
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmorphous cobalt silicate nanobelts@carbon composites as a stable anode material for lithium ion batteries†
Wei
Cheng
a,
Felix
Rechberger
 a,
Gabriele
Ilari
ab,
Huan
Ma
c,
Wan-Ing
Lin
d and
Markus
Niederberger
*a
a,
Gabriele
Ilari
ab,
Huan
Ma
c,
Wan-Ing
Lin
d and
Markus
Niederberger
*a
aLaboratory for Multifunctional Materials, Department of Materials, ETH Zurich, Vladimir-Prelog-Weg 5, 8093 Zurich, Switzerland. E-mail: markus.niederberger@mat.ethz.ch
bElectron Microscopy Center, Empa, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, 8600 Dübendorf, Switzerland
cLaboratory for Nanometallurgy, Department of Materials, ETH Zurich, 8093 Zurich, Switzerland
dDepartment of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland
First published on 26th August 2015
Abstract
During the past decade, tremendous attention has been given to the development of new electrode materials with high capacity to meet the requirements of electrode materials with high energy density in lithium ion batteries. Very recently, cobalt silicate has been proposed as a new type of high capacity anode material for lithium ion batteries. However, the bulky cobalt silicate demonstrates limited electrochemical performance. Nanostructure engineering and carbon coating represent two promising strategies to improve the electrochemical performance of electrode materials. Herein, we developed a template method for the synthesis of amorphous cobalt silicate nanobelts which can be coated with carbon through the deposition and thermal decomposition of phenol formaldehyde resin. Tested as an anode material, the amorphous cobalt silicate nanobelts@carbon composites exhibit a reversible high capacity of 745 mA h g−1 at a current density of 100 mA g−1, and a long life span of up to 1000 cycles with a stable capacity retention of 480 mA h g−1 at a current density of 500 mA g−1. The outstanding electrochemical performance of the composites indicates their high potential as an anode material for lithium ion batteries. The results here are expected to stimulate further research into transition metal silicate nanostructures for lithium ion battery applications.
Introduction
Lithium ion batteries are nowadays one of the most popular electrical energy storage technologies on the market, because of their relatively high energy density as well as good cycling stability.1–3 Regarding the anode in lithium ion batteries, intercalating graphite is the mostly used commercial material. However, graphite offers a low theoretical capacity of only 372 mA h g−1, and also a relatively poor rate performance. These limitations restrict the use of lithium ion batteries in large scale applications such as in electrical vehicles, electric grids and renewable power plants.3,4 Consequently, extensive research efforts have been dedicated to finding new anode materials with high capacity.5–9 Among all the emerging high capacity anode materials, Si-based inorganic materials, including Si and SiO2, have received most of the attention due to their extremely high theoretical capacities (4200 mA h g−1 for Si and 1965 mA h g−1 for SiO2) and the abundance of the element silicon in the Earth’s crust.10,11 However, both compounds suffer from drastic capacity fading during cycling. This issue can be addressed by a careful engineering of the nanostructure and by depositing a conductive coating.10,11 In addition to Si and SiO2, metal silicates represent another type of potential electrode material. In contrast to lithium metal silicates (Li2MSiO4, M = Fe, Co, Mn), which have been extensively investigated as cathodes,12 metal silicates such as M2SiO4 (M = Co, Fe, Mn) have rarely been studied as electrode materials for lithium ion batteries. Only very recently, Mueller et al. reported that cobalt silicate can be used as an anode which undergoes a conversion reaction during lithiation and delithiation, providing a reversible specific capacity of about 600 mA h g−1.13 However, this bulky cobalt silicate exhibited limited capacity and poor cycling stability and rate performance due to the large particle size. Accordingly, there are still many possibilities to study metal silicates as anode materials and to improve their electrochemical performances by downscaling the particle size to the nm range and by coating them with a conductive layer of carbon.Nanostructured materials as electrode materials possess several advantages: improved kinetics of lithium ions due to their small size, large surface area ensuring sufficient contact between electrode materials and electrolyte and reduced strain during lithiation and delithiation.14 Among the many kinds of morphologies, one dimensional nanostructures are particularly interesting, because they additionally offer enhanced transport of electrons along the long axial direction, favoring the current collection during cycling, and thus leading to an improvement of the electrochemical performance.15,16 Moreover, one dimensional nanostructures are promising building blocks for flexible batteries.17 Therefore, substantial efforts have been devoted to developing strategies for the synthesis of one dimensional electrode materials with high performance in lithium ion batteries.16 On the other hand, one dimensional nanostructures suffer from structure degradation and self-aggregation during cycling.16 These issues can be overcome, if the one dimensional nanostructures are coated with a conductive material like carbon. The carbon coating not only improves the conductivity, but also acts as an efficient elastic buffer layer to accommodate the volume expansion and contraction during lithium ion insertion and extraction, and thus significantly improves the electrochemical performance.18,19
It has been reported that amorphous electrode materials can be electrochemically more active than the corresponding crystalline phase, because the high atomic/ionic mobility in amorphous structures favors lithium ion diffusion and also alleviates the strain arising from the volume changes during cycling.20 However, the electron conductivity in amorphous structures is often poor, which is detrimental to the collection of electrons. Again, carbon coating provides an efficient approach to resolve this issue.
Herein, we report the synthesis of a highly anisotropic and amorphous silicate nanostructure with a carbon coating and its lithium storage properties. The synthesis of these cobalt silicate nanobelts@carbon composites involves the hydrothermal treatment of cobalt-based organic–inorganic nanobelts as a template with an alkaline solution in a glass tube. Under hydrothermal conditions, the OH− ions react with the glass, forming silicate ions which transform the organic–inorganic nanobelts into amorphous cobalt silicate with preservation of the one dimensional particle morphology. The carbon coating is realized by the deposition of phenol formaldehyde resin on the nanobelts, followed by annealing under N2 atmosphere. As a result of the unique structural and compositional design, the composite electrode exhibits outstanding electrochemical performance when tested as an anode material. It can deliver a reversible high capacity of about 745 mA h g−1 at a current density of 100 mA g−1. At a higher current density of 500 mA g−1, the composite exhibits an extraordinary long life of 1000 cycles with a stable capacity retention of about 480 mA h g−1, which is much higher than the theoretical capacity of the commercialized graphite anode. This is the first time that it is demonstrated that such a high capacity and long term cycling stability can be accomplished for metal silicates M2SiO4 (M = Co, Fe, Mn, etc.) as anode materials.
Results and discussion
The anisotropic cobalt silicate nanostructures were synthesized in the presence of cobalt-based organic–inorganic hybrid nanobelts as a template. The hybrid nanobelts were prepared according to our previous work, in which their composition was determined to be Co(OH)0.8(C6H5COO)1.2(2C6H5COOH)0.04·1.86H2O.21 The nanobelts have lengths up to hundreds of micrometers, widths of about 50–200 nm, and thicknesses of about 10–20 nm (Fig. S1†). The hydrothermal treatment of these hybrid nanobelts in a glass tube under alkaline conditions reproducibly results in the formation of amorphous cobalt silicate nanobelts. As shown in the scanning electron microscopy (SEM) images (Fig. 1a and b), the products preserve the one dimensional morphology of the template. The sample looks homogeneous, exclusively consisting of nanobelts. In the transmission electron microscopy (TEM) images, we can clearly distinguish between areas of brighter and darker contrast (Fig. 1c–h). The light contrast stems from the silicate nanobelts, while the dark spots represent Co3O4 nanoparticles. Some of these dark nanoparticles are embedded in the core (Fig. 1e) or are located on the surface of selected nanobelts (Fig. 1f). However, most of the nanobelts are just of light contrast without any dark nanoparticles (Fig. 1c, d, g and h). The nanobelts can reach lengths of 5–10 μm with widths of 150–250 nm. On the basis of atomic force microscopy (AFM) analysis, the thickness of the nanobelts is determined to be about 20–50 nm (Fig. S2†). Therefore, the nanobelts have a width-to-height ratio of 3 to 12.5. The reflections in the X-ray diffraction (XRD) pattern (Fig. S3†) can be assigned to cubic Co3O4 (JCPDS 42-1467), indicating that the dark nanoparticles in the TEM images are Co3O4. Energy-dispersive X-ray spectroscopy (EDX) analysis (Fig. S4a†) reveals that besides cobalt and oxygen, there is a significant amount of elemental Si in the nanobelts. Additionally, the EDX element mapping (Fig. S4b–e†) suggests that Si is homogeneously distributed in the sample. The infrared (IR) spectrum (Fig. S5†) of the product shows typical bands at 577 cm−1 and 664 cm−1 belonging to Co3O4 (ref. 22a) and also a pronounced band at 986 cm−1, typical of the Si–O stretching vibration.22b Based on all these data, it is reasonable to assume that the nanobelts are either composed of cobalt silicate or silica. To determine the exact composition, we performed TEM-EDX analysis (Fig. 2a) and acquired Energy-Filtered Transmission Electron Microscopy (EFTEM) element maps of an individual nanobelt without Co3O4 nanoparticles. The EFTEM maps in Fig. 2b–f are recorded using a conventional three-window technique from the Co L-edge, the Si L-edge and the O K-edge. The results show that the light contrast nanobelts are comprised of all three elements Co, Si and O (Fig. 2a–f). Most importantly, the three elements are homogenously distributed within the nanobelt, proving that their composition is cobalt silicate rather than silica. The XRD pattern (Fig. 3) after annealing in air at 800 °C can unambiguously be assigned to Co2SiO4 (JCPDS 01-079-0791) and Co3O4 (JCPDS 42-1467). The high intensity of the Co3O4 reflections indicates that no Co3O4 is consumed during annealing, making a reaction between SiO2 and Co3O4 improbable. Instead, Co2SiO4 is the result of the crystallization of the amorphous nanobelts, indirectly confirming their composition as cobalt silicate. | ||
| Fig. 1 (a and b) SEM and (c–h) TEM images of the as-synthesized cobalt silicate nanobelts. | ||
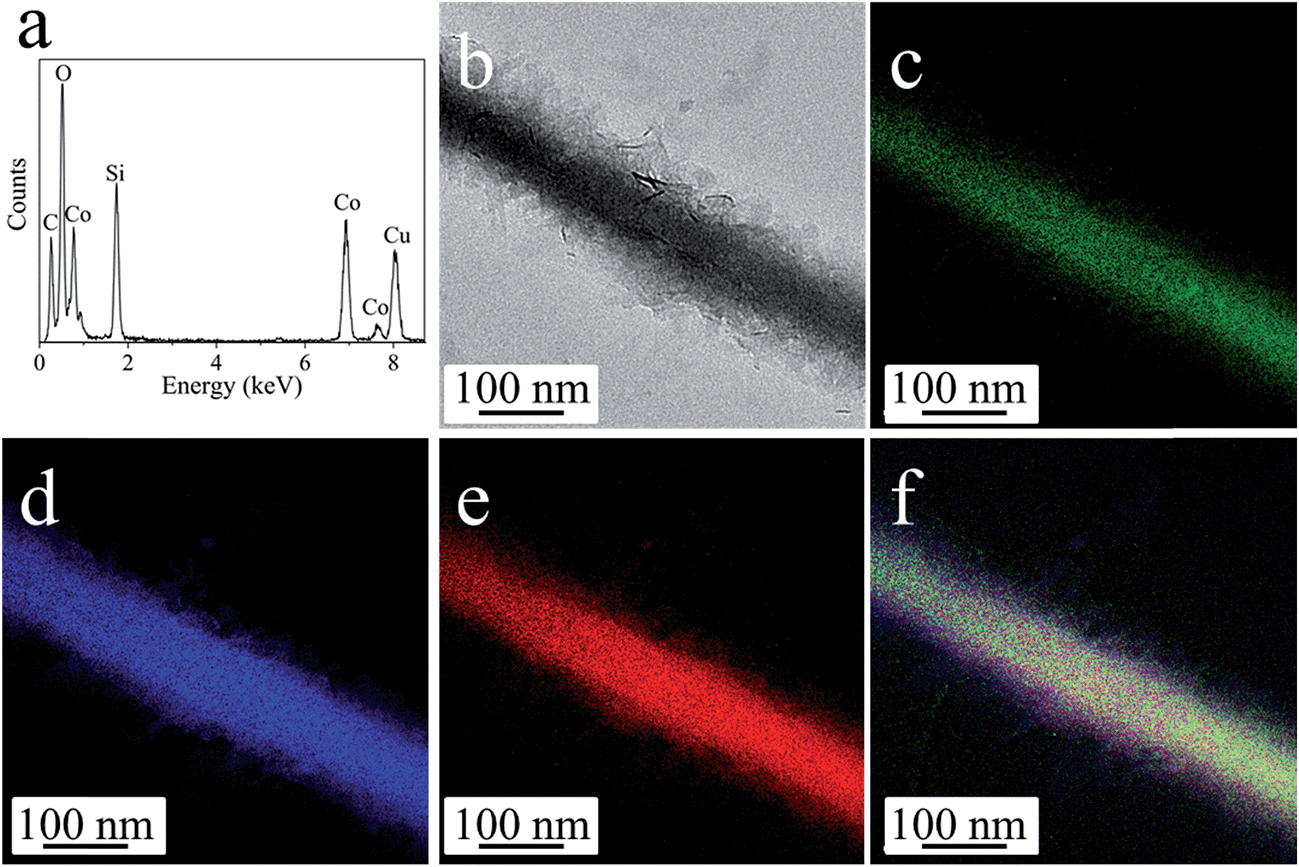 | ||
| Fig. 2 (a) EDX spectrum taken from a single nanobelt, in which the C and Cu signals originate from the TEM grid. (b–f) EFTEM images: (b) zero-loss image, (c) Co L-edge, (d) Si L-edge, (e) O K-edge, (f) overlapping of Co, Si and O maps. | ||
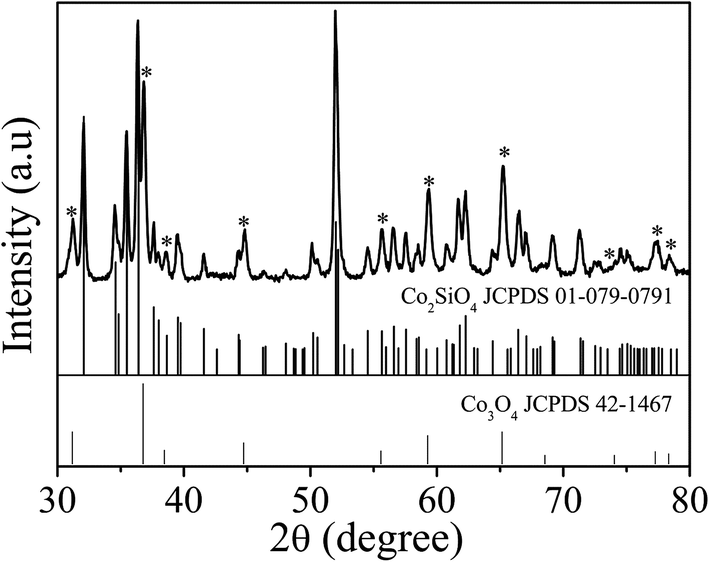 | ||
| Fig. 3 XRD pattern of the nanobelts@Co3O4 nanoparticles after annealing in air at 800 °C. The reflections marked with asterisks can be assigned to Co3O4 (JCPDS 42-1467). All other reflections match with Co2SiO4 (JCPDS 01-079-0791). | ||
Based on the above characterization results, we propose the following mechanisms for the reaction of Co(OH)0.8(C6H5COO)1.2(2C6H5COOH)0.04·1.86H2O into cobalt silicate and Co3O4. During the hydrothermal treatment in aqueous NaOH, the hydroxyl ions react with the Co-based precursor particles as well as with the glass tube inside the Teflon container. The Co-based hybrid transforms into cobalt hydroxide, which subsequently reacts with oxygen to give Co3O4 (eqn (1))23 and with SiO32− (product of the reaction of OH− with SiO2 from the glass tube, eqn (2)) to give Co2SiO4 (eqn (3)).
| 6Co(OH)2 + O2 → 2Co3O4 + 6H2O | (1) |
| SiO2 + 2OH− → SiO32− + H2O | (2) |
| 2Co(OH)2 + SiO32− → Co2SiO4 + H2O + 2OH− | (3) |
The freshly synthesized cobalt silicate nanobelts can be well dispersed in water, as shown in Fig. S6.† The high stability is ascribed to the relatively large zeta potential of about −32.6 mV, originating from the OH− groups on the surface of the nanobelts, as verified by IR measurement (Fig. S5†). The good dispersibility of the nanobelts in water allows us to uniformly deposit phenol formaldehyde resin (PFR) on them (Fig. S7†) by hydrothermal processing of the cobalt silicate nanobelts with phenol and hexamethylenetetramine (HMT).24 Under hydrothermal conditions, HMT decomposes into NH3 and formaldehyde (Fig. S8,† reaction (1)). Then, the newly formed formaldehyde and phenol polymerize into PFR on the surface of the cobalt silicate nanobelts (Fig. S8,† reaction (2)). The PFR coating can be transformed into amorphous carbon by annealing under protection of N2 at 500 °C. The XRD pattern (Fig. S9†) indicates that during the carbonization process the Co3O4 nanoparticles present in the sample are reduced to CoO (JCPDS 01-070-2856). The absence of any other reflections suggests that the cobalt silicate is still amorphous. SEM analysis (Fig. 4a–d) shows that the nanobelt morphology was well-preserved after carbon coating. The coated carbon can be clearly detected by EDX (Fig. S10a†). In addition, the element maps (Fig. S10b–f†) show that Co, Si, O and C are homogeneously distributed in the sample, indicating a uniform carbon coating on the nanobelts. According to the TEM images (Fig. 4e–g), some CoO nanoparticles, identified by XRD and shown as dark dots, are distributed around the nanobelts. Due to the similar low contrast of the thin sheet-like carbon and the amorphous cobalt silicate nanobelts, it is difficult to distinguish the carbon shell from the nanobelt cores. Nevertheless, the carbon is uniformly coated on the nanobelts, as confirmed by the EDX element maps (Fig. S10f†). The presence of carbon can be further confirmed by Raman spectroscopy analysis of the composite. In Fig. S11,† we can observe two pronounced peaks. One is located at 1342 cm−1, which can be identified as the D band of carbon, indicating disordered carbon.25 The other one at 1582 cm−1 can be assigned to the G band of carbon, suggesting the presence of graphitic carbon.25 The higher intensity of the G band in comparison to the D band implies a high ratio of sp2 graphitic carbon in the carbon coating. A higher content of graphitic carbon gives rise to better electronic conductivity, which is crucial to improve the electrochemical performance of amorphous electrode materials. It is important to mention that the amount of carbon coating can be tuned by using different concentrations of phenol and HMT for the polymerization of PFR on the nanobelts. In a typical synthesis using 6 mM phenol and 3 mM HMT, we obtain cobalt silicate nanobelts@carbon with a weight ratio of 20.9 wt%, as determined by thermogravimetric analysis (TGA) (Fig. S12†). By reducing the concentrations of phenol and HMT by half, the weight ratio of carbon decreases to 9.9 wt%, while a doubling of their concentrations increases the carbon content to 36.7 wt%.
 | ||
| Fig. 4 (a–d) SEM and (e–g) TEM images of cobalt silicate nanobelts@carbon. | ||
We investigated the potential application of cobalt silicate nanobelts@carbon as an anode material in lithium ion batteries. The CoO impurities, although in principle electrochemically active, were not removed, because they only represent the minor phase in the cobalt silicate nanobelts@carbon. Fig. S13† shows the first three cyclic voltammetry (CV) scans of the composite in the voltage range from 0.01 to 3 V. Except for the initial cathodic scan, the current peak positions in the rest of the CV curves are the same as for bulk cobalt silicate,13 indicating that our composite undergoes a similar conversion reaction. In this mechanism, the Co2+ is initially reduced to metallic Co nanograins distributed in a Li4SiO4 matrix and can be restored to cobalt silicate in the subsequent delithiation step.13 The mechanism is comparable with the reversible electrochemical reaction of lithium with transition metal oxides, such as CoO, Co3O4, CuO and Fe3O4.8,26 The peaks in the CV curves of our composites are, however, less defined than in the case of bulk cobalt silicate, probably due to the amorphous nature of our nanobelts. Clearly, the details of the reaction mechanism need further clarification. Nevertheless, after the irreversible reaction occurring in the initial discharge, all additional CV curves almost overlap, indicating highly reversible lithiation and delithiation reactions for the composite.
Fig. 5a displays representative charge–discharge profiles of cobalt silicate nanobelts@carbon at a current density of 500 mA g−1 in the voltage window from 0.01 V to 3.0 V. In the initial discharge, the composite electrode delivers a high capacity of 1159.4 mA h g−1. Unless otherwise stated, the capacity is calculated based on the total weight of the composite. Considering that the composite contains 20.9 wt% carbon, the capacity with respect to the cobalt silicate nanobelts is actually as high as 1465.7 mA h g−1, while the bulk Co2SiO4 exhibits a first discharge capacity of only 906.0 mA h g−1 at 80 mA g−1.13 In the subsequent charging step, the composite can only deliver a capacity of 620.9 mA h g−1, corresponding to a coulombic efficiency of 53.3%. The large capacity loss in the first cycle can be ascribed to irreversible electrochemical reactions occurring during the initial discharge, the inevitable formation of the solid electrolyte interphase (SEI) and also electrolyte decomposition, all of which are commonly observed in most anode materials.10,11,20,26,27 After the first discharge–charge cycle, the shapes of the profiles remain basically the same upon further cycling, indicating highly reversible electrochemical reactions, which is in good agreement with the CV analysis. The coulombic efficiency significantly increases to 87.6% at the second, 97.3% at the 10th and 99.1% at the 20th cycle, then maintains this level upon further cycling.
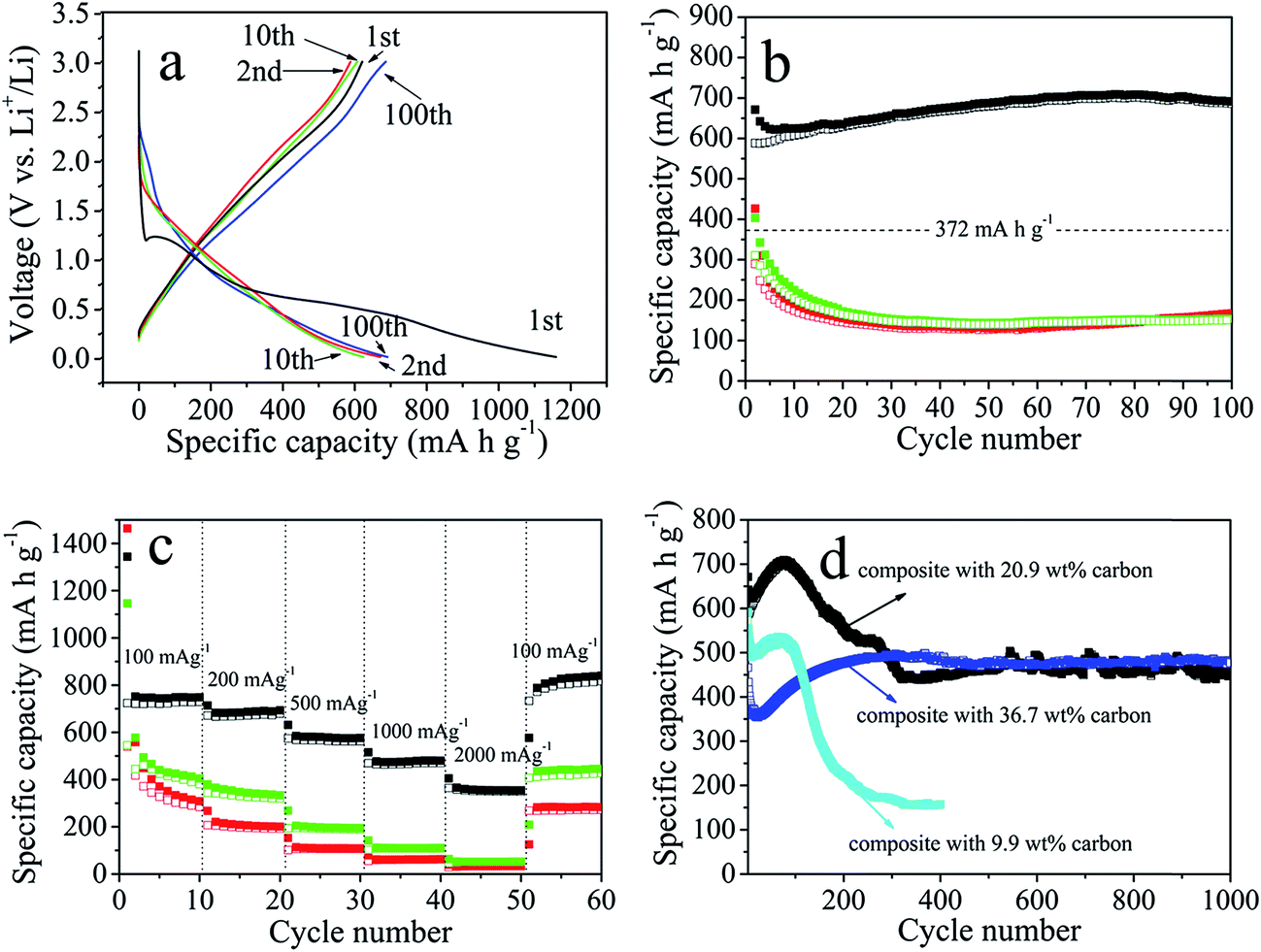 | ||
| Fig. 5 Electrochemical performance of the cobalt silicate nanobelts@carbon composites: (a) charge–discharge voltage profiles for the 1st, 2nd, 10th and 100th cycle at 500 mA h g−1; (b) cycling performance at 500 mA h g−1: cobalt silicate nanobelts@carbon composite with 20.9 wt% carbon (black), as-synthesized cobalt silicate nanobelts (red), and cobalt silicate nanobelts annealed at 500 °C under N2 (green); (c) rate performance of cobalt silicate nanobelts@carbon composite with 20.9 wt% carbon (black), as-synthesized cobalt silicate nanobelts (red), and cobalt silicate nanobelts annealed at 500 °C under N2 (green); (d) long term cycling stability at 500 mA h g−1: cobalt silicate nanobelts@carbon composite with 20.9 wt% carbon (black), composite with 9.9 wt% carbon (cyan), composite with 36.7 wt% carbon (blue). Voltage window 0–3.0 V. In (b–d), the solid and empty squares represent the discharge and charge steps, respectively. | ||
Fig. 5b (black curve) shows the cycling performance of the composite electrode. Starting from the second cycle, the charge capacity gradually increases from 588.4 mA h g−1 to 701.0 mA h g−1 at the 80th cycle, then slowly decreases to 686.4 mA h g−1 at the 100th cycle. The capacity increase in the first 80 cycles is presumably due to the reversible formation of polymeric layers on the electrode materials, which has already been observed and also confirmed in other transition metal oxide electrode materials with conversion mechanisms.25a,b As a comparison, we also measured the cycling performance of freshly synthesized amorphous cobalt silicate nanobelts before and after annealing at 500 °C under N2. The amorphous cobalt silicate before annealing can only deliver a very low initial charge capacity of about 427.9 mA h g−1, which quickly drops to a low capacity of about 140 mA h g−1 (Fig. 5b, red). The annealing step does not improve the electrochemical performance (Fig. 5b, green). The bad performance is anticipated to be associated with the poor electronic conductivity of the amorphous cobalt silicate nanobelts. The carbon coated sample shows a more than 5 times higher capacity than the non-coated sample, indicating that carbon coating leads to a dramatic enhancement of the electrochemical performance of the amorphous electrode materials.
The rate performance of the cobalt silicate nanobelts@carbon has also been investigated. Fig. 5c shows that at 100 mA g−1 the capacity of the composite electrode can reach a stable reversible value of about 745 mA h g−1, while bulk Co2SiO4 provides a reversible specific capacity of about 600 mA h g−1 at 80 mA g−1.13 Increasing the current density to 200 mA g−1, 500 mA g−1, 1000 mA g−1 and 2000 mA g−1, the specific capacity is reduced to 688 mA h g−1, 580 mA h g−1, 478 mA h g−1 and 360 mA h g−1, respectively. Lowering the current density again to 100 mA g−1 results in a capacity of 830 mA h g−1, which is considerably higher than the 745 mA h g−1 achieved in the initial 10 cycles at 100 mA g−1. In comparison, the nanobelts without carbon coating display very poor rate performance. At a very high current density of 2000 mA g−1, almost no capacity can be delivered for the non-coated sample, further confirming the importance of the carbon coating for the enhancement of the electrochemical properties of the amorphous electrode materials.
To further demonstrate the suitability of this unique electrode design as an anode material for lithium ion batteries, we tested the long term stability up to 1000 cycles at 500 mA g−1. As displayed in Fig. 5d, for the composite with 20.9 wt% carbon, in the first 80 cycles the capacity gradually increases to 701.0 mA h g−1, followed by a decrease until the 330th cycle. Afterwards, the capacity is very stable up to 1000 cycles with a capacity retention of about 450 mA h g−1. This value is considerably higher than the theoretical capacity of a graphite anode of about 372 mA h g−1. It is important to note that the amount of carbon coating has a crucial influence on the cycling performance of the composite electrode materials. As shown in Fig. 5d, for the composite with 9.9 wt% carbon, the capacity is much lower than that of the composite with 20.9 wt%. It experiences a slow increase in the initial 80 cycles to 530 mA h g−1, followed by a drastic decrease to 223 mA h g−1 after 200 cycles. The low capacity and the poor stability could be due to the insufficient carbon coating, which fails to protect the structural integrity during cycling. Upon increasing the carbon content to 36.7 wt%, the capacity is less than that of both other samples in the initial 110 cycles. However, in contrast to the other two samples with less carbon, the capacity does not show an obvious drop after the initial increase, but continues to rise to 480 mA h g−1 at the 300th cycle. Then, the value remains at this level until 1000 cycles are reached. Obviously, the large amount of carbon coating compromises the capacity in the initial cycles, but also promotes the cycling stability. The tunable carbon coating allows us to obtain a composite electrode with optimized electrochemical performance. Such an outstanding long term cycling performance has never been achieved for cobalt silicate and silica.11,13 In fact, such a good cyclability has also been seldom observed for other conversion-type transition metal oxide anodes.25–27
The attractive electrochemical properties of our cobalt silicate nanobelts@carbon composite can be interpreted from the following perspectives. First of all, based on the aforementioned results, the carbon coating plays a central role in achieving the extraordinary electrochemical performance. The carbon layer can significantly increase the electronic conductivity, which solves the issue of the poor conductivity as a result of the amorphous nature of the cobalt silicate nanobelts. Additionally, it keeps the electrode structure intact and prevents particle aggregation typically caused by the volume expansion and contraction during cycling, greatly improving the cycling stability. We performed TEM analysis of the electrode materials after cycling. For the carbon coated sample, the one dimensional structure is well preserved after long term cycling. No apparent structure damage or collapse was observed (Fig. S14†). The nanobelts without carbon coating, on the other hand, suffer severely from structural fracture and particle agglomeration (Fig. S15†), giving evidence for the critical role of the carbon coating for the preservation of the structural integrity of the nanobelts. Moreover, it is known from the literature that the carbon coating is able to limit the thickness of the SEI layer and prevents the cracking of the SEI layer, hence effectively avoiding irreversible capacity during charge and discharge.18,19 Secondly, the thin nanobelt structures offer a short lithium diffusion pathway along the direction perpendicular to the nanobelts, while the electron transport along the long axial direction of the composite facilitates the current collection. The composite exhibits a large Brunauer–Emmett–Teller (BET) surface area of about 440 m2 g−1 (Fig. S16†), which ensures an optimal contact between the electrode and the electrolyte, and thus supports the lithium ion flux from the electrolyte into the electrode material. Thirdly, the cobalt silicate nanobelts here are amorphous, and the relatively high atomic and ionic mobility in amorphous materials with homogeneous, isotropic texture favors the lithium ion diffusion and mitigates the strain caused by the volume change during cycling.20 The synergistic effect of these advantageous features contributes to the final substantial improvement of the electrochemical performance of the composite electrode.
Conclusions
In summary, we developed a template method for the synthesis of amorphous cobalt silicate nanobelts with high aspect ratios. Their good dispersibility in water enables us to deposit phenol formaldehyde resin on their surface, which can easily be transformed into a carbon layer. The resulting cobalt silicate nanobelts@carbon composites exhibit excellent lithium ion storage properties. Specifically, they can deliver a highly reversible capacity of 745 mA h g−1 at a current rate of 100 mA h g−1. The long term cycling test shows that at a current density of 500 mA g−1, the composite can be cycled 1000 times with a high remaining capacity of 480 mA h g−1. Our study reveals that the carbon coating holds the key to the excellent electrochemical behavior. In addition, the one dimensional and amorphous nature of the composite might also contribute to the superior electrochemical properties. This study marks the first example to demonstrate that by nanostructure engineering and carbon nanocoating a transition metal silicate is able to deliver a high reversible capacity with an extremely long life span, which makes them a new type of potential anode material for lithium ion batteries. Our work may trigger further research efforts into other nanostructured metal silicates with conductive coatings for energy storage applications.Acknowledgements
We thank ETH Zurich, China Scholarship Council and the Swiss National Science foundation (Project No. 200020_144437) for financial support. We acknowledge the use of equipment in the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zurich and in the Electron Microscopy Center of EMPA Dübendorf.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664–2680 CrossRef CAS.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 CAS.
- S. Goriparti, E. Miele, F. de Angelis, E. Di Fabrizio, R. Proietti Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS.
- (a) C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS; (b) M. H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS; (c) A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS; (d) N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS; (e) D. Chen, X. Mei, G. Ji, M. Lu, J. Xie, J. Lu and J. Y. Lee, Angew. Chem., Int. Ed., 2012, 51, 2409–2413 CrossRef CAS; (f) H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS; (g) H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed; (h) N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS.
- (a) M. Miyachi, H. Yamamoto, H. Kawai, T. Ohta and M. Shirakata, J. Electrochem. Soc., 2005, 152, A2089–A2091 CrossRef CAS; (b) B. Guo, J. Shu, Z. Wang, H. Yang, L. Shi, Y. Liu and L. Chen, Electrochem. Commun., 2008, 10, 1876–1878 CrossRef CAS; (c) M. Sasidharan, D. Liu, N. Gunawardhana, M. Yoshio and K. Nakashima, J. Mater. Chem., 2011, 21, 13881–13888 RSC; (d) Y. Yao, J. Zhang, L. Xue, T. Huang and A. Yu, J. Power Sources, 2011, 196, 10240–10243 CrossRef CAS; (e) N. Yan, F. Wang, H. Zhong, Y. Li, Y. Wang, L. Hu and Q. Chen, Sci. Rep., 2013, 3, 1568 Search PubMed; (f) Z. Favors, W. Wang, H. H. Bay, A. George, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 4605 Search PubMed; (g) M. Li, Y. Yu, J. Li, B. Chen, X. Wu, Y. Tian and P. Chen, J. Mater. Chem. A, 2015, 3, 1476–1482 RSC.
- M. S. Islam, R. Dominko, C. Masquelier, C. Sirisopanaporn, A. R. Armstrong and P. G. Bruce, J. Mater. Chem., 2011, 21, 9811–9818 RSC.
- F. Mueller, D. Bresser, N. Minderjahn, J. Kalhoff, S. Menne, S. Krueger, M. Winter and S. Passerini, Dalton Trans., 2014, 15013–15021 RSC.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS.
- A. I. Hochbaum and P. D. Yang, Chem. Rev., 2010, 110, 527–546 CrossRef CAS.
- L. Mai, X. Tian, X. Xu, L. Chang and L. Xu, Chem. Rev., 2014, 114, 11828–11862 CrossRef CAS.
- G. Zhou, F. Li and H. M. Cheng, Energy Environ. Sci., 2014, 7, 1307–1338 CAS.
- L. Su, Y. Jing and Z. Zhou, Nanoscale, 2011, 3, 3967–3983 RSC.
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC.
- (a) Z. Wang, Z. Wang, W. Liu, W. Xiao and X. W. Lou, Energy Environ. Sci., 2013, 6, 87–91 RSC; (b) W. S. Chang, C. M. Park, J. H. Kim, Y. U. Kim, G. Jeong and H. J. Sohn, Energy Environ. Sci., 2012, 5, 6895–6899 RSC; (c) L. F. Cui, R. Ruffo, C. K. Chan, H. Peng and Y. Cui, Nano Lett., 2008, 9, 491–495 CrossRef; (d) R. Yi, J. Feng, D. Lv, M. L. Gordin, S. Chen, D. Choi and D. Wang, Nano Energy, 2013, 2, 498–504 CrossRef CAS; (e) X. L. Wang, W. Q. Han, H. Chen, J. Bai, T. A. Tyson, X. Q. Yu, X. J. Wang and X. Q. Yang, J. Am. Chem. Soc., 2011, 133, 20692–20695 CrossRef CAS.
- M. Cao, I. Djerdj, Z. Jaglicic, M. Antonietti and M. Niederberger, Phys. Chem. Chem. Phys., 2009, 11, 6166–6172 RSC.
- (a) C.-W. Tang, C.-B. Wang and S.-H. Chien, Thermochim. Acta, 2008, 473, 68–73 CrossRef CAS; (b) M. Stoia, M. Stefanescu, T. Dippong, O. Stefanescu and P. Barvinschi, J. Sol-Gel Sci. Technol., 2010, 54, 49–56 CrossRef CAS.
- Y. Dong, K. He, L. Yin and A. Zhang, Nanotechnology, 2007, 18, 435602 CrossRef.
- S. R. Guo, J. Y. Gong, P. Jiang, M. Wu, Y. Lu and S. H. Yu, Adv. Funct. Mater., 2008, 18, 872–879 CrossRef CAS.
- (a) D. Bresser, E. Paillard, P. Niehoff, S. Krueger, F. Mueller, M. Winter and S. Passerini, ChemPhysChem, 2014, 15, 2177–2185 CrossRef CAS; (b) D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513–523 CrossRef CAS; (c) C. He, S. Wu, N. Zhao, C. Shi, E. Liu and J. Li, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS.
- (a) G. Zeng, N. Shi, M. Hess, X. Chen, W. Cheng, T. Fan and M. Niederberger, ACS Nano, 2015, 9, 4227–4235 CrossRef CAS; (b) H. Zhang, L. Zhou, O. Noonan, D. J. Martin, A. K. Whittaker and C. Yu, Adv. Funct. Mater., 2014, 24, 4337–4342 CrossRef CAS; (c) A. Ponrouch, P. L. Taberna, P. Simon and M. R. Palacín, Electrochim. Acta, 2012, 61, 13–18 CrossRef CAS; (d) Y. Sun, X. Hu, W. Luo and Y. Huang, J. Mater. Chem., 2012, 22, 13826–13831 RSC; (e) Y. Qi, H. Zhang, N. Du and D. Yang, J. Mater. Chem. A, 2013, 1, 2337–2342 RSC; (f) L. Wang, H. Gong, C. Wang, D. Wang, K. Tang and Y. Qian, Nanoscale, 2012, 4, 6850–6855 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, AFM images, XRD patterns, SEM images, SEM-EDX element mapping, TEM images, Raman spectrum, TGA analysis, IR spectra, electrochemical characterization and N2 gas sorption measurements. See DOI: 10.1039/c5sc02525g |
| This journal is © The Royal Society of Chemistry 2015 |