
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systematic chemoenzymatic synthesis of O-sulfated sialyl Lewis x antigens†
Abhishek
Santra
a,
Hai
Yu
a,
Nova
Tasnima
a,
Musleh M.
Muthana‡
a,
Yanhong
Li
a,
Jie
Zeng
ab,
Nicholas J.
Kenyon
c,
Angelique Y.
Louie
d and
Xi
Chen
*a
aDepartment of Chemistry, University of California, Davis One Shields Avenue, Davis, CA 95616, USA. E-mail: xiichen@ucdavis.edu; Fax: +1-530-752-8995; Tel: +1-530-754-6037
bSchool of Food Science, Henan Institute of Science and Technology, Xinxiang, 453003, China
cDivision of Pulmonary, Critical Care and Sleep Medicine, Department of Internal Medicine, University of California, Davis, CA 95616, USA
dDepartment of Biomedical Engineering, University of California, Davis, CA 95616, USA
First published on 17th December 2015
Abstract
O-Sulfated sialyl Lewis x antigens play important roles in nature. However, due to their structural complexity, they are not readily accessible by either chemical or enzymatic synthetic processes. Taking advantage of a bacterial sialyltransferase mutant that can catalyze the transfer of different sialic acid forms from the corresponding sugar nucleotide donors to Lewis x antigens, which are fucosylated glycans, as well as an efficient one-pot multienzyme (OPME) sialylation system, O-sulfated sialyl Lewis x antigens containing different sialic acid forms and O-sulfation at different locations were systematically synthesized by chemoenzymatic methods.
Introduction
O-Sulfated sialyl Lewis x structures play important roles in immune regulation, inflammation, and cancer metastasis.1 For example, 6-O-sulfo-sialyl Lewis x [6-O-sulfo-sLex (1), Neu5Acα2-3Galβ1-4(Fucα1-3)GlcNAc6SβOR] with an O-sulfate group at the carbon-6 of the N-acetylglucosamine (GlcNAc) residue (Fig. 1) is a well known ligand for L-selectin, a C-type (Ca2+-dependent) carbohydrate-binding protein (lectin) expressed broadly in most leukocytes in the blood.1,2 The interaction of 6-O-sulfo-sLex (1) and L-selectin plays a critical role in lymphocyte homing to the peripheral lymph nodes2 and in chronic inflammation.3 It has also been shown that human sialic acid-binding immunoglobulin-like lectin4 Siglec-9 binds strongly5,6 to 6-O-sulfo-sLex, but the biological importance of this interaction is less well understood.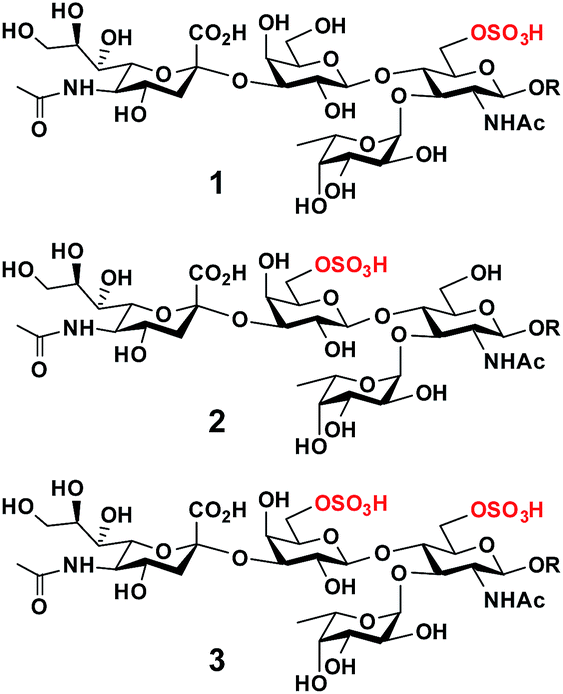 | ||
| Fig. 1 Structures of O-sulfated sialyl Lewis x including 6-O-sulfo-sLex (1), 6′-O-sulfo-sLex (2), and 6′, 6-di-O-sulfo-sLex (3). | ||
On the other hand, 6′-O-sulfo-sialyl Lewis x [6′-O-sulfo-sLex (2), Neu5Acα2-3Gal6Sβ1-4(Fucα1-3)GlcNAcβOR] with an O-sulfate group at the carbon-6 of the galactose (Gal) residue (Fig. 1),7 in addition to 6′-O-sulfo-sialyl-N-acetyllactosamine (6′-O-sulfo-sLacNAc, Neu5Acα2-3Gal6Sβ1-4GlcNAcβOR),8 was shown by glycan microarray studies to be a preferred glycan ligand for Siglec-8 and for its paralog mouse Siglec-F.9 Siglec-8 is expressed in human allergic inflammatory cells including eosinophils, mast cells, and basophils.5,10 Reducing the number of eosinophils, such as by soluble 6′-O-sulfo-sLex synthetic polymer induced apoptosis,11 has been suggested as an approach for asthma therapies.12 Furthermore, 6′-O-sulfo-sLex (2), in addition to 6′-O-sulfo-sLacNAc and 6′-O-sulfo-sialyl-lacto-N-neotetraose (6′-O-sulfo-sLNnT, Neu5Acα2-3Gal6Sβ1-4GlcNAcβ1-3Galβ1-4GlcβOR), was shown to bind to langerin,13 a C-type (Ca2+-dependent) lectin specific to Langerhans cells (immature antigen-presenting specific T cell immunity initiating dendritic cells of epidermis and mucosal tissues).14
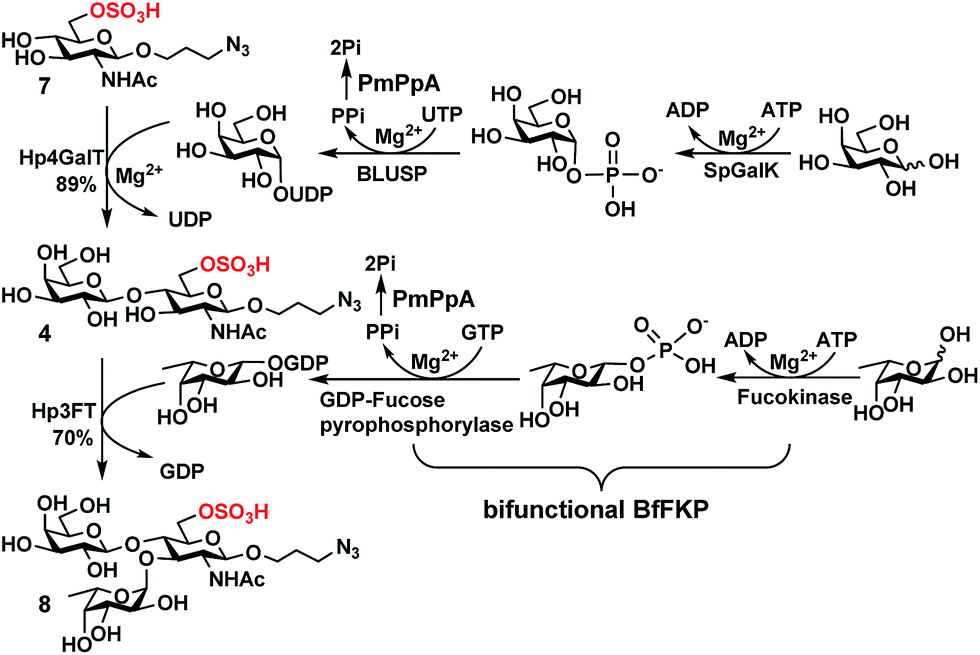 | ||
| Scheme 1 Sequential OPME synthesis of 6-O-sulfo-LexβProN3 (8) from 6-O-sulfo-GlcNAcβProN3 (7) using an OPME β1-4-galactosyl activation and transfer system for the formation of 6-O-sulfo-LacNAcβProN3 (4) followed by an OPME α1-3-fucosyl activation and transfer system for the formation of 6-O-sulfo-LexβProN3 (8). Enzymes and abbreviations: SpGalK, Streptococcus pneumoniae TIGR4 galactokinase;44 BLUSP, Bifidobacterium longum UDP-sugar pyrophosphorylase;45 PmPpA, Pasteurella multocida inorganic pyrophosphorylase;43 Hp4GalT, Helicobacter pylori β1-4-galactosyltransferase;43 BfFKP, Bacteroides fragilis bifunctional L-fucokinase/GDP-fucose pyrophosphorylase;42 and Hp3FT, Helicobacter pylori α1-3-fucosyltransferase.39,41 | ||
Although less efficient than Neu5Acα2-8Neu5Acα2-3LacNAc, both 6-O-sulfo-sLex (1) and 6′-O-sulfo-sLex (2) bound moderately to human Siglec-7.5 Both are present in glycosylation-dependent cell adhesion molecule 1 (GlyCAM-1), an L-selectin ligand,15 with 6′-O-sulfo-sLex (2) as the major sulfated form.16–18 Gal-6-O-sulfotransferase and GlcNAc-6-O-sulfotransferase have been found to synergistically produce L-selectin ligands. This indicates either the potential synergistic involvement of both 6-O-sulfo-sLex (1) and 6′-O-sulfo-sLex (2) or the involvement of 6′,6-di-O-sulfo-sLex (3) (Fig. 1) with O-sulfate groups at both the Gal and GlcNAc residues of sLex in L-selectin-binding.19 Human Siglec-7 and -8 have also been shown to bind more strongly to 6′,6-di-O-sulfo-sLex (3) than their mono-O-sulfated derivatives (1 and 2), while mouse Siglec-F has been shown to bind with similar strength to 6′,6-di-O-sulfo-sLex (3) and 6′-O-sulfo-sLex (2).6
The biological importance of O-sulfated sLex structures makes them attractive synthetic targets. However, the structures of these compounds are relatively complex and include synthetically challenging α2-3-linked sialic acid, which suffers from low stereoselectivity and a high 2,3-elimination rate in chemical synthesis,20–22 as well as the acid labile O-sulfate group.23,24 Chemically20,25,26 or chemoenzymatically27 synthesized Neu5Acα2-3Gal building blocks have been used as effective synthons for constructing more complex sialosides including sLex and 6-O-sulfo-sLex (1).20 Several examples of the chemical22,28 or chemoenzymatic29 synthesis of 6-O-sulfo-sLex (1) as well as the chemical synthesis of 6′-O-sulfo-sLex (2)22,30,31 and 6′,6-di-O-sulfo-sLex (3)32 have been reported. All these examples are, however, limited to compounds with the most abundant sialic acid form, N-acetylneuraminic acid (Neu5Ac). Despite the presence of more than 50 different sialic acid forms identified in nature,33,34O-sulfated sLex containing a sialic acid form other than Neu5Ac has not been synthesized.
We report here the development of efficient chemoenzymatic methods for the systematic synthesis of O-sulfated sLex containing different sialic acid forms. The methods are demonstrated for representative examples of 6′-O-sulfo-sLex (1), 6-O-sulfo-sLex (2) and/or 6′,6-di-O-sulfo-sLex (3) containing the most abundant Neu5Ac form and N-glycolylneuraminic acid (Neu5Gc), a sialic acid form commonly found in mammals other than humans, but which can be incorporated into the human glycome from dietary sources.35
One efficient approach for the synthesis of O-sulfated sLex with different sialic acid forms would be by direct sialylation of O-sulfated Lex using one-pot multienzyme (OPME) sialylation systems36 containing an α2-3-sialyltransferase and a CMP-sialic acid synthetase (CSS),37 with or without a sialic acid aldolase.38 Such an approach has been successfully demonstrated for direct sialylation of non-sulfated Lex for the synthesis of sLex containing a diverse array of naturally occurring and non-natural sialic acid forms, using OPME systems containing a recombinant viral α2-3-sialyltransferase vST3Gal-I39 or a bacterial multifunctional sialyltransferase mutant, Pasteurella multocida α2-3-sialyltransferase 1 (PmST1) M144D.40 The latter, with a high expression level (98 mg L-1 culture, >1000-fold higher than that of vST3Gal-I) and high promiscuity in tolerating different modifications on the sialic acid in the substrates, is a superior choice for the synthesis.40 However, it is not clear whether O-sulfated Lex structures could be used with PmST1 M144D as suitable acceptors in the OPME sialylation process to produce the desired O-sulfated sLex with different sialic acid forms.
Results and discussion
Synthesis of O-sulfated disaccharides and O-sulfated Lex
In order to obtain O-sulfated Lex as potential acceptor substrates for PmST1 M144D, enzyme-catalyzed α1-3-fucosylation of the corresponding O-sulfated disaccharides was tested as a potential strategy. A one-pot three-enzyme (OP3E) α1-3-fucosylation system (Scheme 1)39,41 containing Bacteroides fragilis bifunctional L-fucokinase/GDP-fucose pyrophosphorylase (BfFKP),42Pasteurella multocida inorganic pyrophosphorylase (PmPpA),43 and Helicobacter pylori α1-3-fucosyltransferase (Hp1-3FTΔ66 or Hp3FT) was used for this purpose. The O-sulfated disaccharides tested were 6-O-sulfo-LacNAcβProN3 (4) (Scheme 1), 6′-O-sulfo-LacNAcβProN3 (5), and 6,6′-di-O-sulfo-LacNAcβProN3 (6) (Fig. 2). LacNAcβProN3 (ref. 43) without any O-sulfate groups was used as a positive control. | ||
| Fig. 2 Structures of chemically synthesized 6′-O-sulfo-LacNAcβProN3 (5) and 6,6′-di-O-sulfo-LacNAcβProN3 (6). | ||
6-O-Sulfo-LacNAcβProN3 (4) was synthesized from 6-O-sulfo-GlcNAcβProN3 (7)43 using an improved OPME galactosyl activation and transfer system (Scheme 1) containing Streptococcus pneumoniae TIGR4 galactokinase (SpGalK),44Bifidobacterium longum UDP-sugar pyrophosphorylase (BLUSP),45 PmPpA, and a Helicobacter pylori β1-4-galactosyltransferase (Hp1-4GalT or Hp4GalT).43 The EcGalK, BLUSP, and PmPpA allowed in situ formation of the donor substrate of Hp4GalT, uridine 5′-diphosphate-galactose (UDP-Gal), from monosaccharide galactose (Gal).45 It was previously shown that Hp4GalT, but not Neisseria meningitidis β1-4-galactosyltransferase (NmLgtB), was able to use 6-O-sulfated GlcNAc and derivatives as acceptor substrates for the synthesis of β1-4-linked galactosides.43 The activity of Hp4GalT in synthesizing 6-O-sulfo-LacNAcβProN3 (4) was confirmed again here using the improved OPME approach.45,46 An excellent 89% yield was obtained, comparing favourably to the previous Hp4GalT-dependent OPME β1-4-galactosylation approach (70% yield) which used Escherichia coli K-12 glucose-1-P uridylyltransferase (EcGalU), Escherichia coli UDP-galactose-4-epimerase (EcGalE), and PmPpA to produce UDP-Gal in situ from glucose-1-phosphate.43 6′-O-Sulfo-LacNAcβProN3 (5) and 6,6′-di-O-sulfo-LacNAcβProN3 (6) (Fig. 2) were chemically synthesized (see ESI†).
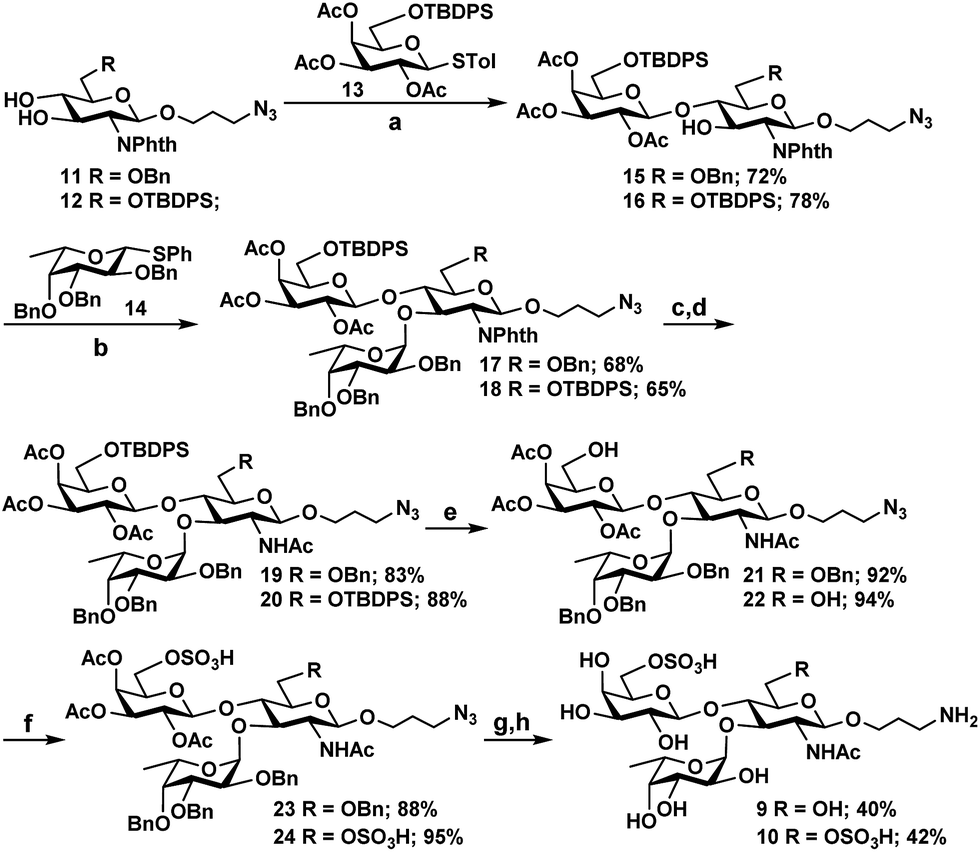 | ||
Scheme 2 Chemical synthesis of 6′-O-sulfo-LexβProNH2 (9) and 6,6′-di-O-sulfo-LexβProNH2 (10). Reagents and conditions: (a) N-iodosuccinimide (NIS), TMSOTf, MS 4 Å, CH2Cl2, −40 °C, 30 min; (b) N-iodosuccinimide (NIS), TMSOTf, MS 4 Å, CH2Cl2–Et2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), −18 °C, 45 min; (c) H2N(CH2)2NH2, n-BuOH, 90 °C, 8 h; (d) pyridine, Ac2O, r.t., 10 h; (e) HF pyridine, 0 °C to r.t., overnight; (f) SO3 pyridine, pyridine, 0 °C to r.t.; (g) 0.1 M NaOMe, MeOH, r.t., 3 h; (h) Pd(OH)2/C, H2, CH3OH, 48 h. :1), −18 °C, 45 min; (c) H2N(CH2)2NH2, n-BuOH, 90 °C, 8 h; (d) pyridine, Ac2O, r.t., 10 h; (e) HF pyridine, 0 °C to r.t., overnight; (f) SO3 pyridine, pyridine, 0 °C to r.t.; (g) 0.1 M NaOMe, MeOH, r.t., 3 h; (h) Pd(OH)2/C, H2, CH3OH, 48 h. | ||
Among the three O-sulfated disaccharides tested, only 6-O-sulfo-LacNAcβProN3 (4) was a suitable acceptor for Hp3FT to produce the desired 6-O-sulfo-LexβProN3 (8). In contrast, 6′-O-sulfo-LacNAcβProN3 (5) and 6,6′-di-O-sulfo-LacNAcβProN3 (6) were not used efficiently by Hp3FT for the synthesis of the corresponding O-sulfated Lex derivatives. With the positive outcome in small scale reactions for fucosylation of 6-O-sulfo-LacNAcβProN3 (4), the preparative-scale synthesis of 6-O-sulfo-LexβProN3 (8) was carried out using the OP3E α1-3-fucosyl activation and transfer system (Scheme 1). A yield of 70% was obtained. The combined sequential OPME β1-4-galactosylation and OPME α1-3-fucosylation (Scheme 1) was an effective approach for obtaining 6-O-sulfo-LexβProN3 (8) from a simple monosaccharide derivative 6-O-sulfo-GlcNAcβProN3 (7) in an overall yield of 62%.
As Hp3FT was not able to use 6′-O-sulfo-LacNAcβProN3 (5) or 6,6′-di-O-sulfo-LacNAcβProN3 (6) efficiently as acceptors for fucosylation to obtain the desired Lex trisaccharides, the target trisaccharides 6′-O-sulfo-LexβProNH2 (9) and 6,6′-di-O-sulfo-LexβProNH2 (10) were chemically synthesized (Scheme 2) from monosaccharide synthons 11, 12,2713, and 14.27 Notable features of the synthetic strategy include: (a) application of an efficient general protection strategy47 for the synthesis of the two trisaccharides (i.e. similar protecting groups were used in the syntheses and the same reagents were used for their removal); (b) use of similar thioglycoside derivatives as glycosyl donors in all glycosylations; (c) high regio- and stereoselectivity in product formation; (d) one step removal of benzyl ethers and reduction of the azido group using 20% Pd(OH)2/C (Pearlman's catalyst) and H2.48 More specifically, for the synthesis of 9 and 10, two N-phthalimide glucosamine derivatives 11 and 12 selectively protected at C6 with benzyl and tert-butyldiphenylsilyl ether (TBDPS), respectively, were coupled stereoselectively with thioglycoside donor 13, which was selectively protected with TBDPS at C6, in the presence of N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf)49 in dichloromethane. Disaccharide derivatives 15 and 16 were obtained in 72% and 78% yields, respectively. The bulky N-phthalimido protecting group in acceptors 11 and 12 provides steric hindrance to the neighboring C-3 hydroxyl group and decreases the reactivity of the C-3 hydroxyl group. Therefore, glycosylation occurs regioselectively at the C-4 hydroxyl group.27 Initial attempts to glycosylate acceptors 15 and 16 in dichloromethane with 1.2 equivalents of thiophenyl fucoside 14 produced trisaccharides in alpha and beta mixtures. In contrast, stereospecific formation of trisaccharides was achieved when a mixed solvent of diethylether and dichloromethane (1:1)50,51 was employed. The reaction of acceptors 15 and 16 with 1.2 equivalents of fucosyl donor 14 produced compounds 17 and 18 in 68% and 65% yields, respectively. Compounds 17 and 18 were then subjected to a series of synthetic transformations: (a) conversion of the N-phthaloyl group to an acetamido group by removing the phthaloyl group using ethylenediamine, followed by N- and O-acetylation using acetic anhydride and pyridine; (b) HF-pyridine-mediated selective removal of the TBDPS group;52 (c) O-sulfation of the primary hydroxyl group by SO3 pyridine complex;52,53 (d) deacetylation by NaOMe in MeOH;54 and (e) hydrogenation using Pd(OH)2/C and H2 (ref. 55) to obtain the desired 6′-O-sulfo-LexβProNH2 (9) and 6,6′-di-O-sulfo-LexβProNH2 (10).
Enzymatic synthesis of O-sulfated sLex
With chemoenzymatically synthesized 6-O-sulfo-LexβProN3 (8) as well as chemically synthesized 6′-O-sulfo-LexβProNH2 (9) and 6,6′-di-O-sulfo-LexβProNH2 (10) in hand, a one-pot two-enzyme (OP2E) sialylation system (Scheme 3) was used to test the tolerance of PmST1 M144D40 for using these O-sulfated Lex compounds as potential acceptor substrates. PmST1 M144D was previously engineered by protein crystal structure-assisted design. It has 20-fold reduced CMP-sialic acid (donor) hydrolysis activity and significantly (5588-fold) decreased α2-3-sialidase activity compared to the wild-type enzyme. It was used efficiently in a one-pot three-enzyme (OP3E) sialylation system for the synthesis of non-sulfated sLex tetrasaccharides containing diverse sialic acid forms from Lex.40 To our delight, PmST1 M144D also tolerated O-sulfated Lex containing O-sulfate at C-6, C-6′, or both. In addition to N-acetylneuraminic acid (Neu5Ac), N-acetylneuraminic acid (Neu5Gc) was also successfully introduced to compounds 8–10. O-Sulfated sLex tetrasaccharides 6-O-sulfo-Neu5Acα2-3LexβProN3 (1a, 80 mg, 85%), 6-O-sulfo-Neu5Gcα2-3LexβProN3 (1b, 22 mg, 47%), 6′-O-sulfo-Neu5Acα2-3LexβProNH2 (2a, 75 mg, 82%), 6′-O-sulfo-Neu5Acα2-3LexβProNH2 (2b, 45 mg, 60%), 6,6′-di-O-sulfo-Neu5Acα2-3LexβProNH2 (3a, 42 mg, 64%), and 6,6′-di-O-sulfo-Neu5Gcα2-3LexβProNH2 (3b, 40 mg, 38%) were successfully obtained using this highly efficient one-pot two-enzyme system containing Neisseria meningitidis CMP-sialic acid (NmCSS)37 and PmST1 M144D40 from the corresponding acceptors 8–10 and Neu5Ac or Neu5Gc, respectively. In general, Neu5Gc was used less efficiently by the OPME sialylation system, leading to lower yields for 1b–3b (38–60%) compared to their Neu5Ac-counterparts 1a–3a (64–85%). O-Sulfated sLex glycans with a propyl amine aglycone (compounds 2a, 3a, 2b and 3b) were found to be more challenging for column purification compared to the ones with a propyl azide aglycone (compounds 1a and 1b). When a desired sialic acid is readily available such as in the case presented here, a one-pot two-enzyme (OP2E) system is sufficient. When only the 6-carbon precursors of the desired sialic acid forms are available, the one-pot three-enzyme (OP3E) sialylation system including an aldolase in addition to NmCSS and PmST1 M144D40 should be used.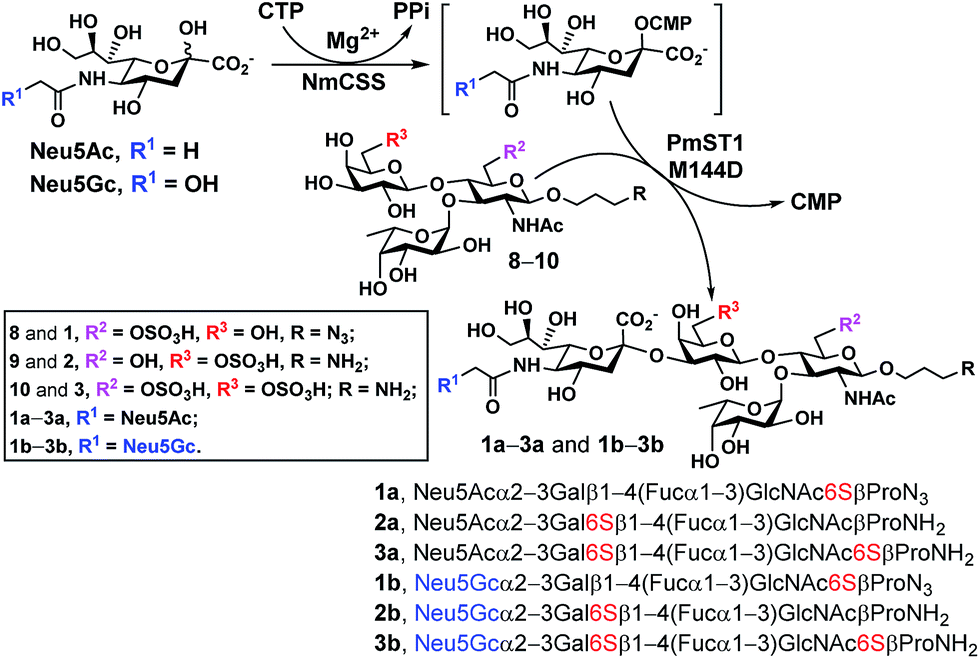 | ||
| Scheme 3 PmST1 M144D-mediated one-pot two-enzyme (OP2E) sialylation of O-sulfo analogues of Lewisx. Yields obtained for O-sulfated sLex tetrasaccharides: 1a, 85%; 1b, 47%; 2a, 82%; 2b, 60%; 3a, 64%; 3b, 38%. Enzymes and abbreviations: NmCSS, Neisseria meningitidis CMP-sialic acid;37 PmST1 M144D, Pasteurella multocida α2-3-sialyltransferase 1 (PmST1) M144D mutant.40 | ||
Conclusions
In conclusion, we have successfully developed an efficient chemoenzymatic method for the systematic synthesis of synthetically challenging O-sulfated sLex (1a–3a and 1b–3b) containing different sialic acid forms (Neu5Ac or Neu5Gc) by direct sialylation of the corresponding O-sulfated Lex structures 8–10 using an efficient one-pot two-enzyme (OP2E) system containing NmCSS and PmST1 M144D. The method can be extended to the synthesis of O-sulfated sLex structures containing other sialic acid forms. We have also shown here that a relatively complex trisaccharide 6-O-sulfo-LexβProN3 (8) can be efficiently produced from a simple monosaccharide derivative 6-O-sulfo-GlcNAcβProN3 (7) by a sequential OPME β1-4-galactosylation and OPME α1-3-fucosylation process. PmST1 M144D has been demonstrated to be a powerful catalyst not only for synthesizing non-sulfated sLex structures as shown previously,40 but also for producing biologically important but difficult-to-obtain O-sulfated sLex.Acknowledgements
This work was supported by NIH grants R01GM094523 (to X. C.) and R21AI097354 (to A. Y. L.) as well as NSF grant CHE-1300449 (to X. C.). M. M. M. acknowledges UC-Davis and USDE for a GAANN Fellowship (P200A120187). A Bruker Avance-800 NMR spectrometer was funded by NSF grant DBIO-722538.References
- S. D. Rosen, Annu. Rev. Immunol., 2004, 22, 129 CrossRef CAS PubMed.
- H. Kawashima and M. Fukuda, Ann. N. Y. Acad. Sci., 2012, 1253, 112 CrossRef CAS PubMed.
- M. Kobayashi, H. Lee, J. Nakayama and M. Fukuda, Curr. Drug Metab., 2009, 10, 29 CrossRef CAS PubMed.
- M. S. Macauley, P. R. Crocker and J. C. Paulson, Nat. Rev. Immunol., 2014, 14, 653 CrossRef CAS PubMed.
- T. Kiwamoto, N. Kawasaki, J. C. Paulson and B. S. Bochner, Pharmacol. Ther., 2012, 135, 327 CrossRef CAS PubMed.
- M. A. Campanero-Rhodes, R. A. Childs, M. Kiso, S. Komba, C. le Narvor, J. Warren, D. Otto, P. R. Crocker and T. Feizi, Biochem. Biophys. Res. Commun., 2006, 344, 1141 CrossRef CAS PubMed.
- B. S. Bochner, R. A. Alvarez, P. Mehta, N. V. Bovin, O. Blixt, J. R. White and R. L. Schnaar, J. Biol. Chem., 2005, 280, 4307 CrossRef CAS PubMed.
- T. Kiwamoto, M. E. Brummet, F. Wu, M. G. Motari, D. F. Smith, R. L. Schnaar, Z. Zhu and B. S. Bochner, J. Allergy Clin. Immunol., 2014, 133, 240 CrossRef CAS PubMed.
- H. Tateno, P. R. Crocker and J. C. Paulson, Glycobiology, 2005, 15, 1125 CrossRef CAS PubMed.
- B. S. Bochner, Clin. Exp. Allergy, 2009, 39, 317 CrossRef CAS PubMed.
- S. A. Hudson, N. V. Bovin, R. L. Schnaar, P. R. Crocker and B. S. Bochner, J. Pharmacol. Exp. Ther., 2009, 330, 608 CrossRef CAS PubMed.
- T. Kiwamoto, T. Katoh, C. M. Evans, W. J. Janssen, M. E. Brummet, S. A. Hudson, Z. Zhu, M. Tiemeyer and B. S. Bochner, J. Allergy Clin. Immunol., 2015, 135, 1329 CrossRef CAS PubMed.
- C. Galustian, C. G. Park, W. Chai, M. Kiso, S. A. Bruening, Y. S. Kang, R. M. Steinman and T. Feizi, Int. Immunol., 2004, 16, 853 CrossRef CAS PubMed.
- J. Valladeau, O. Ravel, C. Dezutter-Dambuyant, K. Moore, M. Kleijmeer, Y. Liu, V. Duvert-Frances, C. Vincent, D. Schmitt, J. Davoust, C. Caux, S. Lebecque and S. Saeland, Immunity, 2000, 12, 71 CrossRef CAS PubMed.
- L. A. Lasky, M. S. Singer, D. Dowbenko, Y. Imai, W. J. Henzel, C. Grimley, C. Fennie, N. Gillett, S. R. Watson and S. D. Rosen, Cell, 1992, 69, 927 CrossRef CAS PubMed.
- S. Hemmerich and S. D. Rosen, Biochemistry, 1994, 33, 4830 CrossRef CAS PubMed.
- S. Hemmerich, H. Leffler and S. D. Rosen, J. Biol. Chem., 1995, 270, 12035 CrossRef CAS PubMed.
- S. Hemmerich, C. R. Bertozzi, H. Leffler and S. D. Rosen, Biochemistry, 1994, 33, 4820 CrossRef CAS PubMed.
- A. Bistrup, S. Bhakta, J. K. Lee, Y. Y. Belov, M. D. Gunn, F. R. Zuo, C. C. Huang, R. Kannagi, S. D. Rosen and S. Hemmerich, J. Cell Biol., 1999, 145, 899 CrossRef CAS PubMed.
- G. V. Pazynina, M. A. Sablina, A. B. Tuzikov, A. A. Chinarev and N. V. Bovin, Mendeleev Commun., 2003, 13, 245 CrossRef.
- C. H. Lai, H. S. Hahm, C. F. Liang and P. H. Seeberger, Beilstein J. Org. Chem., 2015, 11, 617 CrossRef CAS PubMed.
- A. K. Misra, Y. Ding, J. B. Lowe and O. Hindsgaul, Bioorg. Med. Chem. Lett., 2000, 10, 1505 CrossRef CAS PubMed.
- A. Liang, J. N. Thakkar and U. R. Desai, J. Pharm. Sci., 2010, 99, 1207 CrossRef CAS PubMed.
- R. A. Al-Horani and U. R. Desai, Tetrahedron, 2010, 66, 2907 CrossRef CAS PubMed.
- L. Krock, D. Esposito, B. Castagner, C.-C. Wang, P. Bindschadler and P. H. Seeberger, Chem. Sci., 2012, 3, 1617 RSC.
- D. Esposito, M. Hurevich, B. Castagner, C. C. Wang and P. H. Seeberger, Beilstein J. Org. Chem., 2012, 8, 1601 CrossRef CAS PubMed.
- H. Cao, S. Huang, J. Cheng, Y. Li, S. Muthana, B. Son and X. Chen, Carbohydr. Res., 2008, 343, 2863 CrossRef CAS PubMed.
- S. Komba, C. Galustian, H. Ishida, T. Feizi, R. Kannagi and M. Kiso, Angew. Chem., Int. Ed., 1999, 38, 1131 CrossRef CAS.
- M. R. Pratt and C. R. Bertozzi, Org. Lett., 2004, 6, 2345 CrossRef CAS PubMed.
- S. Komba, H. Ishida, M. Kiso and A. Hasegawa, Bioorg. Med. Chem., 1996, 4, 1833 CrossRef CAS PubMed.
- R. K. Jain, R. Vig, R. Rampal, E. V. Chandrasekaran and K. L. Matta, J. Am. Chem. Soc., 1994, 116, 12123 CrossRef CAS.
- S. Komba, H. Ishida, M. Kiso and A. Hasegawa, Carbohydr. Res., 1996, 285, C1 CrossRef CAS PubMed.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163 CrossRef CAS PubMed.
- T. Angata and A. Varki, Chem. Rev., 2002, 102, 439 CrossRef CAS PubMed.
- A. Varki, Am. J. Phys. Anthropol., 2001, 116(S33), 54 CrossRef PubMed.
- H. Yu, H. A. Chokhawala, S. Huang and X. Chen, Nat. Protoc., 2006, 1, 2485 CrossRef CAS PubMed.
- H. Yu, H. Yu, R. Karpel and X. Chen, Bioorg. Med. Chem., 2004, 12, 6427 CrossRef CAS PubMed.
- Y. Li, H. Yu, H. Cao, K. Lau, S. Muthana, V. K. Tiwari, B. Son and X. Chen, Appl. Microbiol. Biotechnol., 2008, 79, 963 CrossRef CAS PubMed.
- G. Sugiarto, K. Lau, H. Yu, S. Vuong, V. Thon, Y. Li, S. Huang and X. Chen, Glycobiology, 2011, 21, 387 CrossRef CAS PubMed.
- G. Sugiarto, K. Lau, J. Qu, Y. Li, S. Lim, S. Mu, J. B. Ames, A. J. Fisher and X. Chen, ACS Chem. Biol., 2012, 7, 1232 CrossRef CAS PubMed.
- H. Yu, K. Lau, Y. Li, G. Sugiarto and X. Chen, Current Protocols in Chemical Biology, 2012, 4, 233 Search PubMed.
- W. Yi, X. Liu, Y. Li, J. Li, C. Xia, G. Zhou, W. Zhang, W. Zhao, X. Chen and P. G. Wang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4207 CrossRef CAS PubMed.
- K. Lau, V. Thon, H. Yu, L. Ding, Y. Chen, M. M. Muthana, D. Wong, R. Huang and X. Chen, Chem. Commun., 2010, 46, 6066 RSC.
- M. Chen, L. L. Chen, Y. Zou, M. Xue, M. Liang, L. Jin, W. Y. Guan, J. Shen, W. Wang, L. Wang, J. Liu and P. G. Wang, Carbohydr. Res., 2011, 346, 2421 CrossRef CAS PubMed.
- X. Chen, J. W. Fang, J. B. Zhang, Z. Y. Liu, J. Shao, P. Kowal, P. Andreana and P. G. Wang, J. Am. Chem. Soc., 2001, 123, 2081 CrossRef CAS PubMed.
- H. Yu, K. Lau, V. Thon, C. A. Autran, E. Jantscher-Krenn, M. Xue, Y. Li, G. Sugiarto, J. Qu, S. Mu, L. Ding, L. Bode and X. Chen, Angew. Chem., Int. Ed., 2014, 53, 6687 CrossRef CAS PubMed.
- L. P. Miranda and M. Meldal, Angew. Chem., 2001, 113, 3767 CrossRef.
- E. J. Corey and J. O. Link, J. Am. Chem. Soc., 1992, 114, 1906 CrossRef CAS.
- G. H. Veeneman, S. H. van Leeuwen and J. H. van Boom, Tetrahedron Lett., 1990, 31, 1331 CrossRef CAS.
- T. Ghosh, A. Santra and A. K. Misra, RSC Adv., 2014, 4, 54 RSC.
- A. Si and A. K. Misra, ChemistryOpen, 2015 DOI:10.1002/open.201500129.
- B. Yang, K. Yoshida, Z. Yin, H. Dai, H. Kavunja, M. H. El-Dakdouki, S. Sungsuwan, S. B. Dulaney and X. Huang, Angew. Chem., Int. Ed., 2012, 51, 10185 CrossRef CAS PubMed.
- A. Santra, G. Guchhait and A. K. Misra, Green Chem., 2011, 13, 1345 RSC.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010 Search PubMed.
- W. M. Pearlman, Tetrahedron Lett., 1967, 8, 1663 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental details of the synthesis, analytical data of 4–10, 1a–3a, and 1b–3b, and NMR spectra of synthesized compounds. See DOI: 10.1039/c5sc04104j |
| ‡ Current address: Children's National Medical Center, Washington DC, 20010, USA. |
| This journal is © The Royal Society of Chemistry 2016 |