
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unified approach to prenylated indole alkaloids: total syntheses of (−)-17-hydroxy-citrinalin B, (+)-stephacidin A, and (+)-notoamide I†
Eduardo V.
Mercado-Marin
and
Richmond
Sarpong
*
Department of Chemistry, University of California–Berkeley, Berkeley, CA 94720, USA. E-mail: rsarpong@berkeley.edu
First published on 18th June 2015
Abstract
A unified strategy for the synthesis of congeners of the prenylated indole alkaloids is presented. This strategy has yielded the first synthesis of the natural product (−)-17-hydroxy-citrinalin B as well as syntheses of (+)-stephacidin A and (+)-notoamide I. An enolate addition to an in situ generated isocyanate was utilized in forging a key bicyclo[2.2.2]diazaoctane moiety, and in this way connected the two structural classes of the prenylated indole alkaloids through synthesis.
Introduction
Historically, the undertaking of total syntheses of natural products has focused on ‘target-oriented’ syntheses whereby a single compound is targeted for synthesis to investigate its biological relevance or aspects of its structure.1 This practice has inspired many new synthesis developments. Recently, however, exercises in complex molecule total synthesis are placing a growing emphasis on the preparation of diverse molecular skeletons from a common intermediate.2 This practice, which mirrors the biological production of many secondary metabolites but does not necessarily follow along biosynthetic lines, maximizes the opportunities for, and efficiency of, accessing molecular diversity to facilitate structure–activity relationship studies. Over the last 30 years, this concept has led to remarkable unified strategies for the syntheses of various families of natural products.3 Here, we present the extension of this idea to the syntheses of congeners in the prenylated indole alkaloid family which features a powerful Dieckmann-type cyclization to forge a key [2.2.2]bicycle.The prenylated indole alkaloids include some of the most structurally diverse secondary metabolites isolated to date (see Fig. 1 for selected examples). Many congeners such as stephacidin A (1), notoamide I (2), mangrovamide A (3) and paraherquamide A (4) contain a bicyclo[2.2.2]diazaoctane structural moiety.4 Over the last decade however, additional members of the family that lack the bicyclo[2.2.2]diazaoctane core have begun to emerge. This includes the citrinalins (e.g., 5), citrinadins (e.g., 9) and the cyclopiamines (e.g., 8 – albeit isolated in 1979).5 While myriad bioactivity has been discovered for various prenylated indole alkaloids (especially anthelmintic activity),4 the recent emergence of the citrinadins and related compounds6 that lack the bicyclo[2.2.2]diazaoctane structural motif as potent anti-tumor compounds has heightened interest in the whole family of secondary metabolites.
 | ||
| Fig. 1 Selected prenylated indole alkaloids. | ||
Results and discussion
From our perspective, a unified synthetic approach that affords prenylated indole alkaloid congeners bearing the bicyclo[2.2.2] diazaoctane core as well as those lacking this structural moiety would provide the most strategically efficient approach to these natural products. However, to date, such an approach has not been reported. All the existing syntheses of this family of molecules have targeted either the subset that contains the [2.2.2] diazaoctane bicycle or those molecules that lack this structural feature.7,8 In this manuscript, we present our studies toward identifying a common intermediate that can be advanced to natural products representative of both prenylated indole alkaloid structural motifs. These studies have led to the identification of 10 (Scheme 1) as such a common intermediate, which now provides the first total synthesis of (−)-17-hydroxy-citrinalin B (7) as well as a synthesis of (+)-stephacidin A (1) and (+)-notoamide I (2). Our synthetic strategy to these two natural products, which rests on ‘network analysis’9 considerations, diverges only at a late stage. Thus, strategic bond disconnection of the maximally bridging ring in, for example, 1 (i.e., the 2,5-diketopiperazine ring) leads back to carbamate 10, where a bond can be formed at a late stage between C16 and the carbamate carbonyl group.10 In this way, the two sub-families of the prenylated indole alkaloids (e.g., 1 and 7) can be connected by a synthesis sequence characterized by a progressive increase in structural complexity, which distinguishes this approach from prior syntheses of related prenylated indole alkaloids.7 Hexacycle 10 can in turn arise from tricycle 11 using an indole annulation reaction, which would provide opportunities to prepare other natural products such as paraherquamide A (4) that differ in their indole substitution pattern. | ||
| Scheme 1 Unified retrosynthetic approach. | ||
A general strategy for the synthesis of the prenylated indole alkaloids that encompasses the two main structural types (of which 1 and 7 are representative) has not been explored before. In all of the previous syntheses of the bicyclo[2.2.2] diazaoctane bearing congeners, the tetrasubstituted center at the bicyclo[2.2.2] bridgehead (e.g., C4 – stephacidin numbering – in 1) is constructed at an early stage or through C4–C5 bond formation, which would necessitate its late-stage cleavage (in a complexity minimizing manipulation) in order to form compounds such as 7 from 1. In this latter scenario, an amide hydrolysis of the bicyclo[2.2.2]diazaoctane, decarboxylation, and a diastereoselective protonation at the ring junction (the diastereoselectivity of which is not certain outside of an enzyme pocket)11 would be required for a conversion of 1 to the sub-family that lacks the diazaoctane structural motif. Our approach to this collection of molecules is complementary.
Synthesis of divergent intermediate 23
Our studies commenced with the preparation of 11 (Scheme 2), which is available in 6 steps from known enantioenriched alcohol 12.12 Analogous to our previously established sequence,8d oxidation of the alcohol group of 12 and alkynylative homologation of the resulting aldehyde using the Ohira–Bestmann reagent (13) affords an alkyne, which upon Boc-cleavage and acylation with α-cyano acetylchloride gives alkyne 14.13 A formal cycloisomerization of 14, presumably proceeding by anti-Markovnikov hydration of the terminal alkyne followed by Knoevanagel condensation of the incipient aldehyde, was effected using the Grotjahn complex (15)14 to yield bicycle 16. Diels–Alder cycloaddition of 16 with diene 17, facilitated by SnCl4 gives enone 11 upon basic workup. Even though we had previously accomplished the analogous synthesis of a tricycle lacking the benzyloxy group at C17 (see numbering in 11), it was unclear what influence this added substituent would exert on the diastereoselectivity of the cycloaddition step and so we were gratified to obtain diastereomer 11 in good yield. The structure of 11 is supported by the X-ray structure of keto-alcohol 18 (see CYLview in Scheme 2), which we obtained following hydrogenation and BBr3-mediated cleavage of the benzyl group of 11.15 | ||
| Scheme 2 Preparation of 11. (a) DMP (1.1 equiv.), NaHCO3, CH2Cl2, 0 °C to rt, 2.5 h, >87%; (b) 13 (1.5 equiv.), NaOMe (5.0 equiv.), THF, −78 °C to −50 °C, 2 h, 91%; (c) 4 N HCl/dioxane (4.0 equiv.), 0 °C to rt, 30 min; concentrate; then cyanoacetyl chloride (2.5 equiv.), Et3N (2.5 equiv.), 0 °C to rt, 3 h, 75%; (d) 15 (8 mol%), acetone/H2O, 70 °C, 24 h, 92%; (e) 17 (2.66 equiv.), SnCl4 (1.2 equiv.), CH2Cl2, −78 °C to −42 °C, 30 min, then rt, 30 min, 75%. DMP = Dess–Martin periodinane; Bn = benzyl. | ||
A Johnson iodination16 of 11 (Scheme 3), followed by hydration of the nitrile group using the Ghaffar–Parkins complex (19)17 and subsequent Hofmann rearrangement of the resulting carboxamide using phenyliodoso trifluoromethyl acetate (PIFA) in the presence of methanol provided carbamate 20.18 Following the precedent of Myers and Herzon,7d Suzuki cross-coupling of iodide 20 with pincacol boronic ester 21 followed by reductive cyclization yielded indole annulated hexacycle 22. At this stage, Wacker oxidation19 of the chromene moiety and treatment of the resulting chromanone with dimethyl sulfide in the presence of methane sulfonic acid unveiled the amine and hydroxyl groups to provide 23, which would serve as the common intermediate to access both 17-hydroxy-citrinalin B as well as stephacidin A. Of note, while the synthesis of 17-hydroxy-citrinalin B would take advantage of the chromanone unit, a synthesis of stephacidin A from 23 would require a reconstitution of the chromene moiety. However, in our hands, the chromanone moiety proved to be more robust (as compared to the chromene) in many of the subsequent steps and so 23 served as a more effective intermediate even to stephacidin A.
 | ||
| Scheme 3 Synthesis of fused hexacycle 23. (a) Iodine (3.0 equiv.), DMAP (3.0 equiv.), pyridine/CCl4, 60 °C, 22 h, 77%; (b) 19 (30 mol%), EtOH/H2O, rt, 4 days, 82%; (c) PIFA (1.1 equiv.), MeOH, 0 °C to rt, 16 h, 89%; (d) 21 (1.5 equiv.), dppfPdCl2 (10 mol%), K3PO4 (3.75 equiv.), DMF, 40 °C, 16 h, 94%; (e) Zn (excess), NaCNBH3 (5.0 equiv.), sat. aq. NH4Cl, MeOH, rt, 2 h, 71%; (f) Pd(OAc)2 (0.40 equiv.), benzoquinone (1.5 equiv.), cat. H2SO4, MeCN/H2O, rt, 17 h, 77%; (g) Me2S (20 equiv.), MsOH, 40 °C, 15 h, 93%. DMAP = 4-dimethylaminopyridine; PIFA = [bis(trifluoroacetoxy)iodo]benzene; dppf = 1,1′-bis(diphenylphosphino)ferrocene; DMF = dimethylformamide; Ms = methanesulfonyl; Bn = benzyl. | ||
Synthesis of (−)-17-hydroxy-citrinalin B (7) from 23
To complete a synthesis of 17-hydroxy-citrinalin B, 23 was subjected to oxidation using oxone as previously described in the synthesis of citrinalin B by our group (Scheme 4).8d The ensuing series of remarkable chemoselective oxidations accomplished the conversion of the indole to the spirooxindole (corresponding to the desired diastereomer for 7) as well as the oxidation of the amino group to the nitro group – all in the presence of the free secondary hydroxy group.20 | ||
| Scheme 4 Completion of the synthesis of 17-hydroxy-citrinalin B (7). (a) Oxone® (11.4 equiv.), NaHCO3, acetone/H2O, 0 °C to rt, 2 h, 50%; (b) Me3OBF4 (12 equiv.), CH2Cl2, 4 Å MS, 45 °C, 16 h, then NaCNBH3 (excess), MeOH, 0 °C, 30 min, 48%. Oxone® = potassium peroxomonosulfate; MS = molecular sieves. | ||
With 24 in hand, chemoselective reductive removal of the tertiary amide carbonyl group (in the presence of several other groups that are susceptible to reduction) was accomplished following an adaptation of a procedure first reported by Borch.21 Thus, subjection of 24 to Me3OBF4 followed by NaCNBH3 proceeded in respectable yield to give (−)-17-hydroxy-citrinalin B.22 Of note, in our hands, it was the TFA salt of 7 that provided analytical data identical in all respects to that of the natural isolate, which had been reported as the neutral compound.8d
Although our synthetic sequence for the preparation of 17-hydroxy-citrinalin B mirrors closely our previous synthesis of citrinalin B (6),23 it required a much more stringent level of chemoselectivity in the endgame. The success of this route is a testament to the robustness of our synthetic plan, which proceeded without event (especially in the endgame) even in the presence of a free hydroxyl group at C17 from intermediate 23 onwards.
Synthesis of (+)-stephacidin A (1) and (+)-notoamide I (2) from 23
In an initial demonstration of the utility of 23 as an intermediate for the synthesis of prenylated indole alkaloid congeners possessing the bicyclo[2.2.2]diazaoctane structural motif, we have completed a synthesis of (+)-stephacidin A and (+)-notoamide I as outlined in Scheme 5. Thus chemoselective carbamoylation of the primary amine of 23 was achieved in high yield in the presence of the secondary hydroxyl to afford phenyl carbamate 25. At this point, oxidation of the hydroxy group and treatment of the resulting ketone with K2CO3 forges the bicyclo[2.2.2]diazaoctane framework of stephacidin A, presumably through a Dieckmann condensation via isocyanate/enolate intermediate 26.24 These exceedingly simple conditions effectively accomplish the task of synthetically connecting the two major sub-families of the prenylated indole alkaloids. From our perspective, polycycle 27 represents a versatile framework that may be advanced to myriad prenylated indole alkaloids including mangrovamide A (3) and paraherquamide A (4). To complete the synthesis of (+)-stephacidin A, removal of the ketone group in the pyrrolidine ring using a Wolff–Kishner protocol followed by reduction of the chromanone carbonyl group and elimination of the resulting hydroxyl gave (+)-stephacidin A (1) in 40% yield over the two steps.25 Analytical data for synthetic stephacidin A prepared by us matched perfectly previously reported spectra.7a,e Furthermore, our X-ray analysis of a single crystal of 1 provided unambiguous support for the structure of the natural isolate (see CYLview in Scheme 5). Stephacidin A is a versatile starting point for the preparation of other prenylated indole alkaloids. For example, 1 was easily converted to (+)-notoamide I (2) upon treatment with MnO2 in EtOAc (32% yield). Our synthesis of 1 also constitutes formal syntheses of (−)-notoamide B, (+)-avrainvillamide and (−)-stephacidin B.26 | ||
| Scheme 5 Completion of the synthesis of (+)-stephacidin A (1) and (+)-notoamide I (2). (a) ClCO2Ph (excess), K2CO3, acetone, rt, 16 h, 90%; (b) DMP (1.5 equiv.), NaHCO3, CH2Cl2, rt, 20 min; (c) K2CO3, acetone, 50 °C, 2 h, 42% over two steps; (d) NH2NH2 (1.1 equiv.), ethylene glycol, 70 °C, 17 h, then t-BuOK (5.0 equiv.), 170 °C, 2 h, 57%; (e) NaBH4 (10.0 equiv.), THF, 16 h, then 0.6 N HCl/H2O, 60 °C, 30 min, 71%. (f) MnO2 (excess), EtOAc, rt, 30 min, 32%. DMP = Dess–Martin periodinane; THF = tetrahydrofuran. | ||
Conclusions
In conclusion, we have achieved the first unified approach to the two sub-families of the prenylated indole alkaloids (i.e., that either lack or possess the bicyclo[2.2.2]diazaoctane structural motif). Our strategy has been exemplified with the first preparation of the natural product (−)-17-hydroxy-citrinalin B and of (+)-stephacidin A. Key to the success of the approach was the identification of a late-stage common intermediate (23), which could be advanced to either subclass of the prenylated indole alkaloids using a remarkably diastereoselective spiro-oxindole formation attended by a chemoselective oxidation of an amino group to a nitro group. Our synthesis of stephacidin A also featured a complexity building isocyanate capture to forge a [2.2.2]bicycle. Our studies now set the stage for the broad-ranging syntheses of congeners of the prenylated indole alkaloid family to facilitate in-depth studies on their biosynthesis and biological activity.Acknowledgements
We are grateful to the NIH (NIGMS RO1 086374) for support. We thank the NSF GRFP, the ACS Division of Organic Chemistry, and the Hellman Graduate Awards Program (UC Berkeley) for graduate fellowships to support E.V.M.-M. We are thankful to A. DiPasquale for solving the crystal structures of 1 and 18 (displayed with CYLview; CCDC 1400756 and 1400755, respectively), supported by NIH Shared Instrumentation Grant (S10-RR027172).Notes and references
- T. Hudlický and J. W. Reed, The Way of Synthesis, Wiley, Weinheim, Germany, 2007, part 1, pp. 1–47 Search PubMed.
- (a) S. B. Jones, B. Simmons, A. Mastracchio and D. W. C. MacMillan, Nature, 2011, 475, 183 CrossRef CAS PubMed; (b) E. Gravel and E. Poupon, Eur. J. Org. Chem., 2008, 27 CrossRef.
- E. E. Anagnostaki and A. L. Zografos, Chem. Soc. Rev., 2012, 41, 5613 RSC.
- For an in-depth review on the isolation, biosynthesis and bioactivity of prenylated indole alkaloids containing the bridging bicyclo[2.2.2]diazaoctane ring system see: J. M. Finefield, J. C. Frisvad, D. H. Sherman and R. M. Williams, J. Nat. Prod., 2012, 75, 812 CrossRef CAS PubMed . For the isolation of mangrovamides see: B. Yang, J. Dong, X. Lin, X. Zhou, Y. Zhang and Y. Liu, Tetrahedron, 2014, 70, 3859 CrossRef.
- (a) Citrinalins see: E. F. Pimenta, A. M. Vita-Marques, A. Tinimis, M. H. R. Seleghim, L. D. Sette, K. Veloso, A. G. Ferreira, D. E. Williams, B. O. Patrick, D. S. Dalisay, R. J. Andersen and R. G. S. Berlinck, J. Nat. Prod., 2010, 73, 1821 CrossRef CAS PubMed; (b) Citrinadins see: M. Tsuda, Y. Kasai, K. Komatsu, T. Sone, M. Tanaka, Y. Mikami and J. Kobayashi, Org. Lett., 2004, 6, 3087 CrossRef CAS PubMed; T. Mugishima, M. Tsuda, Y. Kasai, H. Ishiyama, E. Fukushi, J. Kawabata, M. Watanabe, K. Akao and J. Kobayashi, J. Org. Chem., 2005, 70, 9430 Search PubMed; (c) Cyclopiamines see: R. F. Bond, J. C. A. Boeyens, C. W. Holzapfel and P. S. Steyn, J. Chem. Soc., Perkin Trans. 1, 1979, 1751 RSC; P. S. Steyn, R. Vleggaar and C. J. Rabie, Phytochemistry, 1981, 20, 538 Search PubMed.
- N. Kushida, N. Watanabe, T. Okuda, F. Yokoyama, Y. Gyobu and T. Yaguchi, J. Antibiot., 2007, 60, 667 CrossRef CAS PubMed.
- For selected total syntheses of natural products containing the bicyclo[2.2.2]diazaoctane core see: (a) P. S. Baran, C. A. Guerrero, N. B. Ambhaikar and B. D. Hafensteiner, Angew. Chem., Int. Ed., 2005, 44, 606 CrossRef CAS PubMed; (b) P. S. Baran, C. A. Guerrero, B. D. Hafensteiner and N. B. Ambhaikar, Angew. Chem., Int. Ed., 2005, 44, 3892 CrossRef CAS PubMed; (c) P. S. Baran, B. D. Hafensteiner, N. B. Ambhaikar, C. A. Guerrero and J. D. Gallagher, J. Am. Chem. Soc., 2006, 128, 8678 CrossRef CAS PubMed; (d) S. B. Herzon and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 5342 CrossRef CAS PubMed; (e) T. J. Greshock, A. W. Grubbs, S. Tsukamoto and R. M. Williams, Angew. Chem., Int. Ed., 2007, 46, 2262 CrossRef CAS PubMed; (f) G. D. Artman III, A. W. Grubbs and R. M. Williams, J. Am. Chem. Soc., 2007, 129, 6336 CrossRef PubMed; (g) B. M. Trost, N. Cramer and H. Bernsmann, J. Am. Chem. Soc., 2007, 129, 3086 CrossRef CAS PubMed; (h) F. Frebault, N. S. Simpkins and A. Fenwick, J. Am. Chem. Soc., 2009, 131, 4214 CrossRef CAS PubMed; (i) S. W. Laws and J. R. Scheerer, J. Org. Chem., 2013, 78, 2422 CrossRef CAS PubMed; (j) N. S. Simpkins, I. Pavlakos, M. D. Weller and L. Male, Org. Biomol. Chem., 2013, 11, 4957 RSC. For reviews see: (k) K. A. Miller and R. M. Williams, Chem. Soc. Rev., 2009, 38, 3160 RSC; (l) C. F. Nising, Chem. Soc. Rev., 2010, 39, 591 RSC.
- For selected total syntheses of natural products that lack the bicyclo[2.2.2]diazaoctane core see: (a) Z. Bian, C. C. Marvin and S. F. Martin, J. Am. Chem. Soc., 2013, 135, 10886 CrossRef CAS PubMed; (b) K. Kong, J. A. Enquist Jr, M. E. McCallum, G. M. Smith, T. Matsumaru, E. Menhaji-Klotz and J. L. Wood, J. Am. Chem. Soc., 2013, 135, 10890 CrossRef CAS PubMed; (c) Z. Bian, C. C. Marvin, M. Pettersson and S. F. Martin, J. Am. Chem. Soc., 2014, 136, 14184 CrossRef CAS PubMed; (d) E. V. Mercado-Marin, P. Garcia-Reynaga, S. Romminger, E. F. Pimenta, D. K. Romney, M. W. Lodewyk, D. E. Williams, R. J. Andersen, S. J. Miller, D. J. Tantillo, R. G. S. Berlinck and R. Sarpong, Nature, 2014, 509, 318 CrossRef CAS PubMed.
- (a) E. J. Corey, W. J. Howe, H. W. Orf, D. A. Pensak and G. Petersson, J. Am. Chem. Soc., 1975, 97, 6116 CrossRef CAS; (b) C. H. Heathcock, Angew. Chem., Int. Ed., 1992, 31, 665–681 CrossRef; (c) R. W. Hoffmann, Elements of Synthesis Planning, Springer, Berlin, Germany, 2009, ch. 6, pp. 81–112 Search PubMed.
- For a Dieckmann cyclization route to form 2,5-diketopiperazines see: (a) C. L. Aboussafy and D. L. Clive, J. J. Org. Chem., 2012, 77, 5125 CrossRef CAS PubMed. For a review on the synthesis of 2,5-diketopiperazines see: (b) A. D. Borthwick, Chem. Rev., 2012, 112, 3641 CrossRef CAS PubMed.
- These sequence of events is the proposed biosynthetic origin of congeners that lack the bicyclo[2.2.2]diazaoctane ring, see ref. 5b and 8d.
- T. K. Chakraborty, P. Srinivasu, R. V. Rao, S. K. Kumar and A. C. Kunwar, J. Org. Chem., 2004, 69, 7399 CrossRef CAS PubMed.
- S. Ohira, Synth. Commun., 1989, 19, 561 CrossRef CAS.
- D. B. Grotjahn and D. A. Lev, J. Am. Chem. Soc., 2004, 126, 12232 CrossRef CAS PubMed.
- See ESI† for details.
- C. R. Johnson, J. P. Adams, M. P. Braun, C. B. W. Senanayake, P. M. Wovkulich and M. R. Uskoković, Tetrahedron Lett., 1992, 33, 917 CrossRef CAS.
- T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657 CrossRef CAS.
- R. W. Moriarty, C. J. Chany, R. K. Vaid, O. Prakash and S. M. Tuladhar, J. Org. Chem., 1993, 58, 2478 CrossRef CAS.
- D. G. Miller and D. D. M. Wayner, J. Org. Chem., 1990, 55, 2924 CrossRef CAS.
- For oxidative rearrangements of indoles to oxindoles with DMDO see: X. Zhang and C. S. Foote, J. Am. Chem. Soc., 1993, 115, 8867 CrossRef CAS.
- R. F. Borch, Tetrahedron Lett., 1968, 9, 61 CrossRef.
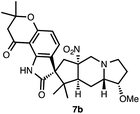
- We also isolated the methyl ether (7b) as a minor product in 19% yield, see the ESI†for more details.
 .
. - Our preliminary attempts to effect direct oxygenation of 6 at C17 to give 7 have been unsuccessful.
- Carbamates are commonly used in industry as precursors for generating isocyanates in situ. For reviews see: (a) D. A. Wicks and Z. W. Wicks Jr, Prog. Org. Coat., 1999, 36, 148 CrossRef CAS; (b) D. A. Wicks and Z. W. Wicks Jr, Prog. Org. Coat., 2001, 41, 1 CrossRef CAS.
- For a similar chromanone to chromene conversion see: K. K. Gruner and H.-J. Knolker, Org. Biomol. Chem., 2008, 6, 3902 CAS.
- (−)-Notoamide B is accessible in one step from 1 according to the procedure of Williams, et al. (ref. 7e). (+)-Avrainvillamide and (−)-stephacidin B are available (as an equilibrium mixture) in two steps from 1 according to the procedure of Baran et al. (ref. 7b)..

Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1400755 and 1400756. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01977j |
| This journal is © The Royal Society of Chemistry 2015 |