
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of salt–hydrogel marbles and hollow-shell microcapsules by an aerosol gelation technique
Marius
Rutkevičius
a,
Georg H.
Mehl
a,
Jordan T.
Petkov
b,
Simeon D.
Stoyanov
c and
Vesselin N.
Paunov
*a
aDepartment of Chemistry, University of Hull, Cottingham Road, Hull HU6 7RX, UK. E-mail: V.N.Paunov@hull.ac.uk; Fax: +44 (0)1482466410; Tel: +44 (0)1482465660
bUnilever Discover Port Sunlight, Quarry Road East, Bebington, Wirral, CH63 3JW, UK
cUnilever R&D Vlaardingen, Olivier van Noortlaan 120, 3133 AT, Vlaardingen, The Netherlands
First published on 4th November 2014
Abstract
We designed a new method for preparation of liquid marbles by using hydrophilic particles. Salt–hydrogel marbles were prepared by atomising droplets of hydrogel solution in a cold air column followed by rolling of the collected hydrogel microbeads in a bed of micrometre sized salt particles. Evaporation of the water from the resulting salt marbles with a hydrogel core yielded hollow-shell salt microcapsules. The method is not limited to hydrophilic particles and could potentially be also applied to particles of other materials, such as graphite, carbon black, silica and others. The structure and morphology of the salt–hydrogel marbles were analysed by SEM and their particle size distributions were measured. We also tested the dissolution times of the dried salt marbles and compared them with those of table salt samples under the same conditions. The high accessible surface area of the shell of salt microcrystals allows a faster initial release of salt from the hollow-shell salt capsules upon their dissolution in water than from the same amount of table salt. The results suggest that such hollow-shell particles could find applications as a table salt substitute in dry food products and salt seasoning formulations with reduced salt content without the loss of saltiness.
Introduction
Liquid marbles consist of liquid drops stabilised by adsorbed layers of solid particles at their air–liquid interface.1,2 These fall into the category of liquid-in-air systems which also include dry water3 and dry oils.4 Such systems have attracted a lot of attention with ongoing applications in microfluidics,2 cosmetics, reactors, gas-sensing elements,5 microencapsulation,6 water evaporation control,7 and others.8,9 Liquid marbles have very low adhesion and low friction at solid surfaces10 and depending on the solid particles' properties they can be made pH responsive,11,12 photo-responsive13 and magneto-responsive.14Hollow-shell microcapsules have been fabricated for a range of applications in electronics,15 drug and nutrient delivery,16 beauty products17 and catalyst formulations.18,19 Recently, liquid marbles were recognised as a viable new way for the encapsulation of liquids1 and formation of microcapsules.8 Aqueous marbles are typically produced by rolling millimetre sized water droplets in a bed of hydrophobic solid microparticles,3–5,7,10 which leads to the formation of a protective particulate shell on the drop surface. Smaller particles from the liquid marble shell have been reported to attach to the liquid drop surface, whereas larger ones tend to form the outer layers of the liquid marbles.20 Liquid marbles coated with hydrophobic model drug particles were dried and their structure showed a hollow core as determined by X-ray tomography.21 Liquid marbles of aqueous solutions of relatively low viscosity have to be stabilised with highly hydrophobic particles. Using hydrophilic particles22 with aqueous drops usually leads to the formation of granules.23,24 This poses a challenge if liquid marbles are to be used in food formulations, as the stabilising microparticles must be edible,25 hydrophobic and approved for use in foods.
Here we propose a new approach to fabricate liquid marbles which involves pre-gelling of the aqueous drops which form the cores of the liquid marbles with a suitable gelling agent. We obtain an aerosol of a hot solution of the gelling agent and then pass it through a cold air column which sets the aerosol drops. The obtained hydrogel microbeads are then collected and rolled onto a bed of microparticles which can be either hydrophobic or hydrophilic. Fig. 1 illustrates this process schematically for salt microcrystals as particles. We demonstrate this technique with salt microcrystals which form layers around the hydrogel microbeads due to the partial syneresis of water from the gel which promotes the salt particle adhesion to the beads due to capillary bridge forces. The marbles are separated from the excess of salt particles by sieving. Using salt-tolerant hydrogels we applied this aerosol gelation method to fabricate salt–hydrogel marbles which upon drying yielded hollow-shell salt microcapsules. We studied the size distribution of salt–hydrogel marbles and their morphology as well as the inner structure of the obtained salt microcapsules. The method could potentially be applied for a range of solid microparticles of various wettabilities at the air–water interface26,27 without the need to be hydrophobic. In addition, Xanthan28 and κ-carrageenan29 hydrogel core also allows encapsulation of hydro-soluble materials such as flavours.30
We demonstrated the usefulness of the obtained salt–hydrogel marbles for preparation of table salt particles with reduced salt content. It has been recognised that salt intake from consumption of processed foods is currently well above the recommended doses.31–33 High salt intake causes many health issues, typically hypertension, increase in the risk of stroke and others.34,35 Reduction in the dietary salt leads to a lower blood pressure and reduction in the risk of developing a cardiovascular disease.34 Saltiness is perceived by the momentary concentration of salt ions dissolving on the tongue, rather than the overall amount of consumed salt.36 Reduction in the dissolution time of salt whilst chewing would boost the local concentration of the salt ions triggering a saltier perception. However, ordinary table salt crystals have a crystalline structure taking a relatively long time to dissolve. Substituting the liquid phase with a hydrogel and replacing the hydrophobic particles with salt microcrystals allowed us to produce salt–hydrogel marbles and hollow-shell salt microcapsules. We tested the dissolution kinetics of such hollow-shell salt microcapsules in water and compared the initial boost in the salt concentration with the same amount of control samples of table salt.
Experimental
Materials
Xanthan gum (Vanzan® NF) was kindly supplied as a free sample by R. T. Vanderbilt Company, Inc., Norwalk, CT, USA, Konjac gum (Nutricol® GP 312) and κ-carrageenan (Gelcarin® GP911 NF) were gifts from FBC BioPolymer, Philadelphia, PA, USA. Agar (bacteriological, no. 1) was purchased from Oxoid Ltd., England. Table salt was supplied by 3663, UK, with an added anti-caking agent (sodium hexacyanoferrate II).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, 0.4–1.5 wt%) or κ-carrageenan (1.0–2.0 wt%), or agar (1.0–3.0 wt%) were prepared by hydrating the gelling agent at 90 °C. The hot solution of the gelling agent was brought to 50 °C and was sprayed using a hand held atomiser through a cold air column over a bed of salt microcrystals. The typical size of atomized droplets increased with increase in viscosity, i.e. for water the diameter was ≈10 μm and for κ-carrageenan (1.5 wt% gelling agent) or Xanthan–Konjac hydrogels (0.5 wt% gelling agent) it was <250 μm. The salt bed was shaken to facilitate the rolling and covering of the collected hydrogel microbeads with salt microcrystals. The powder was then sieved through 0.6 mm and 0.3 mm sieves, respectively, to remove the unattached salt microcrystals and to separate the very large and small salt–hydrogel marbles, followed by drying the separated particles for 2 days at 30 °C to produce hollow-shell salt microcapsules (Fig. 1).
:1 ratio, 0.4–1.5 wt%) or κ-carrageenan (1.0–2.0 wt%), or agar (1.0–3.0 wt%) were prepared by hydrating the gelling agent at 90 °C. The hot solution of the gelling agent was brought to 50 °C and was sprayed using a hand held atomiser through a cold air column over a bed of salt microcrystals. The typical size of atomized droplets increased with increase in viscosity, i.e. for water the diameter was ≈10 μm and for κ-carrageenan (1.5 wt% gelling agent) or Xanthan–Konjac hydrogels (0.5 wt% gelling agent) it was <250 μm. The salt bed was shaken to facilitate the rolling and covering of the collected hydrogel microbeads with salt microcrystals. The powder was then sieved through 0.6 mm and 0.3 mm sieves, respectively, to remove the unattached salt microcrystals and to separate the very large and small salt–hydrogel marbles, followed by drying the separated particles for 2 days at 30 °C to produce hollow-shell salt microcapsules (Fig. 1).
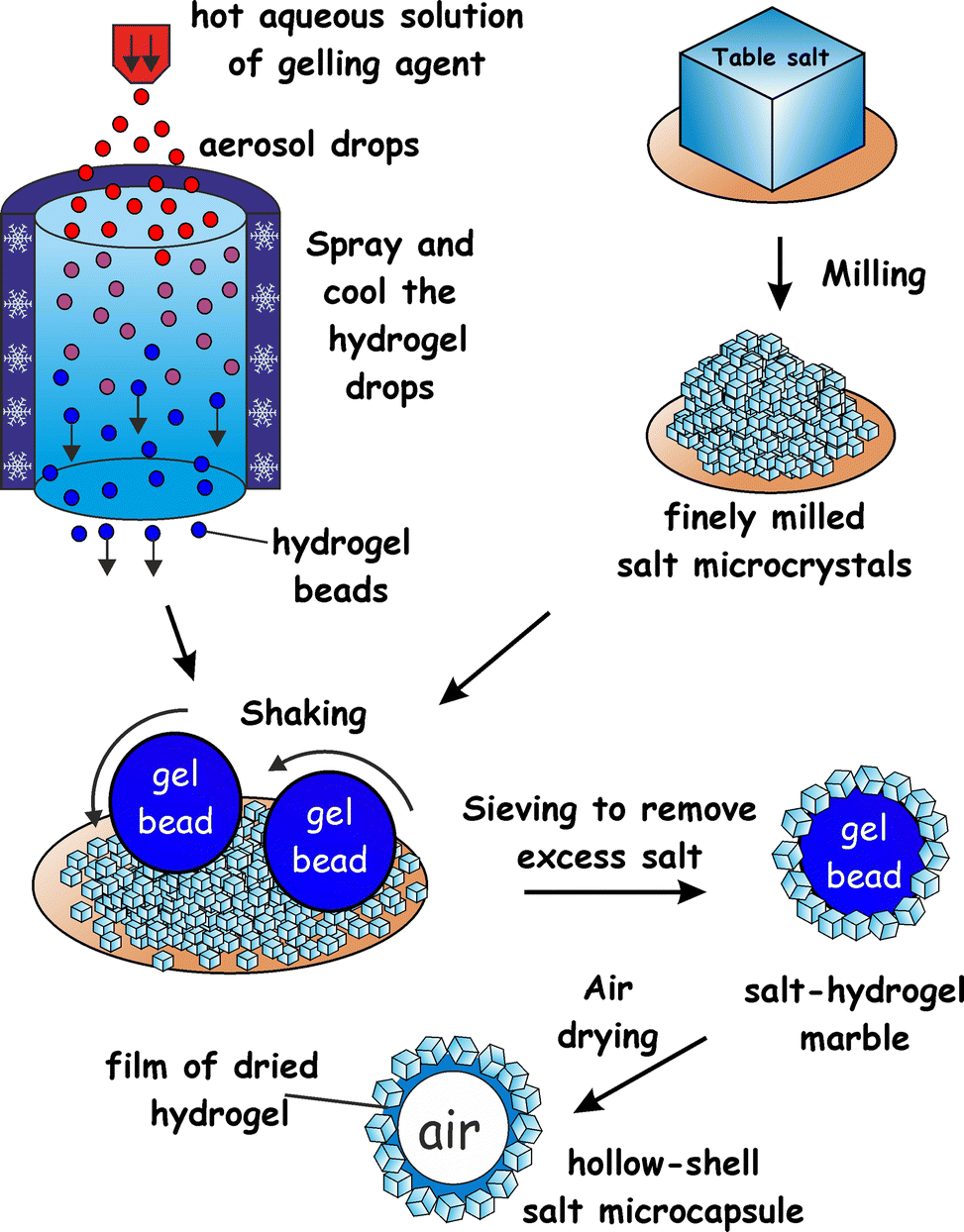 | ||
| Fig. 1 Schematic representation of the method used to produce salt–hydrogel marbles and hollow-shell particles. A hot hydrogel solution is atomized, producing aerosol droplets, which are flash-gelled by passing through the cold air column. The formed aerosol hydrogel beads are collected over a bed of finely milled salt microcrystals. As the salt bed is vibrated, a layer of salt crystals is deposited on the rolling hydrogel beads which forms hydrogel–salt marbles. These are then removed from the excess salt by sieving, and further dried to produce hollow-shell salt marbles. | ||
The samples were dissolved at room temperature by agitation with a magnetic stirrer at a fixed rotation speed (typically 50–200 rpm). The conductance was measured using a Jenson 4510 Bench Conductivity Meter and plotted as a function of the time elapsed from adding the salt sample to the water. The bottom of the probe was adjusted to 1 cm above the bottom of the beaker. From these data, the optimum stirring speed was selected (60 rpm) and used to measure the dissolution time of salt marbles.
Results and discussion
Salt marble size distribution
The salt–hydrogel marbles were imaged with a digital camera prior to drying at 30 °C for 2 days. The produced marbles were polydisperse, therefore a sieving step was introduced to separate them in different particle size fractions, which are shown in Fig. 2. | ||
| Fig. 2 Optical photographs of salt marbles produced from gelled aerosol drops from the following gelling agents: (a) 3 wt% κ-carrageenan; (b) 1.5 wt% κ-carrageenan; (c) 0.5 wt% Xanthan–Konjac 2:1 hydrogel solution. All scale bars are 500 μm. | ||
For Xanthan–Konjac (2:1 ratio) above the overall concentration of 1.2 wt%, the atomiser did not produce an aerosol into individual droplets, rather than a jet of a hydrogel solution, which yielded salt marbles of higher average diameter, polydispersity and a thicker shell of deposited salt microcrystals on the marble surface. We envisage that this is because of the increased viscosity of the solution upon increasing the gelling agent concentration. This changed the size distribution of the produced aerosol drops, and correspondingly the size of the produced salt marbles. Salt–hydrogel marbles from hydrogel microbeads with different gelling agent concentrations were prepared and analysed.
The salt–hydrogel marble size distribution was analysed using optical photographs and image analysis software. The results are shown in Fig. 3 and 4 for salt marble samples made with Xanthan–Konjac (2:1 ratio) and κ-carrageenan hydrogels, respectively. A broad particle size distribution was observed in the majority of the analysed samples, especially upon increasing the concentration of the gelling agent in the atomised solution. The average size of the salt–hydrogel marbles was around 600 ± 200 μm for both κ-carrageenan and Xanthan–Konjac hydrogel based salt marbles, varying slightly due to the different gelling agent concentrations used. As a comparison, ordinary table salt crystals used here had an average size of 280 ± 100 μm. Due to the high polydispersity of the produced particles and the difference in the number of deposited salt microcrystal layers between the marble particles, we have not been able to accurately measure the masses of salt and the remaining gelling agent. In future studies, we will focus our attention on minimising the polydispersity in the particle size and thickness of deposited salt layers in order to accurately determine the mass of salt per individual particle.
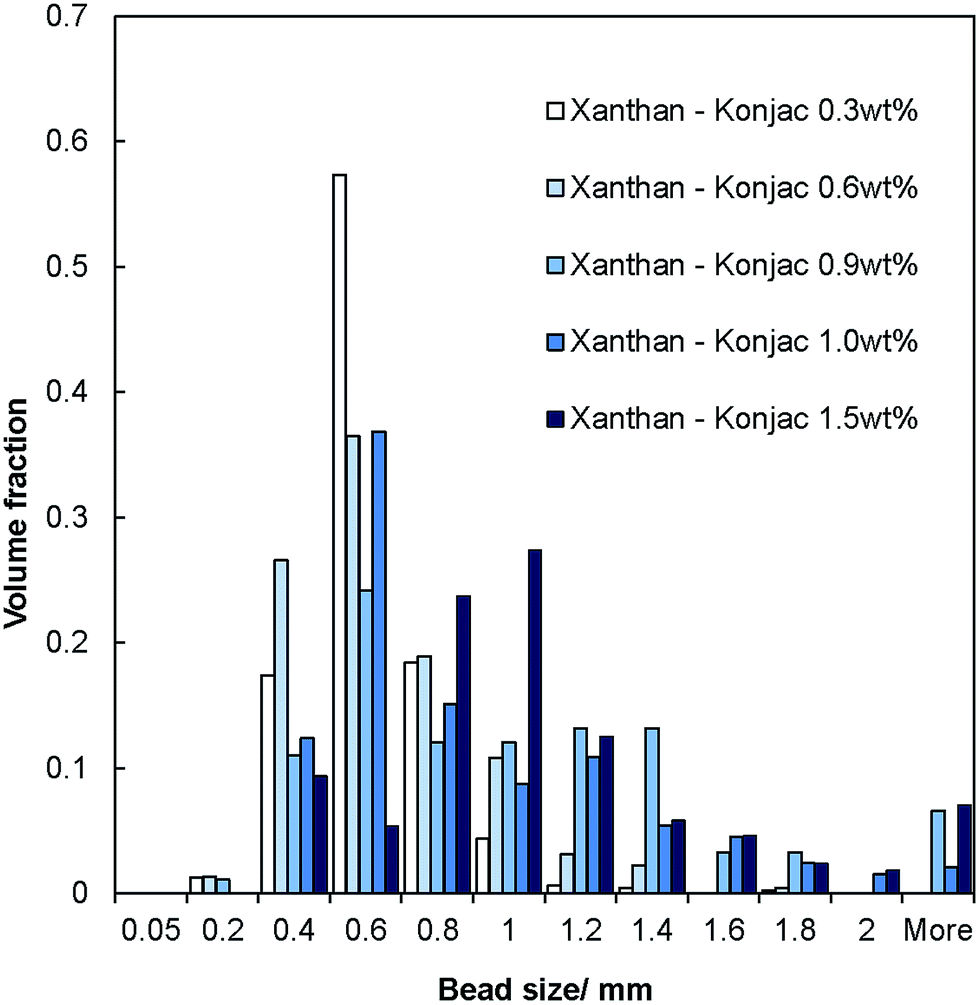 | ||
| Fig. 3 Size distribution diagram for samples of dried salt–hydrogel marbles produced from aerosolised Xanthan–Konjac 2:1 solution as the core of the marbles at different overall concentrations of the gelling agent. The histogram represents the fraction of the marbles of various sizes in the produced samples. | ||
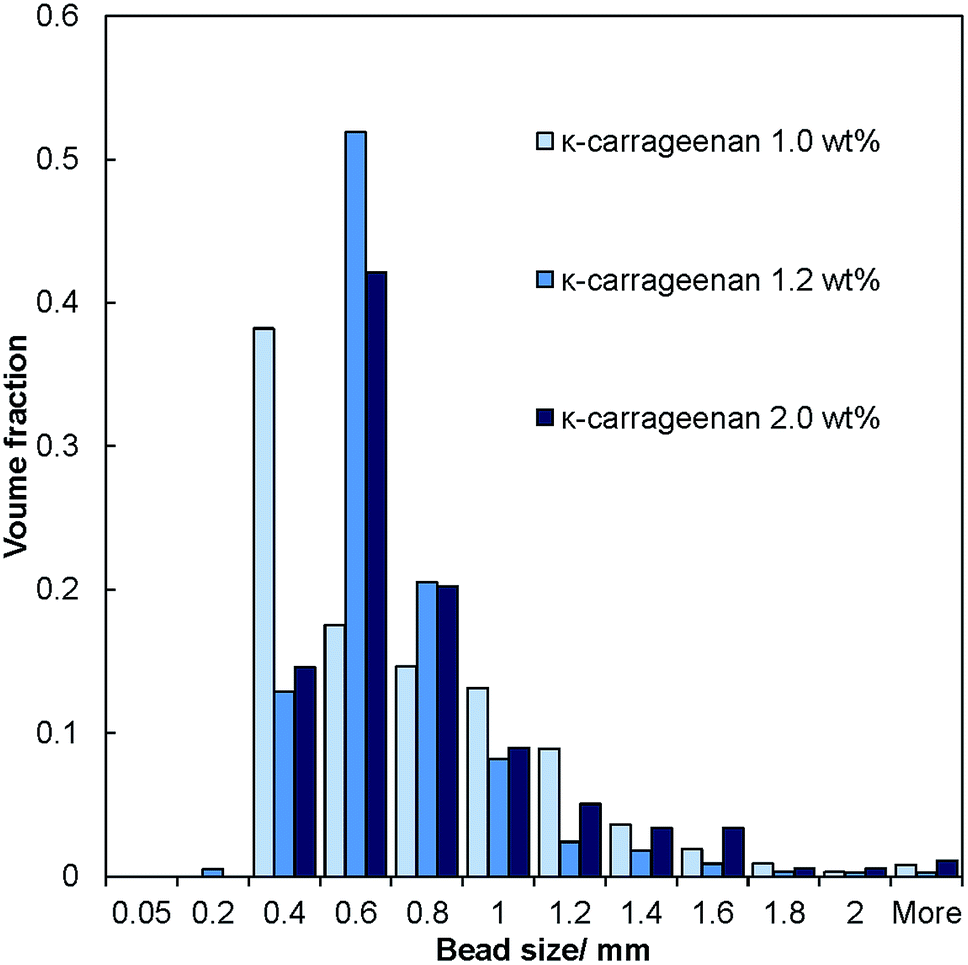 | ||
| Fig. 4 Size distribution diagram for samples of dried salt–hydrogel marbles made with κ-carrageenan hydrogel beads as the core of the marble at different concentrations of the gelling agent. The histogram represents the fraction of the marbles of various sizes in the produced samples. | ||
The size of the hollow-shell salt microcapsules produced after drying of the marble hydrogel cores was much larger than the typical table salt microcrystals (30 ± 20 μm in this case), which varies according to the used milling technique.
Particle morphology analysis
We analysed the morphology of the dried salt marbles by studying their SEM images which are shown in Fig. 5 and 6 below. The dried samples were sectioned by a scalpel and imaged to reveal the approximate thickness of the layer of salt microcrystals and residue produced from the drying of the hydrogel core. We observed the formation of two typical microstructures: (i) for salt marbles prepared using Xanthan–Konjac hydrogels, which are known for their film forming properties,37,38 the images of cracked open samples show particles with a film of dried gelling agent within the inner part of the salt layer (core–shell interface, see e.g.Fig. 5, 6 and 7b); in this case, a layer of salt microcrystals adheres onto the hydrogel bead surface and forms a marble where there is only partial transfer of the water from the hydrogel to the surrounding layer of salt microcrystals. This occurred due to the hydrogel syneresis upon contact with the salt particles and the capillary penetration of the released water into the bed of salt microcrystals. In this case, a large fraction of the hydrogel core remains intact and creates a hollow-shell structure upon drying. Note that the microparticles do not need to be hydrophilic in order to form a core–shell structure around the gel beads. (ii) When the aerosolised drops of hydrogel solution were not fully gelled upon contact with salt microcrystals, they did not form salt marbles, but produced granules with a microporous core of salt (Fig. 7a). We envisage that in this case the liquid fully wets the salt microcrystals and penetrates in between them in the form of capillary bridges. The drying of the later formed salt granules rather than salt marbles. Fig. 8 summarises the differences between the formation of salt marbles and salt granules as a result of the two different processes (i) and (ii). | ||
| Fig. 5 SEM images of dried salt marbles produced with 1.0 wt% Xanthan–Konjac 2:1 hydrogel beads. (a and b) Column inner surface temperature was ∼6 °C. Samples were cracked open with a scalper whilst on the SEM stub. Larger marbles were selected for the analysis for easier sample preparation. (a) and (b) correspond to images from the same batch. | ||
 | ||
| Fig. 6 SEM images of dried salt–hydrogel marbles produced by spraying a hot solution of 0.5 wt% Xanthan–Konjac in 2:1 ratio. Samples have been cracked open with a scalper whilst on the SEM stub. Column inner surface temperature was ∼6 °C. (a)–(d) correspond to images from the same batch. | ||
 | ||
| Fig. 7 SEM images of (a and b) salt granules produced by spraying aqueous solution of 1.5 wt% agar and (c) 3.0 wt% agar solution. In both cases the cold column inner temperature was maintained at 1 °C. | ||
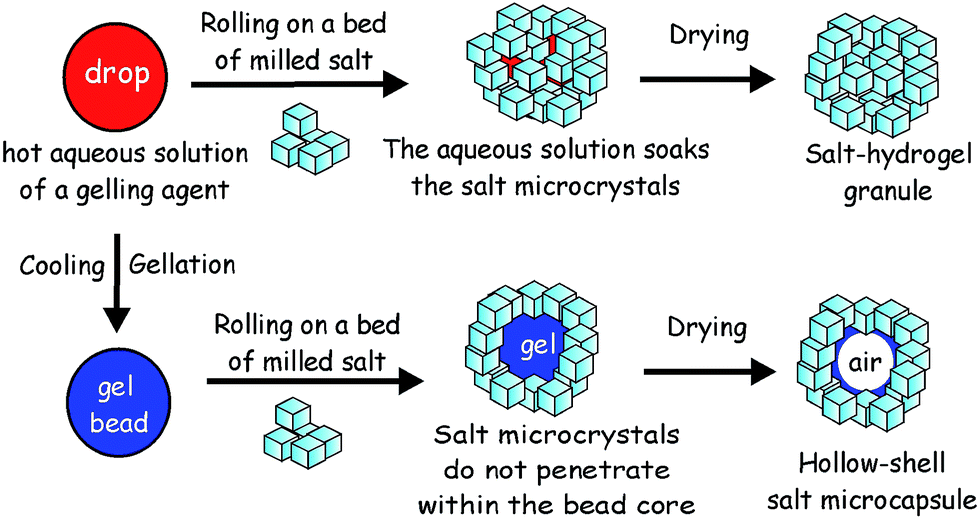 | ||
| Fig. 8 Possible outcomes in the fabrication of salt marbles: the top mechanism shows production of the salt granules, where the liquid drop wets the hydrophilic salt particles to form salt granules. The bottom mechanism shows the preparation of the salt–hydrogel marbles, where a gel bead is formed before rolling onto the bed of salt microcrystals. Here, the salt crystals cannot penetrate into the hydrogel core and form a shell around it. Hollow-shell salt microcapsules are produced upon drying of the salt–hydrogel marbles. | ||
One way to reduce the syneresis could be by increasing the gelling agent concentration within the hydrogel. This would make the hydrogel stronger and less prone to syneresis. Another way to control the surface granulation process in the salt marble production could be saturating the solution of the gelling agent with salt in order to balance the osmotic pressure between the hydrogel core and its surface layer, as it comes into contact with the salt crystals. However, in this case the salt-compatible gelling agent, like Xanthan–Konjac 2:1, would be a better choice.
Another possible approach would be to reduce further the temperature of the cold column and freeze the hydrogel drops before they come into contact with the bed of salt microcrystals. We expect that the amount of salt per salt–hydrogel marble may vary with the size of the hydrogel beads and would also depend on the salt microcrystal size. The water in this case would be physically captured within the core in the form of a frozen hydrogel and a smaller amount of it would be able to be released through melting and gel syneresis into the surrounding layer of microcrystals, thus keeping the salt shell thinner. Our column cooling setup did not allow us to maintain sub-zero temperatures on the column inner surface due to condensation. Therefore, this can be a subject of future improvement which may require a different column design. However, such a syneresis effect in the fabrication of hydrogel marbles by aerosol gelation is likely to be less pronounced for other insoluble particulate materials, such as carbon black, silica, calcium carbonate and others as such particles would not dissolve as much as the salt microcrystals in the hydrogel media.
Note that the driving force of the encapsulation of the hydrogel bead by the salt microparticles is different from the classic liquid marbles. In the salt–hydrogel marbles prepared in this study, the particles (NaCl microcrystals) adhere onto hydrogel bead surfaces. That is the reason why hydrophilic particles can coat the hydrogel drops. On the other hand, in the formation of classic liquid marbles, hydrophobic particles are adsorbed at air–liquid surfaces. The driving force of the adsorption is elimination of an area of bare air–liquid surfaces. If the particles are too hydrophilic, they would not effectively adsorb at the liquid surface. However, for our case of hydrogel beads this is not an issue. The formation of a hydrogel core in the salt marbles and granules could be used to encapsulate flavour or taste enhancing compounds if they are introduced into the hydrogel solution before its atomisation. This could potentially be used in the food industry to produce salt marbles with added flavours, or other nutrient contents for the development of a range of salt-based seasonings and taste enhancing products. Here we demonstrate the usefulness of our approach for preparation of salt marbles in the development of hollow-shell salt seasoning particles with a reduced salt content due to the presence of a cavity. We also demonstrate that the salt marbles have a higher initial dissolution rate which would be useful in maintaining the same or higher degree of “saltiness” taste compared to the one produced by table salt crystals of similar size.
Salt marble dissolution kinetics
We conducted a dissolution kinetics experiment aimed at testing the rate of release of salt from the produced salt marbles. Samples of salt marbles were added to a fixed amount of water under constant stirring and the solution conductance was monitored over a period of time and compared with the control samples of table salt and finely milled salt microcrystals. The experimental data directly represent the dissolved NaCl content, as the conductivity of sodium chloride increases linearly with the increase of its concentration up to 1 M.39 The conductance of the solution was monitored for a set amount of salt samples in a fixed volume of water for different speeds of agitation. Fig. 9a shows the solution conductance plotted vs. time which demonstrated a much faster initial increase in the salt concentration for the salt marbles compared to the table salt sample under the same conditions. This graph also shows the conductance of the salt microcrystals (finely milled salt, particle size 30 ± 20 μm), used to produce the salt marbles. The difference in the solution conductance for the pre-milled salt and the salt marble samples upon dissolution was small, with the former having a slightly higher initial conductance due to the crystals being loose powder rather than trapped on the marble surface. Also, as the gelling agent concentration was increased within the hydrogels, the conductance of the salt marbles seemed to change slower, suggesting that these samples would be perceived as less salty by the consumer. Other test results, shown in Fig. 9b, show the solution conductance for salt marbles, salt microcrystals and table salt samples, 40 seconds after the start of their dissolution under the same conditions. The conductance of the salt marbles solution was higher than non-milled table salt (crystals sizes, 280 ± 100 μm) at this time point of the dissolution experiment. We envisage that 40 seconds is a typical time for development of the taste of saltiness. One may speculate that salt marbles would produce a stronger taste of saltiness, as the concentration of the dissolved salt ions in contact with the receptors for saltiness would be higher at the same time since start of the dissolution of the salt marbles.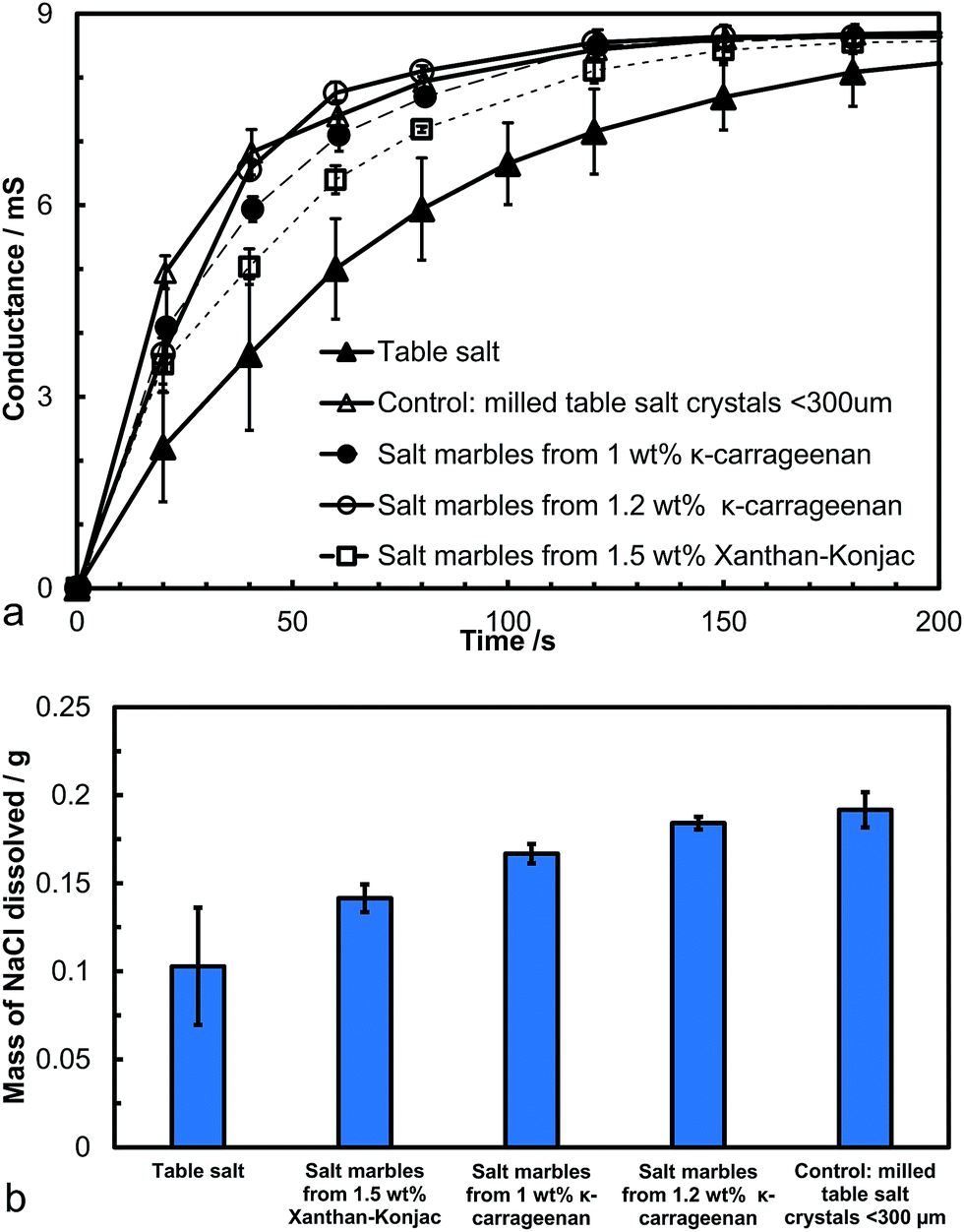 | ||
| Fig. 9 (a) Conductance of the aqueous medium vs. time after the addition of a fixed amount (0.2500 g) of table salt or hollow-shell salt marbles. The table salt particles had an average particle diameter of 280 ± 100 μm and the milled salt crystals were 30 ± 20 μm in diameter. (b) The mass of NaCl dissolved upon dissolution of different samples of salt or salt marbles 40 seconds after the addition of the sample (0.2500 g) into a fixed amount of water (50 cm3), derived from the results in (a). The higher the mass of the dissolved salt the higher the salt concentration and the perception of saltiness. The graph shows that compared with the ordinary non-milled table salt, higher mass and possibly saltiness feel could be delivered using salt marbles produced by the hydrogel bead templating instead the equivalent amount of table salt. Error bars represent the standard deviation of at least two measurements. | ||
However, at the same time a smaller amount of salt marbles is needed to achieve the same conductance over the same period of time compared to the table salt crystals. This could be beneficial for formulating salt seasoning products with a reduced amount of salt without reduction of the taste of saltiness. Both κ-carrageenan and Xanthan–Konjac hydrogel solutions could be used in the production of the salt marbles for food grade products, providing additional advantages of reduced overall salt content and the possibility for additional flavour encapsulation in the hydrogel cores in the marble fabrication process.
Conclusions
We designed a new method for preparation of salt–hydrogel marbles from salt microcrystals and an aqueous solution of gelling agents. The method is based on gelling of drops of an aerosolised hot solution of a gelling agent in a cold air column and the deposition of the obtained hydrogel microbeads over a bed of microcrystalline salt powder which forms a particle layer around them.The produced salt–hydrogel marbles were separated from the excess of salt microcrystals by sieving, and drying of the hydrogel cores yielded hollow-shell salt microcapsules. This method allows fabrication of liquid marbles from aqueous solutions and hydrophilic particles which is a challenge for other methods of preparation of liquid marbles, where hydrophobic particles are used as stabilisers. We show that when hydrogel microbeads are used instead of liquid drops, hollow-shell salt microcapsules can be produced upon drying of the salt marbles. In contrast, liquid drops of the gelling agent yield porous salt granules without apparent cavities due to the complete capillary penetration of the liquid within the bed of the hydrophilic salt microcrystals. We demonstrate the application of this method for fabrication of salt–hydrogel marbles by producing edible hollow-shell salt microcapsules which could be used in the food industry for the development of salt-based seasonings of reduced salt content. The presence of a hollow-shell coated with salt microcrystals provides an advantage of such microcapsules compared to the use of table salt. The methodology of hollow-core marble production is not limited to hydrophilic particles for the shell structures, like the sodium chloride microcrystals, and potentially could also be applied to many other organic and inorganic particles, both hydrophilic and hydrophobic. The present method for preparation of salt marbles could be further improved by using more powerful atomizers instead of conventional sprayers for aerosol generation from aqueous solution of higher viscosity and higher concentration of the gelling agent, which would further reduce the size of the hydrogel beads. We demonstrate that dried salt marbles have a superior rate of salt release compared to ordinary table salt upon dissolution in water. Therefore salt marbles hold promise for possible replacement of the conventional table salt. We show that despite the reduced salt content, salt marbles yield the same or higher rate of salt release upon dissolution which would create the same perception of “saltiness”. Hence such salt marbles could be used as a healthier alternative for reducing salt consumption. Salt marbles can potentially be used to reduce the salt intake by consumers, through reduced amounts of salt used in salt seasonings and the development of healthier snack products.
Acknowledgements
M.R acknowledges EPSRC Industrial CASE award and the financial support from Unilever R&D Vlaardingen for his PhD studies.Notes and references
- P. Aussillous and D. Quéré, Nature, 2001, 411, 924–927 CrossRef CAS PubMed.
- P. Aussillous and D. Quéré, Proc. R. Soc. A, 2006, 462, 973–999 CrossRef CAS.
- B. P. Binks and R. Murakami, Nat. Mater., 2006, 5, 865–869 CrossRef CAS PubMed.
- R. Murakami and A. Bismarck, Adv. Funct. Mater., 2010, 20, 732–737 CrossRef CAS.
- J. Tian, T. Arbatan, X. Li and W. Shen, Chem. Commun., 2010, 46, 4734–4736 RSC.
- J. M. Chin, M. R. Reithofer, T. T. Y. Tan, A. G. Menon, E. Y. Chen, C. A. Chow, A. T. S. Hor and J. Xu, Chem. Commun., 2013, 49, 493–495 RSC.
- M. Dandan and H. Y. Erbil, Langmuir, 2009, 25, 8362–8367 CrossRef CAS PubMed.
- K. Ueno, S. Hamasaki, E. J. Wanless, Y. Nakamura and S. Fujii, Langmuir, 2014, 30, 3051–3059 CrossRef CAS PubMed.
- S. Fujii and R. Murakami, KONA Powder Part. J., 2008, 26, 153–166 CrossRef CAS.
- C. Planchette, A. L. Biance and E. Lorenceau, Europhys. Lett., 2012, 97, 14003 CrossRef.
- D. Dupin, S. P. Armes and S. Fujii, J. Am. Chem. Soc., 2009, 131, 5386–5387 CrossRef CAS PubMed.
- D. Dupin, K. L. Thompson and S. P. Armes, Soft Matter, 2011, 7, 6797–6800 RSC.
- T. T. Y. Tan, A. Ahsan, M. R. Reithofer, S. W. Tay, S. Y. Tan, T. S. A. Hor, J. M. Chin, B. K. J. Chew and X. Wang, Langmuir, 2014, 30, 3448–3454 CrossRef CAS PubMed.
- L. Zhang, D. Cha and P. Wang, Adv. Mater., 2012, 24, 4756–4760 CrossRef CAS PubMed.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- R. S. Underhill, A. V. Jovanovic, S. R. Carino, M. Varshney, D. O. Shah, D. M. Dennis, T. E. Morey and R. S. Duran, Chem. Mater., 2002, 14, 4919–4925 CrossRef CAS.
- S. H. Im, U. Jeong and Y. Xia, Nat. Mater., 2005, 4, 671–675 CrossRef PubMed.
- P. M. Arnal, M. Comotti and F. Schüth, Angew. Chem., 2006, 118, 8404–8407 CrossRef.
- F. Caruso, Adv. Mater., 2001, 13, 11–22 CrossRef CAS.
- T. H. Nguyen, in Engineering, Monash University, 2009 Search PubMed.
- K. P. Hapgood, L. Farber and J. N. Michaels, Powder Technol., 2009, 188, 248–254 CrossRef CAS PubMed.
- Y. Yuan and T. R. Lee, in Surface Science Techniques, Springer, 2013, pp. 3–34 Search PubMed.
- S. Gaunt, S. J. Minter, E. E. Best and W. L. Nehmer, in WO Patent 2,007,085,609, W. I. P. Organization, US, 2013.
- S. J. Minter and S. Maude, in WO 2009/133409 A1, W. I. P. Organization, World Intellectual Property Organization, US, 2009.
- V. N. Paunov, O. J. Cayre, P. F. Noble, S. D. Stoyanov, K. P. Velikov and M. Golding, J. Colloid Interface Sci., 2007, 312, 381–389 CrossRef CAS PubMed.
- V. N. Paunov, Langmuir, 2003, 19, 7970 CrossRef CAS.
- E. L. Sharp, H. Al-Shehri, T. S. Horozov, S. D. Stoyanov and V. N. Paunov, RSC Adv., 2014, 4, 2205–2213 RSC.
- C. G. da Rosa, C. D. Borges, R. C. Zambiazi, M. R. Nunes, E. V. Benvenutti, S. R. d. Luz, R. F. D'Avila and J. K. Rutz, Ind. Crops Prod., 2013, 46, 138–146 CrossRef PubMed.
- D. Krishnaiah, R. Sarbatly, S. Ram Mohan Rao and R. Rajesh Nithyanandam, J. Appl. Sci., 2009, 9, 3062–3067 CrossRef CAS.
- A. Gharsallaoui, G. Roudaut, O. Chambin, A. Voilley and R. Saurel, Food Res. Int., 2007, 40, 1107–1121 CrossRef CAS PubMed.
- C. Ayala, E. V. Kuklina, J. Peralez, N. L. Keenan and D. R. Labarthe, MMWR. Morbidity and mortality weekly report, 2009, vol. 58, pp. 281–283 Search PubMed.
- A. Moshfegh, J. Goldman, J. Ahuja, D. Rhodes and R. Lacomb, What we eat in America: NHANES 2005-2006: Usual nutrient intakes from food and water compared to 1997, US Department of Agriculture, Agricultural Research Service, 2009 Search PubMed.
- L. A. Wyness, J. L. Butriss and S. A. Stanner, Publ. Health Nutr., 2012, 15, 254–261 CrossRef PubMed.
- F. J. He and G. A. MacGregor, The Cochrane database of systematic reviews, 2004, cd004937 Search PubMed.
- F. J. He, J. F. Li and G. A. MacGregor, BMJ, 2013, 346, 15 CrossRef PubMed.
- H. L. Meiselman and B. P. Halpern, Physiol. Behav., 1973, 11, 713–716 CrossRef CAS.
- Y.-q. Zhang, B.-j. Xie and X. Gan, Carbohydr. Polym., 2005, 60, 27–31 CrossRef CAS PubMed.
- R. M. D. Soares, A. M. F. Lima, R. V. B. Oliveira, A. T. N. Pires and V. Soldi, Polym. Degrad. Stab., 2005, 90, 449–454 CrossRef CAS PubMed.
- G. Carta and A. Jungbauer, in Protein Chromatography: Process Development and Scale-Up, John Wiley & Sons, Weinheim, Germany, 2010, p. 133 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |