
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioactive organic/inorganic hybrids with improved mechanical performance
Ailing
Li
a,
Hong
Shen
a,
Huihui
Ren
a,
Chen
Wang
a,
Decheng
Wu
a,
Richard A.
Martin
*b and
Dong
Qiu
*a
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Polymer Physics and Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: dqiu@iccas.ac.cn
bSchool of Engineering & Applied Science and Aston Research Centre for Healthy Ageing, University of Aston, Birmingham, B4 7ET, UK. E-mail: r.a.martin@aston.ac.uk
First published on 22nd December 2014
Abstract
New sol–gel functionalized poly-ethylene glycol (PEGM)/SiO2–CaO hybrids were prepared with interpenetrating networks of silica and PEGM through the formation of Si–O–Si bonds. Bioactive and mechanical properties were investigated for a series of hybrids containing varying organic/inorganic ratios and PEG molecular weights. In contrast to the unmodified PEG/SiO2–CaO hybrids, which rapidly dissolved and crumbled, the epoxy modified hybrids exhibited good mechanical properties and bioactivity. The compressive strength and Young's modulus were greater for higher molecular weight PEGM hybrids (PEGM600 compared to PEGM300). Compressive strengths of 138 MPa and 81 MPa were found for the 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 and 60:40 organic/inorganic hybrid samples respectively, which are comparable with cortical bone. Young's modulus values of ∼800 MPa were obtained for the 50:50 and 60:40 organic/inorganic hybrids. Bioactivity tests were conducted by immersing the hybrids into simulated body fluid and observing the formation of apatite. Apatite formation was observed within 24 hours of immersion. PEGM600 hybrids showed enhanced apatite formation compared to PEGM300 hybrids. Increased apatite formation was observed with increasing organic/inorganic ratio. 70:30 and 60:40 hybrids exhibited the greatest apatite formation. All PEGM hybrids samples had good cell viability and proliferation. The 60:40 PEGM600 hybrids displayed the optimal combination of bioactivity and mechanical strength. The bioactivity of these hybrids, combined with the enhanced mechanical properties, demonstrate that these materials have significant potential for bone regeneration applications.
:50 and 60:40 organic/inorganic hybrid samples respectively, which are comparable with cortical bone. Young's modulus values of ∼800 MPa were obtained for the 50:50 and 60:40 organic/inorganic hybrids. Bioactivity tests were conducted by immersing the hybrids into simulated body fluid and observing the formation of apatite. Apatite formation was observed within 24 hours of immersion. PEGM600 hybrids showed enhanced apatite formation compared to PEGM300 hybrids. Increased apatite formation was observed with increasing organic/inorganic ratio. 70:30 and 60:40 hybrids exhibited the greatest apatite formation. All PEGM hybrids samples had good cell viability and proliferation. The 60:40 PEGM600 hybrids displayed the optimal combination of bioactivity and mechanical strength. The bioactivity of these hybrids, combined with the enhanced mechanical properties, demonstrate that these materials have significant potential for bone regeneration applications.
1 Introduction
Bone is the most widely transplanted material, with the exception of blood.1 Each year more than 2.2 million bone graft operations are performed to repair bone defects in orthopaedics and dentistry.2,3 Bioactive glasses represent one of the most promising synthetic bone replacement materials; these materials have the potential to be more bioactive than calcium phosphate-based materials. Bioactive glasses, developed by Larry Hench in 1969 show class A bioactivity; they bond to bone and stimulate new bone growth even away from the glass–bone interface.4,5Melt-derived Bioglass® (46.1% SiO2, 24.4% Na2O, 26.9% CaO, and 2.6% P2O5 mol%) is used commercially in products such as Novabone® and NovaMin® for repairing bone defects and re-mineralizing dental enamel.5,6 Under physiological conditions the glass slowly dissolves releasing controlled amounts of Ca, P and Si. The soluble Ca and P then precipitate onto the surface to form an amorphous calcium phosphate layer which then crystallizes into apatite (the mineral component of bone).7 Studies have shown that the controlled release of Ca, P and Si can stimulate gene transcription in osteoblasts.8,9 The glass surface also supports cell adhesion and proliferation. There are however limitations; Bioglass® requires very high processing temperatures (∼1400 °C10), is difficult to process into scaffolds, fibres or coatings, and has a very small processing window which can result in crystallization. In addition, melt-quench glasses have low surface areas and low porosity which is not ideal for cell ingrowth or vascularization. Sol–gel derived bioactive glasses have been developed to overcome many of these obstacles.11 Sol–gels, prepared using a low-temperature hydrolysis and condensation process, have many advantages compared with their melt-quench derivatives.12–14 The sol–gels process has greater versatility; bioactive particles, foams, coatings and fibres can all be prepared using this methodology. The resultant gels have a nanometer-scale textural porosity, high specific surface area and abundant exposed silanol groups on surface.15–17 Apatite formation occurs in the macropores resulting in a much greater degree of apatite growth.18,19 Despite their excellent bioactivity and acceptable compressive strength their poor tensile strength and brittle nature has severally limited their applications.
Sol–gel derived glasses can be combined with organic polymers to improve their mechanical strength and toughness.20,21 Bioactive glass particles can be dispersed in a polymer matrix to form composites.22 However, the bioactive particles are generally covered by the polymer matrix and are therefore not in direct contact with body fluid before polymer degradation, resulting in slow bonding to bone tissue in vivo.23 In addition, the bioactive glass particles are usually on the micrometre-scale, thus likely to cause inhomogeneity. Alternatively, inorganic/organic hybrids can be synthesised by introducing the polymer at an early stage e.g. around the hydrolysis stage. Organic/inorganic hybrids show similar improvements in mechanical properties compared to the composites but also show much better bioactivity due to the mixing of glass and polymer on molecular scale. Therefore, hybrids have attracted much attention owing to these advantages.
Hybrids can be classified as class I and class II hybrids according to the interaction between inorganic chains and organic chains. In class I hybrids inorganic chains entangle with organic chains with only weak (hydrogen and/or van der Waals bonding) interactions, whereas by modifying the polymer to react with the inorganic phase strong covalent bonds can be formed (class II hybrids).24 Class II hybrids have previously been fabricated using alkoxysilane functionalized polymer as precursors in the form of scaffolds, electrospun fibres or monoliths.25–27 These hybrids have been shown to exhibit mechanical properties analogous to human cancellous bones.28–30 For example Salinas et al.31 have prepared dense and homogeneous crack-free monoliths of PDMS-modified CaO–SiO2 system. These hybrids are silicate networks covalently bonded to PDMS which show apatite formation when immersed in simulated body fluid (SBF). It was also shown that their elastic modulus decreased with PDMS content but their apatite forming ability increased with calcium content.32 Biodegradable silica/poly(ε-caprolactone) (PCL) have also been prepared, in which hydroxyl groups at either end of the PCL polymer chains were modified with 3-isocyanatopropyl triethoxysilane (IPTS), resulting in a polymer end capped with a triethoxysilyl group. The end capped PCL then was introduced into a sol to synthesize bioactive glass/PCL hybrids.33,34 Bioactive silica–poly(γ-glutamic acid) hybrids have also been obtained by covalently bonding the γ-PGA and bioactive silica through an organosilane coupling agent, glycidoxypropyl trimethoxysilane (GPTMS),24 and calcium was confirmed to be incorporated in the silica network by using calcium alkoxide precursor. However, sometimes the resulting hybrids are not sufficiently hydrophilic for ion exchange with the surrounding physiological fluids. Hydrophilic organic components have been introduced to prepare organic/inorganic hybrids. For example, natural polymer gelatin was functionalized using GPTMS, which was then introduced into a sol of hydrolyzed TEOS to covalently link gelatin to silica with –Si–O–Si–. Then the wet gels were freeze dried to form porous scaffolds, the mechanical properties of which are similar to the cancellous bones.35 Granqvist et al. have developed PEG/siloxane fibres by functionalising PEG end groups with 3-isocyanatopropyl triethoxysilane.25 The functionalised PEG was incorporated into the siloxane network through covalent bonds resulting in fibres with much higher elasticity compared with non-functionalised fibres. The fibres were shown to be effective substrates for the nucleation and growth of bone-like apatite.
Traditionally calcium has always been incorporated in sol–gels in the form of calcium nitrate which then requires the sol–gel to be heated >400 °C to burn off the toxic nitrates and for calcium to be incorporated into the silicate network.19,36 Nitrates are therefore unsuitable for hybrid materials containing polymers which decompose at high temperatures. However, it has recently been shown that the precursor calcium methoxyethoxide (CME) is suitable introducing calcium into hybrids.36,37 Calcium has been shown to enter the silicate network at processing temperatures as low as 60 °C when using CME.
The purpose of the present study was to prepare homogeneous organic/inorganic hybrid materials for bone repair using low molecular weight (300, 600) functionalized PEG. The inorganic phase consisted of (SiO2)70–(CaO)30 (S70C30, the archetypal bioactive sol–gel). The organic phase consisted of polyethylene glycol (PEG), a biocompatible polymer (and bioresorbable for low molecular weight polymers38) which is completely soluble in both water and ethanol, easy to modify and can be incorporated into typical sols for sol–gel hybrid synthesis. PEG ends were first modified with epoxy ethane, then ammoniapropyltriethoxysilane was added to react with epoxy ethane to form PEG end capped siloxane (PEGM). The organic/inorganic hybrid materials were prepared by introducing PEGM into the sol–gel process, using TEOS and calcium methoxyethoxide as precursors, so that the silica network and organic polymer form interpenetrating networks at the nanoscale. The aim was to investigate whether this system can improve the mechanical properties of the hybrid materials whilst retaining the apatite forming bioactivity of the inorganic sol–gel phase.
2 Materials and methods
2.1 Materials and sample preparation
The following precursors were used in the sol–gel preparation: PEG300 (285–315), PEG600 (585–615), ammoniapropyltriethoxysilane (97%) and methoxyethanol (99.8%), (3-aminopropyl)triethoxysilane (APTES) (97%) (Sigma Aldrich), epichlorohydrin, metallic calcium and tetraethyl orthosilicate (TEOS, ≥99.0%) (Sinopharm Chemical Reagent Co., Ltd).The preparation was carried out in a 4 step process as shown in Fig. 1.
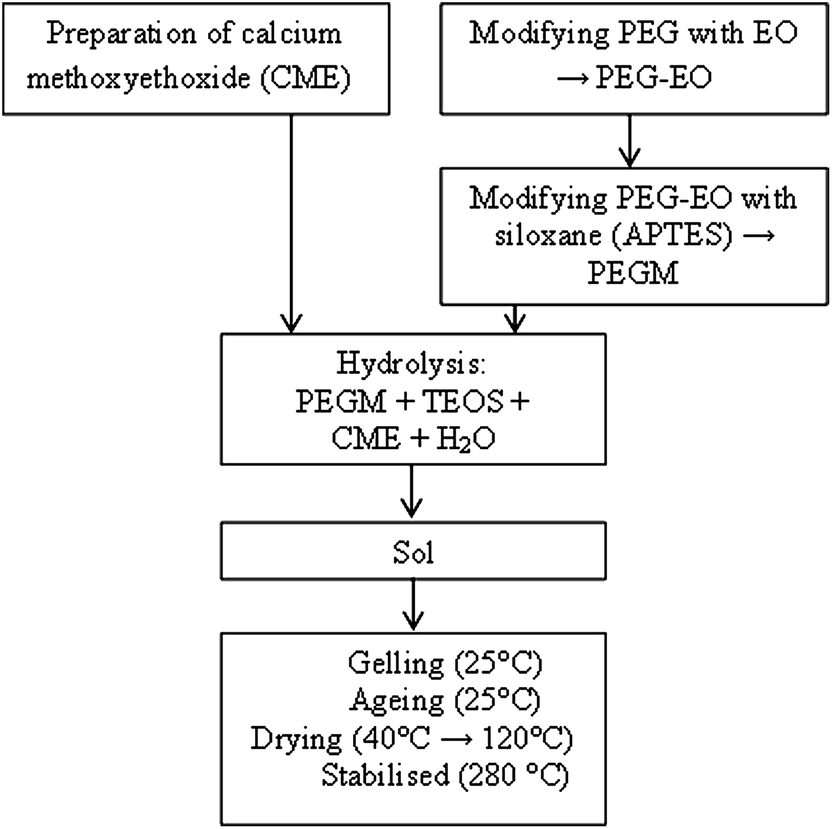 | ||
| Fig. 1 Flow diagram illustrating the hybrid preparation. | ||
:30, 60:40, 50:50 and 40:60 whilst maintaining the inorganic composition of S70C30. After 30 min, water (H2O/TEOS = 2, molar ratio) was added drop wise into the solution and the solution gels within an hour. The resultant gels were aged at ambient temperature for two days then dried in an oven, firstly at 40 °C for two weeks and then at 120 °C for a further week. Finally the samples were dried at 280 °C for 1 h to ensure full condensation and complete evaporation of the solvent. A control sample of PEG600 with a 60:40 organic:inorganic ratio was prepared using unmodified PEG to act as a reference. 29Si solid state MAS NMR spectroscopy experiments were carried out on the dried materials using a Bruker Avance III 400 MHz instrument providing 29Si Larmor frequencies of 79.30 MHz. 4 mm MAS BB probes spinning at 12 kHz were used. A 2.5 μs pulse (∼60° tip angle) was used, and the recycle delay was 90 s. 29Si NMR spectra were referenced using tetramethylsilane (δSi = 0 ppm).
2.2 Bioactivity testing
Bioactivity was tested in vitro by immersing 150 mg monoliths in 100 mL of simulated body fluid at 36.5 ± 0.5 °C. SBF was prepared by dissolving reagents NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·6H2O, CaCl2, and Na2SO4 in deionized water.40 The solution was buffered to pH 7.4 with Tris-(hydroxymethyl)-aminomethane ((CH2OH)3CNH2) and hydrochloric acid to give a resultant ion concentration similar to human plasma. After immersing in SBF for different times the samples were washed gently with pure water and left to dry in a desiccator without heating. Apatite formation was evaluated using scanning electron microscopy (SEM), Energy Disperse X-ray Spectroscopy (EDXS), Fourier transform infrared (FTIR) and X-ray diffraction (XRD).2.3 Mechanical testing
Compressive mechanical properties of the specimens were evaluated using general a purpose-testing machine (Instron3365, Instron Co., Canton, MA, USA) at 23 ± 2 °C and 50 ± 5% relative humidity conditions. Specimens were cut from homogeneous regions of the organic/inorganic hybrids to form blocks measuring 4 mm × 4 mm × 8 mm. These samples were positioned between parallel plates and compressed with a crosshead speed of 0.5 mm min−1 and a 5.0 kN load cell. A minimum of five repeats were taken per sample to provide an average. The mean and standard deviation were reported. The Young's modulus was determined from the slope of the initial linear elastic portion of the stress–strain curve. Compressive strength was taken as the maximum strength of the sample until failure and elongation to failure was the percentage strain at fracture.2.4 Cell evaluation
2.5 Degradation studies
Hybrids (75 mg) in triplicates were immersed in 50 mL of SBF at 36.5 ± 0.5 °C and stirred at 120 rpm. At various time points (1, 4, 24, 72, 168, 336, 504 and 672 h), the hybrids were removed from their respective containers and dried under vacuum prior to weighing. To obtain the rate of weight loss, the initial weight (M0) of each sample was measured as well as the weight loss at time t (Mt) to give a weight loss%: weight loss% = (M0 − Mt)/M0 × 100%. pH values of the extracts were recorded after 1, 3 and 6 days immersion.2.6 Statistical analysis
The one-way analysis of variance (ANOVA) was used to evaluate significant differences between means in the measured data. Each experiment was repeated three times. All quantitative data are presented as mean ± standard deviation.3 Results
3.1 Sample characterisation
PEG end groups were successfully modified with epoxy (EO) as shown in Fig. 2. The 1H NMR spectra confirms the addition of the epoxy functional groups. The spectra are assigned as follows: 1H NMR (400 MHz, CDCl3) δ (ppm), 3.58 (m, 1H, He), 3.43 (m, 28H, Hf), 3.19 (m, 1H, Hd), 2.92 (m, 1H, Hc), 2.56 (m, 1H, Hb), 2.38 (m, 1H, Ha).42 The reaction process of PEG–EO to PEGM and corresponding FTIR spectra are shown in Fig. 3. A feature at 914 cm−1 appears for PEG–EO which is consistent with an epoxy group.43,44 The epoxy peak then disappears for the PEGM as expected (see Fig. 3a bottom). Clear amorphous monoliths were produced as shown in Fig. 4. The orange colour emanates from the CME precursor which is a dark red/brown colour as previously reported.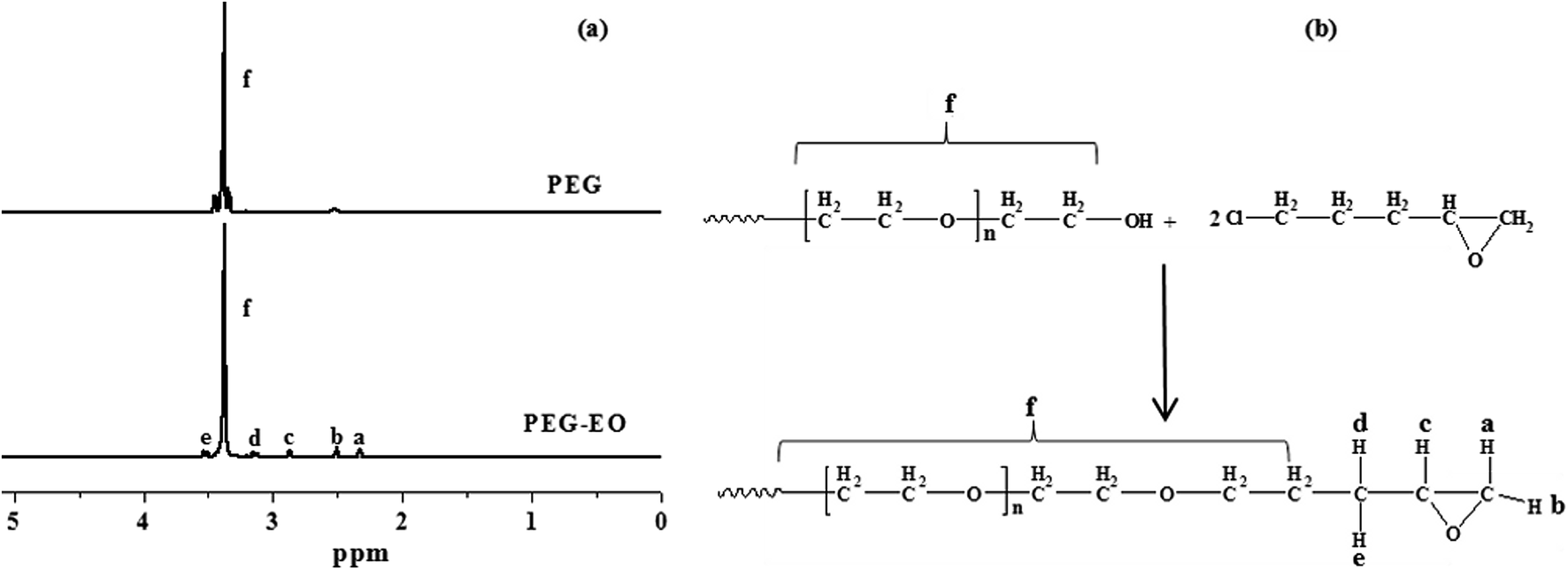 | ||
| Fig. 2 (a) 1H NMR spectra of PEG (top) and the modified PEG–EO polymer (bottom), (b) illustrates the reaction process of PEG to PEG–EO. | ||
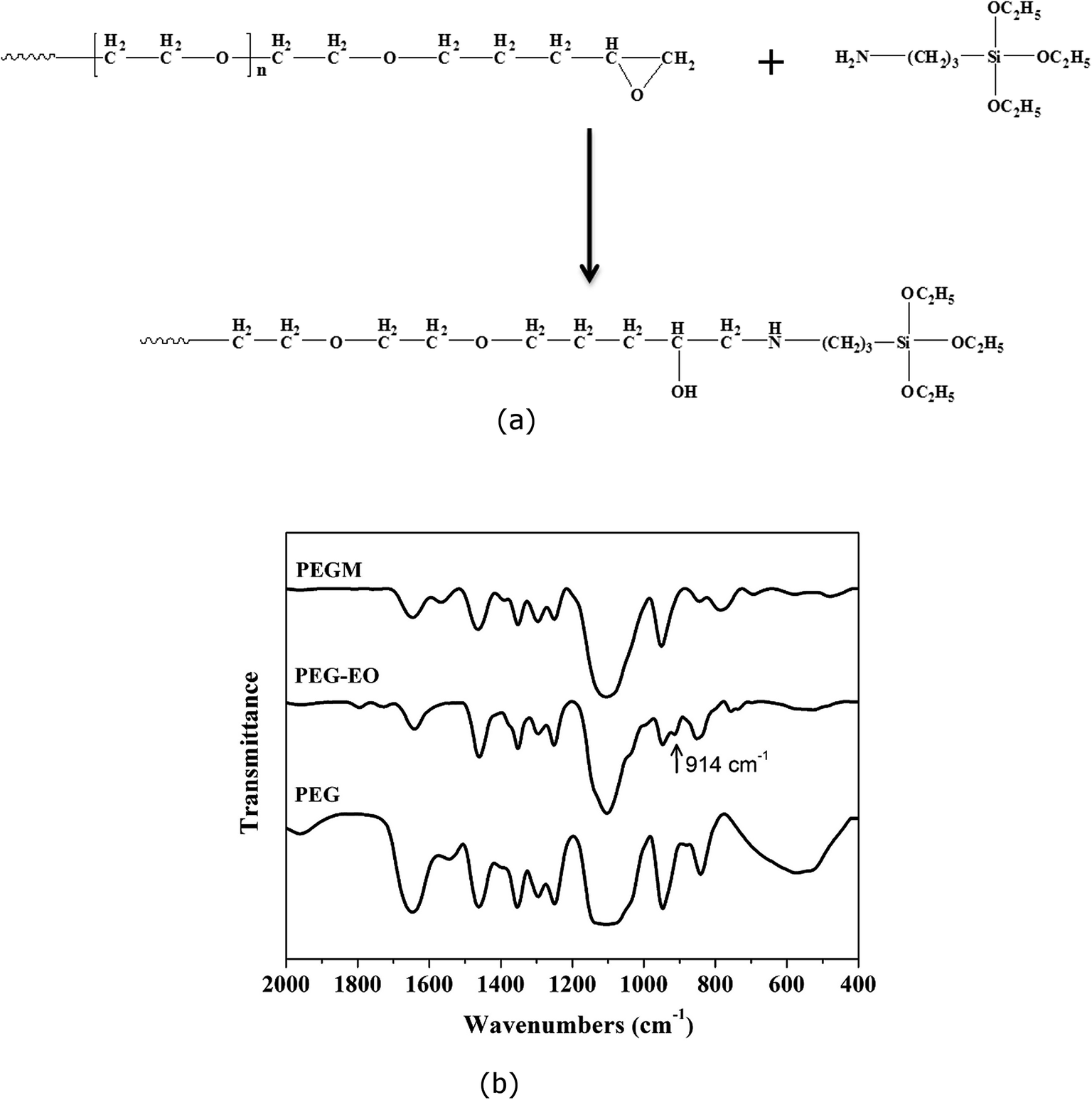 | ||
| Fig. 3 (a) Reaction process illustrating the modification of the PEG–EO end groups, (b) FTIR spectra of PEG, PEG–EO and PEGM. The epoxide group of PEG–EO appears at 914 cm−1. | ||
 | ||
| Fig. 4 Photo of the 60:40 (organic/inorganic) PEGM600 monolith. | ||
3.2 Structural characterisation
Fig. 5 shows the 29Si MAS NMR spectra of (a) PEGM300/TEOS (calcium free) (b) the PEGM/TEOS/CME hybrid and (c) the unmodified PEG300/TEOS/CME hybrid. The modified PEG hybrids (samples a and b) both show the presence of Tn and Qn species (Tn and Qn correspond to the structure of CSi(OSi)n(R)3−n and Si(OSi)n′(R)4−n′ respectively, where R is a non-bridging oxygen). In contrast the unmodified PEG hybrid (sample c) only showed the presence of Qn species. Results of the Gaussian fitting of Tn and Qn distributions of the 29Si MAS NMR spectra are summarised in Table 1. The ratio of T3/Ttot and Q4/Qtot in Table 1 show the degree of condensation of the PEGM and silica network respectively. For the TEOS-derived silica network, 29Si exhibit several overlapping contributions associated with Qn units: three resonances at ∼−95, ∼−101 and ∼−110 ppm were assigned to Q2, Q3 and Q4 units respectively. In addition to these Qn resonances, the 29Si MAS NMR spectra of sample a and b showed two peaks at ∼−60 and ∼−65 ppm characteristic of T2 and T3 units resulting from the hydrolysis/condensation of the terminal triethoxysilane groups of PEGM. Despite poly-condensation between PEGM chains not being excluded, the high occurrence of T3 units suggests that PEGM is successfully grafted to the BG network, thereby coupling the organic and inorganic parts of the hybrid to form a Class II hybrid. Sample a had higher T3/Ttot and Q4/Qtot values compared to samples b and c, which suggests that the silica species were highly condensed.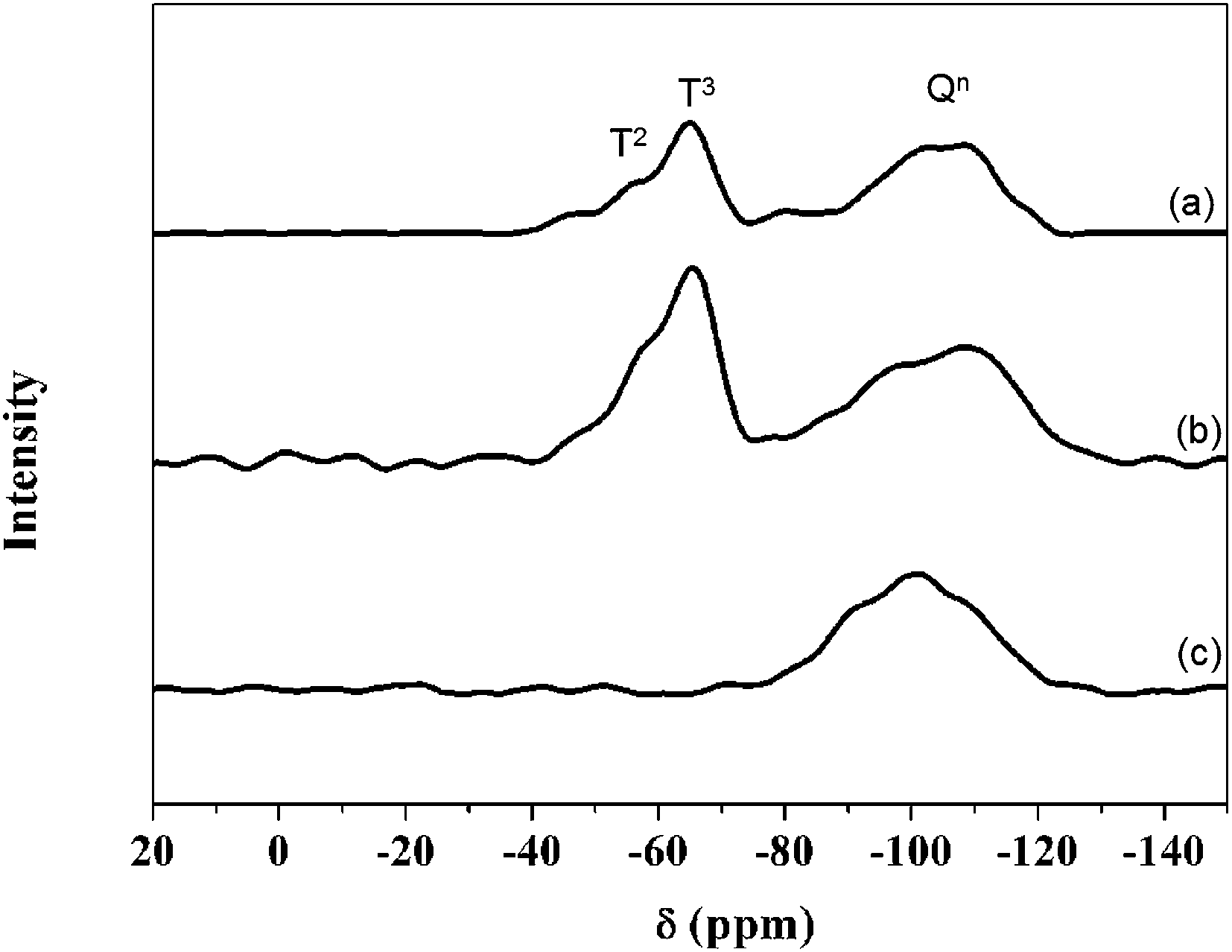 | ||
| Fig. 5 The 29Si solid state MAS-NMR of the sample obtained from different precursors with the ratio of 60/40 (organic/inorganic, molar%): (a) – PEGM300/TEOS; (b) – PEGM300/TEOS/CME; (c) – PEG300/TEOS/CME. | ||
| Sample | T2 | T3 | Q2 | Q3 | Q4 | T3/Ttot | Q4/Qtot | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ [ppm] | [%] | δ [ppm] | [%] | δ [ppm] | [%] | δ [ppm] | [%] | δ [ppm] | [%] | |||
| a | −59.3 | 27.3 | −65.4 | 15.6 | — | 0.0 | 100.8 | 16.4 | −109.6 | 40.7 | 0.4 | 0.7 |
| b | −61.0 | 34.3 | −66.2 | 11.0 | — | 0.0 | −101.0 | 45.1 | −112.1 | 9.6 | 0.2 | 0.2 |
| c | — | 0.0 | — | 0.0 | −95.2 | 60.9 | 100.8 | 3.9 | 108.9 | 35.1 | 0.0 | 0.4 |
3.3 Bioactivity
Apatite formation was observed on the monolith's surface after immersion in SBF. Fig. 6 illustrates a representative SEM image showing both hemispherical and needle like apatite formation. Needle like formations are consistent with previous reports of apatite formation on bioactive sol–gels.45 Apatite formation occurs within 24 hours of immersion and become denser with increasing time. | ||
| Fig. 6 SEM image of hybrid glasses (PEGM600, organic/inorganic = 60:40, molar ratio) after immersion in SBF for 0, 1, 3 and 7 days. | ||
X-ray diffraction results are shown in Fig. 7–9. Fig. 7 illustrates the formation of apatite as a function of SBF immersion time for PEGM600 with an organic/inorganic molar ratio of 60:40. HA diffraction peaks appear after only 24 hours and then increase in magnitude after 3 and 7 days confirming apatite formation on the surface of these materials. Monoclinic HA is shown for reference standard.46Fig. 8 and 9 show the apatite diffraction peaks as a function of organic/inorganic ratio for the PEGM600 and PEGM300 hybrids respectively.
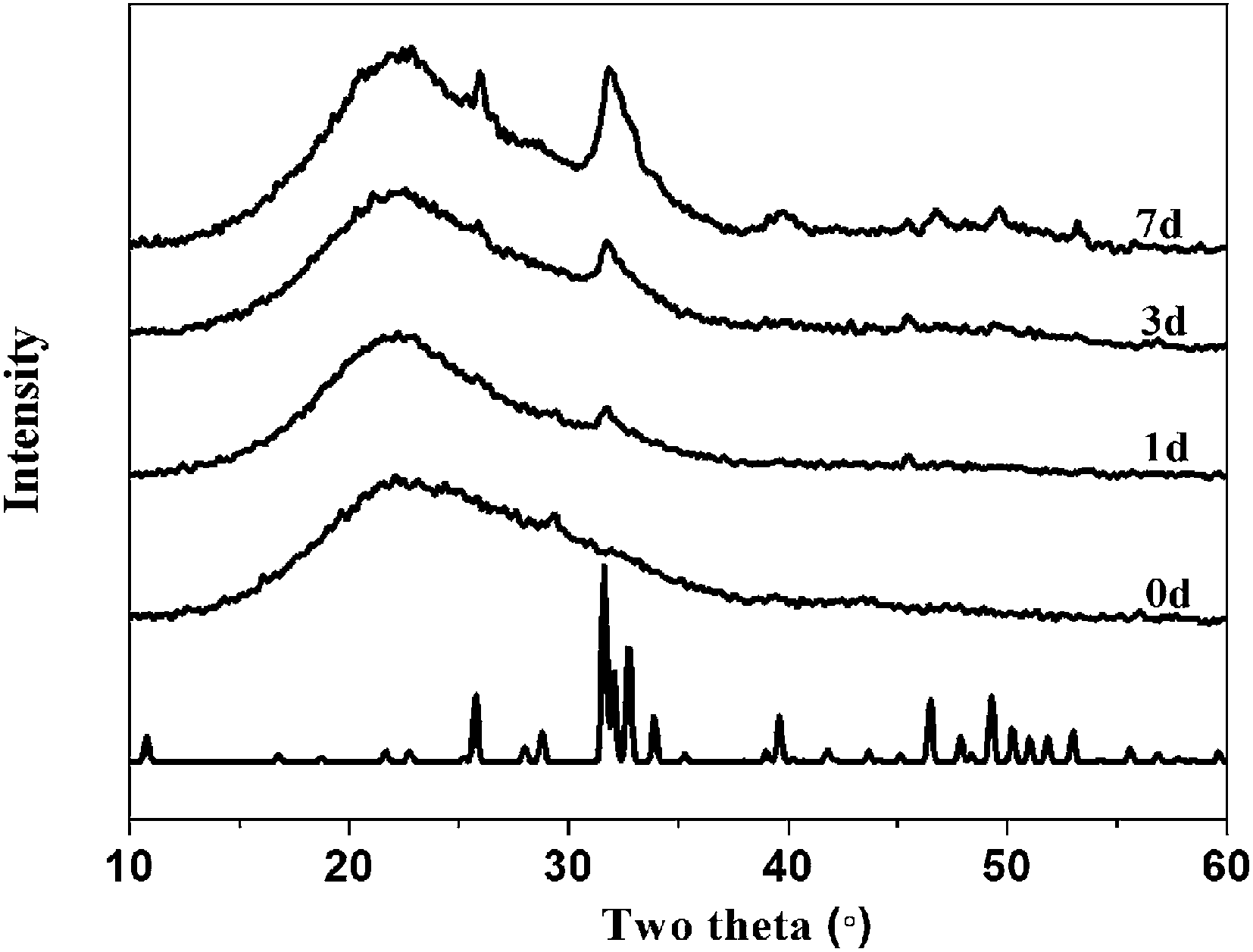 | ||
| Fig. 7 The XRD spectra of hybrid materials (PEGM600, organic/inorganic = 60:40, molar ratio) after immersed in SBF for different times. | ||
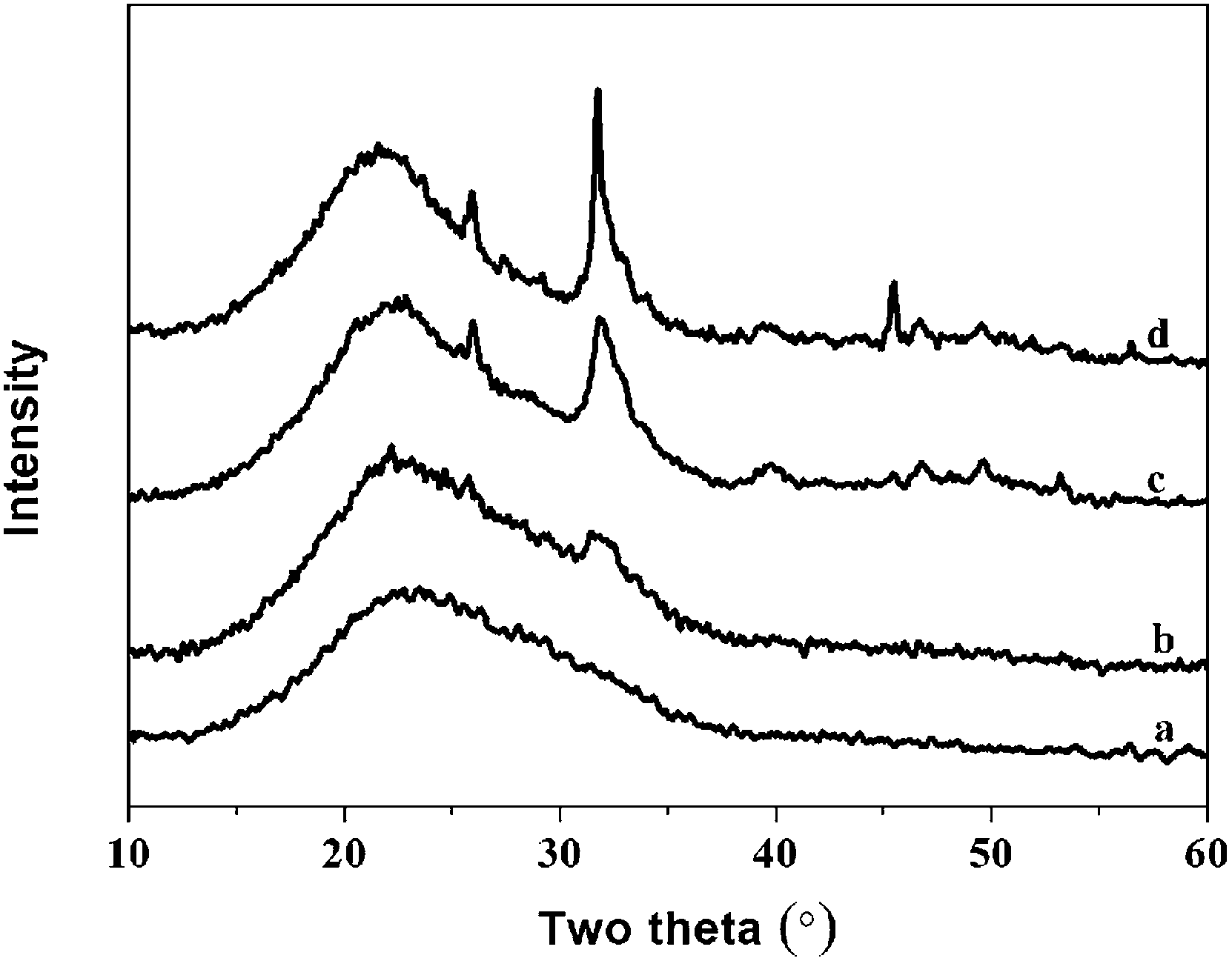 | ||
| Fig. 8 The XRD spectra of hybrid materials with different organic/inorganic (molar ratio, PEGM600) after immersed in SBF for 7 days: (a)-40:60; (b)-50:50; (c)-60:40; (d)-70:30. | ||
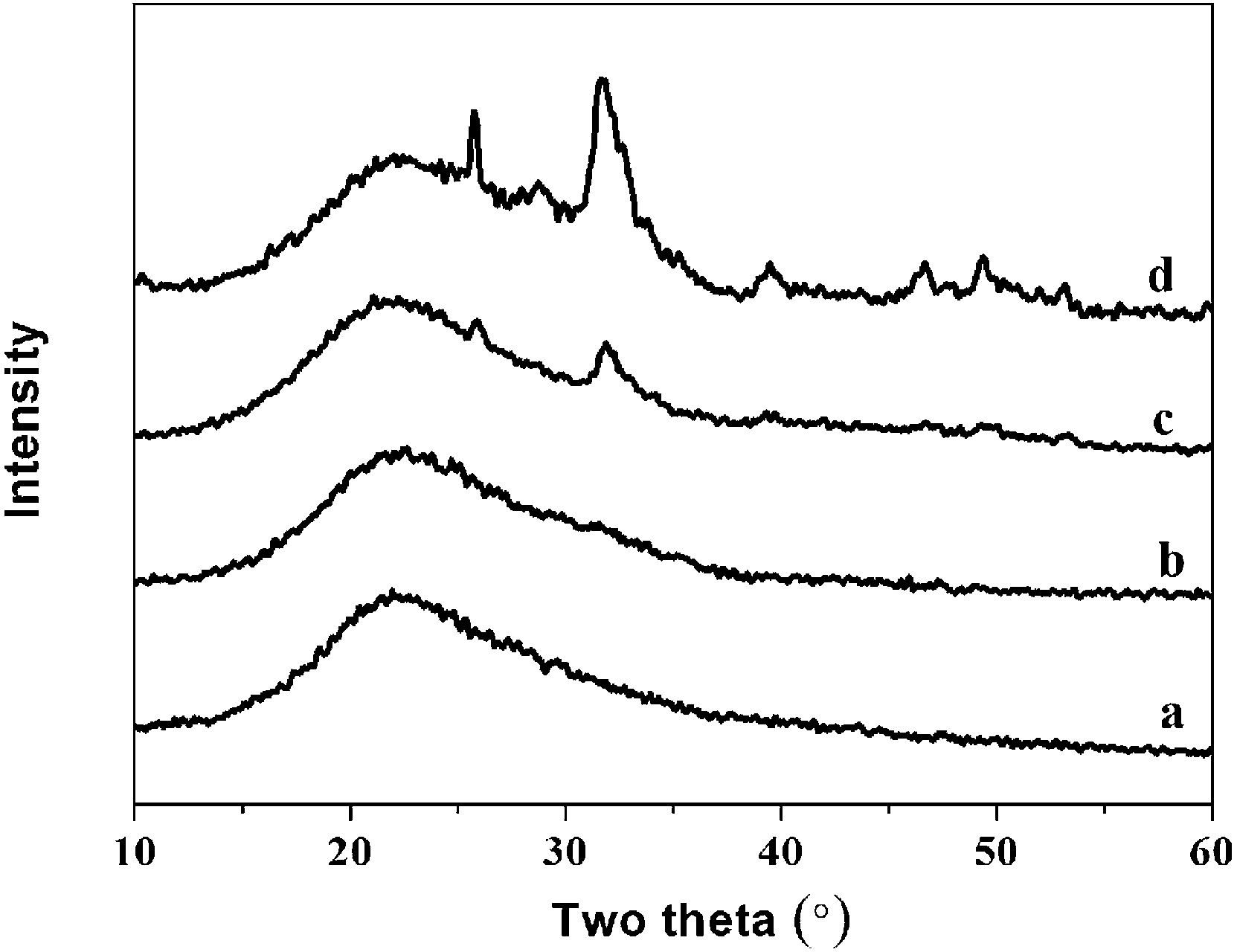 | ||
| Fig. 9 The XRD spectra of hybrid materials with different organic/inorganic (molar ratio, PEGM300) after immersed in SBF for 7 days: (a)-40:60; (b)-50:50; (c)-60:40; (d)-70:30. | ||
The intensity of the apatite diffraction peaks increases with increasing organic/inorganic ratio for both PEGM600 and PEGM300 hybrids. It is evident that the 60:40 and 70:30 hybrids show the greatest bioactivity (apatite formation). For all organic/inorganic ratios the PEGM600 spectra shows significantly more apatite formation demonstrating that PEGM600 hybrids are more bioactive compared to their equivalent PEGM300 counterparts.
EDXS results are shown in Fig. 10 as a function of SBF immersion time. Prior to immersing in SBF, the hybrid surface consisted of Si, C and O as expected. Within a day of immersion in SBF the hybrid surface is rapidly covered with a calcium phosphate layer as expected, in addition the Si and C signal from the bulk hybrid is significantly attenuated by the Ca/P surface formation. The Ca and P signals continue to increase with time (3 and 7 day immersion) and the Si signal is further attenuated.
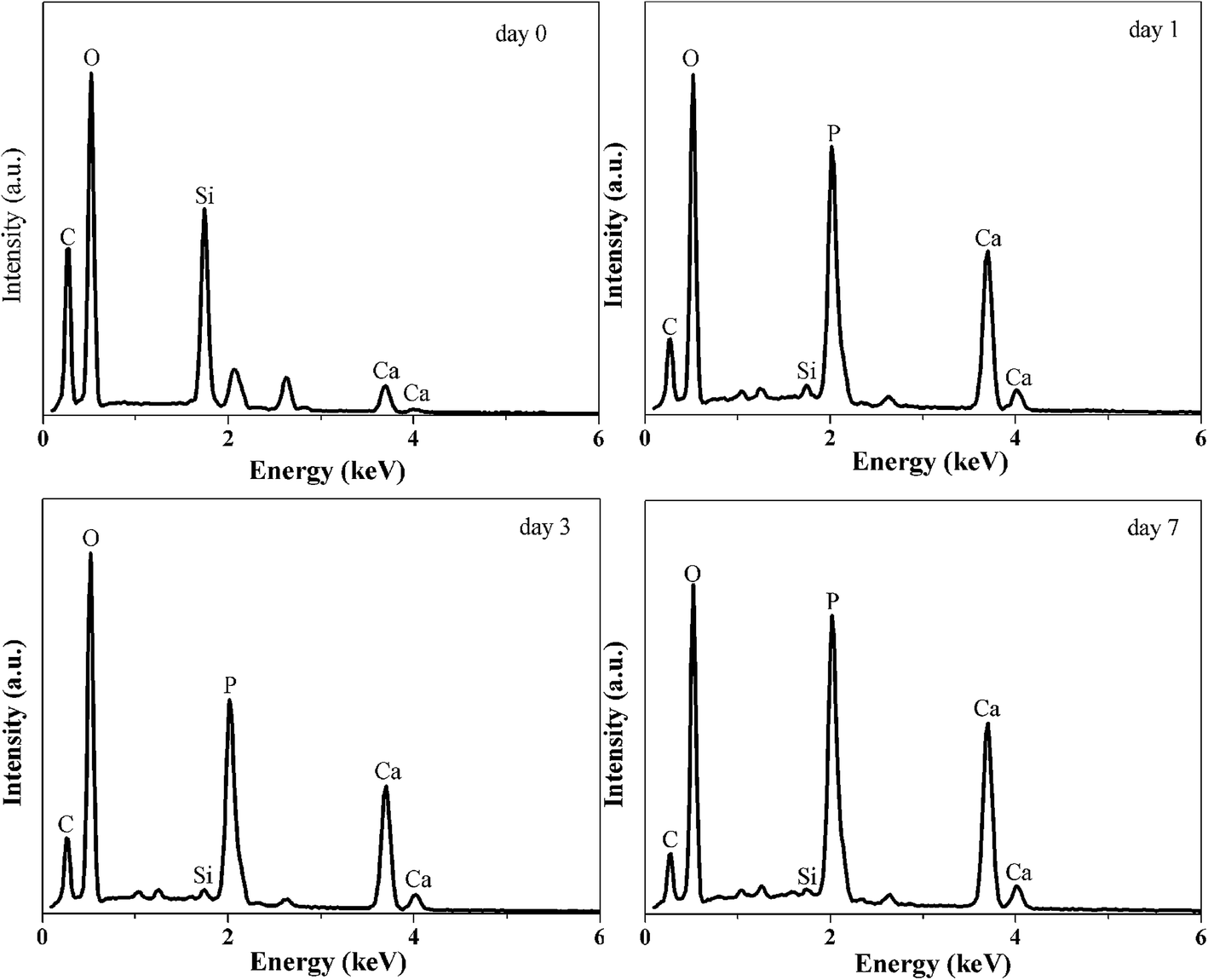 | ||
| Fig. 10 EDXS spectra of the hybrids surface after immersing SBF for 0, 1, 3 and 7 days. | ||
FTIR spectra are shown in Fig. 11. The pronounced band ∼1090 cm−1 and the smaller peak ∼790 cm−1 are attributed to Si–O–Si vibrations.47 The broad band ∼3440 cm−1 and the sharper peak ∼1630 cm−1 are attributed to O–H stretching.47 Peaks at ∼459, 567 and 607 cm−1 are attributed to phosphate absorption bands.45 These peaks evolve as a function of immersion time in SBF further supporting the formation of phosphates on the hybrids surface.
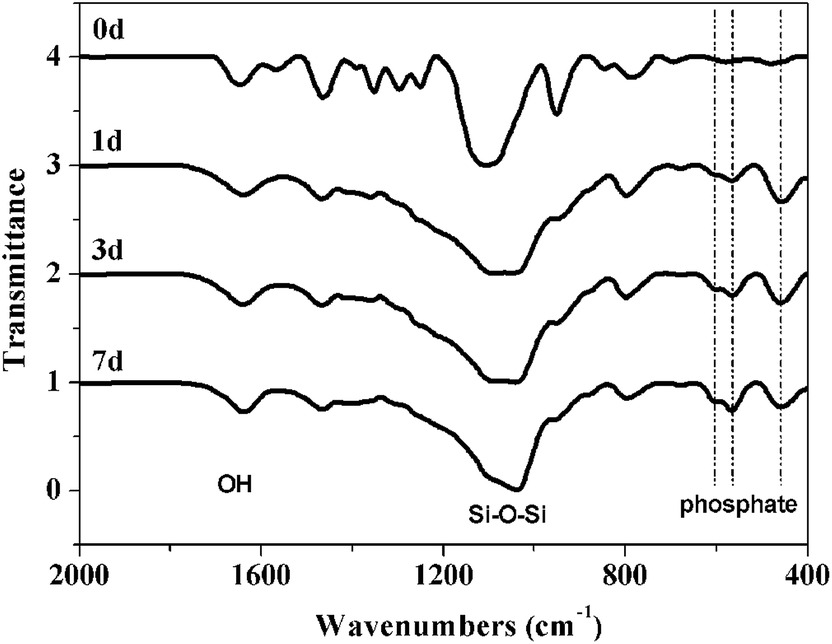 | ||
| Fig. 11 FTIR spectra of the hybrid materials after immersing SBF for 0, 1, 3 and 7 days. | ||
3.4 Mechanical testing
Compressive mechanical testing was performed on the dried hybrid monoliths to test the strength and flexibility of the hybrids. Fig. 12 shows the stress–strain behaviour and effect of organic/inorganic ratio on Young modulus of hybrids, and Table 2 presents the corresponding Young's modulus and compressive stress data for PEGM300 and PEGM600 samples as a function of organic/inorganic ratio. The elongation to failure was greatly increased to 14–35% for the hybrids materials compared to 4.2% for sol–gel glasses (70S30C monoliths, sintered at 800 °C),48 indicating improved strain to failure and therefore increased toughness. Two clear trends are also observed; firstly the mechanical properties increase with increasing inorganic ratio. The Young's modulus and compressive strength are greatest for the 50:50 hybrid with the overall trend 50:50 > 60:40 > 70:30 and this trend is observed for both the PEGM300 and PEGM600 hybrids. The second point is that Young's modulus and compressive stress are greater for the PEGM600 hybrid compared to its equivalent PEGM300 hybrid (for the same organic/inorganic ratio). The PEGM600 hybrid with a 50:50 organic/inorganic ratio was therefore found to have the best mechanical properties with 60:40 PEGM600 having very similar Young's modulus values. The unmodified PEG hybrid had very poor mechanical properties and easily crumbled, it was therefore not possible to measure the modulus or strength of these samples. PEG rapidly dissolved from the unmodified composite resulting in just the inorganic phases. Strong covalent links between the organic and inorganic chains are therefore essential in order to obtain tough composites which degrade uniformly acting as a single phase material.
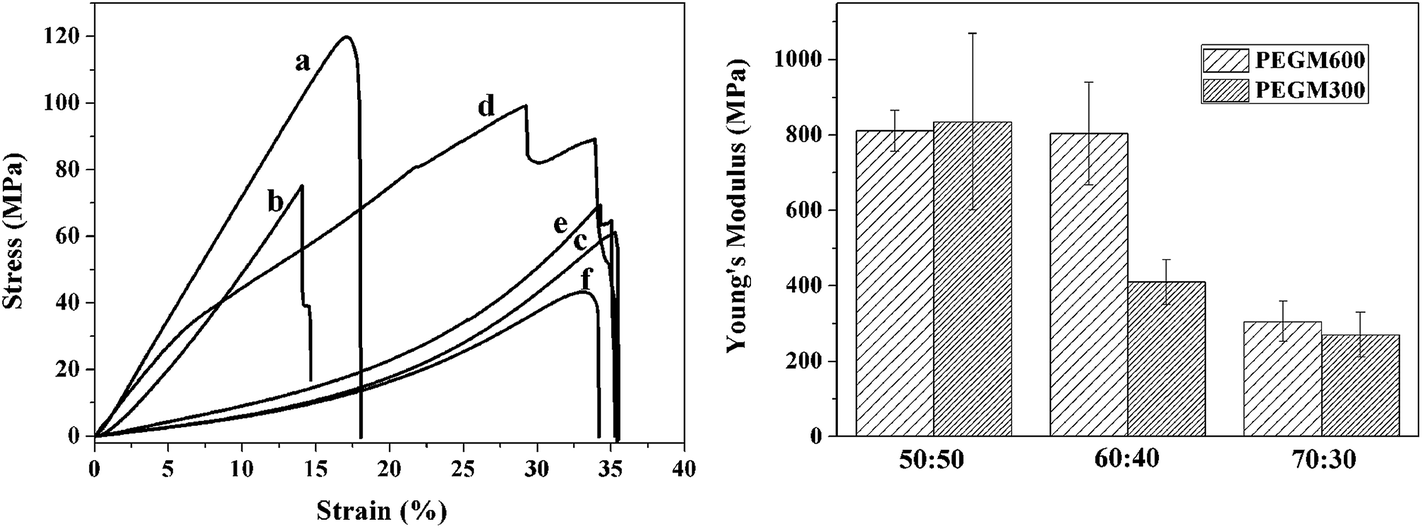 | ||
| Fig. 12 Stress–strain curves (a) and effect of organic/inorganic ratio on Young's modulus (b) of hybrids obtained by compression test for hybrids with different organic/inorganic ratios (molar%): curve (a), PEGM600 50:50; curve (b), PEGM600 60:40; curve (c), PEGM600 70:30; curve (d), PEGM300 50:50; curve (e), PEGM300 60:40; curve (f), PEGM300 70:30. | ||
| PEGM600 | PEGM300 | ||||
|---|---|---|---|---|---|
| Organic/inorganic (molar ratio) | Compressive strength (MPa) | Young's modulus (MPa) | Organic/inorganic (molar ratio) | Compressive strength (MPa) | Young's modulus (MPa) |
| 50:50 |
138.1 ± 36.5 | 811.2 ± 53.6 | 50:50 |
128.3 ± 75.1 | 834.8 ± 234.4 |
| 60:40 |
81.4 ± 5.9 | 804 ± 136.6 | 60:40 |
68.1 ± 12.6 | 410.1 ± 59.6 |
| 70:30 |
52.4 ± 8.98 | 305.6 ± 53.6 | 70:30 |
49.17 ± 15.2 | 270.1 ± 60.3 |
| Cancellous bone | 4–12 | 20–500 | Compact bone | 100–230 | 3 × 103 to 3 × 104 |
3.5 Cell viability test
As shown in Fig. 13, good cell proliferation (96–157%) was observed in the presence of the three extracts (E1, E2 and E3). Good cell viability was observed for all the extracts, no significant difference in viability was observed for the different extracts after 3 days. However after 6 days the cell viability was higher for the both hybrids compared to the control. The cell proliferation for the PEGM600 hybrids extracts is slightly higher than PEGM300 hybrids (Fig. 13a). The cell proliferation increased with culture time in each group (Fig. 13b).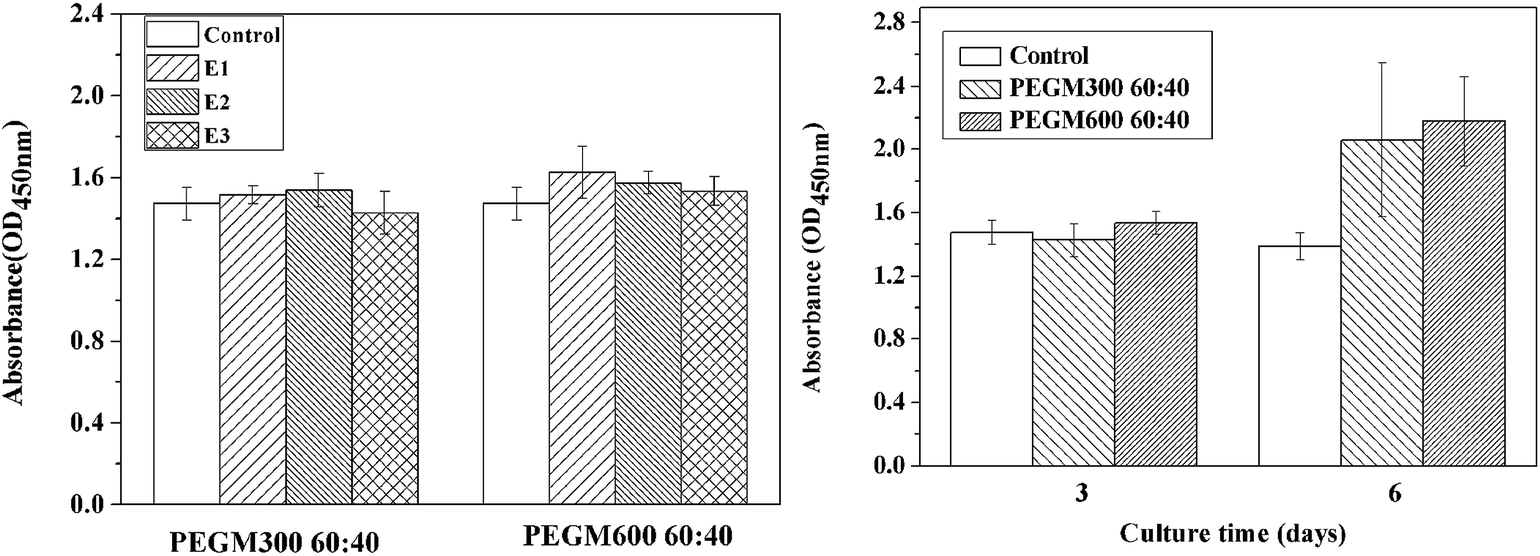 | ||
| Fig. 13 CCK-8 assay for proliferation of MC3T3-E1 cells cultured with (a) extracts (E1, E2 and E3, respectively) of PEGM300 60:40 and PEGM600 60:40 for 3 days and (b) extracts (E3) of PEGM300 60:40 and PEGM600 60:40 for 3 days and 6 days. | ||
3.6 Degradation studies
A preliminary evaluation of the degradation behaviour of the hybrids was conducted through mass loss upon immersion in SBF solution. Results are summarised in Fig. 14. Mass loss increased rapidly for both samples at during the first week before plateauing at ∼43% at 3 weeks. These results are favourable compared to other organic/inorganic hybrids. For example, Russo et al. report mass losses as high as 76% at 3 weeks for dPEG:GPTMS with a 1:2 ratio and losses of 52% at 3 weeks for dPEG:GPTMS with a 1:4 ratio.49
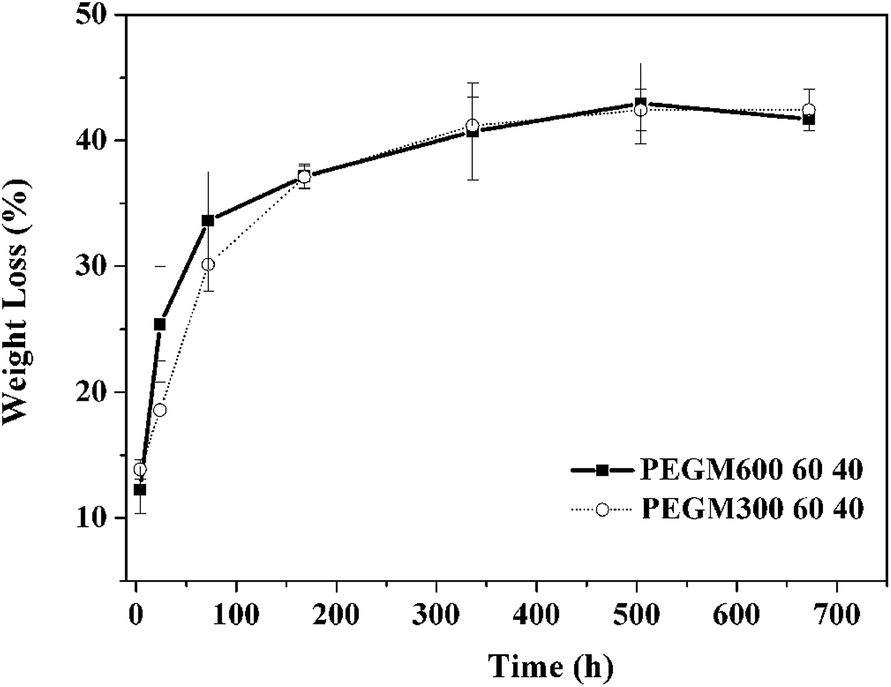 | ||
| Fig. 14 Degradation assay into SBF solution as a function of soaking time for hybrid materials obtained from PEGM600 and PEGM300 with the organic/inorganic ratio of 60:40. | ||
Table 3 presents the pH data for the PEGM600 60:40 and PEGM300 60:40 sample as a function of immersion time. It can be seen that the pH value of the extracts are stable with only a small increase in pH. The pH of these hybrids are lower than values reported for NovaBone (7.64) and S70C30 scaffolds (8.19) after immersion for 72 h.50 It has been reported that higher pH value may be detrimental for cell growth. The PEG modified hybrids exhibit lower pH values compared to the same inorganic composition (S70C30) this indicates that PEG moieties may help stabilize the inorganic phase of the hybrid.
| Sample | 0 day | 1 day | 3 day | 6 day |
|---|---|---|---|---|
| PEGM600 60:40 |
7.30 | 7.36 | 7.41 | 7.48 |
| PEGM300 60:40 |
7.30 | 7.38 | 7.49 | 7.60 |
4 Discussion
PEG was successfully modified, via an epoxide functionalization, to yield PEGM/70S30C bioactive hybrids with covalent (Si–O–Si) bonds linking the organic and inorganic phases. A series of hybrids containing different organic/inorganic molar ratios and PEG molecular weights were prepared and characterised. In all cases the modified PEGM hybrids showed significantly improved mechanical properties compared to the unmodified PEG hybrids. The improvement in mechanical properties was attributed directly to the covalent bonding between the organic and inorganic phases for the modified PEG hybrids. The present samples exhibited favourable bioactivity and mechanical properties compared to equivalent materials previously reported in the literature. In previously studies, Liu et al. prepared PEG/SiO2–CaO–P2O5 hybrid xerogels by introducing PEG during the hydrolysis process to provide a molecular scale interaction between the organic and inorganic phases. The PEG molecular chains were found to penetrate into the SiO2 network forming a semi-IPN structure. Since there was no covalent bonding between inorganic and organic phases, although a significantly increase in both the Young's modulus (320 to 420 MPa) and hardness was achieved compared to the pure inorganic material,47 it still could not reach the level achieved in the present study (in the present study we are achieving Young's modulus values in excess of 800 MPa for PEGM600 50:50 and 60:40 hybrids). Wang et al. have developed nanoparticle-gelatin composites with enhanced mechanical properties;51 the maximum value of Young's modulus reported for these systems was 600 MPa compared to over 800 MPa in the current system.
Rhee has developed poly(ε-caprolactone)/silica hybrids for bone repair applications, with covalent bonding between inorganic and organic phases.33 These materials show good apatite forming ability and Young's moduli of 200–600 MPa. Russo et al. have recently developed silica/bis(3-aminopropyl) polyethylene glycol(dPEG) inorganic/organic hybrids by using 3-glycidopropyltrimethoxysilane (GPTMS) as a coupling agent to provide the covalent links.49 The strain to failure increased from 4.2% for the sol–gel glass to 8–15% for the hybrids and compressive strengths of 15–20 MPa were reported which are comparable to cancellous bone (4–12 MPa) but low compared to cortical bone (100–230 MPa). In the present study we are achieving compressive strength values of 138 and 81 MPa for PEGM600 50:50 and 60:40 hybrids respectively. In addition to the covalent bonding, the improvement in mechanical properties achieved in our study might be partly due to the choice of molecular weight of polymer segments used. Previous studies have employed polymers with molecular weights greater than 1000.33,49 However, the polymer phase is mechanically weaker in compression compared to the inorganic phase. In the present study, PEG600 was found to be better than PEG300 suggesting there is also a lower limit for optimised polymer weight. However, we also note that it was not possible to form monoliths from higher molecular weight PEG (2000, 3000, 4000 and 6000). Therefore, an optimal molecular weight of polymer must to be chosen, in order to maximise the toughness of these hybrids.
Apatite formation was observed for 50:50, 60:40 and 70:30 PEGM600 and 60:40 and 70:30 PEGM300 hybrids. For a given organic/inorganic ratio the apatite formation of PEGM600 hybrids were consistently higher than the equivalent PEGM300 hybrids; this is attributed to the higher the molecular weight of the polymer. Apatite formation increased with increasing organic content with 60:40 and 70:30 (organic/inorganic molar ratio) showing the most promising formation. 50:50 and 40:60 hybrids showed minimal apatite formation. PEG is hydrophilic and relatively flexible, the increase in apatite formation with PEG molecular weight and PEG content are therefore likely to have resulted from better penetration of SBF into the material. This would enable the simulated fluid to react more quickly with the bioactive inorganic phases.
The 60:40 PEGM600 hybrids were the most promising materials demonstrating both good apatite formation and good mechanical properties. Young's modulus values ∼800 MPa are higher than those of cancellous bone and significantly higher than non-functionalised PEG hybrids and other values reported in the literature. Compressive strength of ∼80 MPa is similar to values required for compact bone (100 MPa). The excellent biocompatibility and aqueous solubility of PEG molecules, as well as the bioactivity of the inorganic components, coupled with the improved mechanical properties mean these hybrids exhibit great potential for bone regeneration applications.
The hybrids demonstrate good biocompatibility for both cell viability and proliferation. The hybrids exhibit an initial rapid weight loss, which provides the essential release of Ca and Si that is necessary for apatite and to stimulate cellular activity.8,52 After the initial weight loss observed during the first week which peaked at 37% the weight-loss plateaus and only a further 4% weight-loss is observed during the subsequent 3 weeks. Apatite formation counteracts further weight-loss after the first week such that the total weight loss after 4 weeks in only 41%.
No significant difference was observed between the PEGM300 and PEGM600 samples. This confirms that the molecular weight has little effect on the degradation of hybrids, and the degradability may be controlled by inorganic phase. During the first week, the mass loss was slightly lower for the PEG300M hybrid; this may be attributed to the higher hydrophilicity and the higher cross-link density of lower molecular weight polymer.
5 Conclusions
Functionalised PEGM/70S30C bioactive hybrids were successfully prepared via sol–gel synthesis. PEGM was successfully used as a cross-linking agent between organic and inorganic components at the molecular level. This resulted in strong covalent Si–O–Si bonds between the organic and inorganic phases which significantly improved the mechanical properties of the resultant hybrids. A corresponding increase in the apatite forming ability was observed for increasing molar fraction of the organic phase; at 50% or below the apatite formation was significantly reduced. Hybrids obtained from higher molecular weight PEGM600 had a higher compressive stress and showed enhanced apatite formation compared to the equivalent PEGM300 hybrid. The 60:40 PEGM600 hybrids were the most promising materials demonstrating good apatite formation, cell proliferation and viability and good mechanical properties. These organic/inorganic hybrid materials exhibited improved mechanical properties and excellent apatite forming ability, confirming their potential use as bone regeneration material.
Acknowledgements
This work was supported by MOST (Project no. 2012CB933200, 2013DFG52300), NSFC (Project no. 51173193) and a Royal Society/Natural Science Foundation of China international exchange grant (IE131323, 513111170).References
- A. Van Heest and M. Swiontkowski, Bone-graft substitutes, Lancet, 1999, 353(suppl. 1), SI28–SI29 CrossRef.
- P. V. Giannoudis, H. Dinopoulos and E. Tsiridis, Bone substitutes: An update, Injury, 2005, 36, 20–27 CrossRef PubMed.
- K. U. Lewandrowski, et al., Bioresorbable bone graft substitutes of different osteoconductivities: a histologic evaluation of osteointegration of poly(propylene glycol-co-fumaric acid)-based cement implants in rats, Biomaterials, 2000, 21(8), 757–764 CrossRef CAS.
- L. L. Hench, R. J. Splinter, W. C. Allen and T. K. Greenlee, Bonding mechanisms at the interface of ceramic prosthetic materials, J. Biomed. Mater. Res. Symp., 1971, 5, 25 CrossRef PubMed.
- L. L. Hench, The story of Bioglass (R), J. Mater. Sci.: Mater. Med., 2006, 17(11), 967–978 CrossRef CAS PubMed.
- L. L. Hench and I. Thompson, Twenty-first century challenges for biomaterials, J. R. Soc., Interface, 2010, 7, S379–S391 CrossRef CAS PubMed.
- R. A. Martin, et al., A study of the formation of amorphous calcium phosphate and hydroxyapatite on melt quenched Bioglass(A (R)) using surface sensitive shallow angle X-ray diffraction, J. Mater. Sci.: Mater. Med., 2009, 20(4), 883–888 CrossRef CAS PubMed.
- I. D. Xynos, et al., Gene-expression profiling of human osteoblasts following treatment with the ionic products of Bioglass (R) 45S5 dissolution, J. Biomed. Mater. Res., 2001, 55(2), 151–157 CrossRef CAS.
- I. D. Xynos, et al., Bioglass (R) 45S5 stimulates osteoblast turnover and enhances bone formation in vitro: Implications and applications for bone tissue engineering, Calcif. Tissue Int., 2000, 67(4), 321–329 CrossRef CAS PubMed.
- R. A. Martin, et al., A structural investigation of the alkali metal site distribution within bioactive glass using neutron diffraction and multinuclear solid state NMR, Phys. Chem. Chem. Phys., 2012, 14(35), 12105–12113 RSC.
- R. Li, A. E. Clark and L. L. Hench, An Investigation of Bioactive Glass Powders by Sol–Gel Processing, J. Appl. Biomater., 1991, 2(4), 231–239 CrossRef CAS PubMed.
- R. A. Martin, et al., Characterizing the hierarchical structures of bioactive sol–gel silicate glass and hybrid scaffolds for bone regeneration, Philos. Trans. R. Soc., A, 2012, 370(1963), 1422–1443 CrossRef CAS PubMed.
- D. Arcos and M. Vallet-Regi, Sol–gel silica-based biomaterials and bone tissue regeneration, Acta Biomater., 2010, 6(8), 2874–2888 CrossRef CAS PubMed.
- J. R. Jones, Review of bioactive glass: From Hench to hybrids, Acta Biomater., 2013, 9(1), 4457–4486 CrossRef CAS PubMed.
- P. Sepulveda, J. R. Jones and L. L. Hench, In vitro dissolution of melt-derived 45S5 and sol–gel derived 58S bioactive glasses, J. Biomed. Mater. Res., 2002, 61(2), 301–311 CrossRef CAS PubMed.
- S. Lin, et al., Tailoring the nanoporosity of sol–gel derived bioactive glass using trimethylethoxysilane, J. Mater. Chem., 2010, 20(8), 1489–1496 RSC.
- J. R. Jones, T. F. Kemp, and M. E. Smith, Effect of OH content on the bioactivity of sol–gel derived glass foam scaffolds, Bioceramics 18, 2006. vol. 309–311, pts 1 and 2, pp. 1031–1034 Search PubMed.
- M. Vallet-Regi, D. Arcos and J. Perez-Pariente, Evolution of porosity during in vitro hydroxycarbonate apatite growth in sol–gel glasses, J. Biomed. Mater. Res., 2000, 51(1), 23–28 CrossRef CAS.
- V. FitzGerald, et al., Bioactive glass sol–gel foam scaffolds: Evolution of nanoporosity during processing and in situ monitoring of apatite layer formation using small- and wide-angle X-ray scattering, J. Biomed. Mater. Res., Part A, 2009, 91(1), 76–83 CrossRef CAS PubMed.
- A. R. Boccaccini, et al., Preparation and characterisation of poly(lactide-co-glycolide) (PLGA) and PLGA/Bioglass((R)) composite tubular foam scaffolds for tissue engineering applications, Mater. Sci. Eng., C, 2005, 25(1), 23–31 CrossRef PubMed.
- K. Zhang, et al., Processing and properties of porous poly(L-lactide)/bioactive glass composites, Biomaterials, 2004, 25(13), 2489–2500 CrossRef CAS PubMed.
- K. Rezwan, et al., Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering, Biomaterials, 2006, 27(18), 3413–3431 CrossRef CAS PubMed.
- J. J. Blaker, et al., Mechanical properties of highly porous PDLLA/Bioglass (R) composite foams as scaffolds for bone tissue engineering, Acta Biomater., 2005, 1(6), 643–652 CrossRef CAS PubMed.
- G. Poologasundarampillai, et al., Poly(gamma-glutamic acid)/silica hybrids with calcium incorporated in the silica network by use of a calcium alkoxide precursor, Chem.–Eur. J., 2014, 20(26), 8149–8160 CrossRef CAS PubMed.
- B. Granqvist, et al., Biodegradable and bioactive hybrid organic–inorganic PEG–siloxane fibers. Preparation and characterization, Colloid Polym. Sci., 2004, 282(5), 495–501 CAS.
- O. Mahony, et al., Silica–Gelatin Hybrids with Tailorable Degradation and Mechanical Properties for Tissue Regeneration, Adv. Funct. Mater., 2010, 20(22), 3835–3845 CrossRef CAS.
- M. Manzano, et al., Mechanical properties of organically modified silicates for bone regeneration, J. Mater. Sci.: Mater. Med., 2009, 20(9), 1795–1801 CrossRef CAS PubMed.
- N. Miyata, et al., Apatite-forming ability and mechanical properties of PTMO-modified CaO–SiO2–TiO2 hybrids derived from sol–gel processing, Biomaterials, 2004, 25(1), 1–7 CrossRef CAS.
- Q. Chen, et al., Bioactivity and mechanical properties of PDMS-modified CaO–SiO2–TiO2 hybrids prepared by sol–gel process, J. Biomed. Mater. Res., 2000, 51(4), 605–611 CrossRef CAS.
- M. Kamitakahara, et al., Degradation of bioactive polydimethylsiloxane–CaO–SiO2–TiO2 and poly(tetramethylene oxide)–CaO–TiO2 hybrids in a simulated body fluid, J. Am. Ceram. Soc., 2004, 87(2), 235–239 CrossRef CAS PubMed.
- A. J. Salinas, et al., Microstructure and macroscopic properties of bioactive CaO–SiO2–PDMS hybrids, J. Biomed. Mater. Res., Part B, 2007, 81(1), 274–282 CrossRef CAS PubMed.
- M. Kamitakahara, et al., Bioactivity and mechanical properties of polydimethylsiloxane (PDMS)–CaO–SiO2 hybrids with different calcium contents, J. Mater. Sci.: Mater. Med., 2002, 13(11), 1015–1020 CrossRef CAS.
- S.-H. Rhee, Bone-like apatite-forming ability and mechanical properties of poly(ε-caprolactone)/silica hybrid as a function of poly(ε-caprolactone) content, Biomaterials, 2004, 25(7–8), 1167–1175 CrossRef CAS PubMed.
- S. H. Rhee, et al., Evaluation of a novel poly(epsilon-caprolactone)–organosiloxane hybrid material for the potential application as a bioactive and degradable bone substitute, Biomacromolecules, 2004, 5(4), 1575–1579 CrossRef CAS PubMed.
- X. Dieudonne, et al., Bioactive glass hybrids: a simple route towards the gelatin–SiO(2)–CaO system, Chem. Commun., 2014, 50(63), 8701–8704 RSC.
- B. Yu, et al., Effect of Calcium Source on Structure and Properties of Sol–Gel Derived Bioactive Glasses, Langmuir, 2012, 28(50), 17465–17476 CrossRef CAS PubMed.
- Y.-S. Sun, et al., A low-temperature sol gel route for the synthesis of bioactive calcium silicates, Chin. Chem. Lett., 2013, 24(2), 170–172 CrossRef CAS PubMed.
- R. Marchal, et al., Biodegradability of polyethylene glycol 400 by complex microfloras, Int. Biodeterior. Biodegrad., 2008, 62(4), 384–390 CrossRef CAS PubMed.
- D. M. Pickup, et al., Preparation, structural characterisation and antibacterial properties of Ga-doped sol–gel phosphate-based glass, J. Mater. Sci., 2009, 44(7), 1858–1867 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, How useful is SBF in predicting in vivo bone bioactivity?, Biomaterials, 2006, 27(15), 2907–2915 CrossRef CAS PubMed.
- L. Wang, et al., Differentiation of human bone marrow mesenchymal stem cells grown in terpolyesters of 3-hydroxyalkanoates scaffolds into nerve cells, Biomaterials, 2010, 31(7), 1691–1698 CrossRef CAS PubMed.
- Y. Liu, et al., Shell Cross-Linked Micelle-Based Nanoreactors for the Substrate-Selective Hydrolytic Kinetic Resolution of Epoxides, J. Am. Chem. Soc., 2011, 133(36), 14260–14263 CrossRef CAS PubMed.
- J. M. Yang, et al., Preparation of epoxy–SiO2 hybrid sol–gel material for bone cement, J. Biomed. Mater. Res., Part A, 2003, 64(1), 138–146 CrossRef PubMed.
- P. de Caro, Z. Mouloungui and A. Gaset, Synthesis of alkyloxy (di)alkylamino propanols, hydroxy alkyloxy (di)alkylamino propanols, and the dimer compounds for use as fuel additives, J. Am. Oil Chem. Soc., 1997, 74(3), 235–240 CrossRef CAS PubMed.
- M. Vallet-Regi, et al., XRD, SEM-EDS, and FTIR studies of in vitro growth of an apatite-like layer on sol–gel glasses, J. Biomed. Mater. Res., 1999, 44(4), 416–421 CrossRef CAS.
- J. C. Elliott, P. E. Mackie and R. A. Young, Monoclinic hydroxyapatite, Science, 1973, 180, 1055–1057 CrossRef CAS PubMed.
- W. Liu, et al., Synthesis of bioactive poly(ethylene glycol)/SiO2-CaO–P2O5 hybrids for bone regeneration, Mater. Sci. Eng., C, 2012, 32(4), 707–711 CrossRef CAS PubMed.
- L. L. Hench and J. K. West, Biological applications of bioactive glasses, Life Chem. Rep., 1996, 13, 187 CAS.
- L. Russo, et al., Novel silica/bis(3-aminopropyl) polyethylene glycol inorganic/organic hybrids by sol–gel chemistry, Mater. Chem. Phys., 2013, 140(1), 168–175 CrossRef CAS PubMed.
- S. Midha, et al., Preconditioned 70S30C bioactive glass foams promote osteogenesis in vivo, Acta Biomater., 2013, 9(11), 9169–9182 CrossRef CAS PubMed.
- C. Wang, et al., Bioactive Nanoparticle–Gelatin Composite Scaffold with Mechanical Performance Comparable to Cancellous Bones, ACS Appl. Mater. Interfaces, 2014, 6(15), 13061–13068 CAS.
- I. D. Xynos, et al., Ionic products of bioactive glass dissolution increase proliferation of human osteoblasts and induce insulin-like growth factor II mRNA expression and protein synthesis, Biochem. Biophys. Res. Commun., 2000, 276(2), 461–465 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |