
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into the strong aggregation-induced emission of low-conjugated racemic C6-unsubstituted tetrahydropyrimidines through crystal-structure–property relationship of polymorphs†
Qiuhua
Zhu
 a,
Yilin
Zhang
b,
Han
Nie
c,
Zujin
Zhao
c,
Shuwen
Liu
*a,
Kam Sing
Wong
*b and
Ben Zhong
Tang
*cd
a,
Yilin
Zhang
b,
Han
Nie
c,
Zujin
Zhao
c,
Shuwen
Liu
*a,
Kam Sing
Wong
*b and
Ben Zhong
Tang
*cd
aSchool of Pharmaceutical Sciences, Southern Medical University, 1838 Guangzhou Avenue North, Guangzhou 510515, China. E-mail: liusw@smu.edu.cn
bDepartment of Physics, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong. E-mail: phkswong@ust.hk
cGuangdong Innovative Research Team, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
dDepartment of Chemistry, State Key Laboratory of Molecular Neuroscience, Institute of Molecular Functional Materials, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
First published on 26th May 2015
Abstract
Racemic C6-unsubstituted tetrahydropyrimidines (THPs) are a series of fluorophores with a strong aggregation-induced emission (AIE) effect. However, they do not possess the structural features of conventional AIE compounds. In order to understand their AIE mechanism, here, the influences of the molecular packing mode and the conformation on the optical properties of THPs were investigated using seven crystalline polymorphs of three THPs (1–3). The racemic THPs 1–3 have low-conjugated and highly flexible molecular structures, and hence show practically no emission in different organic solvents. However, the fluorescence quantum yields of their polymorphs are up to 93%, and the maximum excitation (λex) and emission (λem) wavelengths of the polymorphs are long at 409 and 484 nm, respectively. Single-crystal structures and theoretical calculation of the HOMOs and LUMOs based on the molecular conformations of these polymorphs indicate that the polymorphs with the shortest λex and λem values possess a RS-packing mode (R- and S-enantiomers self-assemble as paired anti-parallel lines) and a more twisted conformation without through-space conjugation between the dicarboxylates, but the polymorphs with longer λex and λem values adopt a RR/SS-packing mode (R- and S-enantiomers self-assemble as unpaired zigzag lines) and a less twisted conformation with through-space conjugation between the dicarboxylates. The molecular conformations of 1–3 in all these polymorphs are stereo and more twisted than those in solution. Although 1–3 are poorly conjugated, the radiative rate constants (kr) of their polymorphs are as large as conventional fluorophores (0.41–1.03 × 108 s−1) because of improved electronic conjugation by both through-bond and through-space interactions. Based on the obtained results, it can be deduced that the strong AIE arises not only from the restriction of intramolecular motion but also from enhanced electronic coupling and radiatively-favored inter-crossed local excitation (LE) and intramolecular charge transfer (ICT) excitation states. The abnormal molecular structures, easily-controllable self-assembly of the R- and S-enantiomers, and the strong AIE effect make THPs very useful fluorophores for applications and theoretical research.
Introduction
Solid organic fluorophores have received considerable attention owing to their practical applications, such as organic light-emitting diodes (OLEDs)1 and optical waveguides.2 However, highly emissive organic fluorophores in solution often show weak or even zero emission when aggregated, which has frequently been referred to as the aggregation-caused quenching (ACQ) effect.3 In 2001, a propeller-like fluorophore, 1-methyl-1,2,3,4,5-pentaphenylsilole, was found to be not emissive in solution but highly emissive in aggregates,4 which was termed aggregation-induced emission (AIE). During the last decade, AIE compounds have shown great advantages in many application areas such as OLEDs5 and chemo/biosensors.6 A conjugated moiety with multiple rotatable aryl groups, such as polyarylated ethenes7,8 and siloles,4,9 is considered a structural feature of AIE compounds.10We recently developed a five-component reaction (5CR) for the synthesis of C-6 unsubstituted tetrahydropyrimidines (THPs).11 THPs show practically no emission in solution but strong emission in aggregates with fluorescence quantum yields up to 93%,11 presenting a strong AIE effect. Unlike other reported small organic AIE compounds, THPs do not have a π-conjugated stator connected to multiple rotatable aryl groups. The structural characteristic of THPs is a non-aromatic chiral central ring (tetrahydropyrimidine) connected with three aryl rings that are not conjugated with each other. In addition to their unusual AIE properties, THPs were found to have an abnormal response to copper(II)12 and unique properties in surfactant micelles.13 Since the emission of THPs is attributed to aggregation and as one of the THPs (1 in Fig. 1) was found to form two fluorescent polymorphs (1b and 1c),11 we wonder if other THPs could form different fluorescent polymorphs. If so, it might be possible to study the influences of molecular packing and conformation on their optical properties without a change in the molecular structure, another important factor correlating with optical properties. Fortunately, two polymorphs (2c and 2c′) of THP 2 and three polymorphs (3b, 3c and 3p) of THP 3 (Fig. 1) were obtained. Then, the fluorescence properties and single crystal structures of these polymorphs were investigated. We report our results here.
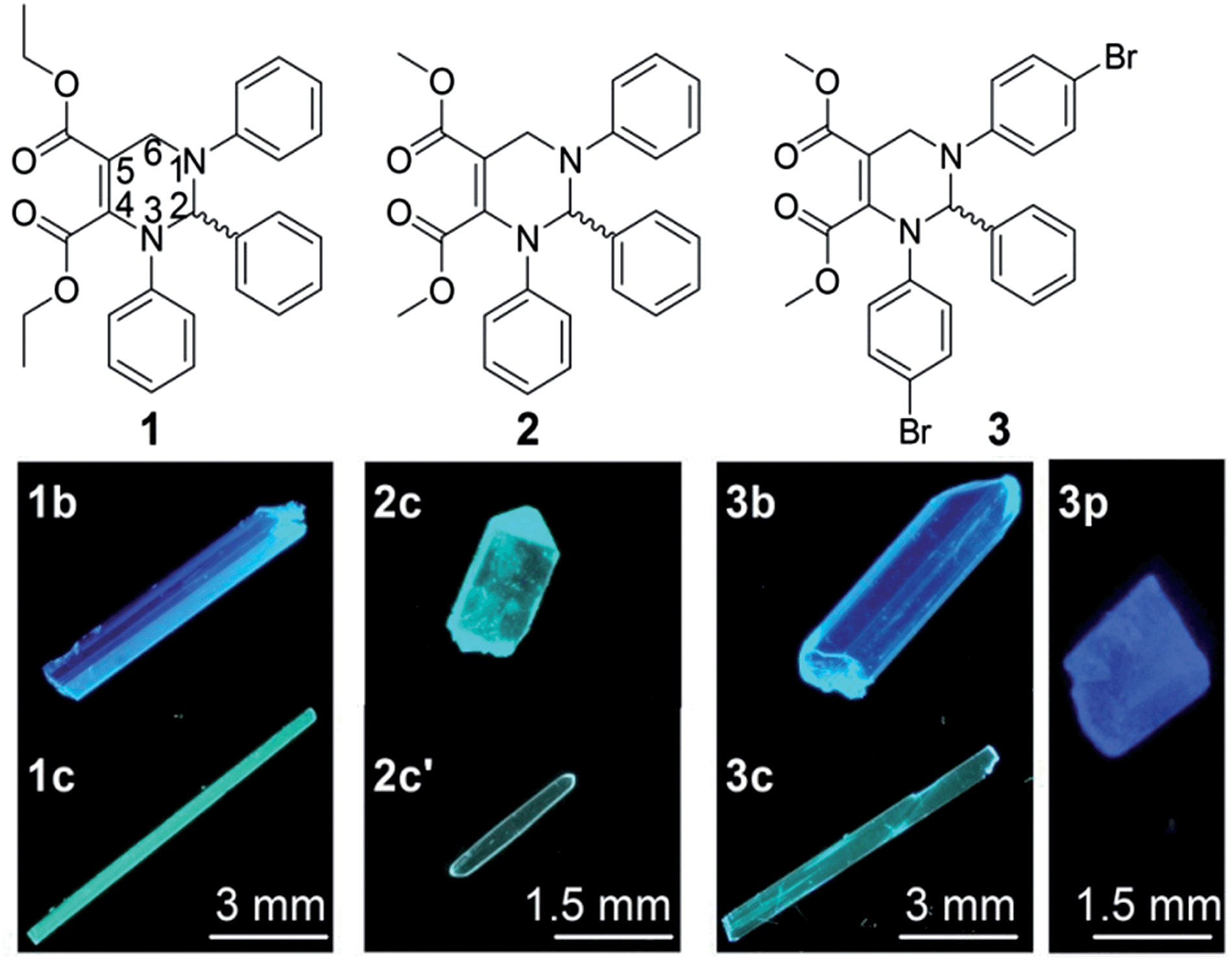 | ||
| Fig. 1 Molecular structures of the racemic THPs 1–3 and their polymorphs under UV light (350 nm). 1b and 1c represent the blue- and cyan-fluorescence polymorphs of 1; 2c and 2c′ represent different cyan-fluorescence polymorphs of 2; 3b, 3c and 3p represent the blue-, cyan- and purple-fluorescence polymorphs of 3, respectively. | ||
Results and discussion
Preparation of the polymorphs (1b, 1c, 2c, 2c′, 3b, 3p and 3c) of THPs 1–3
The polymorphs of THPs 1–3 can be prepared by recrystallization from dichloromethane/n-hexane (for all polymorphs except 3p) or ethyl acetate/n-hexane (for 3p) solution under different conditions. Thermodynamic conditions (high temperature, low concentration and long time) are favorable for the formation of polymorphs 1b and 2c, but kinetic conditions (low temperature, high concentration and short time) favor the formation of polymorphs 1c and 2c′. Polymorphs 3b and 3c were prepared by slow recrystallization at 4 °C, and 3p at room temperature.Absorption spectra of THPs 1–3 in solution and the fluorescence properties of their polymorphs
Fig. 2 shows the absorption spectra of THPs 1–3 in solution as well as the excitation and emission spectra of their polymorphs. As depicted in Fig. 2A, the absorption spectrum of 1 is almost the same as that of 2 except it has a slightly higher absorbance. The absorption peaks at lower energy (317 nm) of 1 and 2 are slightly shorter than that (320 nm) of 3. THPs 1–3 in different organic solvents (cyclohexane, ethanol and acetonitrile etc.) are practically non-emissive (insets in Fig. 2A, their emission spectra are lines parallel to the x-coordinate), but their crystalline polymorphs are highly emissive (Fig. 1). The excitation and emission spectra of these polymorphs are depicted in Fig. 2B–D. The lower energy peak wavelengths in the excitation spectra of these polymorphs, except for 3p, show large red shifts (38–92 nm) with respect to the corresponding absorption peaks in Fig. 2A. The peak emission wavelengths (λem) of these polymorphs vary from 425 to 484 nm (Fig. 2B–D and Table 1), revealing that the molecular packing in different polymorphs has a great impact on the emission wavelengths. | ||
| Fig. 2 Optical properties of THPs 1–3. (A) Absorption spectra of 1–3 in cyclohexane solutions (1.0 × 10−5 M). (B–D) Excitation (left) and emission (right) spectra of the polymorphs of 1–3, respectively. Excitation and emission spectra were detected by emission and excitation at the peak wavelengths marked in (B–D). Insets in (A) are THPs 1–3 in cyclohexane solutions under daylight (top) and UV light at 365 nm (bottom). | ||
| THP | 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|---|
| Polymorph | 1b | 1c | 2c | 2c′ | 3p | 3b | 3c |
| a Peak excitation wavelength at lower energy area. b Peak emission wavelength. c Absolute quantum yield determined via calibrated integrating sphere, excited at 380 nm. d Excited at 360 nm. | |||||||
| λ ex /nm | 355 | 409 | 370 | 387 | 330 | 365 | 390 |
| λ em /nm | 434 | 484 | 469 | 484 | 425 | 445 | 468 |
| Φ F /% | 72 | 93 | 48 | 28 | 30 | 20 | 52 |
| τ /ns | 7.1 | 14 | 11 | 6.8 | 2.9 | 2.3 | 7.6 |
| k r/s−1 × 108 | 1.01 | 0.66 | 0.44 | 0.41 | 1.03 | 0.87 | 0.68 |
| k nr/s−1 × 108 | 0.39 | 0.05 | 0.47 | 1.06 | 2.41 | 3.48 | 0.63 |
The fluorescence quantum yields (ΦF) of the polymorphs of 1–3 were determined by a calibrated integrating sphere. The ΦF values of these polymorphs range from 20 to 93%. The ΦF values of 1c and 3c with longer λem values are higher than those of their corresponding polymorphs 1b and 3p with shorter λem values. However, for 2c and 2c′, 3p and 3b, the opposite is the case (Table 1). These results indicate that the ΦF values are independent of λem values.
The fluorescence lifetimes (τ) of the polymorphs of 1–3 were measured to understand the nature of the excited state. All the fluorescence decay profiles can be well-fitted by a single exponential decay (Fig. S1†). The τ values of these polymorphs are shown in Table 1. Similar to the quantum yields, the τ values are independent of λem values.
Since both τ and ΦF depend on the radiative rate constant (kr) and the non-radiative rate constant (knr), that is, τ = 1/(kr + knr) and ΦF = kr/(kr + knr), the kr (kr = ΦF/τ) and knr (knr = (1 − ΦF)/τ) values were calculated. As shown in Table 1, the kr values of these polymorphs are similar (0.41–1.03 × 108 s−1) and are inversely proportional to the λem values of these polymorphs. However, the knr values are significantly different (0.05–3.48 × 108 s−1) and are unrelated to the λem values.
Molecular stacking modes of the polymorphs of the racemic THPs 1–3
To investigate the influence of enantiomer packing alignments on the fluorescence properties of these polymorphs, their single-crystal structures were determined by X-ray diffraction. Interestingly, all the polymorphs with the shortest λem values, 1b, 2c and 3p, showed a paired packing mode of R- and S-enantiomers (RS packing mode), and all polymorphs with longer λem values, 1c, 2c′, 3b and 3c, show an unpaired packing mode of R- and S-enantiomers (RR/SS packing mode). For instance, the RR/SS packed 1c has an emission peak at 484 nm, being red-shifted by 50 nm compared to that of the RS packed 1b.RS packing mode
Fig. 3 depicts the RS paired packing of 1b, 2c and 3p. The blue and yellow balls, respectively, represent the R- and S-carbon atoms. The three phenyl rings connected to the THP central ring are marked as A, B and C. As shown in Fig. 3A–C, the R- and S-enantiomers of 1b, 2c or 3p are paired with each other in the bc-plane (front view).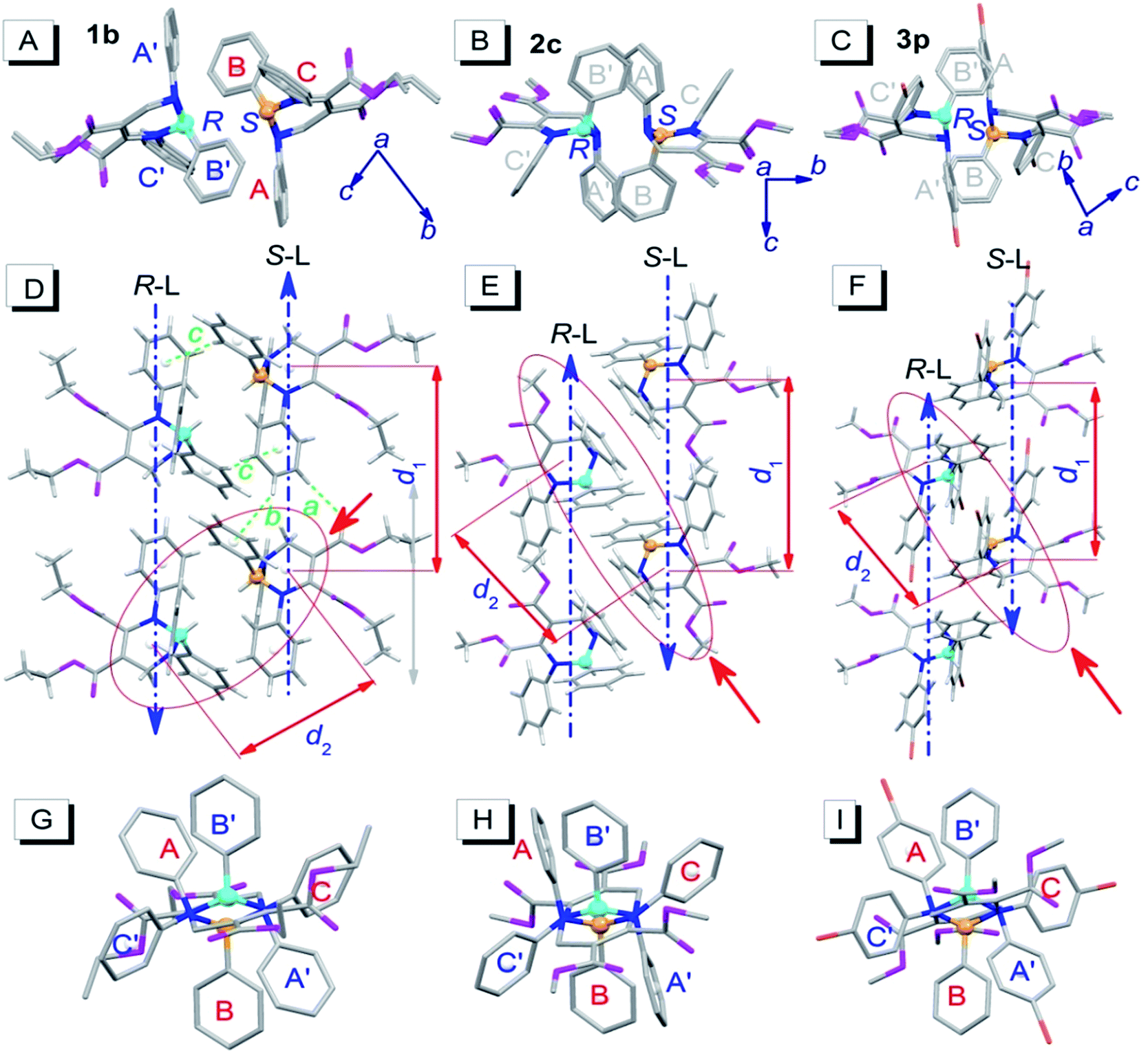 | ||
| Fig. 3 Molecular packing alignments of the R-enantiomers (blue chiral carbon) and the S-enantiomers (yellow chiral carbon) of 1b (left column), 2c (middle column) and 3p (right column).† (A–C) Front view; (D–F) top view; (G–I) side view from the orientation of the red solid arrow in (D–F). Weak hydrogen bonds: a (ary C–H⋯O: 2.487 Å, 177°), b (ary C–H⋯π: 2.930 Å, 146°) and c (ary C–H⋯π: 2.837 Å, 148°). Hydrogen atoms in the front and side views were omitted for clarity. | ||
The top view illustrates that the R- and S-enantiomers of 1b, 2c or 3p align as two parallel lines, R-line and S-line (R-L and S-L), with opposite molecular alignment orientations (represented by two parallel blue dashed lines in Fig. 3D–F). The distances (d1) between the ring centroids of adjacent molecules in the R- or S-lines of 1b, 2c and 3p are 9.726, 9.591 and 10.677 Å, respectively (Fig. 3D–F). All molecules in 1b are connected as a network via weak intermolecular hydrogen bonds (dotted bonds in Fig. 3D and S2†) (the hydrogen bond parameters are listed in Table S1†). However, no weak hydrogen bonds were found in 2c and 3p single crystals. The molecular packing in the unit cell and some crystallographic data of 1b, 2c and 3p are shown in Fig. S2 and Table S2,† respectively.
Very interestingly, the paired enantiomers arranged as flowers with six petals (six phenyls) (Fig. 3G–I). The distances (d2) between the ring centroids of the adjacent R- or S-enantiomers of 1b, 2c and 3p are 7.067, 7.492, and 7.839 Å, respectively. The dihedral angles between adjacent phenyl ring planes (αdih) and the distances between adjacent ring centroids (dring) are 37–76° and 4.294–5.308 Å, respectively (for detailed parameters see Table S1†), which indicate that short-range interactions between the six phenyls of the paired R- and S-enantiomers exist.14 These interactions and the chiral structure of THPs are expected to be the reason for the interesting flower-like arrangement. It is worth mentioning that the short-range ring interactions in 1b and 3p only exist between paired R- and S-enantiomers, but the short-range ring interactions in 2c also exist between another molecule and one of the paired R- and S-enantiomers.
RR/SS packing mode
Fig. 4 depicts the unpaired enantiomer packing mode (RR/SS packing mode) of 1c, 2c′, 3b and 3c. From Fig. 4A–D (front view), it can be seen that the R- or S-enantiomers of 1c, 2c′, 3b or 3c partly overlap.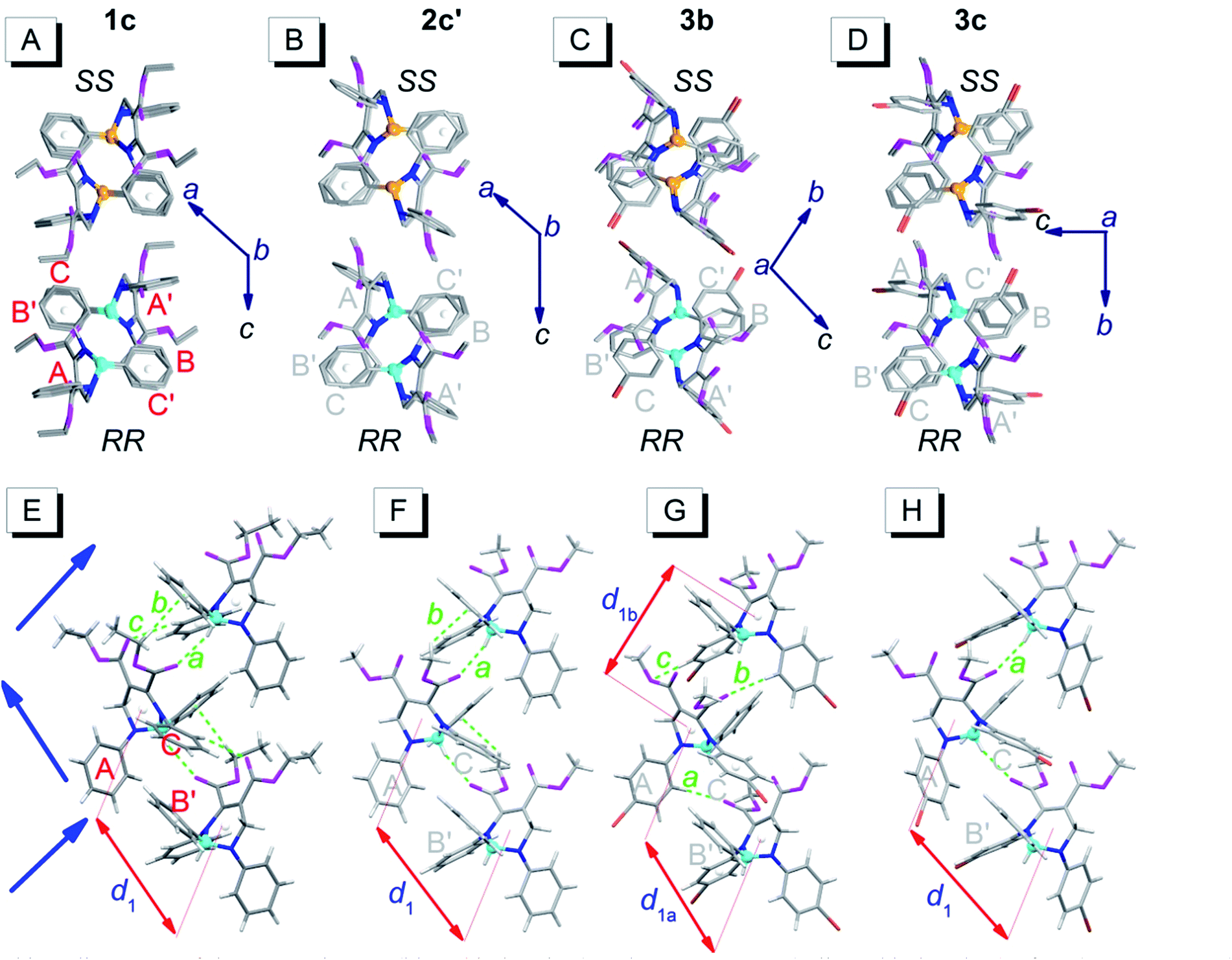 | ||
| Fig. 4 Packing alignments of the R-enantiomers (blue chiral carbon) and the S-enantiomers (yellow chiral carbon) of 1c, 2c′, 3b and 3c (from left to right columns).† (A–D) Front view (hydrogen atoms were omitted for clarity); (E–H) top view. Weak hydrogen bonds: a (C–H⋯O: 2.434 Å, 143°), b (C–H⋯π: 2.634 Å, 144°) and c (ary C–H⋯O: 2.579 Å, 136°) in (E); a (C–H⋯O: 2.407 Å, 145°) and b (C–H⋯π: 2.901 Å, 124°) in (F); a (ary C–H⋯O: 2.569 Å, 176°), b (ary C–H⋯O: 2.553 Å, 165°) and c (ary C–H⋯O: 2.551 Å, 146°) in (G); a (C–H⋯O: 2.362 Å, 151°) in (H). | ||
The top view (only the R-enantiomer alignment is depicted in Fig. 4E–H) illustrates that the R-enantiomers of 1c, 2c′, 3b or 3c arrange in a zigzag line. The d1 values between the ring centroids of the adjacent R(S)- and R(S)-enantiomers of 1c, 2c′, 3b and 3c are 7.53, 7.59, 7.303 (d1a)/7.438 (d1b) and 7.754 Å, respectively, which are similar to the d2 values but shorter than the d1 values in the RS packing modes (Fig. 3). Adjacent molecules in the zigzag lines are connected via six, four, three and two weak hydrogen bonds for 1c, 2c′, 3b and 3c, respectively. In addition to the two hydrogen bonds in Fig. 4H, there are four hydrogen bonds between adjacent R- or S-enantiomers of 3c (Fig. S3E and F†), and hence all molecules in 3c are connected as a network via weak hydrogen bonds. The short-range interactions (αdih = 16–69°, dring = 4.127–5.481 Å, see Table S1†) between the six phenyls of the three adjacent R/S-enantiomers are similar to those in the RS-packed polymorphs. The molecular stacking alignments in the unit cell of 1c, 2c′, 3b and 3c are shown in Fig. S3.† The intermolecular hydrogen bond parameters and some crystallographic data are listed in Tables S1 and S2,† respectively.
Conformations of the polymorphs of THPs 1–3
Since the molecular conformation also influences the optical properties of organic fluorophores, the conformations of these polymorphs were studied. Fig. 5 depicts the molecular conformations of the polymorphs of THP 1. It can be seen that the conformations are stereo with phenyls A–C arranged in completely different spatial directions (up, down and in front of the central ring). There are two significant differences between the conformations of 1b and 1c: (a) the dihedral angle (α) between the phenyl C and the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-plane in 1b is larger than that in 1c (Table 2); (b) the orientation of the carbonyl group at C-5 (marked by red circles) are very different. Two and one intramolecular hydrogen bonds exist in 1b and 1c, respectively (marked as a and b). There are intramolecular short-range ring interactions between rings A and C in 1b and 1c (schematically depicted in Fig. 5, and detailed data are listed in Table S1†).
C-plane in 1b is larger than that in 1c (Table 2); (b) the orientation of the carbonyl group at C-5 (marked by red circles) are very different. Two and one intramolecular hydrogen bonds exist in 1b and 1c, respectively (marked as a and b). There are intramolecular short-range ring interactions between rings A and C in 1b and 1c (schematically depicted in Fig. 5, and detailed data are listed in Table S1†).
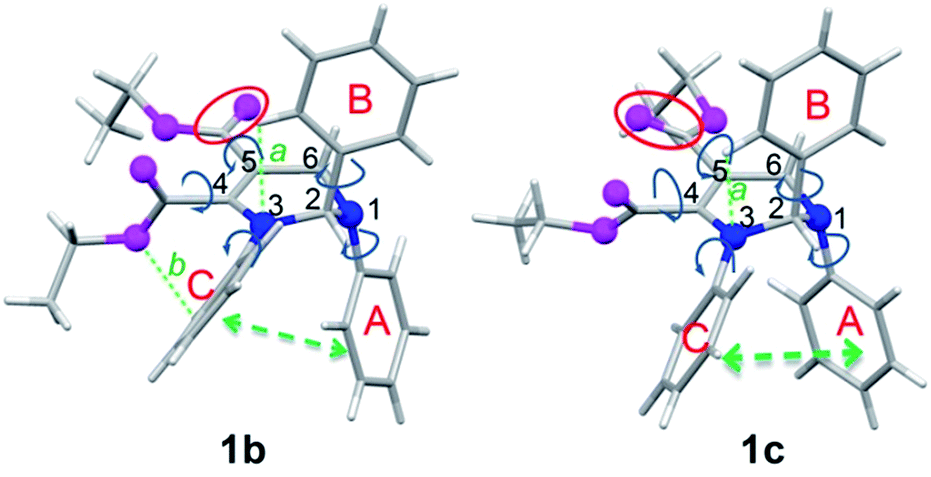 | ||
| Fig. 5 Molecular conformations of polymorphs 1b (left) and 1c (right). Hydrogen bonds a (ary C–H⋯O: 2.505 Å, 102°), b (ary C–H⋯N: 2.578 Å, 116°) in 1b; a (ary C–H⋯N: 2.511 Å, 102°) in 1c. | ||
| THP | 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|---|
| Pola | 1b | 1c | 2c | 2c′ | 3p | 3b | 3c |
|
a Pol: polymorph.
b PM: packing mode.
c Peak emission wavelength.
d The energy of the peak emission wavelength.
e Dihedral angle between phenyl C and the –CC-plane.
f Band gap between the HOMO and LUMO.
g Peak excitation wavelength at lower energy area.
|
|||||||
| PMb | RS | RR/SS | RS | RR/SS | RS | RR/SS | RR/SS |
| λ em /nm | 434 | 484 | 469 | 484 | 425 | 445 | 468 |
| ΔE1d/eV | 2.86 | 2.57 | 2.65 | 2.57 | 2.92 | 2.79 | 2.66 |
| α /° | 49.24 | 36.46 | 48.62 | 31.81 | 53.04 | 43.50 | 34.50 |
| ΔE2f/eV | 4.47 | 4.17 | 4.50 | 4.22 | 4.55 | 4.45 | 4.29 |
| λ bg /nm | 278 | 298 | 276 | 295 | 273 | 279 | 290 |
| λ ex /nm | 355 | 409 | 370 | 387 | 330 | 365 | 390 |
| Δ λ ex−λbg/nm | 77 | 111 | 94 | 92 | 57 | 86 | 100 |
| Δ λ ab−λbg/nm | 39 | 19 | 41 | 22 | 47 | 41 | 30 |
The molecular conformations of other five polymorphs are shown in Fig. S4.† Polymorphs, except 2c, with the same packing modes possess similar conformations, that is, the conformation of the RS-packing of 3p is similar to that of the RS-packing of 1b, and the conformations of 2c′, 3b and 3c are similar to that of the RR/SS-packing of 1c. However, the conformation of the RS-packed 2c is an intermediate case between the conformations of RS-packed and RR/SS-packed polymorphs, that is, the α value between the phenyl C and the –CC-plane of the RS-packed 2c is similar to that of the RS-packed 1b and 3p (Table 2), but the orientation of the carbonyl (CO) at C-5 is similar to that of the RR/SS-packed 1c, 2c′, 3b and 3c. It is worth mentioning that two different conformations of 3b exist (3b1 and 3b2 in Fig. S4†). The intramolecular short-range ring interactions between rings A and C in these polymorphs are shown in Table S1.†
Theoretical calculation of HOMOs and LUMOs
To further understand the different fluorescence between these polymorphs, the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) based on the molecular conformations of the polymorphs of THPs 1–3 were calculated. The plots of the HOMO and LUMO of 1b and 1c are shown in Fig. 6. The HOMO of 1b/1c is mainly located on the CC5–CO moiety, phenyls A and C, but the LUMO is only on two carboxylate groups and the phenyl C, which means that both a local excitation state (LE) and an intramolecular charge transfer excitation state (ICT) exist. The inter-crossed excited states are not only proved to lead to a red shift in absorption/emission but also to favor fluorescence efficiency enhancement.15 In addition, the lone pair of electrons on N1 and the π-electrons conjugate not only through bonds but also through space16 (marked with red circles), which can enhance electronic delocalization and restrict intramolecular motion, leading to a fluorescence efficiency enhancement and red-shifted absorption/emission to some extent. Besides the through-space conjugations marked with red circles, through-space conjugation exists between the dicarboxylates of 1c (marked with a yellow circle), which should be one of the reasons why 1c shows a longer wavelength emission than 1b. The optical band gaps of 1b and 1c are 4.47 and 4.17 eV, respectively, which are wider than that of 1 (3.92 eV) in monomers. This demonstrates that the molecular conformations of 1 in 1b and 1c are more twisted than those in solutions. The plots of the HOMOs and LUMOs of the other polymorphs are similar to those of 1b or 1c (Fig. S5†).
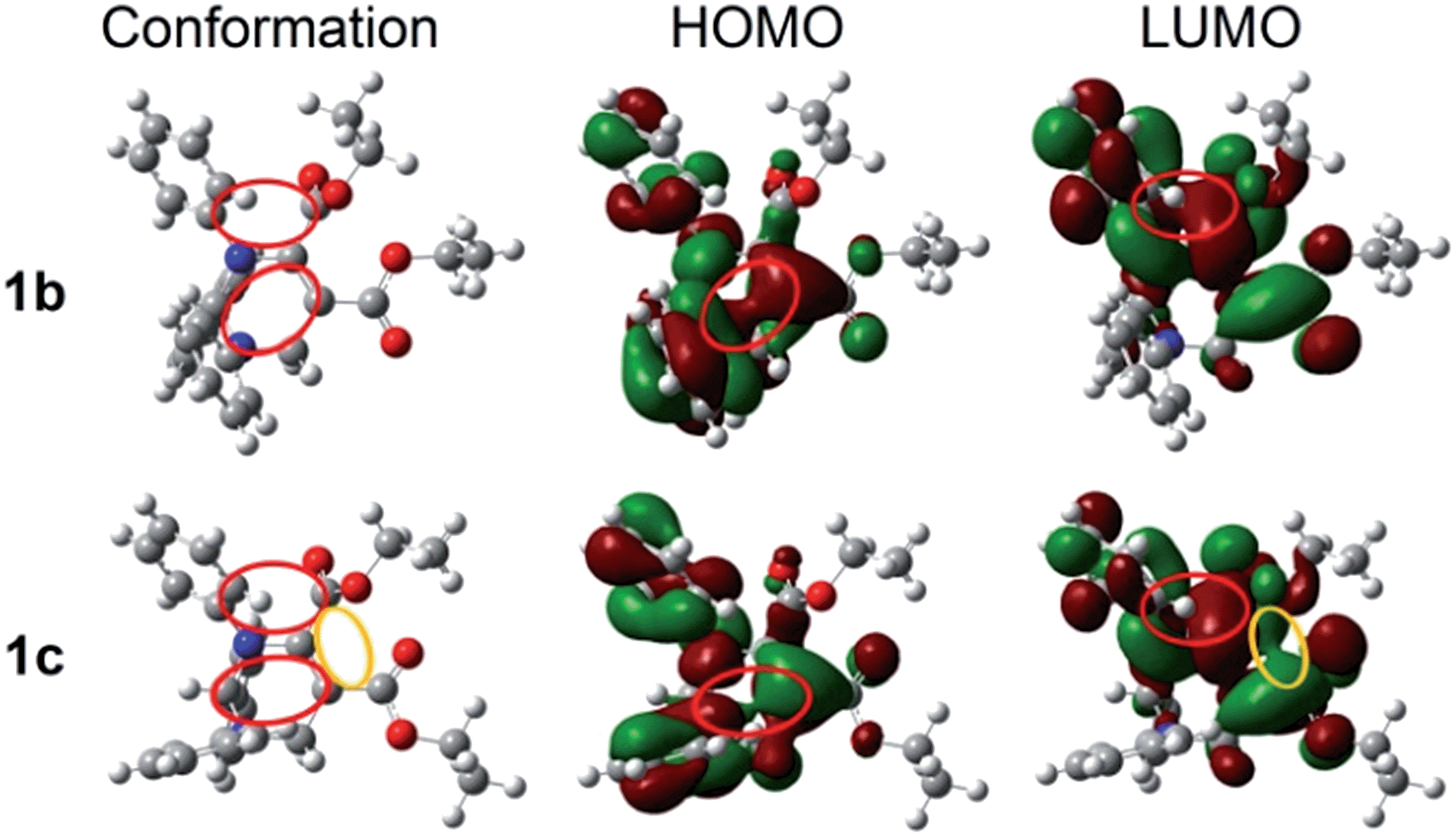 | ||
| Fig. 6 Molecular conformations, HOMOs and LUMOs of polymorphs 1b and 1c. | ||
AIE mechanism
The fluorescence quantum yield of an organic fluorophore depends on its radiative and non-radiative rate constants. Very high fluorescence efficiency can be realized when the kr value is much larger than the knr value, while practically no emission will be observed when knr is much larger than kr. Owing to intramolecular motions (the rotation of single C–C and C–N bonds and the flip of the non-aromatic central ring), the knr values of THPs 1–3 in solution are expected to be very large like those of other AIE compounds.17 In addition, THPs 1–3 have low conjugated molecular structures, that is, very small kr values. Therefore, THPs 1–3 show practically no emission in solution. However, upon aggregation, the knr values of THPs 1–3 dramatically decrease to 0.05–3.48 × 108 s−1 because of the restriction of intramolecular motion (RIM) by intra- and intermolecular interactions, and their kr values increase to 0.41–1.03 × 108 s−1, which are as large as those of conventional π-conjugated fluorophores,18 owing to the improved electronic coupling via through-bond and through-space pathways (Fig. 6 and S5†) as well as the inter-crossed radiative-favorable LE and ICT processes. Consequently, THPs 1–3 show a strong AIE effect and the polymorphs emit intense fluorescence. By comparing the kr and knr values of the polymorphs of THPs 1–3 and the factors influencing these values, it can be deduced that: (a) RS-packing should be more favourable for a radiative process than RR/SS-packing because the kr values of the RS-packing polymorphs are slightly higher than those of their corresponding RR/SS-packing polymorphs even though the RS-packing conformations are more twisted and do not contain through-space conjugation between the dicarboxylates. This might arise from the flower-like packing alignment between R- and S-enantiomers (Fig. 3G–I) because this special packing alignment is similar to the reported cross-stacking mode with a high fluorescence efficiency.19 (b) The differences in ΦF between these polymorphs should mainly result from the different extents of RIM, that is, different knr values, because they possess the same molecular structure and have similar kr values. The short-range interaction between the intramolecular phenyls A and C may be crucial in decreasing the knr value of these polymorphs, as the distances between the intramolecular phenyls A and C in all polymorphs with the lowest knr value (1c, 2c and 3c) are shorter than those of their corresponding polymorphs (Table S1†).The red-shifted absorption and emission spectra of organic fluorophores in aggregates are mainly caused by the planarization of its π-conjugated moiety, intramolecular and intermolecular interactions. The planarization and intramolecular interactions depend on the molecular conformation, but intermolecular interactions depend on the molecular stacking mode. To evaluate the influences of the conformation and the packing mode of the polymorphs of THPs 1–3 on their absorption wavelengths, the lowest energy excitation peaks λex of these polymorphs and the band gap values (λbg) between their HOMOs and LUMOs and their differences (Δλex−λbg) have been listed in Table 2. The differences (Δλab−λbg) between the lowest energy absorption peaks (λab) of THPs 1–3 in solution and the λbg values are also listed in Table 2. The influence of molecular conformation/stacking mode on the absorption spectra of these polymorphs can be evaluated by the Δλab−λbg/Δλex−λbg values. It can be see that the molecular packing modes in these polymorphs can lead to a 57–111 nm (Δλex−λbg) red-shift in their excitation spectra, but their twisted conformation leads to a 19–47 nm (Δλab−λbg) blue-shift. Therefore, the large red-shifts in the absorption and emission spectra of these polymorphs are mainly attributed to intermolecular ring interactions. This is because the ring interactions of adjacent π-conjugated molecules have been demonstrated to cause a larger, non-structural red-shift in the absorption/emission spectra compared to that caused by molecular planarization.20
Then, what is the reason for the differences in the Δλex−λbg and Δλab−λbg values between the polymorphs of THPs 1–3? By comparing the Δλex−λbg values of these polymorphs and the intermolecular interactions in these polymorphs, it can be deduced that the Δλex−λbg values should mainly depend on the short-range (4.1–5.5 Å) interactions between the phenyls of adjacent molecules (the short-range ring interactions in 1c, 1b, 2c, 2c′, 3p, 3b and 3c are depicted in Fig. S6†). Short-range ring interactions in the RR/SS-packing polymorphs 1c and 3c exist among all zigzag-aligned R- or S-enantiomers (Fig. 4E–H), but those in their corresponding RS-packing polymorphs 1b and 3p only exist between two paired R- and S-enantiomers. Therefore, the former can lead to a larger electron delocalization than the latter, and hence the Δλex−λbg values (110 and 100 nm) of the RR/SS-packing polymorphs 1c and 3c are much larger than those (77 and 57 nm) of their corresponding polymorphs 1b and 3p. Although 2c also formed a RS-packing mode, the short-range ring interactions in it are not limited to two paired R- and S-enantiomers (Fig. S6†) and are similar to those in its RR/SS-packing polymorph 2c′, and hence RS-packing 2c and RR/SS-packing 2c′ possess similar Δλex−λbg values (94 and 92 nm). By comparing the Δλab−λbg values of these polymorphs and their conformations, it can be deduced that the differences in the Δλab−λbg values between these polymorphs should mainly be caused by the different dihedral angle α between the phenyl C and the –CC-plane (Table 2) and by the conjugation between the dicarboxylates (with or without through-space conjugation in Fig. 6 and S5†). For example, the Δλab−λbg value of 3b is 6 nm smaller than that of 3p but 11 nm larger than that of 3c, even though the differences in the dihedral angle α between 3b and 3p and between 3b and 3c are similar (9.54 and 9.00°). The reason for this should be that the difference in the Δλab−λbg value between 3b and 3p is mainly caused by their different α values but the difference in the Δλab−λbg value between 3b and 3c arises from their different α values and from conjugation between the dicarboxylates (Fig. S5†).
Conclusions
We have prepared seven polymorphs using the racemic THPs 1–3, and studied the influence of their molecular packing mode and conformation on their optical properties. The experimental results indicate that the polymorphs with the shortest λex and λem values possess a RS-packing mode and a more twisted conformation without through-space conjugation between the dicarboxylates, but the polymorphs with longer λex and λem values adopt RR/SS-packing modes with through-space conjugation between the dicarboxylates. The conformations of these polymorphs are stereo and more twisted than those of their monomers. Although THPs 1–3 show practically no emission in solution owing to their low conjugated molecular structures and high intramolecular motions, they can form emissive polymorphs by through-bond and through-space electron coupling in aggregates. These new fluorophores with unique molecular structures, easily-controllable packing modes of the R- and S-enantiomers, and strong AIE effects are expected to be very useful for practical and theoretical research as well as the design of new highly emissive fluorophores with AIE properties.Acknowledgements
We thank Ian D. Williams and Herman H. Y. Sung for support on the collection and analysis of some XRD data, we thank Zheng S. C. and Yang W. J. for measurement of the optical spectra, and we thank Huang L. for the preparation of two polymorphs. This work was supported by the National Natural Science Foundation of China (21272111 and 31370781), the National Basic Research Program of China (973 Program; 2013CB834701), the Research Grants Council of Hong Kong (604711, 604913, HKUST2/CRF/10 and N_HKUST620/11), Innovation and Technology Commission (ITCPD/17-9), and the University Grants Committee of Hong Kong (AoE/P-03/08).Notes and references
- (a) S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lussem and K. Leo, Nature, 2009, 459, 234–238 CrossRef CAS PubMed; (b) K. H. Lee, Y. S. Kwon, J. Y. Lee, S. Kang, K. S. Yook, S. O. Jeon, J. Y. Lee and S. S. Yoon, Chem.–Eur. J., 2011, 17, 12994–13006 CrossRef CAS PubMed; (c) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., 2014, 126, 2151–2155 CrossRef PubMed.
- (a) Y. S. Zhao, A. Peng, H. Fu, Y. Ma and J. Yao, Adv. Mater., 2008, 20, 1661–1665 CrossRef CAS PubMed; (b) Y. S. Zhao, J. Xu, A. Peng, H. Fu, Y. Ma, L. Jiang and J. Yao, Angew. Chem. Int. Ed., 2008, 47, 7301–7305 CrossRef CAS PubMed; (c) X. Gu, J. Yao, G. Zhang, Y. Yan, C. Zhang, Q. Peng, Q. Liao, Y. Wu, Z. Xu and Y. Zhao, Adv. Funct. Mater., 2012, 22, 4862–4872 CrossRef CAS PubMed.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CAS.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- (a) W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu and N. Xie, Chem. Mater., 2012, 24, 1518–1528 CrossRef CAS; (b) J. Huang, N. Sun, P. Chen, R. Tang, Q. Li, D. Ma and Z. Li, Chem. Commun., 2014, 50, 2136–2138 RSC.
- (a) Y. Liu, Y. Yu, J. W. Y. Lam, Y. Hong, M. Faisal, W. Z. Yuan and B. Z. Tang, Chem.–Eur. J., 2010, 16, 8433–8438 CrossRef CAS PubMed; (b) M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC; (c) Y. Liu, C. Deng, L. Tang, A. Qin, R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2011, 133, 660–663 CrossRef CAS PubMed; (d) X. Li, K. Ma, S. Zhu, S. Yao, Z. Liu, B. Xu, B. Yang and W. Tian, Anal. Chem., 2013, 86, 298–303 CrossRef PubMed; (e) Z. Song, Y. Hong, R. T. K. Kwok, J. W. Y. Lam, B. Liu and B. Z. Tang, J. Mater. Chem. B, 2014, 2, 1717–1723 RSC.
- (a) Z. Zhao, J. W. Lam, C. Y. Chan, S. Chen, J. Liu, P. Lu, M. Rodriguez, J. L. Maldonado, G. Ramos-Ortiz, H. H. Sung, I. D. Williams, H. Su, K. S. Wong, Y. Ma, H. S. Kwok, H. Qiu and B. Z. Tang, Adv. Mater., 2011, 23, 5430–5435 CrossRef CAS PubMed; (b) R. Hu, J. W. Y. Lam, Y. Liu, X. Zhang and B. Z. Tang, Chem.–Eur. J., 2013, 19, 5617–5624 CrossRef CAS PubMed.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- X. Fan, J. Sun, F. Wang, Z. Chu, P. Wang, Y. Dong, R. Hu, B. Z. Tang and D. Zou, Chem. Commun., 2008, 2989–2991 RSC.
- (a) Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (b) T. Nicolini, A. Famulari, T. Gatti, J. Martí-Rujas, F. Villafiorita-Monteleone, E. V. Canesi, F. Meinardi, C. Botta, E. Parisini, S. V. Meille and C. Bertarelli, J. Phys. Chem. Lett., 2014, 5, 2171–2176 CrossRef CAS.
- Q. Zhu, L. Huang, Z. Chen, S. Zheng, L. Lv, Z. Zhu, D. Cao, H. Jiang and S. Liu, Chem.–Eur. J., 2013, 19, 1268–1280 CrossRef CAS PubMed.
- L. Huang, J. Su, D. Zhong, H. Wang, R. Liu, L. Yu, Q. Zhu and S. Liu, RSC Adv., 2013, 3, 13286–13292 RSC.
- Q. Zhu, L. Huang, J. Su and S. Liu, Chem. Commun., 2014, 50, 1107–1109 RSC.
- G. D. Scholes and K. P. Ghiggino, J. Phys. Chem., 1994, 98, 4580–4590 CrossRef CAS.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS PubMed.
- Z.-L. Gong, Y.-W. Zhong and J. Yao, Chem.–Eur. J., 2015, 21, 1554–1566 CrossRef CAS PubMed.
- (a) J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS; (b) G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed.
- (a) S. Choi, J. Bouffard and Y. Kim, Chem. Sci., 2014, 5, 751–755 RSC; (b) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 1–10 RSC.
- (a) Z. Xie, B. Yang, F. Li, G. Cheng, L. Liu, G. Yang, H. Xu, L. Ye, M. Hanif, S. Liu, D. Ma and Y. Ma, J. Am. Chem. Soc., 2005, 127, 14152–14153 CrossRef CAS PubMed; (b) C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS PubMed.
- M. Levitus, K. Schmieder, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2001, 123, 4259–4265 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General method, instruments, preparation of polymorphs, characterization data, 1H and 13C NMR of THP 3, Table S1 and S2, Fig. S1–S4, “cif” files containing single-crystal X-ray diffraction data of 2c, 2c', 3p, 3b and 3c. CCDC 850811, 1011037–1011040. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01226k |
| This journal is © The Royal Society of Chemistry 2015 |