
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective and diastereoselective azo-coupling/iminium-cyclizations: a unified strategy for the total syntheses of (−)-psychotriasine and (+)-pestalazine B†
Qi
Li
ab,
Tingting
Xia
b,
Licheng
Yao
b,
Haiteng
Deng
*c and
Xuebin
Liao
*b
aTsinghua-Peking Centre for Life Sciences, Beijing 100084, China
bDepartment of Pharmacology and Pharmaceutical Sciences, School of Medicine, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Tsinghua University, Beijing 100084, China. E-mail: liaoxuebin@mail.tsinghua.edu.cn
cMOE Key Laboratory of Bioinformatics, School of Life Sciences, Tsinghua University, Beijing 100084, China. E-mail: dht@mail.tsinghua.edu.cn
First published on 5th May 2015
Abstract
We report a unified strategy for the total syntheses of (−)-psychotriasine and (+)-pestalazine B based on the advanced intermediates of 3α-amino-hexahydropyrrolo[2,3-b]indole. To construct these structural motifs, a cascade reaction involving a BINOL-derived phosphoric anion-paired catalyst for enantioselective or diastereoselective azo-coupling/iminium-cyclizations was developed. The remaining key steps of the synthesis involve a sterically hindered amination via hypervalent iodine reagents and the Larock annulation. These transformations enable a general approach to the syntheses of indole alkaloids containing a 3α-amino-hexahydropyrrolo[2,3-b]indole motif and could be further applied to build a natural product-based library.
Introduction
Indole alkaloids containing a 3α-amino-hexahydropyrrolo-[2,3-b]indole scaffold are present in a wide range of natural compounds (Fig. 1).1 Structurally, these indole alkaloids feature the linkage of two or more tryptamines or tryptamine derivatives via C3a–N1 bond formation. Due to their unique structures and versatile biological activities these compounds have attracted significant interest from synthetic chemists. For example, psychotrimine (1) was first isolated by the Takayama group and later synthesized by Takayama, Baran and other groups.2,3 It exhibits antibacterial activity against Gram-positive bacteria through membrane disruption.4 Chetomin (5) is a potential anti-cancer agent against the transcription factor hypoxia inducible factor 1 (HIF-1).5 Williams and co-workers have made great progress in the biosynthesis of chetomin.5e Psychotetramine (3) was the first reported alkaloid bearing C3–C3, C7–N1 and C3a–N1 linkages. It was isolated by the Takayama group and its structure was recently elucidated by Baran et al.6 Among other alkaloids in this family, psychotriasine (2) was isolated by the Hao group,7 while pestalazine B (4) was isolated by the Che group and its total synthesis and structural revision was completed by de Lera and co-workers.8 | ||
| Fig. 1 Compounds containing the 3α-amino-hexahydropyrrolo[2,3-b]indole core. | ||
To develop a unified strategy for the total synthesis of these C–N linked indole alkaloids, we needed to establish a highly efficient method for rapid construction of the 3α-amino-hexahydropyrrolo[2,3-b]indole core structure. To date, a number of synthetic approaches to this core structure have been developed and are summarized in Fig. 2: (a) a well-designed method that involves a BINOL-derived chiral phosphoric acid catalyzing the addition of tryptamine derivatives with DEAD was achieved by Antilla and his colleagues;9 (b) an elegant approach to form a C–N bond through coupling of tryptamine derivatives with aniline in the presence of appropriate oxidants was accomplished independently by the Baran and Ji groups;3b,d,6,10 (c) a nitrene-induced aziridination followed by a ring-opening and imine cyclization cascade reaction was developed by Dauban and co-workers, which readily afforded 3α-amino-hexahydropyrrolo[2,3-b]indole derivatives;11 (d) a recent report by the Rainer group on an expeditious synthesis of 3α-indole-hexahydropyrrolo[2,3-b]indole via the cyclopropane ring-opening process was disclosed;12 (e) an early report on an acid-catalyzed nucleophilic substitution reaction on the indole nitrogen was described by the Somei group.13
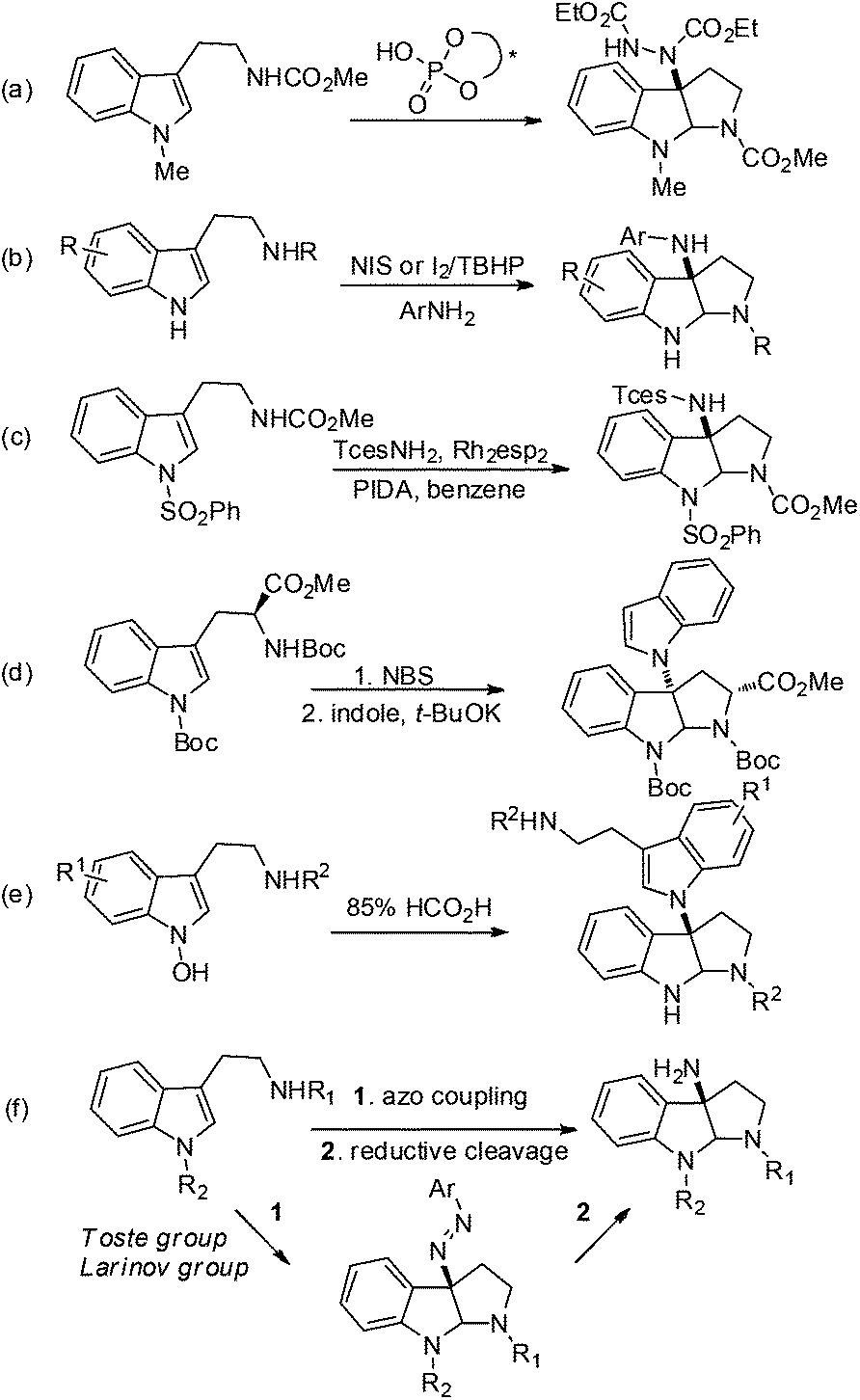 | ||
| Fig. 2 Reported synthetic methods for the 3α-amino-hexahydropyrrolo[2,3-b]indole core structure (a–e) and our approach in this publication and others14 (f). | ||
Most recently, hinging on their signature work on phase transfer catalysis, the Toste group reported a highly enantioselective (up to 96% ee) reaction to construct C3-diazenated pyrroloindolines via a chiral anion phase transfer process (Fig. 2f).14a This elegant work once again demonstrated the beauty of the chiral anion pair concept in asymmetric catalysis. Shortly after, the Larionov group disclosed a racemic version of this reaction.14b However, this capable method to construct the 3α-amino-hexahydropyrrolo[2,3-b]indole scaffold was not available when we started the current investigation of our total syntheses. Thus, we have independently pursued the strategy of stereoselective domino azo-coupling/iminium cyclizations with Na–H tryptomine derivatives as a key step for our total synthesis design. In particular, our methodology development includes an enantioselective azo-coupling catalysis that was very similar to the Toste group's approach, as well as a new process of solvent-enabled diastereo-selective azo-coupling. Both methods allowed us to readily prepare C3-diazenated pyrroloindoline structural motifs via a cascade process. This intermediate could then be subjected to reducing conditions, and the desired 3α-aminohexahydro-pyrrolo[2,3-b]indole scaffold would be synthesized. Herein we report the successful application of these stereoselective cascade processes in the total syntheses of (−)-psychotriasine (2) and (+)-pestalazine B (4). These results demonstrate their utility as a unified strategy to synthesize such indole alkaloids.
Results and discussion
Diazonium salts can be generally utilized as nitrogen sources in organic synthesis. A prominent example of their synthetic application was described in Fukuyama's total synthesis of mersicarpine.15 Similar to Toste's study, we envisioned that the chiral ion pair16–18 which was generated by mixing diazonium salts, Na2CO3 and chiral phosphoric acid,19 would couple with tryptamine derivatives to form iminium intermediates. We anticipated that iminium cyclization would then occur to yield the azo compound in an enantioselective or diastereoselective fashion, which was the precursor of 3α-amino-hexahydropyrrolo[2,3-b]indole (Fig. 2f).We explored the above strategy using Na–H tryptamine derivative 6a (R = CO2Me) and PhN2BF4 as the model substrates (Scheme 1). Initially (S)-TRIP20 was used as the chiral phosphoric acid catalyst (CPA8a) at 0 °C in different solvents (for details see ESI, S-Table 1†). We were encouraged by the formation of desired product 7a in good yields and moderate er (up to 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33) in ethereal solvents such as t-BuOMe. Those results demonstrated that it was possible to construct the target core structure in high enantioselectivity by the chiral ion pair strategy. When switching the N-protecting group from CO2Me to Cbz, Boc or Bz, we found that the Bz-protected product 7d was formed with the highest enantioselectivity (27:73 er). Further screening of the reaction parameters showed a slight increase in enantioselectivity at a lower temperature of −40 °C (25:75 er), while using other CPA (8b–e) catalysts led to similar or lower enantioselectivity. To our surprise, when we used 0.01 equiv. of pyridine as an additive, the enantioselectivity was increased to 20:80 er (CPA: 8a). Replacing pyridine with lutidine gave similar results, and using 2,6-di-t-butylpyridine led to further improvement of enantioselectivity to 14.5:85.5 er. Presumably, a more compact and lipophilic ion pair may result in a higher enantioselectivity, and pyridine-type additives may be involved in the formation of tight ion pairs. Further optimization of the reaction conditions was achieved by applying 4-CF3-PhN2BF4 as the diazonium salt instead of PhN2BF4 and lowering the reaction temperature to −60 °C to obtain product 7f in quantitative yield and 7.5:92.5 er (CPA: 8a). Additional purification of 7f by recrystallization in petroleum ether led to 69% yield and >96% ee. Our findings, in retrospection, are in good agreement with the results of the Toste group.
:33) in ethereal solvents such as t-BuOMe. Those results demonstrated that it was possible to construct the target core structure in high enantioselectivity by the chiral ion pair strategy. When switching the N-protecting group from CO2Me to Cbz, Boc or Bz, we found that the Bz-protected product 7d was formed with the highest enantioselectivity (27:73 er). Further screening of the reaction parameters showed a slight increase in enantioselectivity at a lower temperature of −40 °C (25:75 er), while using other CPA (8b–e) catalysts led to similar or lower enantioselectivity. To our surprise, when we used 0.01 equiv. of pyridine as an additive, the enantioselectivity was increased to 20:80 er (CPA: 8a). Replacing pyridine with lutidine gave similar results, and using 2,6-di-t-butylpyridine led to further improvement of enantioselectivity to 14.5:85.5 er. Presumably, a more compact and lipophilic ion pair may result in a higher enantioselectivity, and pyridine-type additives may be involved in the formation of tight ion pairs. Further optimization of the reaction conditions was achieved by applying 4-CF3-PhN2BF4 as the diazonium salt instead of PhN2BF4 and lowering the reaction temperature to −60 °C to obtain product 7f in quantitative yield and 7.5:92.5 er (CPA: 8a). Additional purification of 7f by recrystallization in petroleum ether led to 69% yield and >96% ee. Our findings, in retrospection, are in good agreement with the results of the Toste group.
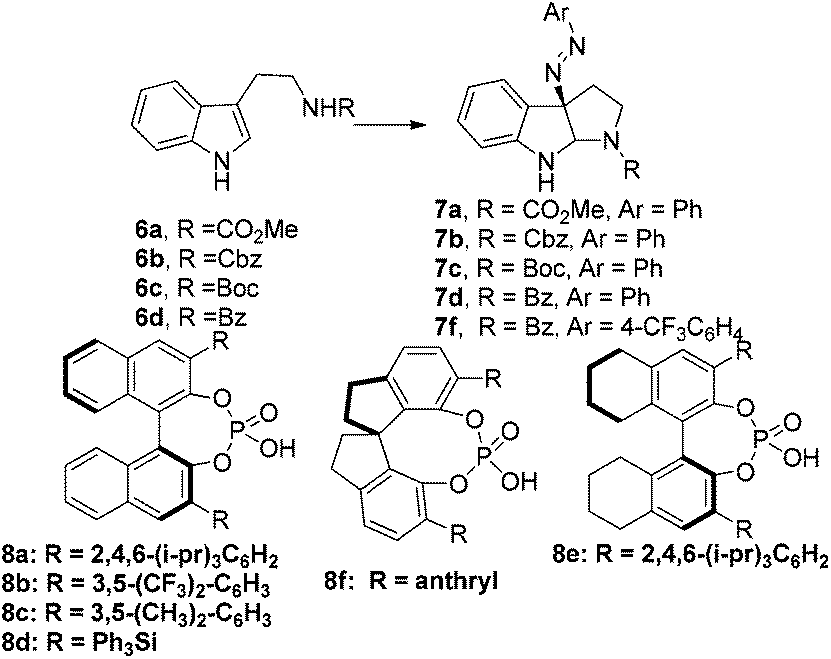 | ||
| Scheme 1 Enantioselective azo-coupling of tryptamine derivatives. | ||
With the core structure of 3α-amino-hexahydropyrrolo[2,3-b]indole 7f obtained in high ee, we strived to complete the enantioselective total synthesis of natural product (−)-psychotriasine (2). Our retrosynthetic analysis is illustrated in Scheme 2; we proposed that (−)-psychotriasine could be readily achieved via several functional transformations from Na-Boc protected compound 9. Compound 9 could be constructed by a Pd-catalyzed Buchwald–Hartwig amination followed by Larock annulation from the amino compound 10. Lastly, amine 10 could be synthesized from compound 7f through a few simple manipulations.
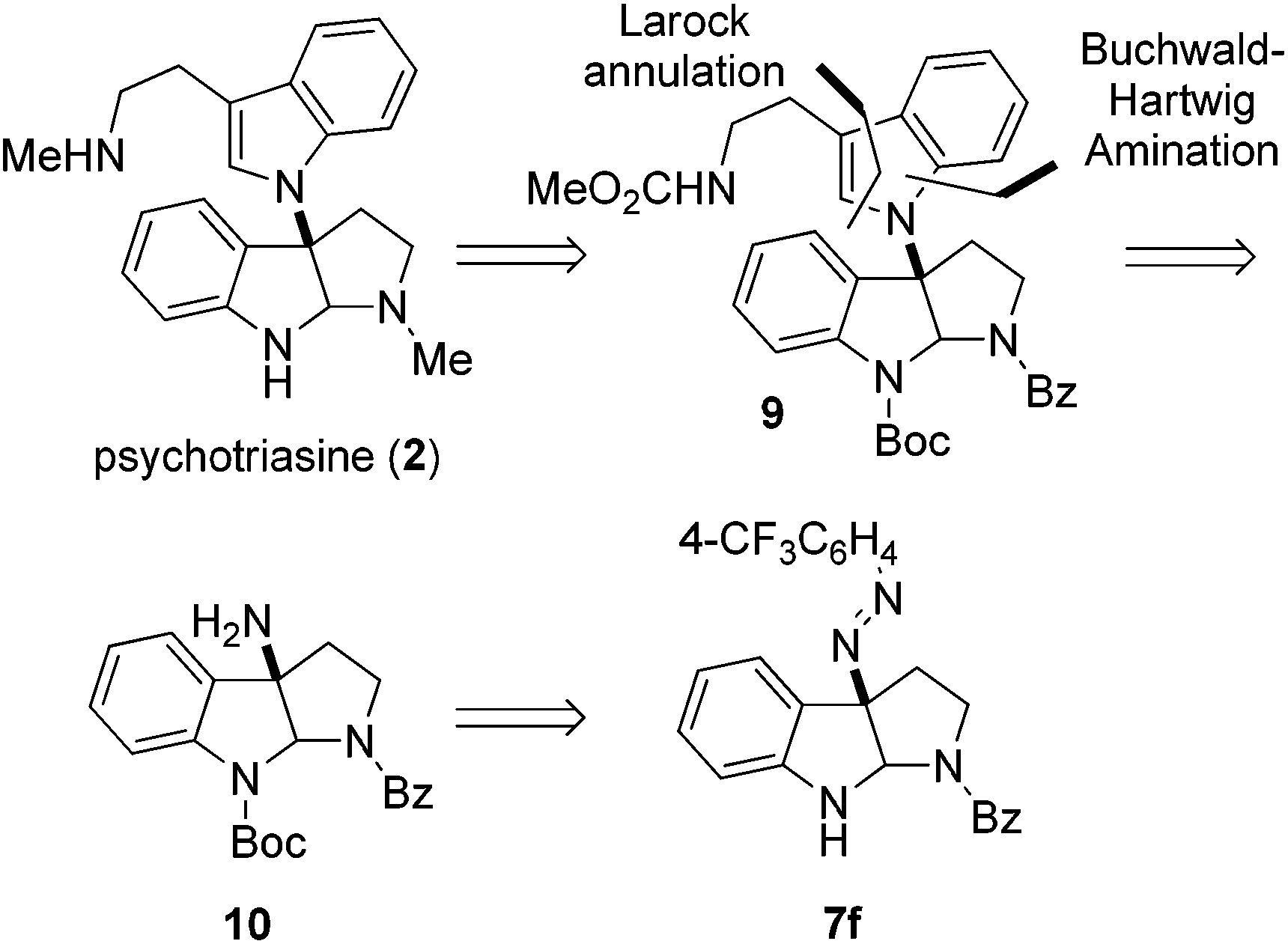 | ||
| Scheme 2 Retrosynthetic analysis of (−)-psychotriasine (2). | ||
Based on the retrosynthetic analysis, our total synthesis of (−)-psychotriasine began with 7f which was readily accomplished using our newly developed method (Scheme 3). The indole nitrogen in compound 7f was first protected with Boc in the presence of NaHMDS and Boc2O at −60 °C under argon. However, subsequent cleavage of the N,N double bond was not trivial, and very poor results were acquired when we tried a variety of reductive cleavage conditions including hydrogenolysis with H2 under different pressures and temperatures, and even reduction with TiCl3/NaOH. We then attempted using hydrazine hydrate in the presence of RANEY® nickel, and it was discovered that only when excess hydrazine hydrate and a catalytic amount of RANEY® nickel were utilized could the desired product 10 be obtained in a satisfactory yield over 2 steps (88%).21,22 Compound 10 was coupled with 1,2-dibromobenzene in the presence of a Pd(OAc)2 catalyst, xantphos ligand and t-BuONa as base additive to afford the Buchwald–Hartwig amination product 11 in 71% yield.23 Next, the tryptamine dimer compound 9 was obtained via Larock cyclization from 11 and a known alkyne building block (N-(methoxycarbonyl)-4-(trimethylsilyl)-3-butynylamine) 13 (ref. 24) using a Pd(OAc)2 catalyst, DtBPF ligand, K2CO3 additive and NMP solvent in 83% yield.25 N-deprotection for Bz removal with DIBAL-H26 followed by carbamation of the amine with ClCO2Me gave 12 in 64% yield over 2 steps. Finally, N-deprotection of 12 to remove the Boc group with CF3CO2H and further reduction with Red-Al provided the desired natural product (−)-psychotriasine (2) in >96% ee.3b With an overall yield of 11.7% over 9 steps, this procedure represents the first enantioselective total synthesis of (−)-psychotriasine.
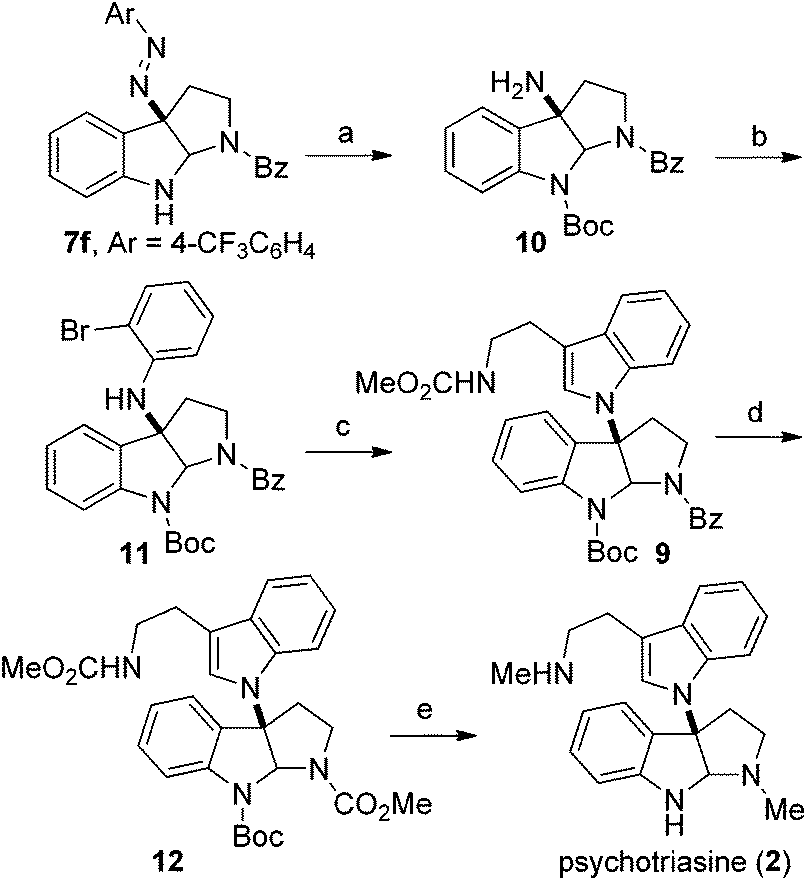 | ||
| Scheme 3 Total synthesis of (−)-psychotriasine (2). Reagents and conditions: (a) (i) NaHMDS, Boc2O, THF, −60 °C; (ii) RANEY® nickel, hydrazine hydrate, MeOH, sealed tube, 70 °C, 88% yield over 2 steps; (b). Pd(OAc)2 (0.2 equiv.), xantphos (0.3 equiv.), t-BuONa (2.0 equiv.), toluene, 80 °C, 71% yield; (c) 13, Pd(OAc)2 (0.2 equiv.), DtBPF (0.3 equiv.), K2CO3 (2.5 equiv.), NMP, 110 °C, 83% yield; (d) (i) DIBAL-H (3.0 equiv.), toluene, −78 °C; (ii). ClCO2Me (3.0 equiv.), Na2CO3 (3.0 equiv.), DCM–H2O (2:1 ratio), rt, 64% yield over 2 steps; (e) (i) CF3CO2H, DCM, 0 °C; (ii) Red-Al (12.0 equiv.), toluene, reflux, 51% yield over 2 steps. Alkyne 13: N-(methoxycarbonyl)-4-(trimethylsilyl)-3-butynylamine. | ||
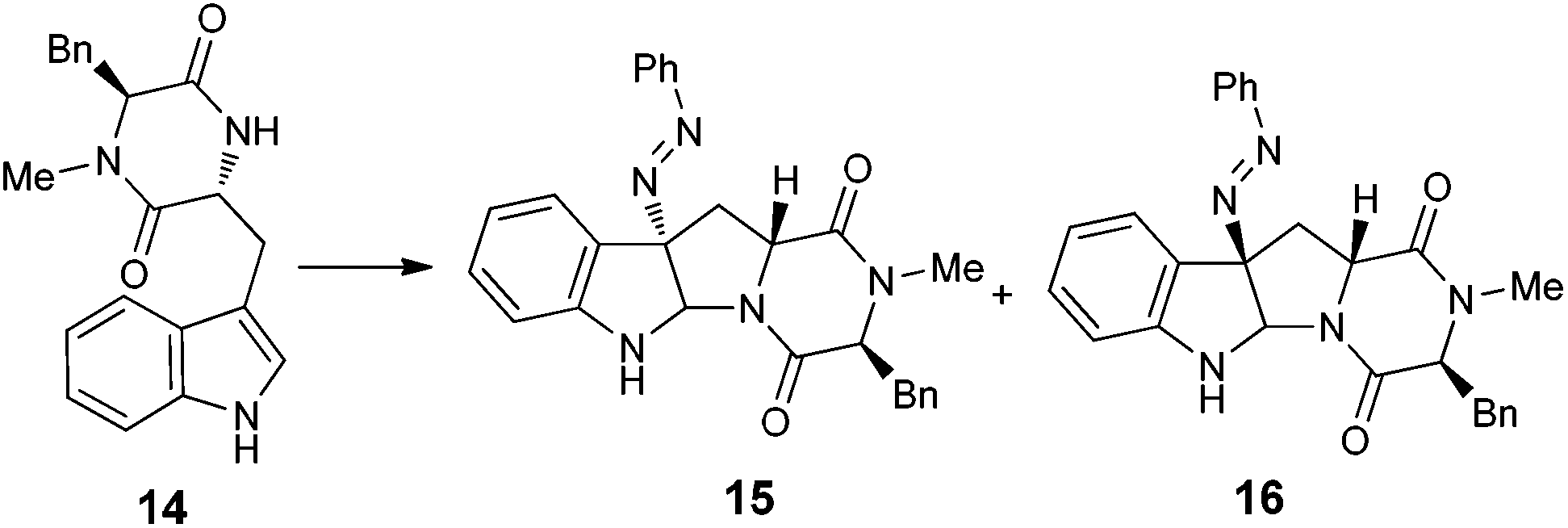
With the successful synthesis of (−)-psychotriasine, we envisioned that other natural products containing the 3α-amino-hexahydropyrrolo[2,3-b]indole core structure could also be synthesized via enantioselective azo-coupling of simple tryptamine derivatives with diazonium salts. However, analogous natural products bearing an extended core structure with the cyclic dipeptide motif, such as (+)-pestalazine B (4) and chetomin (5), couldn't be obtained directly. Baran's group reported an elegant approach to form a C–N bond through tryptamine derivatives coupling with aniline in the presence of appropriate oxidants;3d,6 however, when this method was attempted with 14, only a trace amount of the coupling product was obtained. To address this synthetic challenge, we sought to develop a highly diastereoselective azo-coupling of tryptophan derivatives. To obtain better diasteroselectivity of the azo-coupling reaction, we chose compound 14 as a model substrate to screen different solvents and reaction temperatures (Table 1). Using THF as solvent at 15 °C, diastereomer 16 was formed exclusively but only in 14% yield (entry 5). In comparison, utilizing DCE as solvent led to the formation of both diastereomers in a 82% combined yield and high selectivity for 16 (5:1, 16:15) (entry 6). Evaluation of a mixed THF–DCE solvent system (entries 8–11) led to an optimized solvent ratio of 1.5:1 DCE:THF that promoted a highly diastereoselective azo-coupling to afford 16 in 61% isolated yield and >25:1 diastereoselectivity. The configuration of 16 was confirmed by single crystal X-ray diffraction (Fig. 3). With this result in hand, we decided to pursue the total synthesis of (+)-pestalazine B (4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | Reaction time |
15:16 |
Yieldb (%) |
| a Conditions: 14 (0.1 mmol), PhN2BF4 (0.11 mmol), K2CO3 (0.15 mmol) and solvent (1 mL). b NMR yield with 1,3,5-trimethoxylbenzene as internal standard. c No desired product was detected. d Isolated yield. | |||||
| 1 | MeOH | −78 | 1 h | 1:1 |
66% |
| 2 | MeCN | rt | 24 h | — | —c |
| 3 | Toluene | rt | 24 h | 1:1.5 |
41% |
| 4 | THF | rt | 24 h | 1:1.5 |
74% |
| 5 | THF | 15 | 24 h | Only 16 | 14% |
| 6 | DCE | 15 | 24 h | 1:5 |
82% |
| 7 | DCE | rt | 24 h | 1:5 |
57% |
| 8 | DCE–THF(1:1) |
15 | 48 h | 1:10 |
63% |
| 9 | DCE–THF(1.5:1) |
15 | 48 h | <1:25 |
67% (61d%) |
| 10 | DCE–THF(2:1) |
15 | 96 h | 1:20 |
50% |
| 11 | DCE–THF(4:1) |
15 | 96 h | 1:4 |
43% |
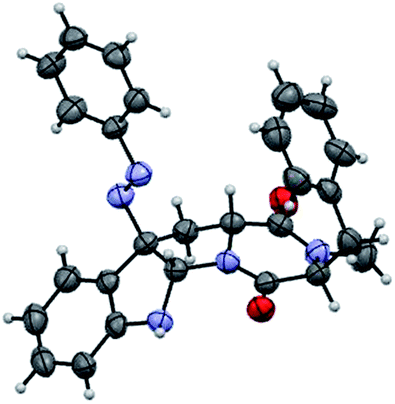 | ||
| Fig. 3 Thermal ellipsoid plot of the X-ray structure of compound 16 at the 50% probability level. | ||
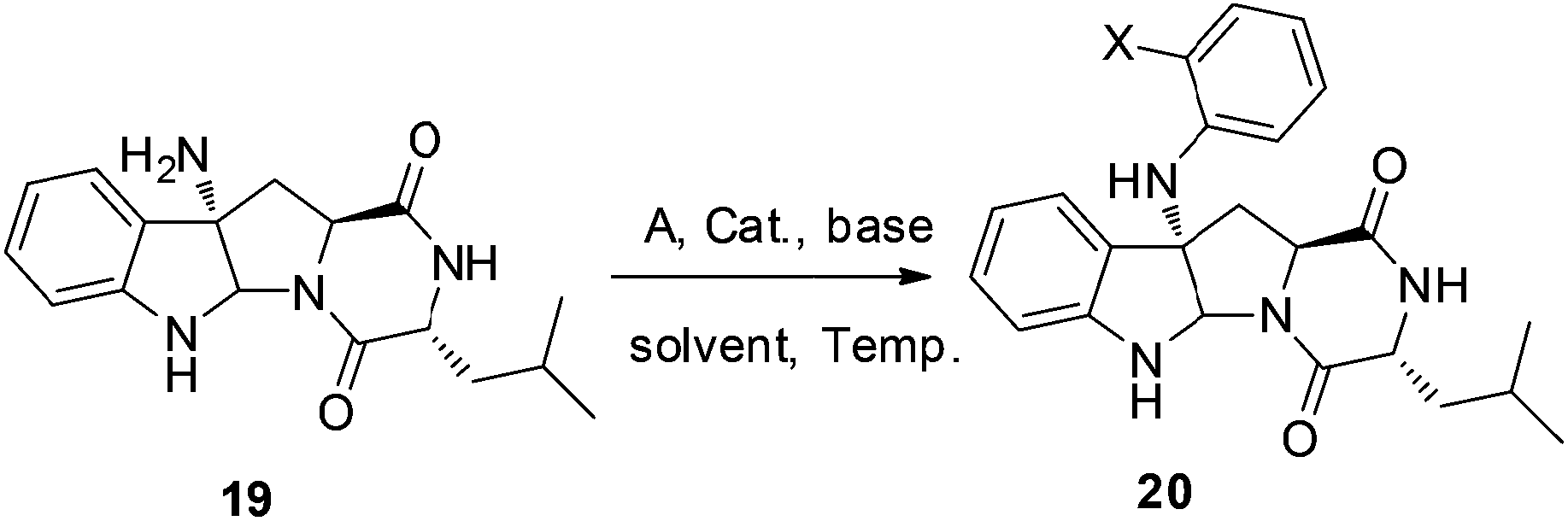
Our synthesis of (+)-pestalazine B (4) began with the diastereoselective azo-coupling using readily available compound 17 (Scheme 4). Under the standard reaction conditions developed with model compound 14 (Table 1), compound 17 coupled with PhN2BF4 to afford compound 18 and its diastereomer in a 6:1 ratio. Changing the base additive from K2CO3 to Na2CO3 while using 0.05 equiv. of CPA8a catalyst, compound 18 was obtained with improved diastereoselectivity (>20:1) and 79% yield. With 18 in hand, reductive cleavage of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond by hydrazine hydrate and Pd/C at 80 °C proceeded smoothly to provide amino compound 19 in 80% yield. The stereochemistry of 19 was consistent with the natural product (confirmed by NOE, see ESI†). Next, we attempted to construct a C–N bond by coupling between 19 and 1,2-dihalobenzene (Table 2) but with minimal success. We initially tried Pd-catalyzed Buchwald–Hartwig amination using various ligands such as xantphos, BINAP, DPPF, Xphos, Brettphos, DPEphos and indole-derived phosphine ligands. However, only trace amounts of amination product 20 were detected by LCMS (entry 1). We then attempted copper-induced Ullmann coupling utilizing CuI or Cu(OAc)2 and ligands such as 2-isobutyrylcyclohexanone or 2,2,6,6-tetramethylheptane-3,5-dione (TMHD). Unfortunately, these yields were still unacceptable (entry 2).27a Success in such reactions would rely on two factors: selective amination on different amine derivatives and the basicity of the reaction solutions. Traditionally, amination with amides as substrates will proceed more easily than with hindered amines. In addition, the strong base used in the reaction mixtures will result in the racemization of compound 19. Since hypervalent iodine reagents were widely used for C–N formation under neutral or weakly basic conditions, we decided to pursue this strategy and explore ortho-brominated hypervalent iodine reagents for formation of the C–N bond of our target (entries 5–11). Under the optimized conditions of using hypervalent iodine reagent A3 (5.0 equiv.), Cu(OAc)2 (1.0 equiv.), and Na2CO3 additive (2.0 equiv.), compound 20 was obtained in 45% yield (entry 10) after heating at 80 °C in DMF solvent for 12 hours.27b Finally, Larock annulation of 20 and 21 with sterically bulky ligand DtBPF provided the target molecule (+)-pestalazine B (4) in 16.2% overall yield over this 4-step total synthesis (from known compound 17).25 The NMR and CD spectral properties of (+)-pestalazine B were in excellent agreement with the natural product.8a
N double bond by hydrazine hydrate and Pd/C at 80 °C proceeded smoothly to provide amino compound 19 in 80% yield. The stereochemistry of 19 was consistent with the natural product (confirmed by NOE, see ESI†). Next, we attempted to construct a C–N bond by coupling between 19 and 1,2-dihalobenzene (Table 2) but with minimal success. We initially tried Pd-catalyzed Buchwald–Hartwig amination using various ligands such as xantphos, BINAP, DPPF, Xphos, Brettphos, DPEphos and indole-derived phosphine ligands. However, only trace amounts of amination product 20 were detected by LCMS (entry 1). We then attempted copper-induced Ullmann coupling utilizing CuI or Cu(OAc)2 and ligands such as 2-isobutyrylcyclohexanone or 2,2,6,6-tetramethylheptane-3,5-dione (TMHD). Unfortunately, these yields were still unacceptable (entry 2).27a Success in such reactions would rely on two factors: selective amination on different amine derivatives and the basicity of the reaction solutions. Traditionally, amination with amides as substrates will proceed more easily than with hindered amines. In addition, the strong base used in the reaction mixtures will result in the racemization of compound 19. Since hypervalent iodine reagents were widely used for C–N formation under neutral or weakly basic conditions, we decided to pursue this strategy and explore ortho-brominated hypervalent iodine reagents for formation of the C–N bond of our target (entries 5–11). Under the optimized conditions of using hypervalent iodine reagent A3 (5.0 equiv.), Cu(OAc)2 (1.0 equiv.), and Na2CO3 additive (2.0 equiv.), compound 20 was obtained in 45% yield (entry 10) after heating at 80 °C in DMF solvent for 12 hours.27b Finally, Larock annulation of 20 and 21 with sterically bulky ligand DtBPF provided the target molecule (+)-pestalazine B (4) in 16.2% overall yield over this 4-step total synthesis (from known compound 17).25 The NMR and CD spectral properties of (+)-pestalazine B were in excellent agreement with the natural product.8a
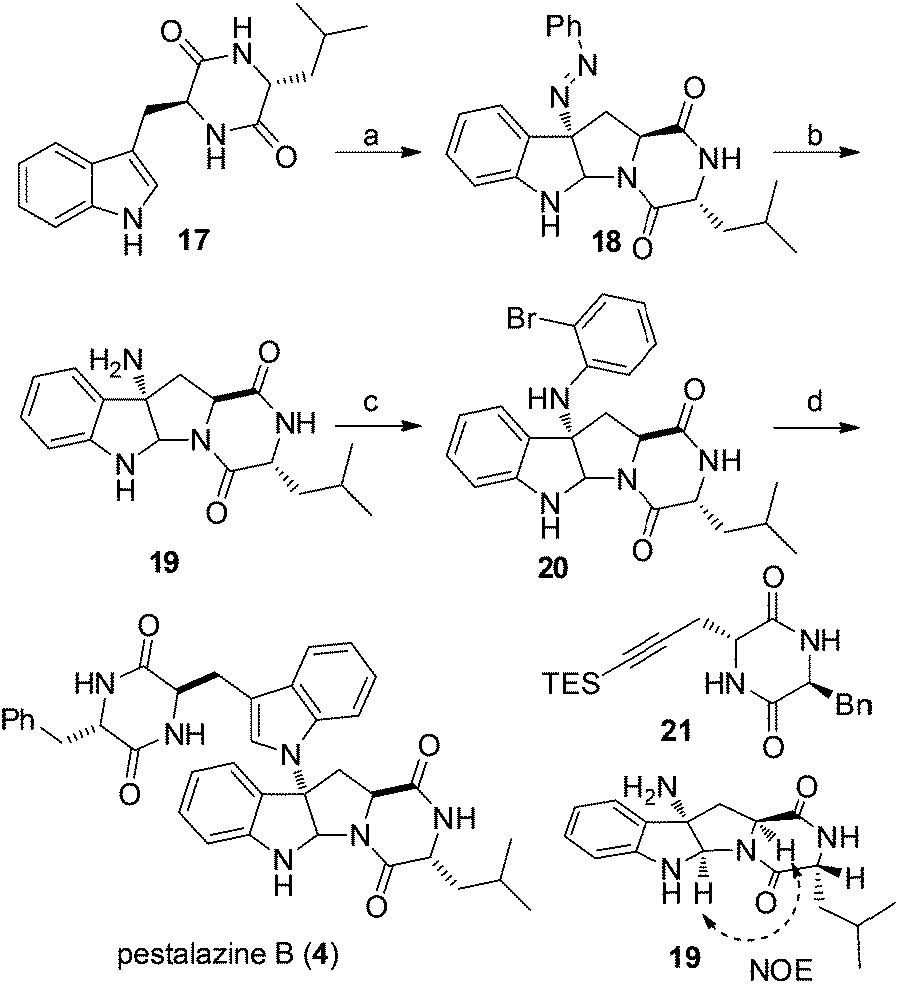 | ||
| Scheme 4 Total synthesis of (+)-pestalazine B (4). Reagents and conditions: (a) PhN2BF4, K2CO3, CPA8a, DCE–THF (1.5:1), 0 °C, 79% yield; (b) hydrazine hydrate, Pd/C, EtOH, 85 °C, 80% yield; (c) A3, Cu(OTf)2, Na2CO3, DMF, 65 °C, 45% yield, (d) 21, Pd(OAc)2, DtBPF, Na2CO3, NMP, 80 °C, 57% yield. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | A | Metal | Base | Solvent | T (°C) | Yield |
| a Ligand tested: xantphos, BINAP, DPPF, Xphos, Brettphos, DPEphos or indole-derived phosphine ligands. b Ligand used: 2-isobutyrylcyclohexanone or TMHD. | ||||||
| 1a | A1 | Pd2dba3 | t-BuONa | Toluene | 80 | Trace |
| 2b | A2 | CuI or Cu(OAc)2 | K2CO3 or Li2CO3 or Cs2CO3 | DMF or THF or MeCN | 60 | Trace |
| 3 | A3 | CuI | Na2CO3 | NMP | 80 | Trace |
| 4 | A3 | CuI | Na2CO3 | DMF | 80 | 22% |
| 5 | A3 | Cu(OAc)2 | Na2CO3 | DMF | 80 | 34% |
| 6 | A3 | Cu(OAc)2 | K2CO3 | DMF | 80 | Trace |
| 7 | A3 | Cu(OAc)2 | Na3PO4 | DMF | 80 | 28% |
| 8 | A3 | Cu(OAc)2 | Na2CO3 | DMF | 65 | 42% |
| 9 | A3 | Cu(OAc)2 | Na2CO3 | DMF | 50 | 26% |
| 10 | A3 | Cu(OTf)2 | Na2CO3 | DMF | 65 | 45% |
| 11 | A4 | Cu(OAc)2 | Na2CO3 | DMF | 80 | 33% |
|
||||||
Conclusions
We have completed the first enantioselective total synthesis of (−)-psychotriasine (2) and the enantioselective total synthesis of (+)-pestalazine B (4) based on a unified strategy with the newly developed method. In particular, to construct the 3α-amino-hexahydropyrrolo[2,3-b]indole core structure, a method involving a ligand-controlled enantioselective azo-coupling of tryptamine derivatives by ion-pairing induction or via mixed solvent-induced diastereoselective azo-coupling of tryptophan derivatives was applied. This cascade reaction sequence provides a rapid synthetic approach to the related indole alkaloids. In addition, during our synthesis of (+)-pestalazine B (4), we utilized a strategy that features a sterically hindered amination process using a hypervalent iodine reagent and copper(I) catalyst. Further total syntheses of other related natural products employing this unified strategy and investigations of their biological activities are underway.Acknowledgements
This work was supported by Tsinghua-Peking Centre for Life Sciences and “1000 Talents Recruitment Program”. We thank Prof. Dr Pinjing Zhao (North Dakota State University) and Dr Alex Cortez (GNF) for helpful discussion during the preparation of this manuscript. We thank Prof. Dr Liansuo Zu (Tsinghua University) for providing HPLC to test ee.Notes and references
- P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Alvarez, Chem.–Eur. J., 2011, 17, 1388 CrossRef CAS PubMed.
- H. Takayama, I. Mori, M. Kitajima, N. Aimi and N. H. Lajis, Org. Lett., 2004, 6, 2945 CrossRef CAS PubMed.
- Leading studies on total synthesis of psychotrimine: (a) Y. Matsuda, M. Kitajima and H. Takayama, Org. Lett., 2008, 10, 125 CrossRef CAS PubMed; (b) T. Newhouse and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 10886 CrossRef CAS PubMed; (c) N. Takahashi, T. Ito, Y. Matsuda, N. Kogure, M. Kitajima and H. Takayama, Chem. Commun., 2010, 46, 2501 RSC; (d) T. Newhouse, C. A. Lewis, K. J. Eastman and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 7119 CrossRef CAS PubMed; (e) T. Araki, T. Ozawa, H. Yokoe, M. Kanematsu, M. Yoshida and K. Shishido, Org. Lett., 2013, 15, 200 CrossRef CAS PubMed; (f) H. Zhang, H. Kang, L. Hong, W. Dong, G. Li, X. Zheng and R. Wang, Org. Lett., 2014, 16, 2394 CrossRef CAS PubMed.
- M. A. Schallenberger, T. Newhouse, P. S. Baran and F. E. Romesberg, J. Antibiot., 2010, 63, 685 CrossRef CAS PubMed.
- (a) S. A. Waksman and E. Bugie, J. Bacteriol., 1944, 48, 527 CAS; (b) A. G. Mcinnes, A. Taylor and J. A. Walter, J. Am. Chem. Soc., 1976, 98, 6741 CrossRef CAS; (c) A. L. Kung, S. D. Zabludoff, D. S. France, S. J. Freedman, E. A. Tanner, A. Vieira, S. Cornell-Kennon, J. Lee, B. Q. Wang, J. M. Wang, K. Memmert, H. U. Naegeli, F. Petersen, M. J. Eck, K. W. Bair, A. W. Wood and D. M. Livingston, Cancer Cell, 2004, 6, 33 CrossRef CAS PubMed; (d) K. M. Cook, S. T. Hilton, J. Mecinovic, W. B. Motherwell, W. D. Figg and C. J. Schofield, J. Biol. Chem., 2009, 284, 26831 CrossRef CAS PubMed; (e) T. R. Welch and R. M. Williams, Tetrahedron, 2013, 69, 770 CrossRef CAS PubMed.
- K. Foo, T. R. Newhouse, I. Mori, H. Takayama and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 2716 CrossRef CAS PubMed.
- H. Zhou, H. He, Y. Wang and X. Hao, Helv. Chim. Acta, 2010, 93, 1650 CrossRef CAS PubMed.
- (a) G. Ding, L. Jiang, L. Guo, X. Chen, H. Zhang and Y. Che, J. Nat. Prod., 2008, 71, 1861 CrossRef CAS PubMed; (b) C. Perez-Balado and Á. R. de Lera, Org. Biomol. Chem., 2010, 8, 5179 RSC.
- Z. Zhang and J. C. Antilla, Angew. Chem., Int. Ed., 2012, 51, 11778 CrossRef CAS PubMed.
- Z. Cai, S. Wang and S. Ji, Org. Lett., 2013, 15, 5226 CrossRef CAS PubMed.
- S. Beaumont, V. Pons, P. Retailleau, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2010, 49, 1634 CrossRef CAS PubMed.
- V. R. Espejo and J. D. Rainier, J. Am. Chem. Soc., 2008, 130, 128 CrossRef PubMed.
- T. Hayashi, W. Peng, Y.-Y. Nakai, K. Yamada and M. Somei, Heterocycles, 2002, 57, 421 CrossRef CAS.
- (a) H. M. Nelson, S. H. Reisberg, H. P. Shunatona, J. S. Patel and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 5600 CrossRef CAS PubMed; (b) D. E. Stephens and O. V. Larionov, Eur. J. Org. Chem., 2014, 3662 CrossRef CAS PubMed.
- R. Nakajima, T. Ogino, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2010, 132, 1236 CrossRef CAS PubMed.
- For reviews on asymmetric ion-pairing catalysis, see: (a) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed; (b) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed; (c) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed.
- Representative reports on asymmetric ion-pairing catalysis: (a) D. B. Llewellyn, D. Adamson and B. A. Arndtsen, Org. Lett., 2000, 2, 4165 CrossRef CAS PubMed; (b) S. Mayer and B. List, Angew. Chem., Int. Ed., 2006, 45, 4193 CrossRef CAS PubMed; (c) J. W. Yang, M. T. H. Fonseca, N. Vignola and B. List, Angew. Chem., Int. Ed., 2005, 44, 108 CrossRef CAS PubMed; (d) S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32 CrossRef CAS PubMed; (e) X. Wang and B. List, Angew. Chem., Int. Ed., 2008, 47, 1119 CrossRef CAS PubMed; (f) M. E. Muratore, C. A. Holloway, A. W. Pilling, R. I. Storer, G. Trevitt and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 10796 CrossRef CAS PubMed; (g) C. A. Holloway, M. E. Muratore, R. I. Storer and D. J. Dixon, Org. Lett., 2010, 12, 4720 CrossRef CAS PubMed; (h) M. J. Wanner, R. N. S. van der Haas, K. R. De Cuba, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2007, 46, 7485 CrossRef CAS PubMed; (i) N. V. Sewgobind, M. J. Wanner, S. Ingemann, R. De Gelder, J. H. van Maarseveen and H. Hiemstra, J. Org. Chem., 2008, 73, 6405 CrossRef CAS PubMed; (j) M. J. Wanner, R. N. A. Boots, B. Eradus, R. De Gelder, J. H. van Maarseveen and H. Hiemstra, Org. Lett., 2009, 11, 2579 CrossRef CAS PubMed; (k) F.-L. Sun, M. Zeng, Q. Gu and S.-L. You, Chem.–Eur. J., 2009, 15, 8709 CrossRef CAS PubMed; (l) Q.-X. Guo, Y.-G. Peng, J.-W. Zhang, L. Song, Z. Feng and L.-Z. Gong, Org. Lett., 2009, 11, 4620 CrossRef CAS PubMed; (m) T. Liang, Z. Zhang and J. C. Antilla, Angew. Chem., Int. Ed., 2010, 49, 9734 CrossRef CAS PubMed; (n) G. Bergonzini, S. Vera and P. Melchiorre, Angew. Chem., Int. Ed., 2010, 49, 9685 CrossRef CAS PubMed; (o) M. Terada, H. Tanaka and K. Sorimachi, J. Am. Chem. Soc., 2009, 131, 3430 CrossRef CAS PubMed; (p) Q.-W. Zhang, C.-A. Fan, H.-J. Zhang, Y.-Q. Tu, Y.-M. Zhao, P. Gu and Z.-M. Chen, Angew. Chem., Int. Ed., 2009, 48, 8572 CrossRef CAS PubMed; (q) U. Hennecke, C. H. Muller and R. Frolich, Org. Lett., 2011, 13, 860 CrossRef CAS PubMed; (r) S. E. Denmark and M. T. Burk, Org. Lett., 2012, 14, 256 CrossRef CAS PubMed; (s) D. Huang, H. Wang, F. Xue, H. Guan, L. Li, X. Peng and Y. Shi, Org. Lett., 2011, 13, 6350 CrossRef CAS PubMed.
- Representative studies by Toste and co-workers on anion phase-transfer catalysis: (a) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed; (b) R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376 CrossRef CAS PubMed; (c) Y.-M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928 CrossRef CAS PubMed; (d) R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268 CrossRef CAS PubMed; (e) H. P. Shunatona, N. Fruh, Y.-M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2013, 30, 7724 CrossRef PubMed; (f) J. Wu, Y.-M. Wang, A. Drljevic, A. V. Rauniyar, R. J. Phipps and F. D. Toste, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13729 CrossRef CAS PubMed; (g) A. J. Neel, J. P. Hehn, P. F. Tripet and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14044 CrossRef CAS PubMed; (h) X. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225 CrossRef CAS PubMed.
- For reviews on chiral phosphate acids, see: (a) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed; (b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed; (c) M. Terada, Synthesis, 2010, 1929 CrossRef CAS PubMed; (d) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed.
- S. Hoffmann, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424 CrossRef CAS PubMed.
- Y. Zhang, Q. Tang and M. Luo, Org. Biomol. Chem., 2011, 9, 4977 CAS.
- A. Job, C. F. Janeck, W. Bettray, R. Peters and D. Enders, Tetrahedron, 2002, 58, 2253 CrossRef CAS.
- For leading books and reviews on catalytic C–N cross-coupling, see: (a) L. Jiang and S. L. Buchwald, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, Germany, 2004, pp. 699–760 Search PubMed; (b) S. Shekhar, Q. Shen, F. Barrios-Landeros and J. F. Hartwig, in The Chemistry of Anilines, ed. Z. Rapoport, Wiley Interscience, New York, 2007, pp. 455–536 Search PubMed; (c) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed; (d) J. P. Wolfe, S. Wagaw, J. F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805 CrossRef CAS.
- J. R. Dunetz and R. L. Danheiser, J. Am. Chem. Soc., 2005, 127, 5776 CrossRef CAS PubMed.
- (a) R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689 CrossRef CAS; (b) M. Shen, G. Li, B. Z. Lu, A. Hossain, F. Roschangar, V. Farina and C. H. Senanayake, Org. Lett., 2004, 6, 4129 CrossRef CAS PubMed; (c) S. P. Breazzano, Y. B. Poudel and D. L. Boger, J. Am. Chem. Soc., 2013, 135, 1600 CrossRef CAS PubMed; (d) M. E. Kieffer, K. V. Chuang and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 5557 CrossRef CAS PubMed.
- J. Gutzwiller and M. Uskokovic, J. Am. Chem. Soc., 1970, 92, 204 CrossRef CAS.
- (a) A. Shafir, P. A. Lichtor and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3490 CrossRef CAS PubMed; (b) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, characterization data, X-ray structures of compound 15, and NMR spectra. CCDC 1040494. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00338e |
| This journal is © The Royal Society of Chemistry 2015 |