
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational synthesis of normal, abnormal and anionic NHC–gallium alkyl complexes: structural, stability and isomerization insights†‡
Marina
Uzelac
,
Alberto
Hernán-Gómez
,
David R.
Armstrong
,
Alan R.
Kennedy
and
Eva
Hevia
*
WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: eva.hevia@strath.ac.uk
First published on 3rd July 2015
Abstract
Advancing the rational design of main-group N-heterocyclic carbene complexes, this study reports the synthesis, X-ray crystallographic and NMR spectroscopic characterisation of a novel series of Ga complexes containing neutral or anionic NHC ligands using the unsaturated carbene IPr (IPr = 1,3-bis-(2,6-di-isopropylphenyl)imidazol-2-ylidene). Starting from normal adduct GaR3·IPr (1) (R = CH2SiMe3), the addition of polar LiR led to the formation of NHC-stabilised gallate species IPr·LiGaR4 (2), resulting from co-complexation of the single-metal species. Contrastingly, reversing the order of addition of these organometallic reagents, by treating unsaturated free IPr, first with LiR followed by GaR3, furnished novel heteroleptic gallate (THF)2Li[:C{[N(2,6-iPr2C6H3)]2CHCGa(CH2SiMe3)3}] (3), which contains an anionic NHC ligand acting as an unsymmetrical bridge between the two metals, coordinating through its abnormal C4 position to Ga and through its normal C2 position to Li. Electrophilic interception studies of 3 using methyl triflate (MeOTf), methanol and imidazolium salt (IMes·HCl) led to the isolation and structural elucidation of the two novel neutral abnormal NHC (aNHC) complexes [CH3C{[N(2,6-iPr2C6H3)]2CHCGa(CH2SiMe3)3}] (4) and aIPr·GaR3 (5) (aIPr = HC{[N(2,6-iPr2C6H3)]2CHC}). These studies disclose the preference of the anionic IPr ligand present in 3 to react with electrophiles via its C2 position, leaving its Ga–C4 bond intact. Abnormal complex 5 can also be accessed by a thermally induced rearrangement of its normal isomer 1. Combining NMR spectroscopic and kinetic studies with DFT calculations, new light has been shed on this intriguing transformation, which suggests that it occurs via a dissociative mechanism, highlighting the importance of the donor ability of the solvent used in these thermal isomerizations as well as the steric bulk of the substituents on the NHC and the Ga reagent. These findings intimate that relief of the steric hindrance around Ga by forming an abnormal complex is a key driving force behind these rearrangements.
Introduction
Over the past two decades, N-heterocyclic carbenes (NHCs), in particular imidazol-2-ylidenes, have progressed from mere curiosities to commodity neutral σ-donor ligands with a multitude of applications in synthesis and materials.1 Typically, the carbene centre is located between the two nitrogen atoms (C2 position) allowing π-donation by both adjacent N-heteroatoms into the empty pπ orbital of the carbene, which makes these ligands remarkably more stable than other non-cyclic, all-carbon counterparts.2,3 Acting as strong σ-donors, these versatile ligands have been pivotal to recent breakthroughs in transition-metal catalysis.4–9 Furthermore, the application of NHCs to main group chemistry has enabled the stabilisation of novel low valent compounds,10–17 as well as the development of several frustrated Lewis pair (FLP) systems.18,19In parallel to these studies, a different type of NHC complex has been developed where the imidazole ring binds to the metal centre through its backbone. These less stabilised carbenes, where there is only one N-atom adjacent to their carbenic-C,20 have been termed as abnormal (or mesoionic) NHCs (aNHCs).21–23 Following Crabtree's seminal report in 2001 of the first transition-metal complex with an aNHC, several other examples have been prepared.24 However, it was only in 2009 when Bertrand succeeded in the isolation of the first stable free aNHC by the elegant deprotonation of a 1,2,3,4-tetraarylated imidazolium chloride.25,26
Interestingly, experimental and theoretical studies point to aNHCs being better donors than their normal counterparts, which is in part attributed to their reduced steric congestion.25,27–29 Thus Layfield has recently reported a thermally induced rearrangement of IPr·Fe(HMDS)2 [IPr = 1,3-bis-(2,6-di-isopropylphenyl)imidazol-2-ylidene, HMDS = 1,1,1,3,3,3-hexamethyldisilazide] which after 3 h in refluxing toluene evolves to its abnormal isomer.30 Within main group chemistry, the number of complexes containing aNHCs remains very limited. Thus the first example of an adduct of this type, a substituted phosphinidene complex, was reported by Carty in 2006.31 Related thermal rearrangements as that mentioned above in iron chemistry have been proposed for tris(pentafluorophenyl)borane NHC systems which exhibit FLP chemistry.32–34 Similarly, within group 13 Dagorne has shown the isomerization of ItBu·AlMe3 [IBu = 1,3-bis(tert-butyl)imidazol-2-ylidene] to its C4 bound isomer (aItBu·AlMe3) at room temperature in THF, although the mechanisms involved in these processes remain unclear.35
In addition to these isomerization studies, Robinson has demonstrated that anionic NHCs, resulting from the lithiation of the imidazole backbone of unsaturated NHCs,36 can be employed as platforms to access aNHC-complexes of B and Zn by quenching the relevant anionic B or Zn complex with a suitable electrophile such as HCl·NEt3 or MeOTf.37–39 More recently, Lavallo et al. have shown that implementing carborane anions as N-imidazolium substituents allows the selective formation of both normal and abnormal NHC constitutional isomers from a single precursor.40 In addition, the authors report a thermal isomerization from abnormal to normal NHC, which can be proton catalyzed.
Building on these precedents, exporting this chemistry to the heavier group 13 metal gallium, and using the unsaturated carbene IPr as a case study, here we report a novel series of normal, anionic and abnormal NHC complexes derived from the same metal fragment, tris(alkyl)gallium Ga(CH2SiMe3)3. Combining X-ray crystallographic, kinetic and spectroscopic studies with theoretical investigations we assess the constitution and stability of these complexes. An insightful comparison between two alternative synthetic methods to access aNHC complexes: namely metallation/electrophilic interception and thermal isomerisation, is also provided. It should be noted that although there are several examples of Ga–NHC adducts reported in the literature,41–45 with some of them finding applications as π-acid catalysts,46,47 the only abnormal NHC complex known to date aIPr·GaCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39 was reported as recently as 2014, although its synthesis is not straightforward as it was obtained by transmetallation of GaCl3 with an anionic NHC mixed Li–B complex.
39 was reported as recently as 2014, although its synthesis is not straightforward as it was obtained by transmetallation of GaCl3 with an anionic NHC mixed Li–B complex.
Results and discussion
NHC-stabilised lithium gallate complexes
We started our studies by reacting equimolar amounts of IPr and trimethylsilylmethylgallium(III)48 (GaR3) [R = CH2SiMe3] at room temperature in non-polar hexane which afforded colourless crystals of the adduct IPr·GaR3 (1) in a 75% isolated yield (Scheme 1). The molecular structure of 1 (Fig. 1) was elucidated by a single crystal X-ray diffraction analysis which revealed the formation of a complex with the four-carbon-coordinated gallium atom attached to three alkyl groups and the C2 (i.e. C1 in Fig. 1) of a neutral carbene. A distorted tetrahedral geometry adopted by Ga centre is evidenced by the C–Ga–C bond angles which range from 96.36(6)° to 119.43(7)° (average angle 108.79°). The Ga–Calkyl distances range from 2.0034(15) Å to 2.0164(16) Å (mean 2.0106 Å) which is only slightly elongated (by ∼2.5%) when compared to parent monomeric GaR3 (Ga–C bonds ranging from 1.952(4) Å to 1.971(3) Å, average 1.959 Å)49 in agreement with the increase in the coordination number of Ga in 1. | ||
| Scheme 1 Synthesis of normal adduct 1, homoleptic tetraalkyl gallate 2 and heteroleptic gallate 3. | ||
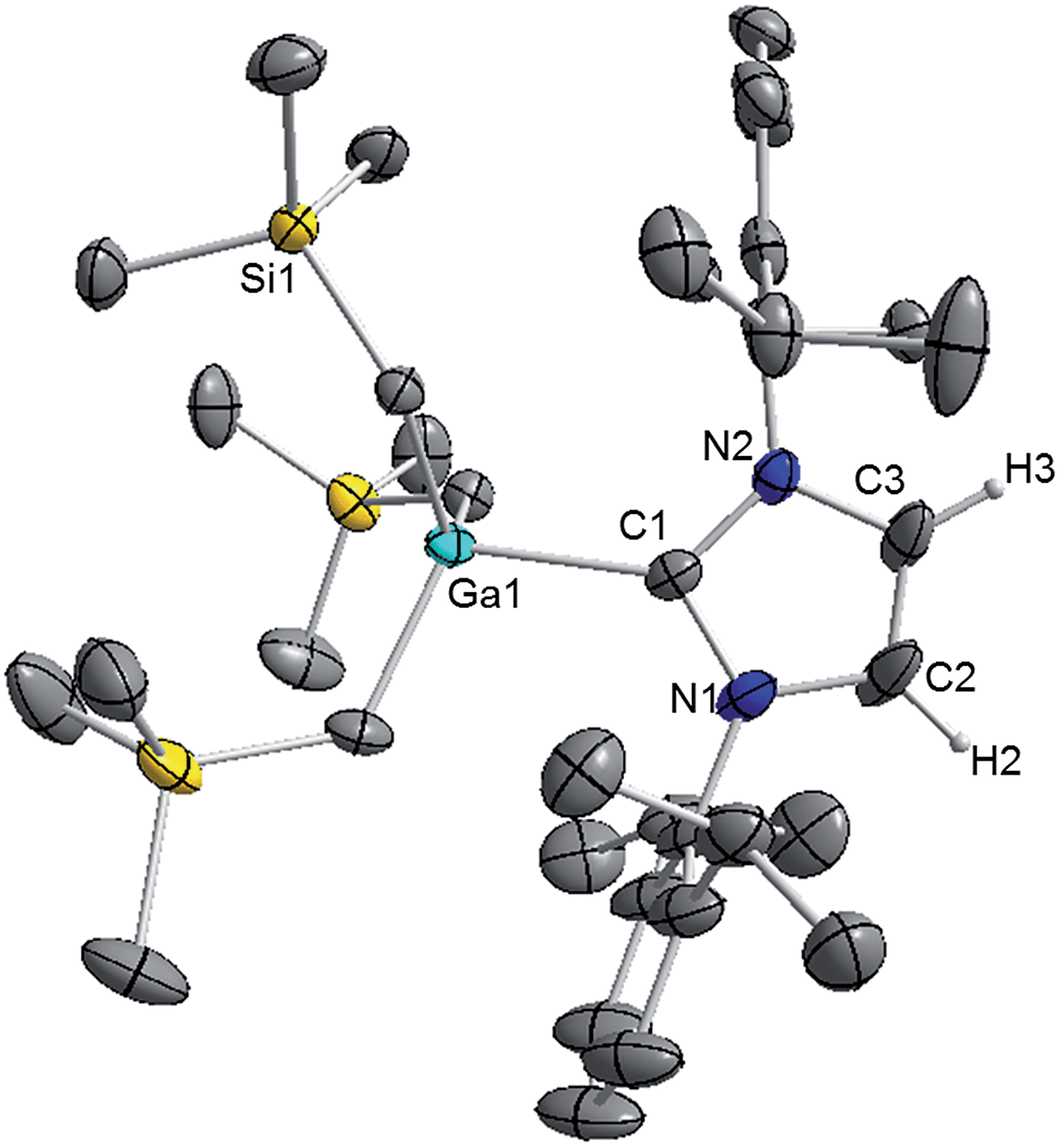 | ||
| Fig. 1 Molecular structure of 1 with 50% probability displacement ellipsoids. All hydrogen atoms except those on the imidazole ring and minor disorder in one isopropyl group and one monosilyl group are omitted for clarity. | ||
Noticeably, the Ga–CNHC distance of 2.1960(16) Å is significantly longer compared to that observed in the related Ga halide complex IPr·GaCl3 (2.016(2) Å).42 This elongation can be rationalised in terms of a combination of the greater steric congestion in 1 imposed by the monosilyl groups as well as the stronger Lewis acidity of GaCl3 compared to GaR3.
Despite the long Ga–C1 distance, it should be noted that 1 retains its integrity in C6D6 solution as evidenced by DOSY NMR studies, which show that the IPr and monosilyl groups belong to the same sized species, as the cross-point for both ligand resonances are aligned in the second dimension (average D value = 6.2 × 10−10 m2 s−1; see Fig. S39 in ESI‡). The most informative resonance in the 13C NMR spectrum is that for the carbenic carbon at 186.6 ppm, a chemical shift consistent with retention of the Ga–C bond in solution.50
Next the reactivity of 1 towards LiCH2SiMe3 was investigated. Previous work by Roesky and Stalke51 has shown that when borane adduct IPr·BH3 is treated with BuLi, lithiation of the C4 position of the imidazole ring takes place affording an anionic NHC which binds through its C4 position to Li, leaving the B–C2 bond untouched.51 Similar reactivity has also been described for the alkylborane IPr·BEt3.36
Interestingly in our studies, the polar organometallic RLi fails to deprotonate the NHC ligand of 1, affording instead lithium gallate [IPr·LiGa(CH2SiMe3)4] (2) in an isolated yield of 48%. Single crystal X-ray diffraction analysis established the molecular structure of [IPr·LiGa(CH2SiMe3)4] which represents to the best of our knowledge the first example of an alkali-metal gallate stabilised by an NHC ligand (Fig. 2).52 Compound 2 exhibits a contacted ion pair (CIP) motif where the two metals are connected by two bridging alkyl groups with the neutral NHC binding via its C2 (i.e. C1) position to lithium.
 | ||
| Fig. 2 Molecular structure of 2 with 30% probability displacement ellipsoids. All 3 CH2SiMe3 groups are disordered. Only one component of the disordered model is shown above. All hydrogen atoms except those on the imidazole ring have been omitted for clarity. | ||
These findings show that under these reaction conditions the polar Li alkyl reagent preferentially co-complexes with GaR3, to yield [LiGaR4]53 which is then trapped and stabilised by the neutral NHC ligand, instead of lithiating the carbene backbone. Clearly the Ga atom favours coordination of another R anion rather than a neutral IPr ligand. It should be noted that a similar reactivity has been reported for IPr·ZntBu2 with tBuLi in hexane, producing zincate complex [IPr·LiZntBu3].54 Gallate 2 can also be prepared by reacting polymeric [{LiGaR4}∞] with free IPr. Solution state studies of 2 were hindered by its poor solubility in arene solvents such as C6D6; whereas in coordinating THF, the adduct dissociates into free IPr and multi-THF-solvated LiGaR4, as evidenced by multinuclear NMR spectroscopy (see ESI‡). Contrastingly, if the order of the monometallic reactants is reversed, by treating first IPr with LiR followed by the addition of gallium alkyl GaR3 in THF, heteroleptic (THF)2Li[:C{[N(2,6-iPr2C6H3)]2CHCGa(CH2SiMe3)3}] (3) was obtained in a 56% isolated yield. As shown in Scheme 1, 3 is the result of an indirect gallation process where IPr is first metallated at the C4 position by polar LiR,36 which in turn undergoes transmetallation to the lower polarity GaR3. X-Ray crystallographic studies established the CIP structure of 3 where now the metals are connected by an anionic NHC which coordinates as an asymmetric bridge via its normal C2 position to Li and its abnormal C4 position to Ga (Fig. 3). The C2–Li (i.e. C1 in Fig. 3) distance is 2.093(5) Å which is similar to those reported for related complexes containing anionic NHC bridged in a similar C2–Li/C4–M fashion to Li/Al and Li/B pairings.36,38
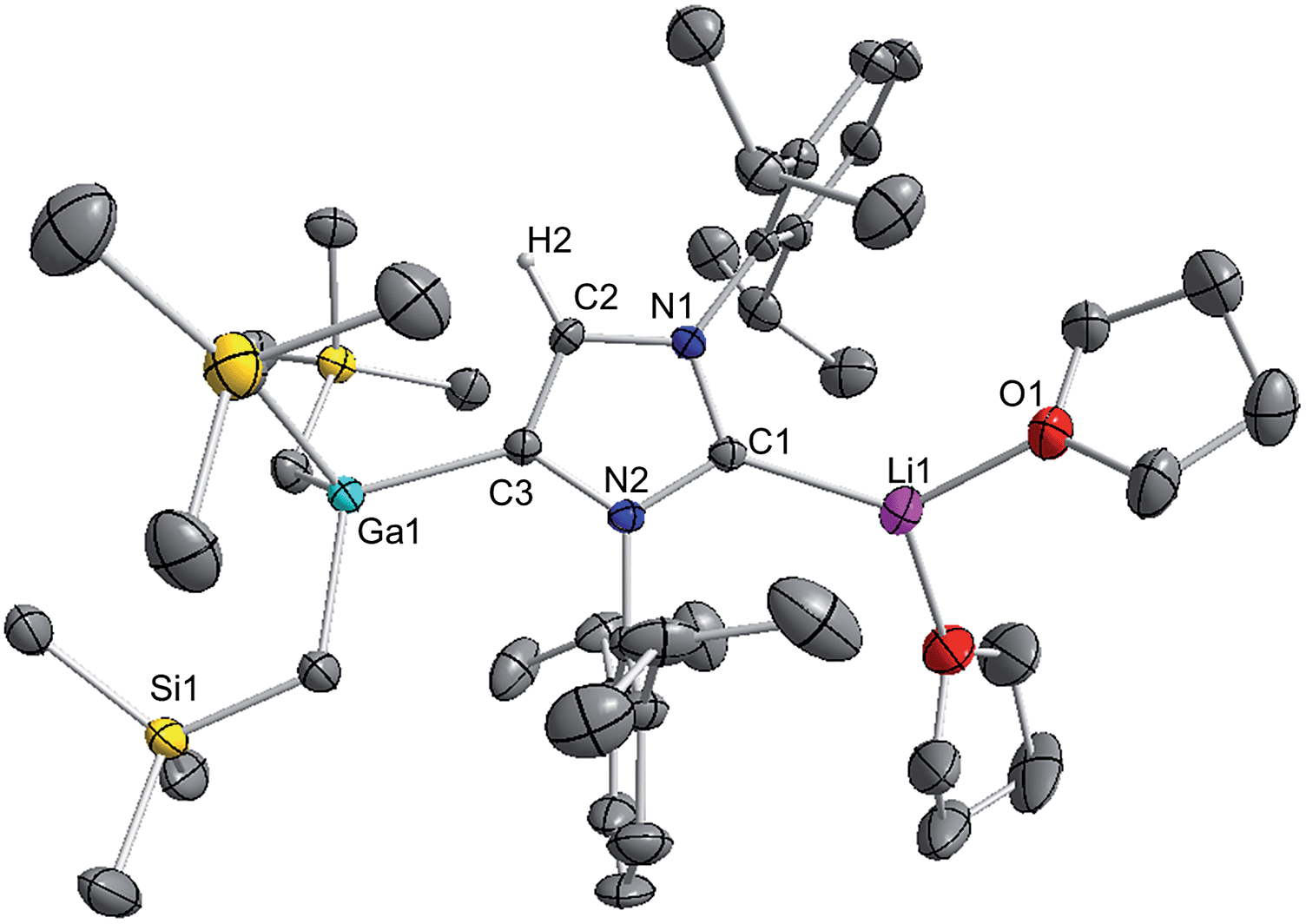 | ||
| Fig. 3 Molecular structure of 3 with 50% probability displacement ellipsoids. All hydrogen atoms except H2 on the imidazole ring and minor disorder of THF ligand have been omitted for clarity. | ||
The Ga–C4 distance (i.e. C3 in Fig. 3) of 2.052(2) Å is close in value with Ga–Calkyl bonds (average 2.028 Å)55 and understandably it is significantly shorter (by 0.144 Å) to that found in the neutral C2 bound IPr adduct 1. It is noteworthy, that unlike in 1 where a pyramidalization of Ga coordination sphere was evident (vide supra), in 3 gallium atom exhibits nearly ideal tetrahedral geometry with the average bond of 2.034 Å and mean angle of 109.46° (angles ranging from 105.68(9)° to 112.99(10)°). This decrease in distortion around the metal centre can be attributed to the relief of the steric congestion of 3 when compared to 1. From the NMR data in d8-THF solutions (see ESI‡), metallation of IPr was demonstrated by the large downfield chemical shift of the C4 resonance in the 13C NMR spectrum (from 122.3 ppm in free IPr to 155.1 in 3), as well as an informative singlet at 6.64 ppm (integral 1H) in the 1H NMR spectrum of the imidazole CH (versus 7.19 ppm in free IPr). In addition, a resonance in the 13C NMR spectrum at 201.4 ppm for carbenic C2 confirms the formation of a NHC complex. The loss of symmetry in the imidazole ring is evidenced in the 1H and 13C NMR spectra with the appearance of two distinct sets of Dipp signals.
Abnormal NHC–Ga complexes via electrophilic interception
Recent studies have shown that certain anionic NHC complexes, when treated with an electrophile can be transformed into neutral abnormal adducts.38,39 To explore this reactivity here, we treated 3 with a molar equivalent of MeOTf in toluene at −78 °C. The reaction occurred with the formation of a white precipitate (presumably LiOTf) furnishing a neutral abnormal NHC–Ga complex [CH3C{[N(2,6-iPr2C6H3)]2CHCGa(CH2SiMe3)3}] (4) in a 68% yield (Scheme 2a). Complex 4 results from the selective C2 methylation of the anionic NHC leaving the Ga–C4 bond intact (see ESI‡ for experimental details and NMR spectroscopic characterisation). | ||
| Scheme 2 Electrophilic interception of anionic NHC complex 3 with (a) MeOTf, (b) MeOH and (c) imidazolium salt IMes·HCl. | ||
The molecular structure of 4 was established by X-ray crystallographic studies (Fig. 4). The bond length of 1.538(6) Å for C2–CMe (i.e. C1–C4 in Fig. 4) is consistent with a single bond, while the Ga–C4 (i.e. C3 in Fig. 4) bond length of 2.087(3) Å is only slightly elongated to that found in the anionic variant 3 (2.052(2) Å), and significantly shorter than the Ga–C2 bond length in 1 (2.1960(16) Å). Reflecting the formation of a neutral abnormal complex, the 13C NMR spectrum of 4 shows a resonance at 161.2 ppm for the C4 attached to Ga (vs. 155.1 ppm in 3) whereas the methylated carbon (originally C2 carbenic position in 3) resonates significantly upfield at 145.0 ppm in comparison with that observed for 3 (at 201.5 ppm).
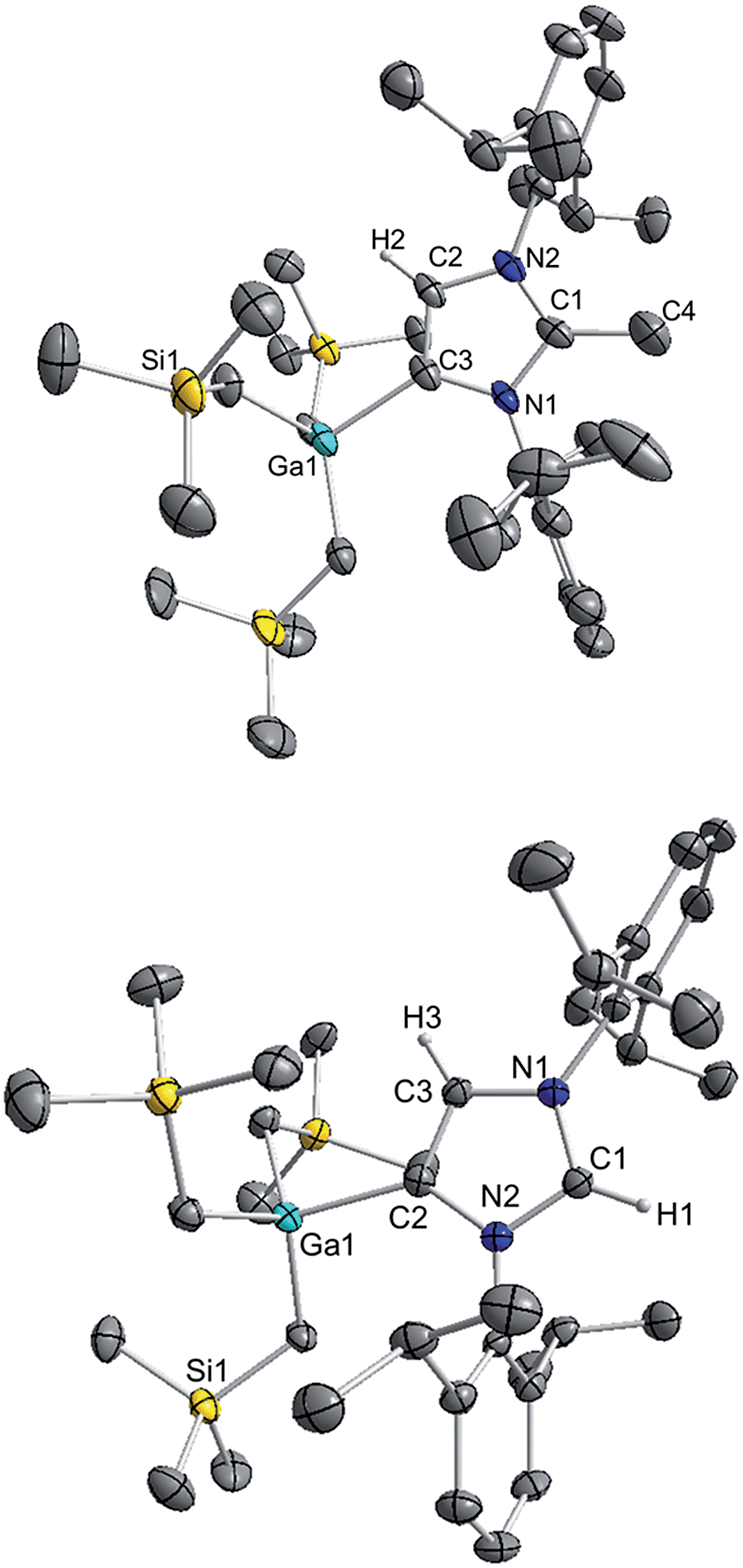 | ||
| Fig. 4 Molecular structure of 4 (top) and 5 (bottom) with 50% probability displacement ellipsoids. Only one component of disordered groups is shown. All hydrogen atoms except those on the imidazole rings have been omitted for clarity. | ||
The use of methanol as a quenching reagent resulted in clean conversion of 3 to the abnormal adduct aIPr·GaR3 (5) (Scheme 2b). Notably, 5 is also formed as a protonation product from the reaction of 3 with the imidazolium salt IMes·HCl (1,3-bis-(2,4,6-trimethylphenyl)imidazolium chloride) (Scheme 2c). These findings not only demonstrate that the C2 position of 3 in the imidazole ring is its preferred basic site, but also the high strength of its Ga–C4 bond as it is retained in 5. Furthermore in view of these results it appears that for the GaR3 fragment aIPr is a better ligand than the related normal IMes carbene (obtained from deprotonation of the imidazolium salt, Scheme 2c), as no ligand exchange occurs. Compound 5 was isolated as a crystalline solid in a 61% yield (see ESI‡ for experimental details and NMR spectroscopic studies) and its molecular structure was established by X-ray crystallography (Fig. 4).
The Ga–C4 bond length (i.e. Ga–C2 in Fig. 4) of 2.0759(16) Å in 5 is significantly shorter than the corresponding bond in the normal congener 1 (by 0.1201 Å), supporting previous studies which suggest that abnormal carbenes are stronger σ-donors, less sterically congested, and consequently are able to form stronger bonds with metal centres. In fact, as previously discussed for 4, despite the neutral constitution of the aNHC ligand, the strength of this interaction is similar to that observed for the Ga–C bond of the anionic carbene present in 3.
As mentioned above, the only example of an abnormal NHC Ga complex prior to this work has been reported by Robinson,39 where aIPr·GaCl3 was formed while attempting the transmetallation of mixed lithium/boron anionic NHC complex with GaCl3. Interestingly the Ga–Ccarbene distance of this complex (1.978(3) Å) differs only by 0.097 Å to that found for 5 (2.0759(16) Å). This contrasts with the markedly different bond distances found when comparing the relevant normal isomer (Ga–Ccarbene bond distance in IPr·GaX3, 2.1960(16) Å when X = R (1); vs. 2.016(2) Å when X = Cl), hinting that in the abnormal systems, due to the increased steric space around the metal centre, the size of the anionic groups attached to Ga has a significantly smaller influence than in the normal adducts.
1H NMR spectrum of 5 in d8-THF showed a diagnostic singlet at 9.00 ppm belonging to the H attached to the C2 position of the carbene, whereas the remaining H in the imidazole ring resonates at 7.20 ppm. Similarly to that found in 4, the 13C NMR spectrum shows two informative singlets at 162.8 and 139.1 ppm which can be assigned to Ga–C4 and NC(H)N respectively.
Compound 5, along with 1 and 3 constitute a rare example of a series of normal, anionic and abnormal complexes incorporating the same metal–coligand partnership.56 Complementary DFT computational studies57 were undertaken on these three compounds employing the B3LYP method58 and the 6-311G** basis set59 (see ESI‡ for details). Natural bond orbital (NBO) analysis60 of the optimised structures of IPr·GaR3 (IIIIPr), aIPr·GaR3 (IVIPr) and (THF)2Li[:C{[N(2,6-iPr2C6H3)]2CHCGa(CH2SiMe3)3}] (VIPr) suggest considerable covalent character of the Ga–C bonds. The Ga natural charges range from +1.43 to +1.35 and the Wiberg bond indices (WBIs) of the Ga–C bonds range from 0.45–0.64. Contrastingly reflecting the more ionic nature of the Li–C contact in VIPr, the natural charge of Li is +0.88 and the WBI of the Li–C is 0.08. In agreement with our experimental findings the estimated Ga–C4 bonds in IVIPr and VIPr are significantly shorter (2.146 and 2.054 Å, WBIs = 0.56 and 0.53 respectively) than the Ga–C2 bond in the normal complex IIIIPr (2.333 Å, WBI = 0.45), although it should be noted that for IIIIPr and IVIPr, the strength of these Ga–C bonds is somewhat underestimated (vide infra). A comparative natural charges analysis of these three models shows that while in bimetallic VIPr the amount of electrons transfer to the GaR3 unit is 0.34, in the case of adducts IIIIPr and IVIPr these values are 0.27 and 0.31 which is consistent with the neutral constitution of the NHC ligands.
Fig. 5 shows the highest occupied molecular orbitals (HOMOs) calculated for models IIIIPr, IVIPr and VIPr which in all cases correspond to the Ga–C bonding orbitals at the CH2 groups of the monosilyl ligands, involving also in the case of VIPr the C4 of the anionic NHC. For this bimetallic system, these calculations contrast with those reported for the related anionic lithium dicarbene [:C{[N(2,6-iPr2C6H3)]2CHCLi(THF)}]n prepared by Robinson,36 whose HOMO and HOMO−2 correspond to the two strongly polarised Li–C bonding orbitals at the C2 and C4 positions of the imidazole ring.
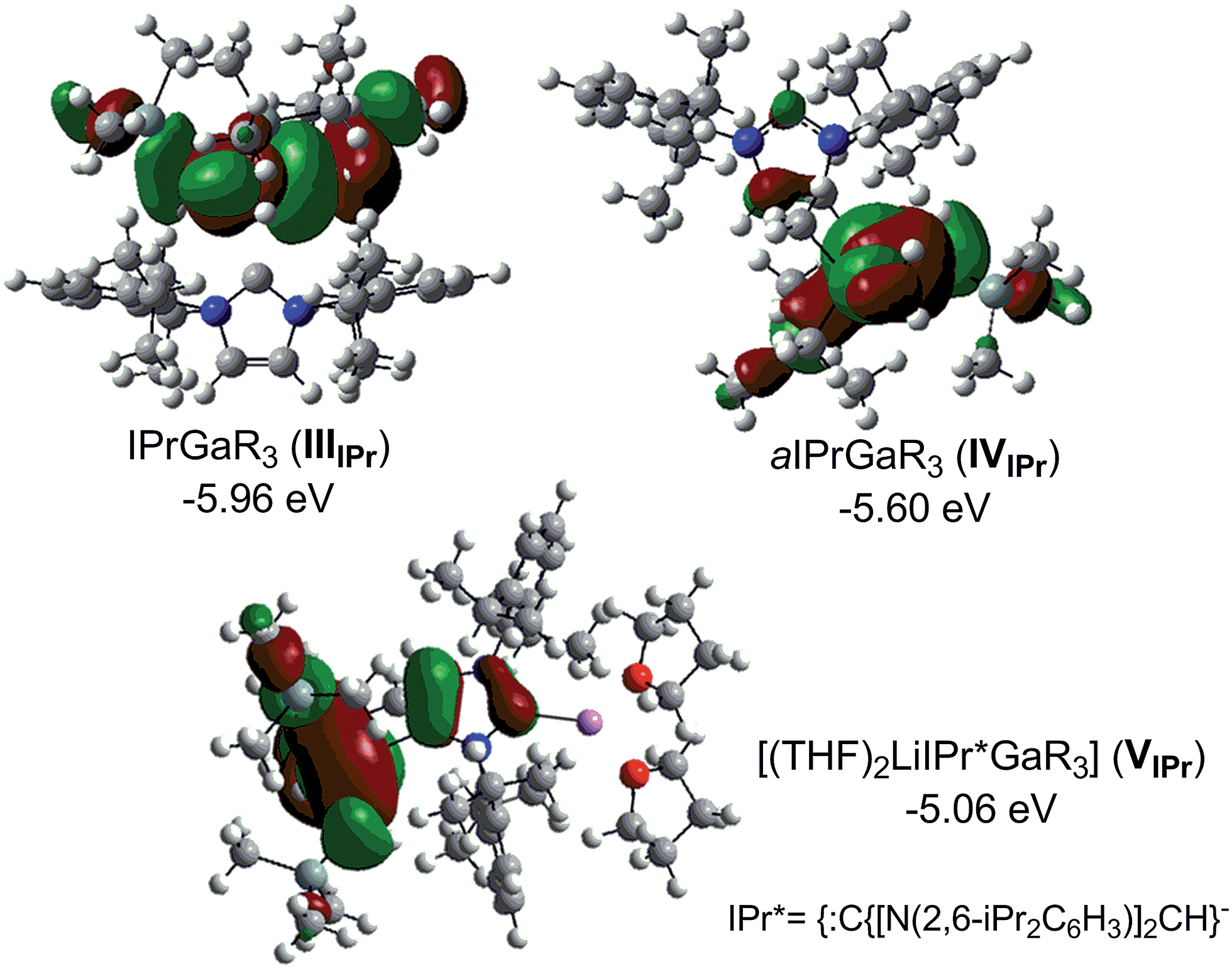 | ||
| Fig. 5 Calculated molecular orbitals HOMO of models IIIIPr, IVIPr and VIPr. | ||
Interestingly, calculations on the regioisomeric structure of VIPr with the positions of the {GaR3} and {Li(THF)2}+ were reversed, giving rise to Ga–C2 and Li–C4 coordination modes (model VIIPr in ESI‡) showed that this model is significantly less stable (by 11.7 kcal mol−1) which is consistent with the formation of a significantly weaker (longer) Ga–C bond (2.272 Å for VIIPrvs. 2.054 Å for VIPr, see ESI‡ for details).
Normal to abnormal isomerization
It is rare to find examples where both normal and abnormal isomers have been structurally characterised.61 Amongst them, intriguing studies from the groups of Layfield30 and Dagorne35 have shown that the systems IPr·Fe(HMDS)2 and ItBu·AlMe3 respectively, thermally isomerize to the relevant abnormal species although the possible reaction pathways for these transformations remain obscure. Similarly to our findings for complexes 1 and 5, in these Fe and Al examples, analysis of the metal–C distances have revealed that the abnormal NHCs bind more strongly to the metal centres than the isomeric normal carbenes. These studies also suggest the formation of the abnormal-NHC complex is thermodynamically controlled with steric factors strongly influencing isomerization processes. Since compound 5 was obtained using an indirect method (metallation/electrophilic interception), we pondered if such types of thermal rearrangement would also be in operation when 1 was heated in solution.Indeed, heating a d6-benzene solution of 1 at 100 °C and monitoring progress by 1H NMR, produced 5 in 77% yield after 10 h. The reaction was greatly accelerated by using the more coordinating solvent d8-THF (75% conversion in only 1 h) Scheme 3.62 Previous studies on ItBu·AlMe3 have shown that the isomerization is much faster using a Lewis donor solvent such as THF, hinting that dissociation (at least partially) of the carbene from the metal must play a significant role in the process. Interestingly no isomerization is observed for IPr·GaCl3 when using smaller and stronger Lewis acid GaCl3, as mentioned above, the carbene binds significantly more strongly to the Ga centre. Related to these findings, reflecting the relevance of the Lewis acidic character of the metal fragment, using the related alkyl compounds of other s-block metals such as MgR2·IPr63 and ZnR2·IPr64 (6) complexes [R = CH2SiMe3] no rearrangements were observed after 72 h at 100 °C.
 | ||
| Scheme 3 Thermally-induced rearrangement of 1 into 5. | ||
The effect of the steric bulk of the substituents on the NHC ligand was assessed. IMes·GaR3 (7), containing the less bulky 1,3-bis-(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes)65 carbene, rearranges at a significantly slower rate than 1, showing after 30 hours at 100 °C in d8-THF a modest 8% conversion to its abnormal isomer. Contrastingly, 1,3-bis(tert-butyl)imidazol-2-ylidene (ItBu) failed to form a normal adduct with GaR3, furnishing instead only the abnormal isomer aItBu·GaR3 (8) at room temperature within one hour. This reactivity contrasts with that reported for the iron complex ItBu·Fe(HMDS)2 which undergoes thermal decomposition,30 and appears to be more in line with that reported by Tamm for the B(C6F5)3/ItBu frustrated Lewis pair system (FLP) which has been used for the activation of small molecules such as H2 or alkynes. Although the two components fail to give an isolable normal complex, in the absence of other reactive substrates, the irreversible formation of the relevant abnormal carbene–borane adduct is observed.32–34
DFT calculations57
Encouraged by the formation of several aNHC–Ga complexes using this approach, we performed theoretical calculations at the DFT level employing the B3LYP method58 and the 6-311G** basis set59 to optimize structures and to gain new insights into the thermodynamics involved in these processes. A comparison of geometrical parameters of optimised structures IPr·GaR3 (IIIIPr) and aIPr·GaR3 (IVIPr) shows in general good agreement with those found experimentally from the X-ray determinations of 1 and 5 respectively (see Fig. 6 and Table S3‡) although for both models there is a slight underestimation of the strength of the Ga–Ccarbene interaction (Δ[d(Ga–C)calc − d(Ga–C)exp) = 0.137 and 0.070 Å for 1 and 5 respectively). Interestingly model IVIPr was computed to be more stable than IIIIPr by just 1.5 kcal mol−1. Using the same level of theory we also found that free aIPr is 16.2 kcal mol−1 less stable than its normal isomer IPr.66 Notably, the dissociation energy of IVIPr was found to be 17.7 kcal mol−1 higher than for IIIIPr (Fig. 7), in agreement with our experimental findings which suggest a greater donor ability of the abnormal NHC ligand for the GaR3 fragment when compared to its normal isomer.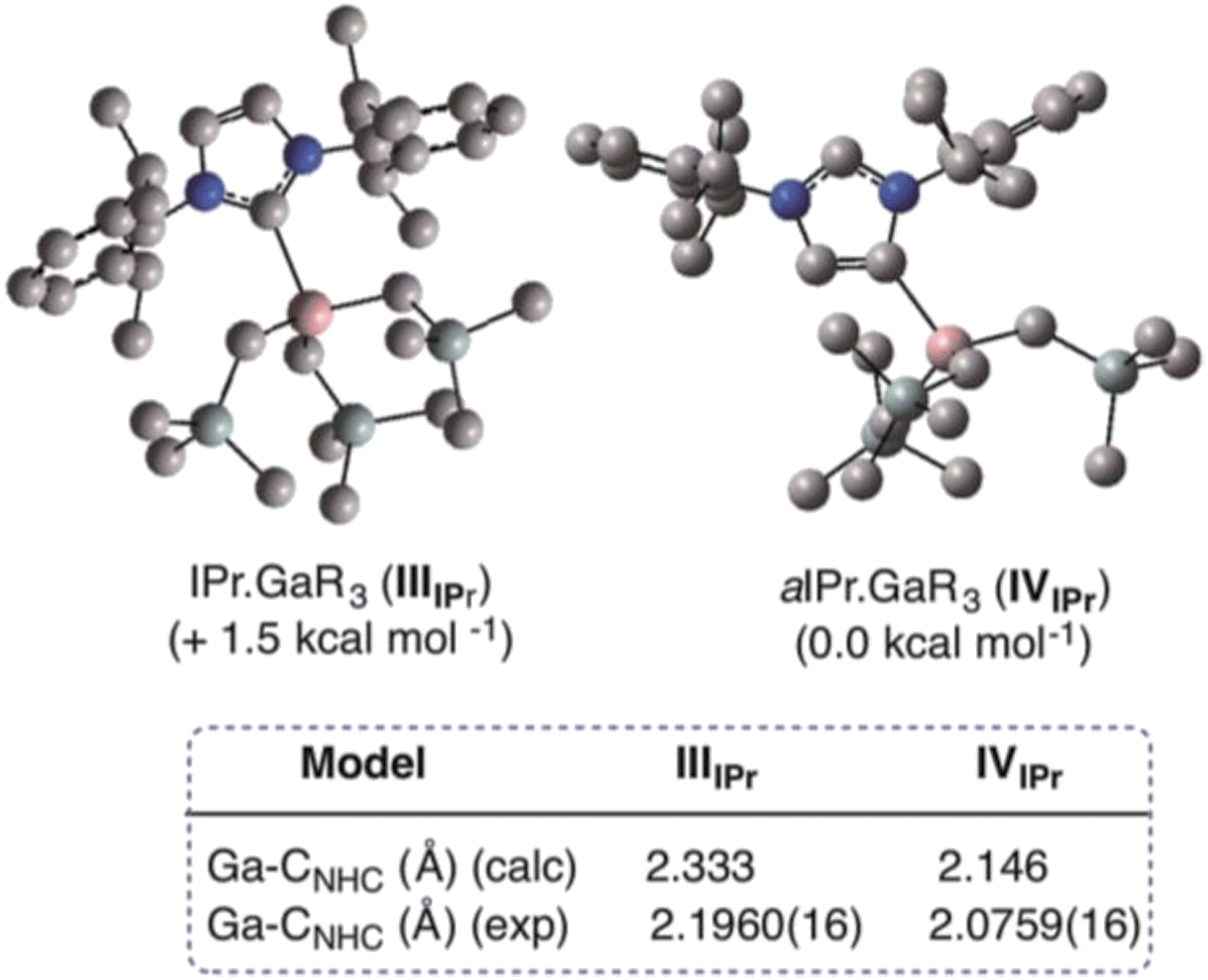 | ||
| Fig. 6 Modelled structures and relative energies of NHC-adducts IIIIPr and IVIPr. | ||
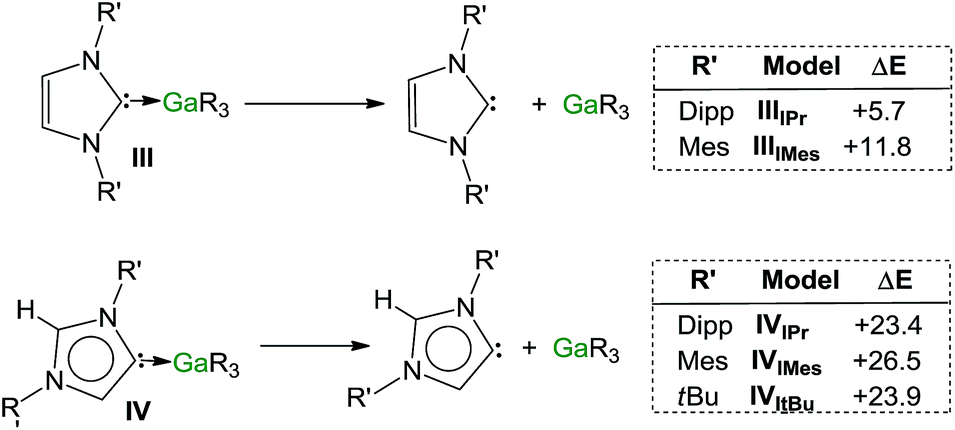 | ||
| Fig. 7 Estimated dissociation energies (kcal mol−1) of complexes III and IV (R = Dipp, Mes, tBu). | ||
These studies were extended to the related carbenes IMes and ItBu. For IMes, which is less sterically demanding than IPr, the order of stability of IIIIMes and IVIMes is reversed, with the normal isomer IIIIMes being 2.1 kcal mol−1 more stable (see ESI‡). Interestingly, the energy difference between free aIMes and IMes of +16.8 kcal mol−1 is almost identical to that for free aIPr and IPr. As shown in Fig. 7, the calculated values for the dissociation energies of IIIIMes and IVIMes follow the same trend as described for the IPr complexes, although now the dissociation of IMes·GaR3 (IIIIMes) is noticeably more endothermic (by 6.1 kcal mol−1) than in IPr·GaR3 (IIIIPr). These subtle but significant changes in the energy values could explain the lower conversions observed experimentally when IMes·GaR3 (7) is heated in d8-THF (max yield 8% for aIMes·GaR3, vide supra), as the dissociation of IMes·GaR3 is more thermodynamically challenging.
A more dramatic effect is observed for ItBu, which containing aliphatic tBu substituents, is significantly bulkier than IPr, more basic and therefore a better donor, from an electronic perspective.67 Notably attempts to optimize the structure of ItBu·GaR3 were unsuccessful, as all the obtained models showed no stabilisation compared to the separate constituents ItBu and GaR3. This lack of coordination between ItBu and GaR3 can best be explained by steric incompatibility of tBu and monosilyl groups and supports our experimental findings that when ItBu and GaR3 are mixed at RT aItBu·GaR3 (8) is formed. The dissociation energy for this abnormal complex was found to be +23.9 kcal mol−1 (Fig. 7).
Collectively these computational results not only offer further support for the greater donor ability of abnormal NHC ligands compared to their normal isomers but also highlight the crucial role that the steric profile plays in these isomerization processes. Thus, in the case of IMes, the less bulky of the carbenes investigated, it becomes slightly endothermic, whereas for IPr and ItBu, the formation of the abnormal complexes is thermodynamically favoured by −1.5 and −6.6 kcal mol−1 respectively, in accord with their sizes.
Mechanistic implications
Both computational and spectroscopic studies suggest that the isomerization of 1 into 5 may involve a dissociative step. Supporting this assumption, when a mixture of 1 and GaR3 (two equivalents) was heated at 100 °C in d8-THF, the formation of 5 becomes significantly slower (46% conversion observed after 1 h), which can be rationalised in terms of the effect that the excess of this reagent will have in the equilibrium depicted in eqn (1), namely shift it towards the left.| GaR3–IPr ⇌ GaR3 + IPr | (1) |
Contrastingly, when the reaction is carried out using an excess of IPr (2 equivalents), the isomerization process occurs significantly faster (90% after 30 minutes). In order to shed some light on the mechanism involved in this isomerization process, kinetic analysis of a NMR scale reaction ([1] = 0.22 M) performed at 100 °C in d8-THF revealed a pseudo-zeroth-order kinetics over a period of two-half lives (64% conversion).62 An identical experiment using IPrD·GaR3 (1D) (IPrD = 1,3-bis(2,6-di-isopropylphenyl)-4,5-dideutero-imidazolin-2-ylidene)68 allowed the comparison of the subsequent zero order rate constants, revealing no observable KIE (see ESI‡ for details). These findings suggest that the bond cleavage of the C4–H in the IPr ligand is unlikely to be rate-determining in the isomerization process. Encouraged by these findings we chose to study further the mechanism of this process using the method of initial rates.69,70Table 1 shows the results of the initial rate experiments conducted in a sealed NMR tube (50 °C, 1, d8-THF). These data were fitted to the approximately linear region of the formation of 5 restricted to conversions of 5–7% in order to calculate the initial rate, ro of the reaction. Entries 1–4 demonstrate a first-order dependence on the concentration of complex 1 while Fig. 8a shows this correlation graphically. A first-order dependence is also observed for the concentration of IPr as shown in entries 2, 5–7 and Fig. 8b, which is consistent with the dissociative step previously discussed and the involvement of free IPr in the isomerization process. By contrast to IPr, a negative order of −1 is observed for the concentration of GaR3 (entries 8–10, Fig. 8c). A plausible interpretation of these results is that the isomerization process takes place by the partial dissociation of 1 (which appears to be the rate-determining step of the reaction)71 to form free IPr that in turn can activate the H atom from the backbone of the NHC ligand coordinated to Ga in complex 1.72 This proposed modus operandi is similar to that described for the activation of small molecules such as acetylenes, H2 or amines using NHC/borane FLP systems, that, as mentioned before, in the absence of another substrate form the relevant abnormal aNHC–BAr3 adducts (Ar = C6F5 or XyF6).32–34,73 This could lead to the formation of the transient ion-pair species [IPr–H]+[IPr*GaR3]− (A) (IPr* = :C{[N(2,6-iPr2C6H3)]2CHC), comprising an imidazolium cation and an NHC-gallate containing an anionic NHC (which on the basis of the constitution of lithium gallate 3, it could be expected to have its Ga center coordinated to the C4 position) (Fig. 9). This reactivity can be interpreted in terms of “thermally induced frustration,” a concept recently introduced by Pápai which refers to the thermal activation of strained dative bonds of bulky Lewis donor–acceptor pairs.74 Intermediate A will evolve fast with the irreversible formation of abnormal complex 5 and the regeneration of free IPr. This proposed behaviour mirrors that described in Scheme 2c, for the protonation of the anionic carbene present in 3 by the imidazolium salt IMes·HCl, which occurs at the C2 position, forming neutral aIPr·GaR3 and free IMes. Since IPr is regenerated at the end of the process, it can then be envisaged that under the conditions studied it acts as a catalyst in the isomerization process.
| Entry | [1] M | [IPr] (M) | [GaR3] (M) | r o × 106 (Ms−1) |
|---|---|---|---|---|
| 1 | 0.17 | 1.95 ± 0.08 | ||
| 2 | 0.29 | 3.26 ± 0.08 | ||
| 3 | 0.37 | 4.81 ± 0.03 | ||
| 4 | 0.42 | 5.04 ± 0.04 | ||
| 5 | 0.29 | 0.07 | 4.67 ± 0.05 | |
| 6 | 0.29 | 0.15 | 5.71 ± 0.15 | |
| 7 | 0.29 | 0.26 | 6.40 ± 0.09 | |
| 8 | 0.32 | 0.16 | 3.39 ± 0.04 | |
| 9 | 0.32 | 0.31 | 2.30 ± 0.02 | |
| 10 | 0.32 | 0.42 | 2.06 ± 0.04 |
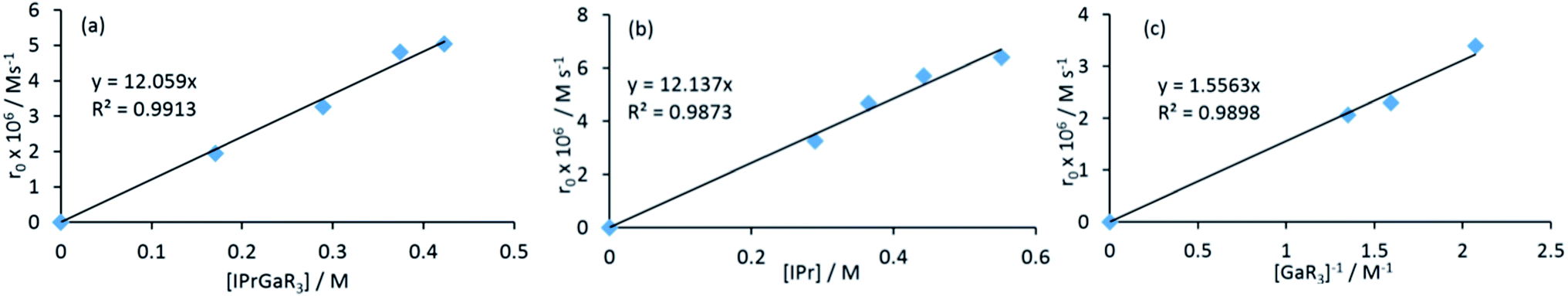 | ||
| Fig. 8 Initial rates versus concentration of (a) [1], (b) [IPr] and (c) [GaR3]−1. | ||
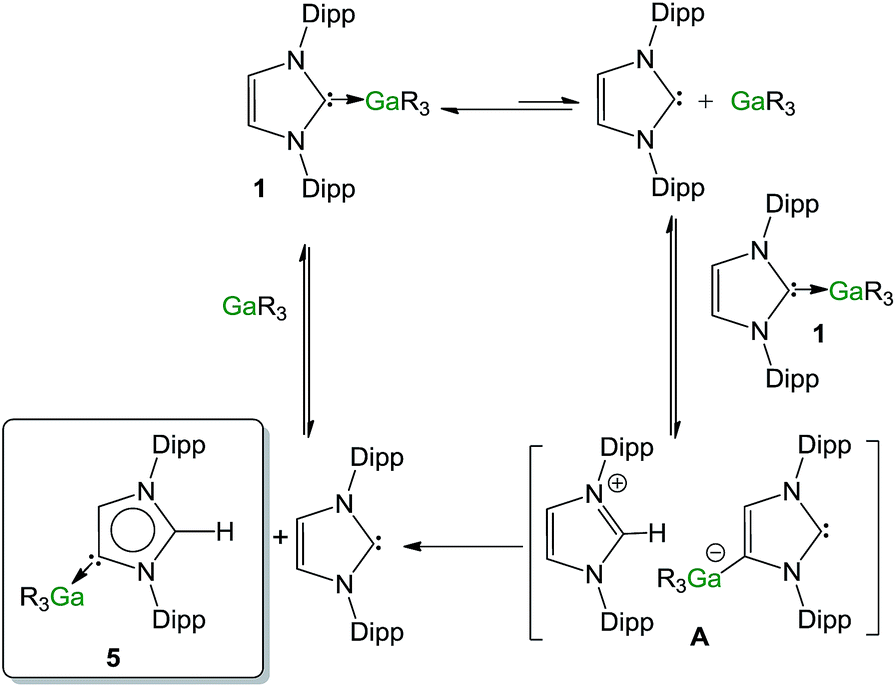 | ||
| Fig. 9 Proposed mechanism for the isomerisation of 1 into 5. | ||
Conclusions
Progressing main-group NHC chemistry, this systematic study of the synthesis and stability of abnormal NHC–gallium complexes has demonstrated two alternative and efficient methodologies to access aIPr·GaR3 (5). Studies investigating the synthesis of anionic NHC complexes have shown that the functionalization of the imidazole backbone can be achieved by sequentially treating IPr with the polar organometallic reagent LiR followed by GaR3 addition (indirect stepwise gallation), to afford heteroleptic gallate (THF)2Li[:C{[N(2,6-iPr2C6H3)]2CHCGa(CH2SiMe3)3}] (3). Electrophilic interception of 3 with MeOTf or the imidazolium salt IMes·HCl led to the isolation of neutral abnormal NHC (aNHC) complexes [CH3C{[N(2,6-iPr2C6H3)]2CHCGa(CH2SiMe3)3}] (4) and aIPr·GaR3 (5). These studies disclose the preference of the anionic IPr ligand present in 3 to react with these electrophiles via its C2 position, leaving its Ga–C4 interaction intact. Compound 5 can also be accessed by a thermally induced rearrangement of its normal isomer 1. NMR spectroscopic studies coupled with theoretical calculations have revealed the importance of the donor ability of the solvent used in these thermal isomerization processes as well as the steric bulk of the substituents on the N atoms of the NHC ligands and the Ga reagent, suggesting that the relief of the steric hindrance by forming an abnormal complex is one of the main driving forces behind these rearrangements and hinting at the potential FLP reactivity that these systems may exhibit. Mechanistic studies intimate that these processes occur via a rate-determining dissociative step, supporting the formation of free NHC, which in turn can catalyse the isomerization process.Acknowledgements
We thank the Royal Society, and the European Research Council (ERC) for their generous sponsorship of this research. We also thank Professor Robert E. Mulvey, Dr David. J. Nelson (University of Strathclyde) and Professor Michael S. Hill (University of Bath) for their insightful comments, Mr Alexander Clunie for his assistance in the CHN analysis of highly air and moisture sensitive compounds and Mr Craig Irving for his help with NMR spectroscopy experiments.Notes and references
- (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2010, 100, 39 CrossRef; (b) M. Albrecht, Science, 2009, 326, 532 CrossRef CAS PubMed; (c) W. Kirmse, Angew. Chem., Int. Ed., 2010, 49, 8798 CrossRef CAS PubMed; (d) N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Netherlands, 2010 Search PubMed; (e) N-Heterocyclic Carbenes: Effective Tools for Organometallics Synthesis, ed. S. P. Nolan, Wiley VCH, 2014 Search PubMed.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- A. J. Arduengo III, H. V. Rasika Dias, D. A. Dixon, R. L. Harlow, W. T. Klooster and T. F. Koetzle, J. Am. Chem. Soc., 1994, 116, 6812 CrossRef.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS PubMed.
- W. Hermann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef.
- C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS PubMed.
- S. P. Nolan, N-Heterocyclic Carbenes in Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed.
- S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef CAS PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069 CrossRef CAS PubMed.
- A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701 CrossRef CAS PubMed.
- R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683 CrossRef CAS PubMed.
- C. Jones, A. Sidiropoulos, N. Holzmann, G. Frenking and A. Stasch, Chem. Commun., 2012, 48, 9855 RSC.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schafer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970 CrossRef CAS PubMed.
- M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Chem.–Eur. J., 2010, 16, 432 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420 CrossRef CAS PubMed.
- For excellent overviews of the area see: (a) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444 CrossRef CAS PubMed; (b) Y. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326 CrossRef CAS PubMed.
- L. J. Hounjet and D. W. Stephan, Org. Process Res. Dev., 2014, 18, 385 CrossRef CAS.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306 CrossRef CAS PubMed.
- R. Tonner, G. Heydenrych and G. Frenking, Chem.–Asian J., 2007, 2, 1555 CrossRef CAS PubMed.
- S. Gründemann, A. Kovacevic, M. Albrecht, J. W. Faller and R. H. Crabtree, Chem. Commun., 2001, 2274 RSC.
- S. Gründemann, A. Kovacevic, M. Albrecht, J. W. Faller and R. H. Crabtree, J. Am. Chem. Soc., 2002, 124, 10473 CrossRef.
- L. N. Appelhans, D. Zuccaccia, A. Kovacevic, A. R. Chianese, J. R. Miecznikowski, A. Macchioni, E. Clot, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2005, 127, 16299 CrossRef CAS PubMed.
- (a) P. L. Arnold and S. Pearson, Coord. Chem. Rev., 2007, 251, 596 CrossRef CAS; (b) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445 CrossRef CAS PubMed; (c) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS.
- E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 326, 556 CrossRef CAS PubMed.
- Related to these findings, Ghadwal et al. have recently reported a Pd catalysed methodology to access C2-arylated imidazolium salts which can be used as precursors to prepare abnormal-NHC Cu complexes, see: R. S. Ghadwal, S. O. Reichmann and R. Herbst-Irmer, Chem.–Eur. J., 2015, 21, 4247 CrossRef CAS PubMed.
- J. B. Waters and J. M. Goicoechea, Coord. Chem. Rev., 2015, 293–294, 80 CrossRef CAS.
- M. Heckenroth, E. Kluser, A. Neels and M. Albrecht, Angew. Chem., Int. Ed., 2007, 46, 6293 CrossRef CAS PubMed.
- The Tolman Electronic Parameter (TEP) has been employed in transition metal complexes to evaluate the electron donating abilities of normal and abnormal NHC's, see for example: R. A. Kely III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202 CrossRef.
- B. M. Day, T. Pugh, D. Hendriks, C. F. Guerra, D. J. Evans, F. M. Bickelhaupt and R. A. Layfield, J. Am. Chem. Soc., 2013, 135, 13338 CrossRef CAS PubMed.
- T. W. Graham, K. A. Udachin and A. J. Carty, Chem. Commun., 2006, 2699 RSC.
- D. Holschummacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428 CrossRef PubMed.
- D. Holschummacher, T. Bannenberg, K. Ibrom, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2010, 39, 10590 RSC.
- E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2012, 18, 16938 CrossRef CAS PubMed.
- A. L. Schmitt, G. Schnee, R. Welter and S. Dagorne, Chem. Commun., 2010, 46, 2480 RSC.
- Y. Wang, Y. Xie, M. Y. Abraham, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2010, 132, 14370 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, M. Y. Abraham, R. G. Gilliard Jr, P. Wei, C. F. Campana, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Angew. Chem., Int. Ed., 2012, 51, 10173 CrossRef CAS PubMed.
- Y. Wang, M. Y. Abraham, R. G. Gilliard Jr, P. Wei, J. C. Smith and G. H. Robinson, Organometallics, 2012, 31, 791 CrossRef CAS.
- M. Chen, Y. Wang, R. J. Gilliard Jr, P. Wei, N. A. Schwartz and G. H. Robinson, Dalton Trans., 2014, 43, 14211 CAS.
- (a) A. El-Hellani and V. Lavallo, Angew. Chem., Int. Ed., 2014, 53, 4489 CrossRef CAS PubMed; (b) M. J. Asay, S. P. Fisher, S. E. Lee, D. Borchardt, F. S. Tham and V. Lavallo, Chem. Commun., 2015, 51, 5359 RSC.
- X.-W. Li, J. Su and G. H. Robinson, Chem. Commun., 1996, 2683 RSC.
- N. Marion, E. C. Escudero-Adan, J. Benet-Buchholz, E. D. Stevens, L. Fensterbank, M. Malacria and S. P. Nolan, Organometallics, 2007, 26, 3256 CrossRef CAS.
- C. Fliedel, G. Schnee, T. Aviles and S. Dagorne, Coord. Chem. Rev., 2014, 275, 63 CrossRef CAS.
- A. Higelin, S. Keller, C. Göhringer, C. Jones and I. Krossing, Angew. Chem., Int. Ed., 2013, 52, 4941 CrossRef CAS PubMed.
- M. L. Cole, S. K. Furfari and M. Kloth, J. Organomet. Chem., 2009, 694, 2934 CrossRef CAS.
- S. Tang, J. Monot, A. El-Hallani, B. Michelet, R. Guillot, C. Bour and V. Gandon, Chem.–Eur. J., 2012, 18, 10239 CrossRef CAS PubMed.
- C. Bour, J. Monot, S. Tang, R. Guillot, J. Farjon and V. Gandon, Organometallics, 2014, 33, 594 CrossRef CAS.
- O. T. Beachley Jr and R. G. Simmons, Inorg. Chem., 1980, 19, 1021 CrossRef.
- M. U. Kramer, D. Robert, Y. Nakajima, U. Englert, T. P. Spaniol and J. Okuda, Eur. J. Inorg. Chem., 2007, 5, 665 CrossRef.
- Two singlets are observed for the monosilyl group at −0.95 (Ga–CH2) and 0.18 ppm (Si(CH3)3) contrasting with the 1H NMR spectrum of GaR3 in the same solvent where both signals accidentally overlap displaying a singlet at 0.13 ppm.
- A. Jana, R. Azhakar, G. Tavčar, H. W. Roesky, I. Objartel and D. Stalke, Eur. J. Inorg. Chem., 2011, 25, 3686 Search PubMed.
- For both 2 and 4 the three monosilyl groups were found to be disordered. This obviously compromises any discussion of structural details such as bond distances, however in each case the crystallographic analysis does establish the gross composition and conformation of the molecular complexes.
- The structure of this unsolvated lithium gallate has been reported recently, see: D. R. Armstrong, E. Brammer, T. Cadenbach, E. Hevia and A. R. Kennedy, Organometallics, 2013, 32, 480 CrossRef CAS.
- D. R. Armstrong, S. E. Baillie, V. L. Blair, N. G. Chabloz, J. Diez, J. Garcia-Alvarez, A. R. Kennedy, S. D. Robertson and E. Hevia, Chem. Sci., 2013, 4, 4259 RSC.
- All four Ga–C bonds are similar in length ranging from 2.013(2) Å to 2.052(2) Å (average 2.034 Å) which is in good agreement with other tetra-coordinated gallate species such as {[K-dibenzo-18-c-6]+[Ga(η1-C3H5)4]−} (2.029 Å mean) see ref: C. Lichtenberg, T. P. Spaniol and J. Okuda, Inorg. Chem., 2012, 51, 2254 CrossRef CAS PubMed.
- As far as we can ascertain there are only two other examples containing the BEt3 (see ref. 38 and 39 and Y. Yamaguchi, T. Kashiwabara, K. Ogata, Y. Miura, Y. Nakamura, K. Kobayashi and T. Ito, Chem. Commun., 2004, 2160) and B(C6F5)3 fragments (see ref. 32 and 34 and S. Kronig, E. Theuergarten, C. G. Daniliuc, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2012, 51, 3240).
- W. Kohn, A. D. Becke and R. G. Parr, J. Phys. Chem., 1996, 100, 12974 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785 CrossRef.
- (a) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS; (b) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- (a) T. Guo, S. Dechert and F. Meyer, Organometallics, 2014, 33, 5145 CrossRef CAS; (b) M. Heckenroth, A. Neels, M. G. Garnier, P. Aebi, A. W. Ehlers and M. Albrecht, Chem.–Eur. J., 2009, 15, 9375 CrossRef CAS PubMed.
- Under these conditions the maximum conversion observed of 1 to 5 was 75%. Extended reaction times (48 h) did not lead to an increase in this conversion.
- A. R. Kennedy, J. Klett, R. E. Mulvey and S. D. Robertson, Eur. J. Inorg. Chem., 2011, 4675 CrossRef CAS.
- ZnR2·IPr was prepared by mixing equimolar amounts of ZnR2 and IPr in hexane. See ESI‡ for experimental details and X-ray crystallographic and spectroscopic characterisation.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- The relative stabilities of IPr/aIPr have previously been computed using a different level of theory (ZORA-BLYP/TZ2P level using COSMO to simulate the solvent effect), with ΔEa–n = 12.5 kcal mol−1, see ref. 30.
- As shown in Table S4‡ and in agreement with the previous calculations for IPr and IMes, normal free ItBu is 17.2 kcal mol−1 more stable than its free abnormal isomer.
- R. M. Stolley, H. A. Duong, D. R. Thomas and J. Louie, J. Am. Chem. Soc., 2012, 134, 15154 CrossRef CAS PubMed.
- Attempts to study transformation of 1 into 5 under pseudo-first-order conditions (using a 10 molar equivalent excess of GaR3 per complex 1) completely inhibited the isomerization process.
- R. B. Jordan, Reaction Mechanisms of Inorganic and Organometallic Systems, Oxford University Press, Oxford, UK, 3rd edn, 2007 Search PubMed.
- Consistent with this interpretation, the formation of aItBuGaR3 (8) from an equimolar mixture of ItBu and GaR3 occurs under much milder conditions (room temperature, 60 minutes) than for 5. In this case ItBu and GaR3 fail to form a stable normal adduct, thus a dissociation step is not required.
- NMR studies of equimolar amounts of IPrD·GaR3 (1D) and IPr·GaR3 (1) in d8-THF were carried out in order to assess if the transformation of 1 to 5 occurs via intra- or intermolecular mechanism. However analysis of the NMR data proved to be inconclusive due to the overlapping of the signals from the protonated and deuterated species.
- In particular the reactivity of 1 can be related to that described by Tamm for ItBu·B(XyF6)3 that forms a stable normal adduct at room temperature but heating at 110 °C isomerizes to aItBu·B(XyF6)3. This system exhibits FLP reactivity and can activate CO2, HCCPh and THF, as reported in ref. 34.
- T. A. Rokob, A. Hamza, A. Stirling and I. Pápai, J. Am. Chem. Soc., 2009, 131, 2029 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of Paul von R. Schleyer. |
| ‡ Electronic supplementary information (ESI) available: CIF files giving crystallographic results, experimental details and copies of the NMR spectra. CCDC 1405459–1405464. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02086g |
| This journal is © The Royal Society of Chemistry 2015 |