
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Arginine–glycine–aspartic acid functional branched semi-interpenetrating hydrogels†
Richard A. Plenderleith a,
Christopher J. Patemanb,
Cornelia Rodenburgc,
John W. Haycockb,
Frederik Claeyssensb,
Chris Sammond and
Stephen Rimmer*a
a,
Christopher J. Patemanb,
Cornelia Rodenburgc,
John W. Haycockb,
Frederik Claeyssensb,
Chris Sammond and
Stephen Rimmer*a
aThe Polymer and Biomaterials Chemistry Laboratories, Department of Chemistry, University of Sheffield, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.rimmer@bradford.ac.uk
bDepartment of Materials and Engineering, Kroto Research Institute, University of Sheffield, North Campus, Broad Lane, Sheffield S3 7HQ, UK
cDepartment of Materials Science and Engineering, University of Sheffield, Mappin Street, Sheffield, S1 3JD, UK
dMaterials and Engineering Research Institute, Sheffield Hallam University, Howard Street, Sheffield, South Yorkshire, S1 1WN, UK
First published on 11th August 2015
Abstract
For the first time a series of functional hydrogels based on semi-interpenetrating networks with both branched and crosslinked polymer components have been prepared and we show the successful use of these materials as substrates for cell culture. The materials consist of highly branched poly(N-isopropyl acrylamide)s with peptide functionalised end groups in a continuous phase of crosslinked poly(vinyl pyrrolidone). Functionalisation of the end groups of the branched polymer component with the GRGDS peptide produces a hydrogel that supports cell adhesion and proliferation. The materials provide a new synthetic functional biomaterial that has many of the features of extracellular matrix, and as such can be used to support tissue regeneration and cell culture. This class of high water content hydrogel material has important advantages over other functional hydrogels in its synthesis and does not require post-processing modifications nor are functional-monomers, which change the polymerisation process, required. Thus, the systems are amenable to large scale and bespoke manufacturing using conventional moulding or additive manufacturing techniques. Processing using additive manufacturing is exemplified by producing tubes using microstereolithography.
Introduction
Interpenetrating polymer networks (IPNs) consist of either two crosslinked polymer networks with the chains of both components entwined (full IPN) or a crosslinked component with an entwined linear non-crosslinked component (semi-IPN) (S-IPN).1 They are finding widespread applications in biotechnology and medicine,2 with more recent applications as scaffolds for adherent cell growth.3 Single component hydrogels have been widely functionalised so that they can act as substrates for cell culture; both bio-functionality and mechanical properties are important parameters.4–8The extracellular matrix in which eukaryotic cells form tissues is a functional multi-component hydrogel. Therefore, it seems reasonable to propose that functional hydrogels can be optimised to provide useful scaffolds for controlling cells as they form regenerated tissues. Formulation of functional hydrogels can provide materials with mechanical properties, swelling and functionality that are optimised for specific cell types and tissues.5–7 However, it is also necessary to devise fabrication strategies that can generate 3-dimensional (3D) constructs that will provide also the spatial organisation for the developing tissues.9 In this respect additive manufacturing techniques are emerging fields with the potential to provide devices with bespoke shapes. However, there are few examples of hydrogel materials that can be fabricated using these techniques and here we propose that a class of functional semi-interpenetrating networks can provide functional hydrogel scaffolds, which can also be fabricated into 3D objects. Additive manufacturing and micro-engineering have also become increasingly important in the manufacture of cell-scaffold constructs and hydrogels can be key scaffold materials in this area.10,11 Controlling the mechanical and swelling properties, together with the presentation of biochemical functionality is important when attempting to influence cell function. In this respect, IPNs offer a way to combine both mechanical and biochemical properties independently by using one of the components to provide the latter, while the other component provides the former. For example, when attempting to provide stem cell niches, Tong and Yang showed that functional IPNs could be a useful way of controlling mechanical properties, using one of the IPN components, independently from biochemical functionality, provided by the second component polymer.12 Also, independent control of stiffness and biochemical functionality was recently achieved using IPNs prepared from reconstituted extracellular matrix components and alginate.13 Recent other developments in the modification of hydrogels for use as biomaterials include preparing conetwork architectures14,15 and the addition of carbon nanotubes.16 However, few attempts have been made to provide IPNs that could be processed by additive manufacturing techniques.
IPNs are distinguishable from blends, as the two polymeric components are mixed at the nanometre length scale.17 Synthesis of full IPNs can involve the synthesis of the first network followed by subsequent swelling with the monomer of the second network, which is then polymerised. This process can be restrictive in manufacturing processes because the swelling and secondary polymerisation can change the dimensions of the device or coating. The problem is most acute in additive manufacturing strategies were swelling and polymerisation with a second monomer feed would lead to gross deformations of the previous well-defined dimensions of the object. In some systems both monomer sets can be mixed before polymerisation. Here, mechanisms that do not interfere with each other are used. S-IPN synthesis is much easier and involves simply adding a preformed linear polymer to the network-forming monomer mixture. S-IPNs are suitable for applications that do not require swelling but they are not suitable as swollen hydrogels because the linear polymer can often be extracted and leached from the device. An alternative that has not been explored previously is to produce a S-IPN with a branched soluble component (BS-IPN). Provided that the chains are mixed at the nanoscale the reptation of an entrapped branched polymer within a swollen network is severely limited. We consider that a substantial fraction of the branched component will not diffuse out of the network, even in the swollen state. Here we report on the first example of BS-IPN hydrogels, describing their use as substrates for supporting the adhesion, spreading and migration of primary human dermal fibroblast cells (HDF) cultured in vitro.
A schematic diagram of the entrapment of a branched polymer within a hydrogel network is shown in Fig. 1. The figure shows how chain reptation is severely limited because this would involve the adoption of unfavorable chain conformations in order to minimize the frequency of clashes between branch points and crosslinks. The system reported here is poly(N-vinyl pyrrolidone-co-diethylene glycol diacrylate) (X-PVP) with a highly-branched poly(N-isopropyl acrylamide) (HB-PNIPAM) entrapped within the network. It is the first example of an S-IPN in which a substantial fraction of a soluble polymer cannot be extracted.
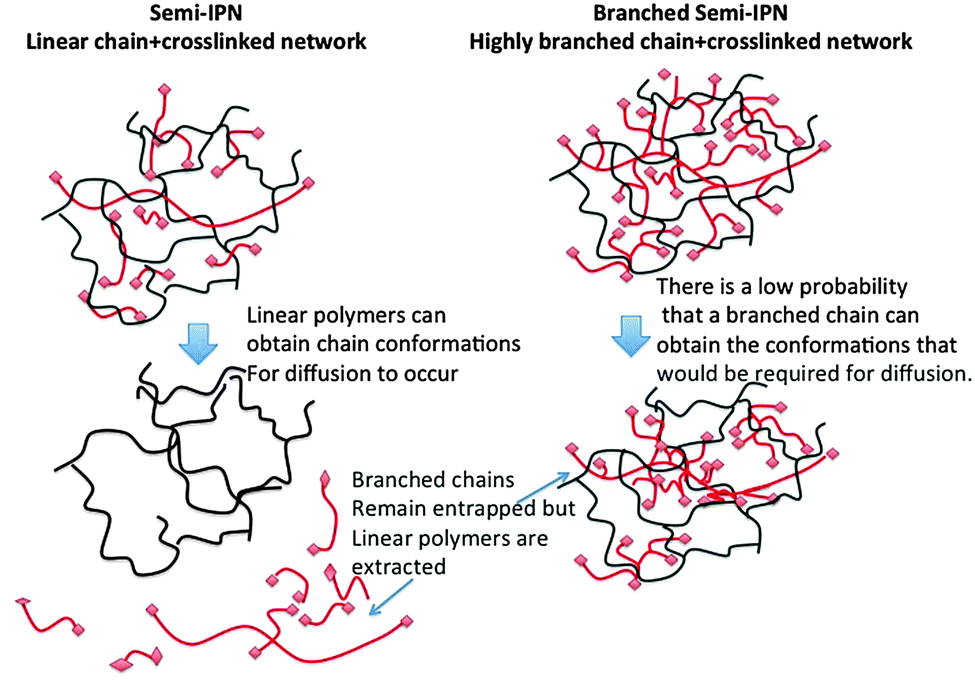 | ||
| Fig. 1 Schematic diagram of a S-IPNs and a BS-IPN composed of linear or highly branched polymer within a crosslinked matrix respectively. | ||
Results and discussion
HB-PNIPAM with either COOH (HB-PNIPAM-COOH) or glycine–arginine–glycine–aspartic acid–serine peptide (HB-PNIPAM-GRGDS) end groups were prepared using our previously reported technique.18,19 These polymers were added to mixtures of N-vinyl pyrrolidone (NVP) and diethylene glycol diacrylate (DEGDA) before photopolymerisation. The fractions of branched polymer (HB-PNIPAM-COOH) that remained in the networks after extraction with 2-propanol, a good solvent for PNIPAM, were obtained using FTIR spectroscopy and the results are shown in Fig. 2. Fig. 2a and b show the data obtained using 10 and 40 wt% (relative to NVP), respectively, of DEGDA in the monomer feed. Both sets of data clearly show that, unlike in conventional S-IPNs prepared with linear polymer, large fractions of the HB-PNIPAM remained within the networks after multiple extraction cycles.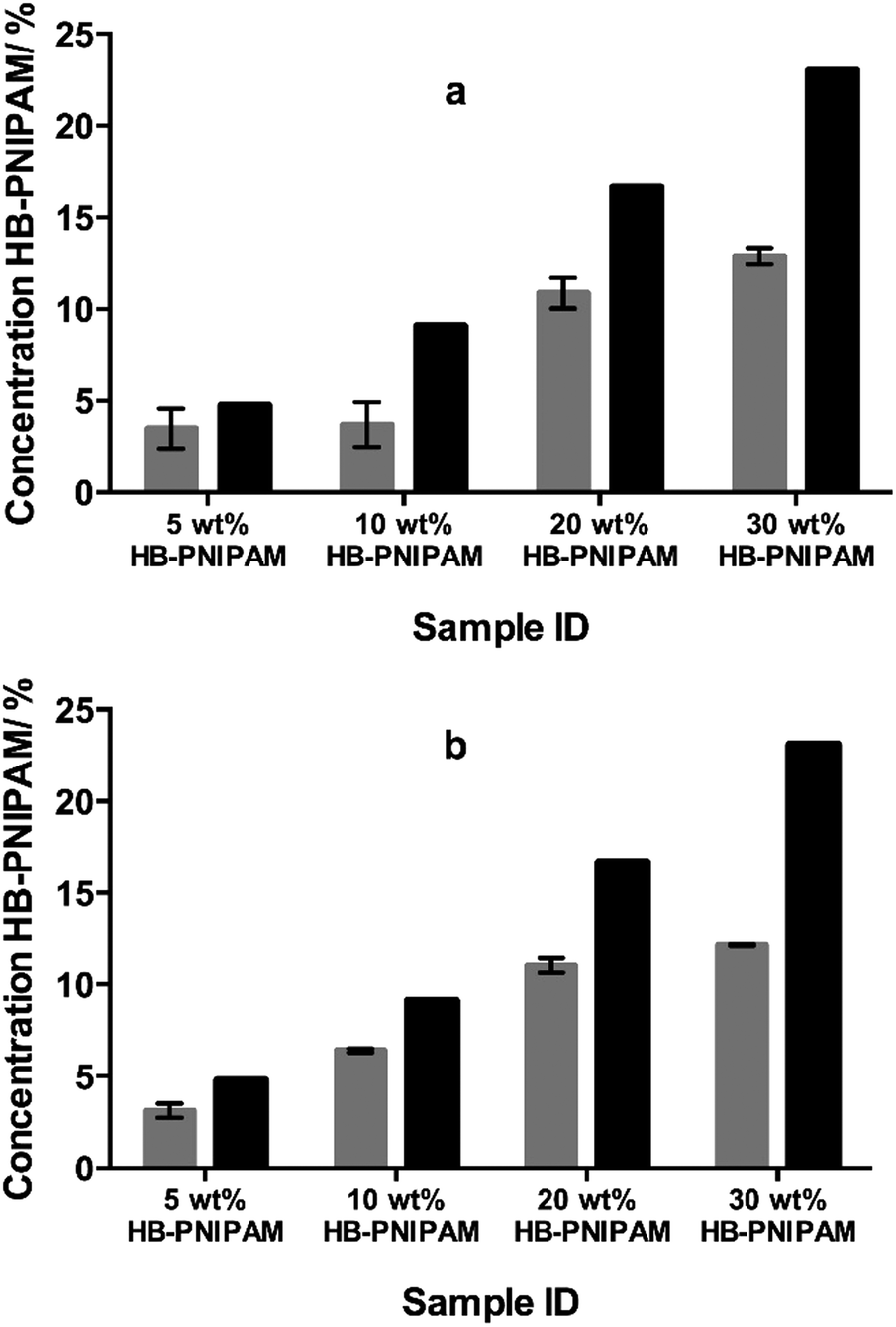 | ||
| Fig. 2 Composition (determined by FTIR spectroscopy) of the networks prior to and after Soxhlet extraction with 2-propanol: (a) 10 wt% DEGDA in the monomer feed and (b) 40 wt% DEGDA in the monomer feed. The grey columns show the percentage of HB-PNIPAM in the BS-IPNs after extraction and the black columns show the percentage of HB-PNIPAM in the feeds. The spectral region used for analysis was 1480–1760 cm−1. | ||
The data support the hypothesis that the branched architecture of the polymers inhibits diffusion of the polymer through the crosslinked network because for such polymers to pass through the network it would be necessary for the chain to take up a series of highly unfavourable conformations. The other results reported here each used the formulation containing 10 wt% of DEGDA.
Much interest in PNIPAM is associated with the thermal behaviour of the material in aqueous solution. The linear polymer passes through a coil-to-globule transition at a lower critical solution temperature (Tc–g). The Tc–g results in desolvation and phase separation and this property is the focus of many applications in health, biological and separation technology.20–27 The branched variants of PNIPAM introduced here show the same temperature dependent solution behaviour so that the HB-PNIPAM-COOH studied here displays a cloud point of 26 °C. In a crosslinked PNIPAM network the Tc–g is manifested as a step change in deswelling but the BS-IPNs produced in this study did not show this behaviour (see Fig. S1 to S4, ESI†). The equilibrium water contents (by weight, in deionised water) were 85 to 95 wt% in each sample with no significant decrease on raising the temperature. In most PNIPAM swollen hydrogels the Tc–g is an endothermic desolvation transition that can be observed by differential scanning calorimetry (DSC). However, the Tc–g was not observed in the BS-IPNs. Fig. 3a shows examples of the typical featureless thermograms derived from BS-IPNs. In contrast Fig. 3a also includes a thermogram showing an endothermic peak around the Tc–g from a conventional crosslinked PNIPAM hydrogel. This appeared to be the first observation of an apparent suppression of the Tc–g phase transition. In order to examine whether the absence of the Tc–g was a feature of the BS-IPN structure other possible combinations of PVP and PNIPAM were examined. Fig. 3b shows thermograms from conventional S-IPNs composed of either crosslinked PVP (X-PVP) with linear PNIPAM or crosslinked PNIPAM (X-PNIPAM) with linear PVP. Note that the amount of PNIPAM in the linear PNIPAM/X-PVP S-IPN (10 wt%) was chosen to be equivalent to the amount of HB-PNIPAM in the extracted HB-PNIPAM/X-PVP BS-IPN with 20% HB-PNIPAM in the feed. This material provided some of the data in Fig. 3a.
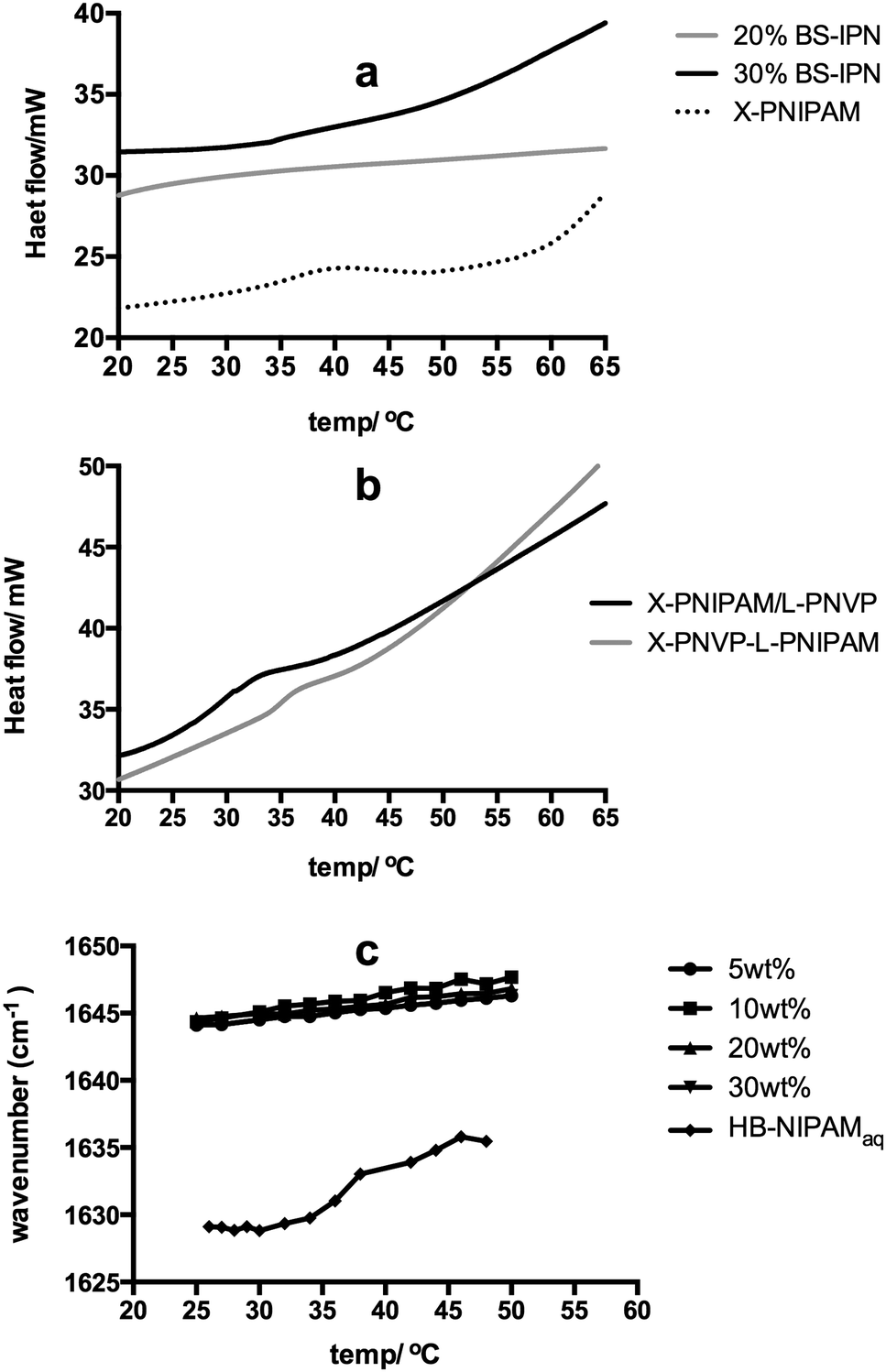 | ||
| Fig. 3 (a) DSC data showing the desolvation event in a crosslinked PNIPAM hydrogel network and the absence of a thermal event in the BS-IPN materials containing 20 or 30 wt% HB-PNIPAM/X-PVP. (b) DSC data showing the endothermic peak associated with the desolvation of PNIPAM in 10 wt% linear PNIPAM/X-NVP S-IPN, 10 wt% linear PVP/X-PNIPAM S-IPN. (c) Vibrational frequency of the amide carbonyl in HB-PNIPAM-COOH of BS-IPN systems with increasing concentration of HB-PNIPAM (5, 10, 20 and 30 wt% HB-PNIPAM-COOH in a water/PVP matrix) and of HB-PNIPAM in water vs. temperature. | ||
These data show that both of the conventional S-IPNs display endothermic events that can be assigned to Tc–g behaviour. Therefore, the data indicated that the absence of an endotherm associated with a Tc–g appears to be a feature of the BS-IPN structure. This was corroborated by swelling data supplied in Fig. S1–S4 (ESI†), which showed no temperature dependant changes in swelling. These results were in contrast to our previous work on hydrogel brushes, where (1,2-propandiol methacrylate-co-ethanediol dimethacrylate) networks with grafts of PNIPAM displayed a clear Tc–g by DSC between 35–40 °C, for a range of polymer compositions.28 The absence of the HB-PNIPAM Tc–g within the PVP hydrogel matrix was also shown by FTIR spectroscopy. The study of the coil-to-globule transitions using FTIR provides information relating the nature of interactions between the solvating medium and the polymer chains by monitoring changes in the peak centres of the bands associated with functional groups such as amide I (∼1630–1640 cm−1), amide II (∼1560 cm−1) and ν(CH3) (∼2930 cm−1).29–33 Typically at Tc–g bands associated with these functional groups exhibit a sharp shift as the local environment changes due to desolvation of polymer chains during the conformational change.
Plotting the measured peak centre as a function of T provides a means of determining the Tc–g. Fig. 3c shows the plot of the amide I peak centre as a function of T for a range of HB-PNIPAM-COOH within the BS-IPN systems with increasing concentration of HB-PNIPAM-COOH (5, 10, 20 and 30 wt% HB-PNIPAM-COOH in a D2O/PVP matrix) and of a plot of the amide I peak from HB-PNIPAM-COOH in water.
The aqueous solution of HB-PNIPAM-COOH shows the profile expected when the polymer undergoes a coil to globule transition: a sharp shift in peak centre (from ∼1630 to ∼1635 cm−1 between 32 and 34 °C). The absolute values of these peak centres are somewhat influenced by the convolution of the amide I peak with the water bending mode (δ(OH)) in this system. Convolution lowered the measured wavenumber position but the position of the δ(OH) has been shown to be independent of temperature.29 Therefore, any change in peak position can be assumed to be derived from changes to the amide I peak of the PNIPAM segments.
Conversely when this polymer was incorporated into a set of BS-IPN materials there was no sharp shift in the measured amide I peak centre (Fig. S5 to S8, ESI†) as the temperature was increased. The peak centre did exhibit a small systematic shift to a higher wavenumber as a function of T (Fig. S9 to S16, ESI†). This is most likely due to a reduction in the hydrogen bonding strength between the D2O and the NIPAM amide I at elevated temperatures, as observed by Maeda et al.33 The absence of an observable Tc–g in both swelling, DSC and FTIR experiments could either be due to an elevation of Tc–g, such that the measurements never reach the required temperature, or a decrease in Tc–g to below the range recorded during each measurement. Examination of the infrared spectra supports the latter hypothesis because the amide I band occurs ∼1646 cm−1 at all temperatures rather than the lower values ∼1635 cm−1 observed for PNIPAM homopolymer in D2O as described by Maeda et al.33 and in the fully dissolved HB-PNIPAM-COOH material shown in Fig. 3c. Individual amide I peak plots of BS-IPN systems, with 10 and 40% crosslinker, can be found in ESI† (Fig. S5–S13). We propose that HB-PNIPAM could have been desolvated at temperatures at which it normally would be solvated in homogeneous solution if the matrix network effectively competed for water. The crosslinked network structure would limit the amount of water available and it seems reasonable that this limited water would solvate the most hydrophilic chain segments in preference to the less hydrophilic segments. This would lead to a deswollen HB-PNIPAM conformation below the solution Tc–g. That is we propose that solvation of the amphiphilic HB-PNIPAM can be limited when it is blended with another more hydrophilic polymer network.
As far as we are aware this phenomenon has not been reported previously. This polymer mediated desolvation minimised the Tc–g behaviour of these polymers but they could provide good surfaces for cell adhesion and the addition of a functional branched polymer to hydrogels in this way allows for the modification, without a significant change in the EWC, of otherwise non-cell adhesive hydrogels.
Changes in surface topography are often correlated with cell behaviour34–36 and we have shown that topography derived from phase separation in amphiphilic conetworks had a large effect on the adhesion and spreading of fibroblasts in 2D culture.15,37 To examine possible phase separation, low voltage SEM (LV-SEM) of the materials was carried out. The use of LV-SEM does not require the coating of materials with a conductive metal layer prior to imaging. Hence, phase distributions are not obscured by the metal coating. Fig. 4 shows the polymer surfaces. The X-PVP hydrogel prepared in the absence of HB-PNIPAM provided a smooth featureless surface whereas a vein-like surface morphology was observed when 10% of HB-PNIPAM was added to the X-PVP. In comparison with previous work on phase imaging in polymers by LV-SEM38,39 the separation of these "veins" can be determined from auto-correlation functions (ACF), which is provided in the ESI† (Fig. S14). From line profiles across the ACF in perpendicular directions it was found that addition of 10% of HB-PNIPAM to X-PVP resulted in linear structures that extended on average to ∼2 μm and were separated by an average distance of 139 ± 17 nm. The latter was identical to the radii of the HB-PNIPAM particles in solution (136 ± 10 nm) as measured by DLS. Increasing the amount of added PNIPAM to 20% lead to a reduction of the strong directionality: the separation distance was less well defined and increased to between 400–500 nm, while the average length was also less well defined and only ∼1 μm.
 | ||
Fig. 4 High resolution electron micrographs of polymer surfaces ×3500 (left column), ×35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (right column).The images show X-PVP hydrogel (top row), 10% HB-PNIPAM/X-PVP S-IPN (central row), 20% HB-PNIPAM/X-PVP S-IPN (bottom row). Left column shows SEM images at ×3500 magnification with 10 μm scale bars, the right column shows SEM images at ×35000 magnification with 1 μm scale bars. 000 (right column).The images show X-PVP hydrogel (top row), 10% HB-PNIPAM/X-PVP S-IPN (central row), 20% HB-PNIPAM/X-PVP S-IPN (bottom row). Left column shows SEM images at ×3500 magnification with 10 μm scale bars, the right column shows SEM images at ×35000 magnification with 1 μm scale bars. | ||
We consider the incorporation of branched polymers into crosslinked polymers can be a useful method for adding other functionalities. In order to examine this, HB-PNIPAM with a cell adhesive peptide, GRGDS (HB-PNIPAM-GRGDS), was introduced into X-PVP. The GRGDS peptide is known to bind to the α5β1 integrin and many polymers containing this motif have been reported to enhance the adhesion and proliferation of various cell types on a wide range of substrates.40 Most non-charged hydrogels, such as PVP, are highly resistant to cell adhesion.41 However, hydrogels have advantages as scaffolds in tissue engineering, notably: (1) nutrients and waste products can freely diffuse; (2) they are resistant to general biofouling and (3) their physical properties can be tuned by controlling swelling and crosslinking.
Three materials were prepared: (1) X-PVP and BS-IPNs composed of either (2) X-PVP/HB-PNIPAM-COOH or (3) X-PVP/HB-PNIPAM-GRGDS. X-PVP/HB-PNIPAM-COOH and X-PVP/HB-PNIPAM-GRGDS contained 5 wt% of the branched material which X-PVP/HB-PNIPAM-GRGDS provided 19.5 μmol g−1 of GRGDS. Fig. 5 shows images of primary human dermal fibroblast cells cultured on each of the materials for 24 hours compared to the same cells cultured on tissue culture plastic. Clear and important differences in the behaviour of the cells on these materials arose. HDFs were labelled for intracellular actin filaments using phalloidin–TRITC (red) and for nuclei using 4′,6-diamidino-2-phenylindole (DAPI, blue). Cells were cultured on the materials for 24 hours before being fixed and labelled. HDFs were observed to adhere equally well to the TCP positive control and X-PVP/HB-PNIPAM-GRGDS, with a similarly high cell density, reaching near confluence after 24 hours and also with similar morphologies. The X-PVP/HB-PNIPAM-GRGDS material was able to support firm adhesion, attachment and spreading of the fibroblasts, with organised actin filaments clearly visible.
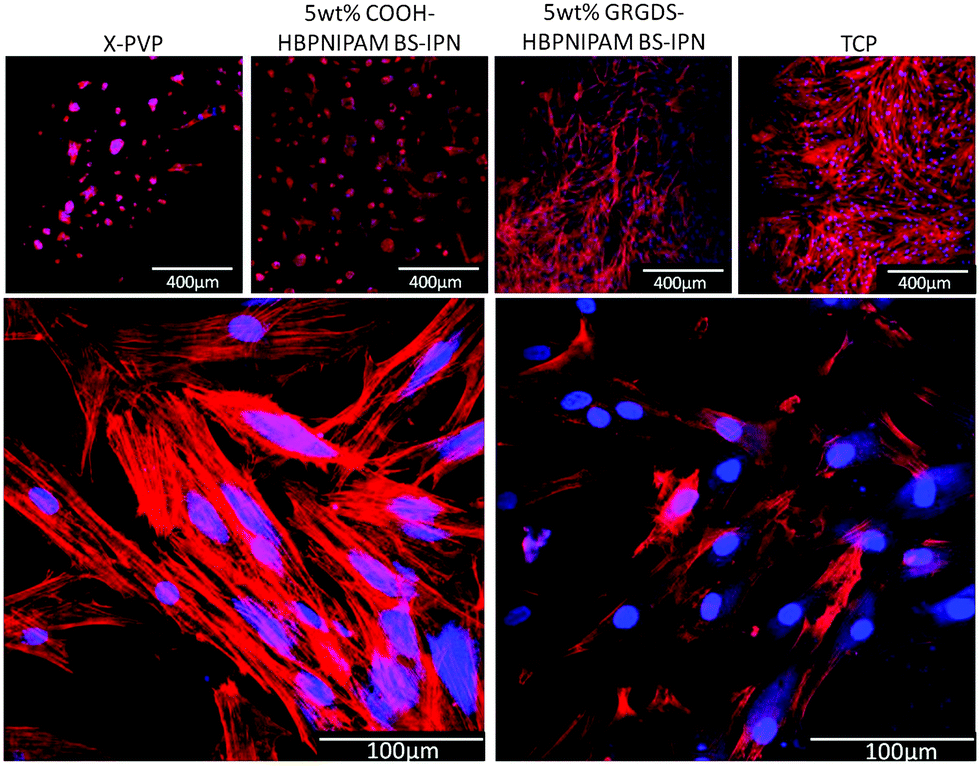 | ||
| Fig. 5 (Top) Confocal micrographs at ×10 magnification (400 μm scale bars) of normal human dermal fibroblasts grown on PVP control (left), 5 wt% HB-PNIPAM-COOH/X-PVP BS-IPN (central left), 5 wt% HB-PNIPAM-GRGDS/X-PVP BS-IPN (central right), TCP (right). (Bottom) Micrographs at ×40 magnification (100 μm scale bars) of normal human dermal fibroblasts on TCP (left) and 5 wt% HB-PNIPAM-GRGDS/X-PVP BS-IPN (right). Actin filaments labelled with phalloidin–TRITC (red), nuclear staining with DAPI (blue). | ||
Actin filaments were observed to run orthogonally within the majority of fibroblast cells showing cytoskeletal organisation running from nuclei to the cell membrane. The convergence of actin filaments was evident when in contact with the cell membrane and at high magnification (Fig. 5) the formation of several focal adhesion contacts were observed for each cell. These were co-located with convergent filaments, indicating firm adhesion. In contrast, X-PVP/HB-PNIPAM-COOH BS-IPN and the X-PVP negative control supported a relatively low cell density, with only a few fibroblast cells observed to attach to these substrates. The cells also had rounded morphologies, which were atypical and predominantly spherical or ellipsoidal in shape, with very few focal contact adhesions formed. The presence of filamentous organised actin structures was noted by positive labelling with phalloidin–TRITC, but very little organisation was observed as indicated by the lack of orthogonal filament formation or filament convergence near the membrane. This observation was consistent with the lack of focal contact adhesion points. Thus, it could be concluded from these data that the non-cell adhesive PVP hydrogel could be modified with HB-PNIPAM-GRGDS to produce a material that supported cell adhesion and proliferation.
Next the effect of varying the amount of HB-PNIPAM-GRGDS was investigated. Micrographs of HDFs cultured on BS-IPNs containing 0% to 2.5%, 5% and 10% HB-PNIPAM-GRGDS (equivalent to 0, 9.75, 19.5 and 39 μmol g−1 of GRGDS) are shown in Fig. 6. It was found that as the GRGDS peptide content was increased the number of fibroblast cells attached also increased. Fibroblast cell density was confluent when the cells were cultured on the 10% HB-PNIPAM-GRGDS material. Labeling of vinculin using anti-vinculin IgG (green) was undertaken and this revealed a relatively high level of expression of this protein in fibroblasts when cultured on 2.5%, 5% and 10% HB-PNIPIM-GRGDS BS-IPNs. Substantial co-localisation was observed with actin filaments on the 2.5% and 5% GRGDS materials (red + green = yellow), with particular co-localisation at the point of focal adhesion contact formation. The cells were observed to be most numerous but less well adhered on the 10% GRGDS material. Thus, on the 10% GRGDS HB-PNIPAM-GRGDS BS-IPN the fibroblasts were clearly adhered and growing but as a consequence of having less space per individual fibroblast this impeded the ability of each to form mature focal adhesion contacts. These data were corroborated by MTT cell viability data (see Fig. 7), which showed a significantly (p < 0.001) higher overall viability on the peptide-functionalised polymers but there was no significant differences between the IPNs with different loadings of the GRGDS loaded polymer.
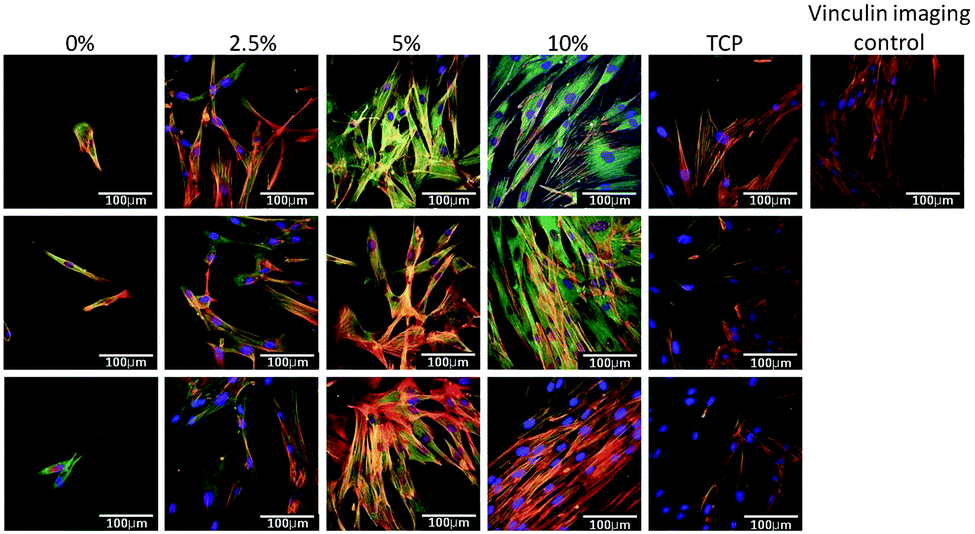 | ||
| Fig. 6 Confocal micrograph images at ×40 magnification (100 μm scale bars) of human dermal fibroblasts on BS-IPNs containing 0, 2.5, 5, and 10% HB-PNIPAM-GRGDS. Actin filaments labelled with phalloidin–TRITC (red), nuclear staining with DAPI (blue) and vinculin labelled green. In the control sample the primary vinculin antibody has been omitted from the staining procedure to negate the possibility of non-specific binding of the fluorophore. | ||
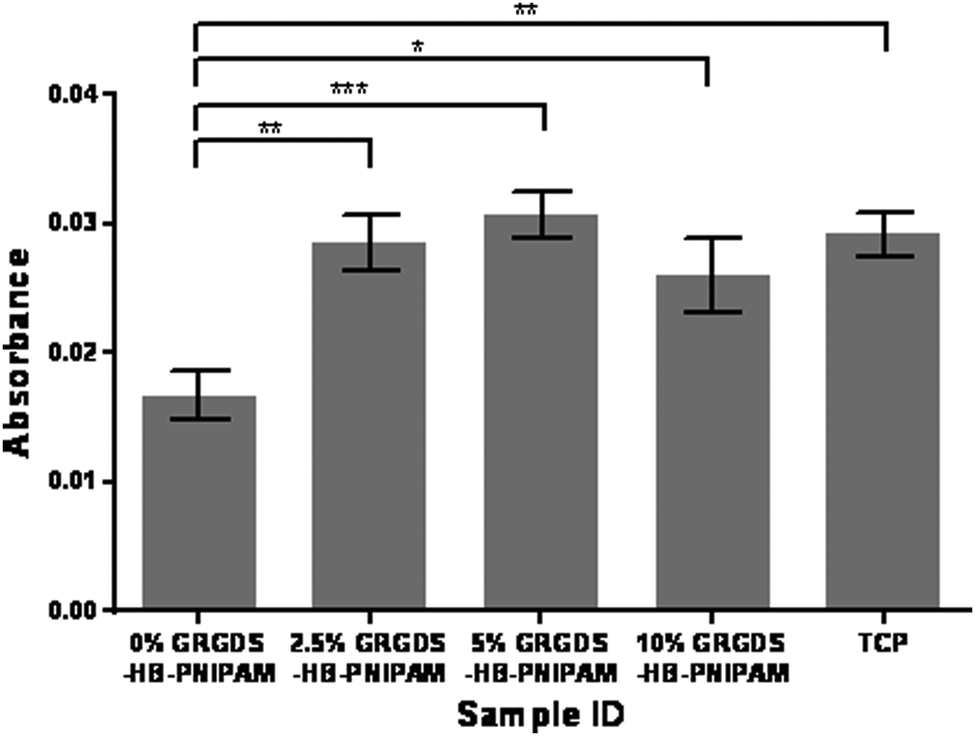 | ||
| Fig. 7 Metabolic viability data (MTT, absorbance at 410 nm) for human dermal fibroblast cells on BS-IPNs containing 0, 2.5, 5 and 10 wt% of HB-PNIPAM-GRGDS. | ||
Degradable versions of these hydrogel materials could be useful in wound healing applications and in this context cell migration is an important parameter. Data on cell migration are also of interest in terms of assessing the fundamental properties of cell supporting materials. Three sets of data can be provided from these experiments: (1) the accumulated distance; (2) the Euclidean distance and (3) the apparent speed at which cells migrate. The accumulated distance, calculated by the sum of the data from the fibroblast cell adhesion experiments, are consistent with the DSC and FTIR data for the HB-PNIPAM-GRGDS BS-IPN, that is the formation of the BS-IPN with a permanently swollen hydrophilic crosslinked component drove the HB-PNIPAM through the Tc–g at lower temperatures than the free solution Tc–g. Therefore, the cells were not released as the temperature was lowered; it was not possible to decrease the temperature sufficiently to produce the swelling required at the Tc–g to release the cells.
The distances moved by the cell in each image, the Euclidean distance also known as displacement is a measure of directionality of cell movement. It measures the distance between the start and end point of the cell over the observed time. The apparent speed is the accumulated distance divided by time.
HDFs were tracked on hydrogel BS-IPN surfaces with a range of concentrations of HB-PNIPAM-GRGDS and the results are shown in Fig. 8. The Euclidean distance travelled by the cells showed a dose dependent response to the concentration of HB-PNIPAM within the BS-IPN sample, while cell speed and accumulated distance showed a significant increase in all HB-PNIPAM-GRGDS containing samples compared to the control sample (0% HB-PNIPAM).
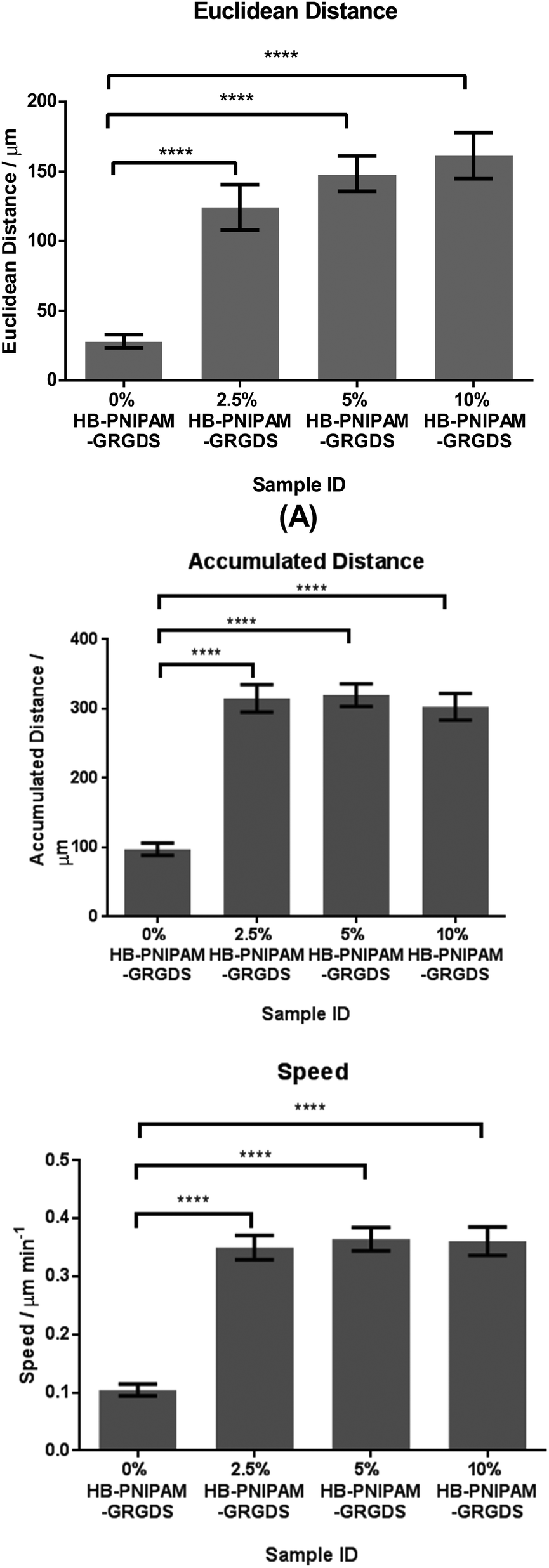 | ||
| Fig. 8 Euclidean distance, accumulated distance and speed of human dermal fibroblasts tracked at 15 minute intervals over 15 hours. | ||
Finally, we examined the feasibility of producing structured hydrogels with an in-house projection stereolithography set-up and objects produced from this additive manufacturing method are shown in Fig. 9. Both the X-PVP and PNIPAM-PVP BS-IPN mixtures provided tube structures with a write speed of 0.03 mm s−1 and thus structures of 5 mm length were produced in 167 seconds. Fabricated tubes displayed good structural integrity with ∼250 μm wall thickness and a variety of physical features could be reliably reproduced, indicating the usability of these photocurable resins for additive manufacturing applications.
 | ||
| Fig. 9 Images of tubes created by microstereolithography Left shows X-PVP hydrogel, right shows X-PVP BS-IPN with 5% HB-GRGDS PNIPAM. | ||
Discussion
Although, S-IPNs containing branched polymers as the soluble component are contained within the IUPAC definition few examples appear to have been reported and as far as we are aware there are no examples of hydrogel BS-IPNs. This is surprising and growing interest in the use of IPNs as medical materials prompted us to examine the feasibility of using the branched structure to prevent extraction of the soluble component and to allow its use as an additive to provide bio-functionality. Highly branched polymers in this context should be difficult to extract from a network. The ability of these polymers to be blended into liquid pre-polymerised monomer mixtures can be useful in reactive processing fabrication methods, such as additive manufacturing processes. Alternatively, linear polymers added to a swollen S-IPN would be easily removed in swelling solvents and these would inevitably be leached from any materials used in vivo. The experiments described confirmed this concept so that the highly branched architecture prevented the extraction in good solvents for both the highly branched and network phases.The concept of a S-IPN hydrogel with a highly branched functional polymer as the soluble component that can provide bio-functionality to an otherwise inert hydrogel is general. The systems can provide a highly flexible way of formulating functional biomaterials as an alternative to functionalisation with either functional monomers or by post-polymerisation modification. Post-polymerisation modifications are often unsatisfactory for producing functional hydrogels because of rapid surface reorganisations that occur after modification. The use of polymer additives is well practiced in a number of technologies and provides the advantage that the safety of the additive can be proven independently of any formulation or modification steps. Our proposition is firstly, that the use of a highly branched polymer with functional end groups can provide processes with fewer toxicological problems than using new functional monomers. Monomers are often toxic before polymerisation and most processes require extensive experimentation to prove that the residues of these new monomers can induce toxic reactions. The other process advantage of this approach is that the functionalisation step (i.e. the use of a highly branched polymer as a functional additive) does not affect the polymerisation by which the device is formed. This provides flexibility in the formulation process, which can focus on optimisation independently of other process variables. With the advantages of this strategy in mind, the potential of using these materials in additive manufacturing strategies was shown by forming a number of 3-dimensional objects using microstereolithography.
In this first example of a BS-IPN hydrogel containing a highly branched bio-functional polymer we focused on a hydrogel matrix composed of polymer with a long record of safe use as a medical material, PVP. This was modified by inclusion of a HB-PNIPAM with the GRGDS peptide at the chain ends with the aim of improving the cell adhesive properties of the PVP hydrogel. The material did support human dermal fibroblasts, and we were able to show that there was a significant benefit of adding the peptide compared to a modification with a similar carboxylic acid functional variant. Also, it was shown that the behavior of the cells was dependant on the amount of GRGDS-functional polymer added. The presence of GRGDS in these materials clearly had a beneficial effect on the ability of the cells to from focal adhesions. A number of studies have indicated the importance of the surface concentration of RGD ligands42 and other studies have pointed to the effects substrate stiffness.43 In this respect recently it was shown that both modulus and RGD spacing (concentration) both affected stem cell differentiation, although the effects of swelling were not considered.44,45 Since the degree of swelling and crosslink density define the modulus of a swollen polymer network it is predicted that there are no significant changes in modulus within these series of constant water content and constant crosslink density materials. In that respect we do not consider the mechanical properties of this set of materials further in this work. Although recent published data shows some indications of possible effects of mechanical properties on the behaviour of fibroblasts cultured on polymer materials46 the effects of swelling are rarely considered. In these multi-component materials careful consideration of the interactions of the key variables are required to establish the structure–property–performance relationships required. Therefore, in our continuing studies a larger set of materials will be presented that will enable elucidation of the effects of mechanical properties, swelling, surface properties and biochemical functionalities in a substantial and unified set of experiments.
The effects of the spacing/density of RGD based ligands on cell behaviour is well documented47–52 and clustering of RGD ligands is known to have a major effect on behaviour of cells in the natural systems.53 With this in mind dendrimers with RGDS functionality have recently been incorporated into hyaluronic acid hydrogels and these materials were used to control mesenchymal stem cells.54 The materials reported on here provide opportunities to provide controlled clustering of ligands to provided higher local ligand densities. However, we are unfortunately unable at this time to provide the quantitative maps of ligand density that would be required to fully characterise these materials and relate the ligand distributions to performance. Not withstanding this comment the technique described here allows us to produce various materials containing clustered ligands, were the clustering is defined by the structure of the incorporated branched polymer, and we have shown that adding these functional polymers directly effects cell adhesion, spreading and migration. As with mechanical characterisation, swelling and assessment of performance substantial further work is underway that involves producing much larger sets of materials and studying multiple cell types.
Many reports have described that PNIPAM above the Tc–g supports cell adhesion but cooling to below Tc–g releases the cells.55 In solution, both HB-PNIPAM-GRGDS and HB-PNIPAM-COOH have Tc–gs below 37 °C. However, as we have shown the interpenetrated structure of a BS-IPN affects the coil-to-globule transition. Also, in our previous work we showed that PNIPAM grafted to a hydrogel supported the adhesion of chondrocyte cells at 37 °C without the requirement of a cell adhesive, integrin binding, peptide.28 In those materials the Tc–g was observed and cell adhesion was possible only above this temperature. As the materials were cooled the cells were released. However, cell spreading on the BS-IPN containing HB-PNIPAM-COOH did not occur, whereas the fibroblasts adhered and were well spread on the BS-IPNs containing HB-PNIPAM-GRGDS. The data from the fibroblast cell adhesion experiments are consistent with the DSC and FTIR data for the HB-PNIPAM-GRGDS BS-IPN, that is the formation of the BS-IPN with a permanently swollen hydrophilic crosslinked component drove the HB-PNIPAM through the Tc–g at lower temperatures than the free solution Tc–g. Therefore, the cells were not released as the temperature was lowered; it was not possible to decrease the temperature sufficiently to produce the swelling required at the Tc–g to release the cells.
Cell migration on and in synthetic biomaterials can be important in a number of wound healing applications and treatments requiring long term stability of implants. Therefore, the migration of these cells on the BS-IPNs was also studied. The large, non-significant disparity between the accumulated and Euclidean distance can be explained by the characteristic of a ‘random walk’ behavior of adherent cells cycling through the process of adhesion, spreading, migration and division, and further suggests no directionality of the fibroblasts on these materials. The significant difference in speed, accumulated distance and Euclidean distance of fibroblasts grown on 0% HB-PNIPAM-GRGDS versus 2.5, 5 and 10% was particularly noteworthy. The relationship between fibroblast response and inter-peptide distance in the form of RGD when covalently grafted to the surface of an otherwise poorly adhesive substrate is well described.56 A spacing of 440 nm is reported as the minimum permissive distance for maximum fibroblast attachment and spreading, whereas a spacing of 140 nm leads to focal contact formation and stress fibre formation. These two conditions equate to 12000 and 120000 ligands per spread cell, respectively. As focal contact and stress fibre formation is necessary for migration, we suggest that the 2.5% GRGDS condition described herein equates to an inter-peptide space of 140 nm or less at the HB-PNIPAM surface (i.e. 120000 ligands per cell or higher). Interestingly, higher HB-PNIPAM-GRGDS percentages did not lead to a significant increase in migration speed or distance and this observation suggested complete binding (or saturation) of available α5β1 integrin receptors.
Conclusions
We show here for the first time that branched polymers can be incorporated into polymer hydrogel networks. In the swollen state substantial fractions of these polymers are entrapped within the polymer matrix and do not leach into the surrounding medium. Incorporation of HB-PNIPAM into the network suppressed the thermally induced coil-to-globule transition in water, which is usually an important feature of polymers composed of the NIPAM repeat unit. Inclusion of a peptide-functional version of this branched polymer was shown to alter the cell-adhesive nature of a non-cell-adhesive crosslinked hydrogel, producing high water content hydrogels that could support the adhesion and proliferation of human fibroblasts.Experimental
Synthesis of 4-vinylbenzyl pyrrolecarbodithioate18
Pyrrole (100 mL) was distilled. NaH (11.98 g, 0.497 mol) was added to a 3 neck flask under N2, with stirring, as a suspension in DMF (240 mL) and the flask allowed to purge with N2. Pyrrole (20 g, 0.298 mol) in DMF (40 mL) was added dropwise to the reaction over 30 min at 0 °C. The solution was stirred at room temperature for 30 minutes. Carbon disulphide (22 g, 0.298 mol) in DMF (40 mL) was added dropwise over 10 minutes at 0 °C. The solution was stirred at room temperature for 30 minutes. 4-Vinylbenzyl chloride (45.48 g, 0.289 mol) in DMF (40 ml) was added dropwise over 20 minutes at 0 °C with stirring. The solution was stirred at room temperature for 30 minutes. Deionised water (400 ml) was added to the reaction mixture and the product was extracted with diethyl ether (4 × 500 ml). The organic layers were combined, then dried with magnesium sulfate and evaporated yielding a dark coloured liquid, which was purified by silica column chromatography in hexane to yield 52 g of a yellow solid.Actual results: carbon 65.3%, hydrogen 5.1%, nitrogen 5.3%, sulphur 22.41%.
1H NMR (400 MHz, CDCl3) (ppm): δ 4.61 (2H, s, RCH2Ar), δ 5.81 (1H, m, RC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), δ 6.35 (1H, m, RCCH), δ 6.76 (2H, m, Ar), δ 7.18 (2H, m, Ar), δ 7.45 (2H, d, Ar), δ 7.59 (2H, d, Ar).
CH), δ 6.35 (1H, m, RCCH), δ 6.76 (2H, m, Ar), δ 7.18 (2H, m, Ar), δ 7.45 (2H, d, Ar), δ 7.59 (2H, d, Ar).
Synthesis of pyrrolecarbodithioate ended HB-PNIPAM18
N-Isopropyl acrylamide (14.416 g, 0.127 mol), recrystallised from 40:60 hexane:toluene, and 4-vinylbenzyl pyrrolecarbodithioate (1.289 g, 4.970 × 10−3 mol) and azobis(cyanovaleric acid) (ACVA) (1.391 g, 4.970 × 10−3 mol) were dissolved in dioxane (50 ml). The solution was transferred to a glass ampoule, 3 cycles of freeze pump thaw were carried out. The ampoule was sealed and placed at 60 °C for 48 hours. The viscous solution was precipitated into diethyl ether and dried under vacuum. Precipitation was repeated three times to give 15 g of an off white solid.
1H NMR (400 MHz, CDCl3) (ppm): δ 0.9–1.3 (6H, s, –N(CH3)2), δ 1.4–1.8 (2H, br, m, –CH2–CH–Ar–), δ 1.9–2.2 (2H, br, m, –CH2–CH–CO–NH–) and (1H, br, m, CH2–CH–CONH–), δ 3.7 (H2O–polymer bound), δ 4.0 (1H, br, s, (CH3)2CH–), δ 6.3 (H2, br, s, N–pyrrole–H), δ 6.6–7.2 (br, m, –Ar–), δ 7.65 (2H, br, s, N–pyrrole–H).
Carboxylic acid functionalisation of pyrrolecarbodithioate ended HB-NIPAM18
The concentration of pyrrole end groups in samples was calculated from 1H NMR spectra. The HB-PNIPAM with pyrrolecarbodithioate end groups (HB-PNIPAM-P) (15 g) was dissolved in DMF (675 mL) and ACVA (26 g, 0.94 mol, 20 eq.) was added. The mixture was heated for 24 hours at 60 °C. The process was repeated twice with a total of 60 eq. of ACVA being added. The solvent was removed by rotary evaporation and the product was precipitated drop wise into diethyl ether. The product was then dried under vacuum and purified by ultrafiltration (using a 3000 g mol−1 cut off membrane) in acetone:ethanol (9:1).
1H NMR (400 MHz, CDCl3) (ppm): δ 0.9–1.3 (6H, br, s, –N(CH3)2), 1.4–1.8 (2H, br, m, –CH2–CH–C6H4–), δ 1.9–2.2 (2H, br, m, –CH2–CH–CO–NH–) and (1H, br, m, CH2–CH–CONH–), δ 3.7 (H2O–polymer bound), 4.0 (1H, br, s, (CH3)2CH–), 6.6–7.2 (br, m, –Ar–), 7.85 (br, s, –NH–CO–)
(GPC/SEC) Mn = 3350 g mol−1, Mw = 24850 g mol−1, Mz = 103300 g mol−1, Mz+1 = 236300 g mol−1, ĐM = 7.5.
Peptide synthesis
A ChemSpeed automated peptide synthesizer was used with 735 sampler software. Serine loaded Wang resin was swollen in DMF for 24 hours. The amino acids were added sequentially. To couple each amino acid; 5 equivalents of each amino acid, 4.9 equivalents of the coupling agent benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) and 0.1 equivalent of diisopropylethylamine (DIPEA) were added. The Fmoc protecting groups were removed by deprotection, three times, with 20% piperidine in DMF and the peptide chain was built up from the Serine on the resin to the final Glycine. This gave the desired peptide sequence GRGDS. The resin bound peptide was washed with DMF then DCM then methanol, dried and stored at −18 °C.A cleavage cocktail containing trifluoroacetic acid (TFA), water, phenol and triisopropyl silane (TIPS) in the ratio 88:5:5:2 was added to the resin at 10 mL per gram of resin with stirring for two hours. The resin was filtered off and washed with the cleavage cocktail and three times with TFA. The TFA was removed by rotary evaporation with a cold trap containing CO2(S). The remaining TFA was removed by triturating with five aliquots of diethyl ether. The remaining ether was removed by rotary evaporation and the product stored at −8 °C.
The peptide was purified by preparative HPLC and freeze-dried yielding a white solid.
MH+ (HPLC-ESI) = 490, (calculated = 490.21 (100.0%), 491.22 (19.1%), 492.22 (3.6%), 491.21 (3.0%) g mol−1).
GRGDS functionalisation of HB-PNIPAM18
HB-PNIPAM-COOH (2.72 g) were dissolved in DMF (20 mL). N,N′-Dicyclohexylcarbodiimide (1.626 g, 7.88 × 10−3 mol, 5 eq.) and N-hydroxysuccinimide (0.909 g, 7.88 × 10−3 mol, 5 eq.) were added to the reaction vessel and stirred under nitrogen for 24 hours at room temperature. The DMF was removed by rotary evaporation and the product was purified by ultrafiltration in acetone with 10% ethanol. Once purified the solvent was removed by rotary evaporation yielding 1.5 g of a buff colored solid. 1.5 g of this HB-PNIPAM with N-hydroxysuccinimide end groups was dissolved at 0 °C in 0.01 M pH 8.5 phosphate buffer. This was added to a 3 neck flask along with GRGDS peptide, (380 mg, 0.775 mmol, 5 eq.) cooled to 0 °C and stirred under nitrogen for 48 hours. The resulting product was purified by ultrafiltration and freeze dried yielding a white fluffy solid. The product contained 3.9 mmol g−1 of GRGDS (NMR).1H NMR (400 MHz, CDCl3) (ppm): δ 0.9–1.3 (6H, br, s, –N(CH3)2), 1.4–1.8 (2H, br, m, –CH2–CH–C6H4–), δ 1.9–2.2 (2H, br, m, –CH2–CH–CO–NH–) and (1H, br, m, CH2–CH–CONH–), δ 3.0–3.4 (2H, br, s –CH2–NH–C(NH)–NH2 arginine), δ 3.7 (H2O–polymer bound), 4.0 (1H, br, s, (CH3)2CH–), 6.6–7.2 (br, m, –Ar–), 7.85 (br, s, –NH–CO–).
Production of interpenetrating polymer networks
Hydrogels were produced from the photoinitiated radical copolymerisation of NVP and DEGDA in the presence of the HB-PNIPAM materials in isopropanol as solvent. 2-Hydroxy-2-methyl-1-phenyl propanone (HMPP) was used as the photoinitiator. Full details of the formulations are contained in the ESI.†Polymer (thickness = 100 μm) sheets were synthesised by injecting the polymer/monomer blends into a quartz mould before irradiating with UV light. The samples were removed from the quartz mould and subjected to a rigorous 48 h soxhlet extraction in isopropanol. When preparing samples for cell culture, 13 mm glass coverslips were methacrylate functionalized by cleaning with piranha solution followed by thorough washing with methanol. The cover slips were added to a 5% solution of 3-methacryloxypropyltrimethoxysilane in toluene, in which they were stored until required. A drop of the polymer/monomer blend was applied to a clean microscope slide and a clean methacrylate functionalized cover slip was placed on top. This was cured by UV light and the resulting coated cover slip was removed from the microscope slide.
Determination of amounts of soluble extractable material
The polymer sheets were subjected to Soxhlet extraction for 48 hours in 2-propanol to remove the fraction of HB-PNIPAM-COOH, which was not held within the crosslinked hydrogel system. FTIR spectra were taken of the samples before and after extraction. Partial least squares analysis was carried out using TQ analyst to determine the concentration of HB-PNIPAM within the system after extraction compared to a calibration of known standards made up of samples before extraction. The points at 0, 5, 10, 20, 30 and 100% HB-PNIPAM were used. The calibrations from the 10% and 40% crosslinker systems gave correlation coefficients of 0.999 respectively. The spectral region used for analysis was 1480–1760 cm−1.Determination of LCST by FTIR
All FTIR spectra were collected using a temperature controlled single reflection diamond ATR sampling accessory (Specac) coupled to a Thermo Nexus FTIR spectrometer with the clean ATR crystal at the same temperature as a background using Omnic 6.1 software. Collected spectra were the result of an average of 128 scans at a spectral resolution of 2 cm−1. The IPN sample was swollen in D2O and loaded onto ATR crystal in an air-tight chamber (1 cm3 volume) and sealed using beeswax. The sample was left at 25 °C overnight to allow the D2O/H2O concentration to equilibrate at the ATR crystal interface. To determine the Tc–g spectra were collected at increments of 2 °C from 25 °C to 55 °C, allowing 7 minutes for the system to equilibrate between each temperature change.Primary human fibroblast cell culture
Human fibroblasts were obtained under local ethical permission and consent from female patients undergoing abdominoplasty or breast reduction operations. The method for the isolation and culture of fibroblasts was as previously described.57 Dermis was separated from the epidermis initially by removing subcutaneous tissue and cutting in to 0.5 cm2 pieces. Fibroblasts were obtained from the dermis after trypsinisation of the skin. Dermis was then washed in PBS and then minced finely with a scalpel and placed in 0.5 wt% collagenase overnight at 37 °C. The collagenase digest obtained was centrifuged (1000 g per 5 minutes) and supernatant discarded. The fibroblast cell pellet was resuspended in of fibroblast culture medium (4 mL): DMEM (440 mL) supplemented with neonatal calf serum (50 mL), glutamine (2 × 10−3 mol L−1, 5 mL), amphotericin B (0.625 μg mL−1, 1.25 mL), penicillin (100 IU mL−1, 2.5 mL) and streptomycin (100 μg mL−1, 0.25 mL). Samples were placed in a T25 flask in an incubator (37 °C/5% CO2) and passaged when 80–90% confluent (using a 1 mL 1:1 mixture of 0.1% w/v trypsin and 0.02% w/v EDTA per T25 flask) transferring to a T75 flask. Cells were used for experimentation between passages 4 and 9.
Immunolabelling and confocal microscopy for fibroblast vinculin, actin and presence of nuclei
Human dermal fibroblasts were cultured on test samples versus control (TCP) surfaces for 24 hours and then fixed with 3.7% formaldehyde and labelled for vinculin, actin and nuclei.Samples containing cultures of primary human dermal fibroblasts were washed three times with PBS and fixed with 4% (v/v) paraformaldehyde for 20 min, permeabilised with 0.1% Triton X-100 for 20 min followed by washing with PBS (×3). Unreactive binding sites were blocked with 10% (v/v) goat serum in PBS for 1 hour. The primary antibody (mouse monoclonal anti-vinculin from Sigma Aldrich) was diluted (1:150) in 1% (v/v) goat serum in PBS and fibroblasts incubated for 1 hour at room temperature. The antibody solution was removed and cells were washed twice with PBS. The secondary antibody (rabbit biotinylated anti-mouse IgG) was diluted (1:1000) in 1% goat serum in PBS. Fibroblasts were incubated with secondary antibody for 1 hour at RT. The cells were washed twice with PBS before adding a tertiary fluorescein-conjugated streptavidin label (diluted 1:100 v/v in 1% goat serum in PBS) and incubated for 1 hour. The tertiary label was removed and cells washed three times with PBS before adding phalloidin–TRITC (1:1000 titre) and 4′,6-diamidino-2-phenylindole dihydrochloride (300 nM; DAPI) (Sigma Aldrich) in PBS, to label actin filaments and nuclei, respectively. Cells were incubated for 30 minutes at room temperature before removing the labels and washing a further 3 times in PBS prior to imaging. Fibroblasts were imaged using an upright Zeiss LSM 510 confocal microscope using an argon ion laser (488 nm) for FITC excitation (λex = 495 nm, λem = 521 nm). Nuclei were visualized by two photon excitation using a Ti:Sapphire (Coherent Ultra III) laser (716 nm) for DAPI (λex = 358 nm λem = 461 nm). Image acquisition was conducted using the Kroto Imaging Facility, University of Sheffield, UK.
Primary human fibroblast cell migration
Triplicate samples were placed into individual wells on a 12 well plate. Samples were held in place with inverted cell crowns (manufactured in house). HDFs were seeded at a density of 10000 per well in of culture medium (1 mL). The plates were incubated at 37 °C with 5% CO2 for 24 hours to allow the HDFs to adhere. HDF were imaged using a AF6000 Time-lapse (Leica, Milton Keyes) with LAS AF software. The incubation chamber of the microscope was set to 5% CO2 and 37 °C the plate was allowed to equilibrate within the chamber to avoid focal drift. Six set positions were found on each sample using the 2-dimensional multiposition stage. Images were taken at each set position every 15 minutes for 15 hours. Time-lapse videos produced in LAS AF were analysed in Image J using the manual tracking plugin to track cell movement. The Chemotaxis tool was used to convert the cell tracking data into a speed and accumulated distance and Euclidean distance for the cells tracked.
Scanning electron microscopy
Scanning electron microscopy was carried out in a FEI-Sirion Field Emission Gun SEM on uncoated materials at very low beam energies (0.75 keV) and beam currents (<60 pA) in TV scan mode (integration over 128 frames) to avoid damage or charging.Microstereolithography
For this experiment a projection stereolithography set-up was used based on a Digital Micromirror Device (DMD). This set-up consisted of (i) a Digital Micromirror Device (DMD) (Texas Instruments Incorporated, TX, USA), (ii) a 100 mW 405 nm laser source (Vortran Laser Technology Inc, Sacramento, CA, USA) and (iii) a computer controlled z-axis translation stage (Thorlabs Ltd, Cambridgeshire, UK). A color inverted image was created in MS paint and uploaded to the DMD by control software (ALP basic). The laser power was set to 10 mV and projected via a beam expander onto the DMD. The image from the DMD was focused via several lenses onto the surface of the monomer. The z-stage was moved in a downwards direction at a speed of 0.03 mm s−1 allowing the polymerisation to occur at the surface of the monomer.Acknowledgements
The authors acknowledge funding from EPSRC for a studentship for Richard Penderleith.Notes and references
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS PubMed.
- M. Shivashankar and B. K. Mandal, Int. J. Pharm. Pharm. Sci., 2012, 4, 1–7 CAS.
- G. C. Ingavle, S. H. Gehrke and M. S. Detamore, Biomaterials, 2014, 35, 3558–3570 CrossRef CAS PubMed.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 1792 Search PubMed.
- J. Zhu, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- R. Zhang, H. K. Mjoseng, M. A. Hoeve, N. G. Bauer, S. Pells, R. Besseling, S. Velugotla, G. Tourniaire, R. E. B. Kishen, Y. Tsenkina, C. Armit, C. R. E. Duffy, M. Helfen, F. Edenhofer, P. A. de Sousa and M. Bradley, Nat. Commun., 2013, 4, 1335 CrossRef PubMed.
- I. T. Ozbolat and Y. Yin, IEEE Trans. Biomed. Eng., 2013, 60, 691–699 CrossRef PubMed.
- T. Billiet, M. Vandenhaute, J. Schelfhout, S. Van Vlierberghe and P. Dubruel, Biomaterials, 2012, 33, 6020–6041 CrossRef CAS PubMed.
- A. Khademhosseini and R. Langer, Biomaterials, 2007, 28, 5087–5092 CrossRef CAS PubMed.
- X. Tong and F. Yang, Biomaterials, 2014, 35, 1807–1815 CrossRef CAS PubMed.
- O. Chaudhuri, S. T. Koshy, C. Branco da Cunha, J.-W. Shin, C. S. Verbeke, K. H. Allison and D. J. Mooney, Nat. Mater., 2014, 13, 970–980 CrossRef CAS PubMed.
- S. F. Grundfest-Broniatowski, G. Tellioglu, K. S. Rosenthal, J. Kang, G. Erdodi, B. Yalcin, M. Cakmak, J. Drazba, A. Bennett, L. Lu and J. P. Kennedy, ASAIO J., 2009, 55, 400–405 CrossRef CAS PubMed.
- Y. Sun, J. Collett, N. J. Fullwood, S. Mac Neil and S. Rimmer, Biomaterials, 2007, 28, 661–670 CrossRef CAS PubMed.
- X. Li, J. Zhou, Z. Liu, J. Chen, S. Lü, H. Sun, J. Li, Q. Lin, B. Yang, C. Duan, M. Xing and C. Wang, Biomaterials, 2014, 35, 5679 CrossRef CAS PubMed.
- L. K. Kostanski, R. Huang, C. D. M. Filipe and R. Ghosh, J. Biomater. Sci., Polym. Ed., 2009, 20, 271–297 CrossRef CAS PubMed.
- S. Hopkins, S. R. Carter, J. W. Haycock, N. J. Fullwood, S. MacNeil and S. Rimmer, Soft Matter, 2009, 5, 4928–4937 RSC.
- S. Rimmer, S. Carter, R. Rutkaite, J. W. Haycock and L. Swanson, Soft Matter, 2007, 3, 971–973 RSC.
- Y. Guan and Y. Zhang, Soft Matter, 2011, 7, 6375–6384 RSC.
- Z. M. O. Rzaev, S. Dinçer and E. Pişkin, Prog. Polym. Sci., 2007, 32, 534–595 CrossRef CAS PubMed.
- B. R. Saunders, N. Laajam, E. Daly, S. Teow, X. Hu and R. Stepto, Adv. Colloid Interface Sci., 2009, 147–148, 251–262 CrossRef CAS PubMed.
- Q. Yu, L. M. Johnson and G. P. López, Adv. Funct. Mater., 2014, 24, 3751–3759 CrossRef CAS PubMed.
- Y. Morita and I. Kaetsu, Radiat. Phys. Chem., 1992, 39, 473–476 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- T. Owaki, T. Shimizu, M. Yamato and T. Okano, Biotechnol. J., 2014, 9, 904–914 CrossRef CAS PubMed.
- L. S. Wang, P. Y. Chow, T. T. Phan, I. J. Lim and Y. Y. Yang, Adv. Funct. Mater., 2006, 16, 1171–1178 CrossRef CAS PubMed.
- J. Collett, A. Crawford, P. V. Hatton, M. Geoghegan and S. Rimmer, J. R. Soc., Interface, 2007, 4, 117–126 CrossRef PubMed.
- C. Sammon, C. Li, S. P. Armes and A. L. Lewis, Polymer, 2006, 47, 6123–6130 CrossRef CAS PubMed.
- S. Malik and A. K. Nandi, J. Phys. Chem. B, 2003, 108, 597–604 CrossRef.
- Y.-l. Su, J. Wang and H.-z. Liu, Macromolecules, 2002, 35, 6426–6431 CrossRef CAS.
- Y.-L. Su, H.-Z. Liu, C. Guo and J. Wang, Mol. Simul., 2003, 29, 803–808 CrossRef CAS PubMed.
- Y. Maeda, T. Nakamura and I. Ikeda, Macromolecules, 2001, 34, 1391–1399 CrossRef CAS.
- E. Martínez, A. Lagunas, C. A. Mills, S. Rodríguez-Seguí, M. Estévez, S. Oberhansl, J. Comelles and J. Samitier, Nanomedicine, 2009, 4, 65–82 CrossRef PubMed.
- A. M. Ross, Z. Jiang, M. Bastmeyer and J. Lahann, Small, 2012, 8, 336–355 CrossRef CAS PubMed.
- K. J. McHugh, M. Saint-Geniez and S. L. Tao, J. Biomed. Mater. Res., Part B, 2013, 101, 1571–1576 CrossRef PubMed.
- S. Rimmer, M. J. German, J. Maughan, Y. Sun, N. Fullwood, J. Ebdon and S. MacNeil, Biomaterials, 2005, 26, 2219–2230 CrossRef CAS PubMed.
- M. T. Postek and A. E. Vladar, Scanning, 2008, 30, 457 CrossRef PubMed.
- V. Kumar, H. Wang and C. Rodenburg, Org. Electron., 2014, 15, 2059–2067 CrossRef CAS PubMed.
- L. Perlin, S. MacNeil and S. Rimmer, Soft Matter, 2008, 4, 2331–2349 RSC.
- L. E. Smith, S. Rimmer and S. MacNeil, Biomaterials, 2006, 27, 2806–2812 CrossRef CAS PubMed.
- K. A. Kilian and M. Mrksich, Angew. Chem., Int. Ed., 2012, 51, 4891–4895 CrossRef CAS PubMed.
- D. E. Discher, P. Janmey and Y.-l. Wang, Science, 2005, 310, 1139–1143 CrossRef CAS PubMed.
- K. Ye, X. Wang, L. Cao, S. Li, Z. Li, L. Yu and J. Ding, Nano Lett., 2015, 15, 4720–4729 CrossRef CAS PubMed.
- T. D. Hansen, J. T. Koepsel, N. N. Le, E. H. Nguyen, S. Zorn, M. Parlato, S. G. Loveland, M. P. Schwartz and W. L. Murphy, Biomater. Sci., 2014, 2, 745–756 RSC.
- I. Hopp, A. Michelmore, L. E. Smith, D. E. Robinson, A. Bachhuka, A. Mierczynska and K. Vasilev, Biomaterials, 2013, 34, 5070–5077 CrossRef CAS PubMed.
- F. R. Maia, A. H. Lourenço, P. L. Granja, R. M. Gonçalves and C. C. Barrias, Macromol. Biosci., 2014, 14, 759–771 CrossRef CAS PubMed.
- H. W. Chien, S. W. Fu, A. Y. Shih and W. B. Tsai, Biotechnol. J., 2014, 9, 1613–1623 CrossRef CAS PubMed.
- L. Y. Koo, D. J. Irvine, A. M. Mayes, D. A. Lauffenburger and L. G. Griffith, J. Cell Sci., 2002, 115, 1423–1433 CAS.
- D. J. Irvine, A. M. Mayes and L. G. Griffith, Biomacromolecules, 2001, 2, 85–94 CrossRef CAS PubMed.
- S. P. Massia and J. A. Hubbell, J. Cell Biol., 1991, 114, 1089–1100 CrossRef CAS.
- S. H. Lee, J. J. Moon and J. L. West, Biomaterials, 2008, 29, 2962–2968 CrossRef CAS PubMed.
- C. H. Yu, N. Rafiq, A. Krishnasamy, K. Hartman, G. Jones, A. Bershadsky and M. Sheetz, Cell Rep., 2013, 5, 1456–1468 CrossRef CAS PubMed.
- R. J. Seelbach, P. Fransen, M. Peroglio, D. Pulido, P. Lopez-Chicon, F. Duttenhoefer, S. Sauerbier, T. Freiman, P. Niemeyer, C. Semino, F. Albericio, M. Alini, M. Royo, A. Mata and D. Eglin, Acta Biomater., 2014, 10, 4340–4350 CrossRef CAS PubMed.
- M. Yamato, Y. Akiyama, J. Kobayashi, J. Yang, A. Kikuchi and T. Okano, Prog. Polym. Sci., 2007, 32, 1123–1133 CrossRef CAS PubMed.
- S. P. Massia and J. A. Hubbell, J. Cell Biol., 1991, 114, 1089–1100 CrossRef CAS.
- D. R. Ralston, C. Layton, A. J. Dalley, S. G. Boyce, E. Freedlander and S. Macneil, Br. J. Dermatol., 1999, 140, 605–615 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sm00695c |
| This journal is © The Royal Society of Chemistry 2015 |