, D. R. Lanctot, C. M. Agrawal and X. Wang, Tissue Eng., 2000, 6, 361–381 CrossRef CAS PubMed.
- D. Desimone, W. Li, J. A. Roether, D. W. Schubert, M. C. Crovace, A. C. M. Rodrigues, E. D. Zanotto and A. R. Boccaccini, Sci. Technol. Adv. Mater., 2013, 14, 045008 CrossRef.
- M. Erol, A. Ozyuguran, O. Ozarpat and S. Kucukbayrak, J. Eur. Ceram. Soc., 2012, 32, 2747–2755 CrossRef CAS PubMed.
- A. Bigi, G. Cojazzi, S. Panzavolta, N. Roveri and K. Rubini, Biomaterials, 2002, 23, 4827–4832 CrossRef CAS.
- C.-H. Yao, B.-S. Liu, C.-J. Chang, S.-H. Hsu and Y.-S. Chen, Mater. Chem. Phys., 2004, 83, 204–208 CrossRef CAS PubMed.
- D. Bellucci, A. Sola, P. Gentile, G. Ciardelli and V. Cannillo, J. Biomed. Mater. Res., Part A, 2012, 100, 3259–3266 CrossRef CAS PubMed.
- H.-W. Sung, R.-N. Huang, L. L. H. Huang and C.-C. Tsai, J. Biomater. Sci., Polym. Ed., 1999, 10, 63–78 CrossRef CAS PubMed.
- E. M. Hetrick and M. H. Schoenfisch, Chem. Soc. Rev., 2006, 35, 780–789 RSC.
- V. Mouriño, J. P. Cattalini, J. A. Roether, P. Dubey, I. Roy and A. R. Boccaccini, Expert Opin. Drug Delivery, 2013, 10, 1353–1365 CrossRef PubMed.
- R. A. Smith, N. M. M'Ikanatha and A. F. Read, Health Commun., 2014, 1–6 CAS.
- G. J. Gabriel, A. Som, A. E. Madkour, T. Eren and G. N. Tew, Mater. Sci. Eng., R, 2007, 57, 28–64 CrossRef PubMed.
- D. Wei, Q. Ma, Y. Guan, F. Hu, A. Zheng, X. Zhang, Z. Teng and H. Jiang, Mater. Sci. Eng., C, 2009, 29, 1776–1780 CrossRef CAS PubMed.
- C. Mattheis, H. Wang, C. Meister and S. Agarwal, Macromol. Biosci., 2013, 13, 242–255 CrossRef CAS PubMed.
- H. Wang, C. V. Synatschke, A. Raup, V. Jerome, R. Freitag and S. Agarwal, Polym. Chem., 2014, 5, 2453–2460 RSC.
- Z. X. Zhou, D. F. Wei, Y. Guan, A. N. Zheng and J. J. Zhong, J. Appl. Microbiol., 2010, 108, 898–907 CrossRef CAS PubMed.
- Z. Zhou, A. Zheng and J. Zhong, Acta Biochim. Biophys. Sin., 2011, 43, 729–737 CrossRef CAS PubMed.
- W. Li, N. Garmendia, U. Perez de Larraya, Y. Ding, R. Detsch, A. Gruenewald, J. Roether, D. Schubert and A. R. Boccaccini, RSC Adv., 2014, 4, 56156–56164 RSC.
- D. F. Wei, Y. Guan, Q. X. Ma, X. Zhang, Z. Teng, H. Jiang and A. N. Zheng, Electroact. Polym., 2012, 12, 848–857 Search PubMed.
- W. Li, M.-I. Pastrama, Y. Ding, K. Zheng, C. Hellmich and A. R. Boccaccini, J. Mech. Behav. Biomed. Mater., 2014, 40, 85–94 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- D. M. Hashim, Y. B. C. Man, R. Norakasha, M. Shuhaimi, Y. Salmah and Z. A. Syahariza, Food Chem., 2010, 118, 856–860 CrossRef CAS PubMed.
- C. Tonda-Turo, P. Gentile, S. Saracino, V. Chiono, V. K. Nandagiri, G. Muzio, R. A. Canuto and G. Ciardelli, Int. J. Biol. Macromol., 2011, 49, 700–706 CrossRef CAS PubMed.
- M. N. Rahaman, D. E. Day, B. Sonny Bal, Q. Fu, S. B. Jung, L. F. Bonewald and A. P. Tomsia, Acta Biomater., 2011, 7, 2355–2373 CrossRef CAS PubMed.
- Y. Zhu and S. Kaskel, Microporous Mesoporous Mater., 2009, 118, 176–182 CrossRef CAS PubMed.
- X. Liu, M. Rahaman and D. Day, J. Mater. Sci.: Mater. Med., 2013, 24, 583–595 CrossRef CAS PubMed.
- S. Sanchez-Salcedo, S. Shruti, A. J. Salinas, G. Malavasi, L. Menabue and M. Vallet-Regi, J. Mater. Chem. B, 2014, 2, 4836–4847 RSC.
- D. Groh, F. Döhler and D. S. Brauer, Acta Biomater., 2014, 10, 4465–4473 CrossRef CAS PubMed.
- W. Tang, Y. Yuan, D. Lin, H. Niu and C. Liu, J. Mater. Chem. B, 2014, 2, 3782–3790 RSC.
- J. M. Qian, Y. H. Kang, Z. L. Wei and W. Zhang, Mater. Sci. Eng., C, 2009, 29, 1361–1364 CrossRef CAS PubMed.
- M. Dressler, F. Dombrowski, U. Simon, J. Börnstein, V. D. Hodoroaba, M. Feigl, S. Grunow, R. Gildenhaar and M. Neumann, J. Eur. Ceram. Soc., 2011, 31, 523–529 CrossRef CAS PubMed.
- M. Peroglio, L. Gremillard, J. Chevalier, L. Chazeau, C. Gauthier and T. Hamaide, J. Eur. Ceram. Soc., 2007, 27, 2679–2685 CrossRef CAS PubMed.
- G. Pezzotti and S. M. F. Asmus, Mater. Sci. Eng., A, 2001, 316, 231–237 CrossRef.
- A. Philippart, A. R. Boccaccini, C. Fleck, D. W. Schubert and J. A. Roether, Expert Rev. Med. Devices, 2015, 12, 93–111 CrossRef CAS PubMed.
- Q. Yao, P. Nooeaid, R. Detsch, J. A. Roether, Y. Dong, O.-M. Goudouri, D. W. Schubert and A. R. Boccaccini, J. Biomed. Mater. Res., Part A, 2014, 102, 4510–4518 Search PubMed.
- B.-S. Liu, C.-H. Yao, Y.-S. Chen and S.-H. Hsu, J. Biomed. Mater. Res., Part A, 2003, 67, 1163–1169 CrossRef PubMed.
- K.-Y. Chen, P.-C. Shyu, Y.-S. Chen and C.-H. Yao, Macromol. Biosci., 2008, 8, 942–950 CrossRef CAS PubMed.
- Q. Z. Chen, A. Efthymiou, V. Salih and A. R. Boccaccinil, J. Biomed. Mater. Res., Part A, 2008, 84, 1049–1060 CrossRef CAS PubMed.
- Q. Zhang, K. Tan, Y. Zhang, Z. Ye, W.-S. Tan and M. Lang, Biomacromolecules, 2013, 15, 84–94 CrossRef PubMed.
- G. Teti, A. Bigi, M. Mattioli-Belmonte, R. Giardino, M. Fini, A. Mazzotti and M. Falconi, J. Life Sci., 2013, 7, 965–970 CAS.
- M. Gioffrè, P. Torricelli, S. Panzavolta, K. Rubini and A. Bigi, J. Bioact. Compat. Polym., 2012, 27, 67–77 CrossRef PubMed.
- S. Baiguera, C. Del Gaudio, E. Lucatelli, E. Kuevda, M. Boieri, B. Mazzanti, A. Bianco and P. Macchiarini, Biomaterials, 2014, 35, 1205–1214 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb00044k |
| ‡ These two authors contributed equally to the experimental part. |
| This journal is © The Royal Society of Chemistry 2015 |