
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of D-α-amino amides from aliphatic aldehydes†
Kenneth E.
Schwieter
and
Jeffrey N.
Johnston
*
Department of Chemistry, Vanderbilt Institute of Chemical Biology Vanderbilt University, Nashville, Tennessee 37235, US. E-mail: jeffrey.n.johnston@vanderbilt.edu
First published on 27th February 2015
Abstract
Peptides consisting of D-amino amides are highly represented among both biologically active natural products and non-natural small molecules used in therapeutic development. Chemical synthesis of D-amino amides most often involves approaches based on enzymatic resolution or fractional recrystallization of their diastereomeric amino acid salt precursors, techniques that produce an equal amount of the L-amino acid. Enantioselective synthesis, however, promises selective and general access to a specific α-amino amide, and may enable efficient peptide synthesis regardless of the availability of the corresponding α-amino acid. This report describes the use of a cinchona alkaloid-catalyzed aza-Henry reaction using bromonitromethane, and the integration of its product with umpolung amide synthesis. The result is a straightforward 3-step protocol beginning from aliphatic aldehydes that provides homologated peptides bearing an aliphatic side chain at the resulting D-α-amino amide.
Introduction
Noncanonical amino acids and D-amino acids are present in a multitude of biologically relevant peptides including many marketed pharmaceuticals.1–5 Current preparative methods that serve the goal of peptide homologation rely almost entirely on the enantioselective synthesis of carboxylic acid feedstock and rely on traditional condensative amide bond formation for peptide synthesis (Fig. 1).6,7 Notable enantioselective approaches to carboxylic acid donors include the preparation of active ester precursors by hydrogenation of dehydro-α-amino acids,8,9 alkylation of masked forms of glycine,10–15 and the Strecker reaction.16,17 Although these approaches generally provide high selectivity, they ultimately require the use of an active ester intermediate to form the amide (simple or peptidic), a species inherently prone to epimerization when bearing an α-C–H bond.7 In order to circumvent the epimerization pathway while minimizing functional group manipulations, we sought an integration of Umpolung Amide Synthesis (UmAS)18,19 and the enantioselective synthesis of α-bromonitroalkane donors necessary to provide the desired amides bearing aliphatic side chains (Fig. 1). Reports detailing the stereoselective synthesis of α-bromonitroalkanes have focused entirely on those that deliver α-aryl amides (aryl glycinamides)16 and α-oxy amides.18 To that end, a route to enantioenriched β-alkyl-β-amino-α-bromonitroalkanes is needed.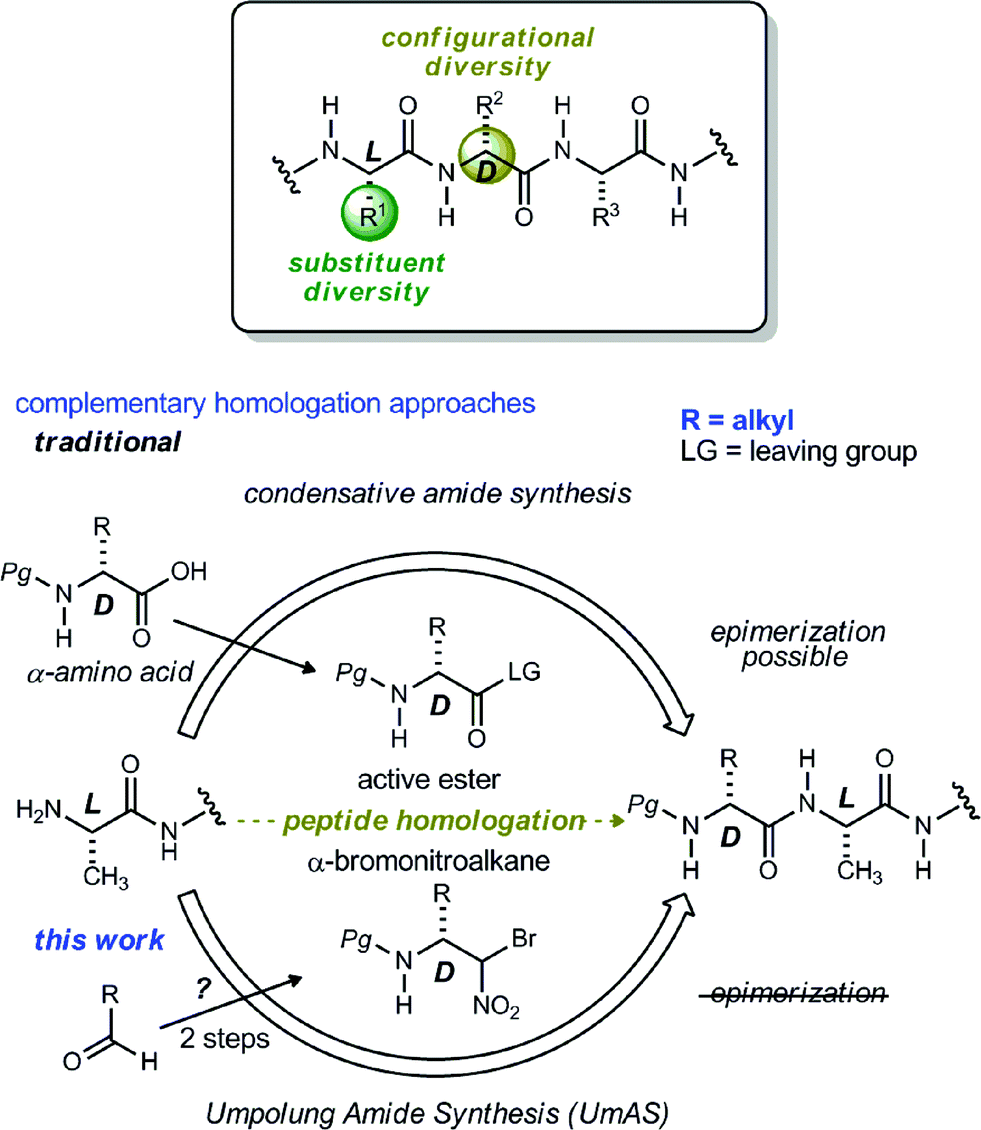 | ||
| Fig. 1 Complementary approaches to peptide homologation with α-alkyl-α-amino amide precursors. | ||
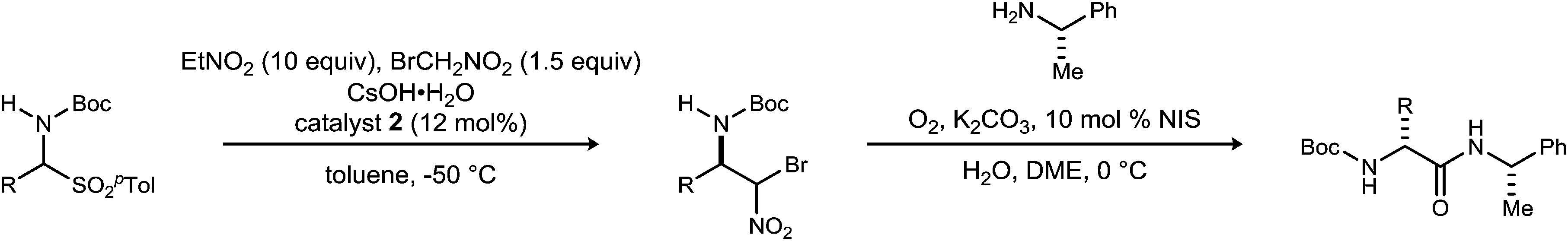
Although bromonitromethane has been used in the enantioselective Henry20–22 and aza-Henry16,23,24 reactions, it has never been successfully employed in an enantioselective aza-Henry addition using alkyl imine electrophiles. Similar enantioselective transformations, however, utilizing a variety of nitroalkanes have been reported.25–28 Of particular note is the absence of an adaptation of the protocol developed by Palomo for nitromethane to bromonitromethane (Scheme 1), as it would provide the desired enantioenriched alkyl β-amino-α-bromonitroalkanes, which would serve as noncanonical alkyl amino acid synthons and potential precursors to D-amino amides.29 This report describes the reason for this, as well as a solution to the problem. In so doing, the application of UmAS to α-amino amide peptide homologation is reduced to practice, and applied to the chemical synthesis of a homochiral D-peptide bearing aliphatic substituents, using entirely enantioselective methods.
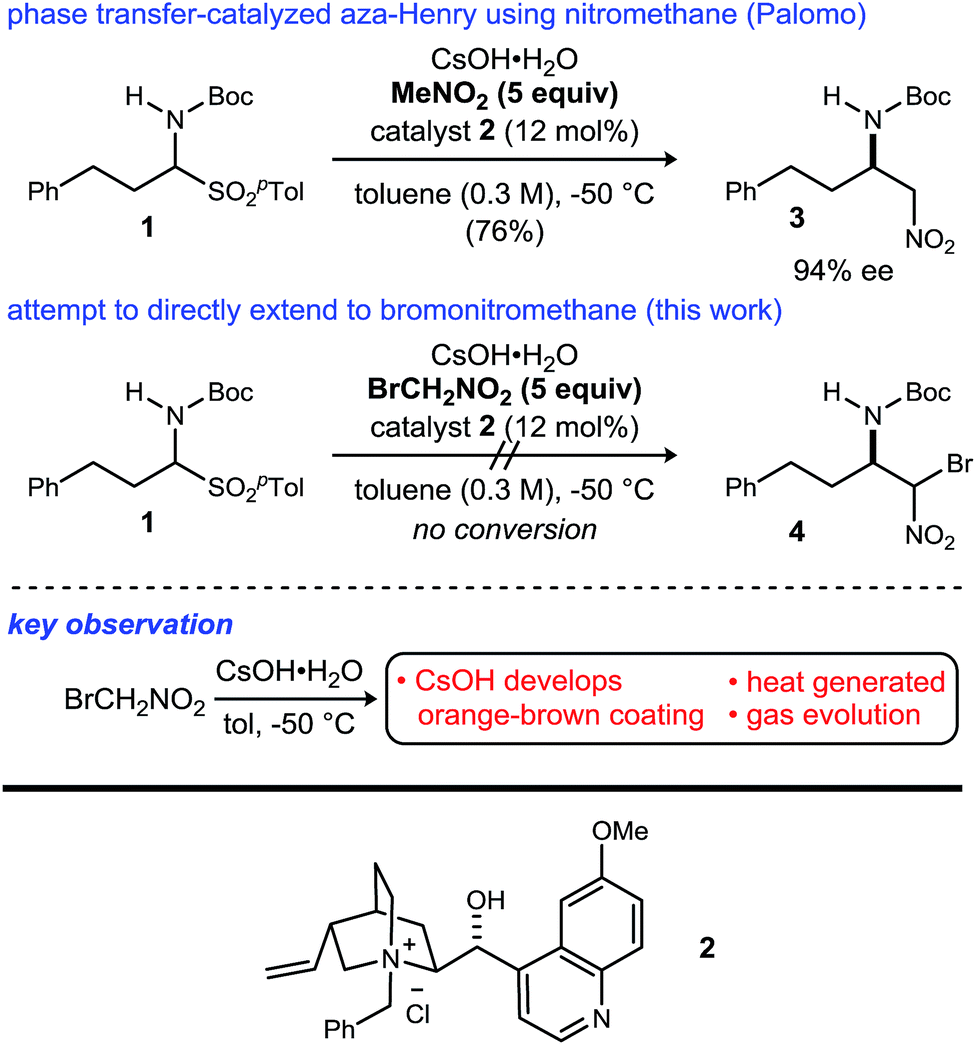 | ||
| Scheme 1 Efforts to translate Palomo's enantioselective phase transfer-catalyzed aza-Henry protocol from nitromethane to bromonitromethane. | ||
Results and discussion
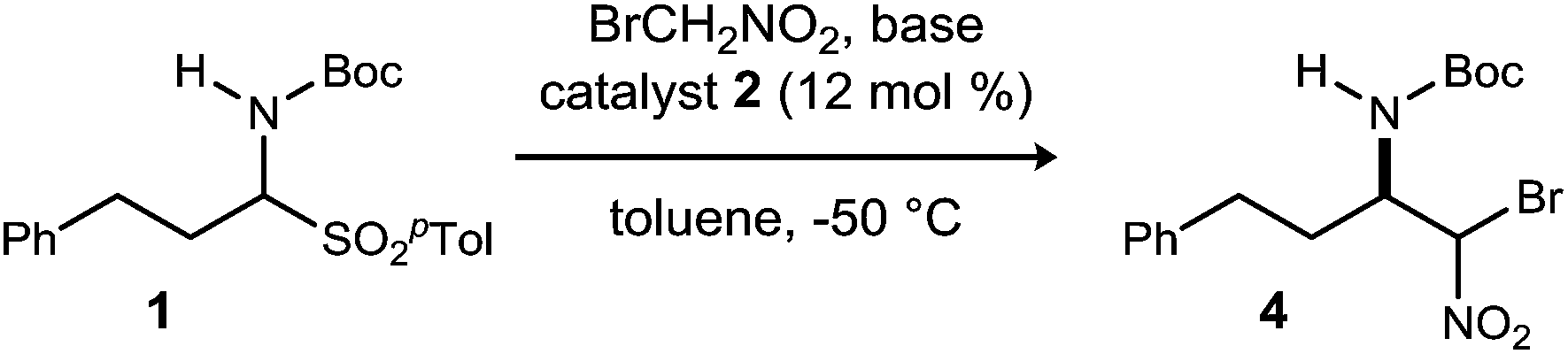
Initial attempts to translate Palomo's protocol to aliphatic N-Boc aldimines focused on N-Boc-α-amido sulfone301 using phase transfer catalyst N-benzylquininium chloride 2.23 In our hands, nitromethane provided results aligned with those reported, providing 3 in 94% ee and 76% yield (Scheme 1), but straightforward substitution of bromonitromethane for nitromethane resulted in recovery of the unreacted α-amido sulfone (Scheme 1). During these attempts, several key observations were made. First, significant heat was generated immediately after addition of CsOH·H2O, concurrent with the release of gas and the formation of an insoluble brown solid that coated the solid cesium hydroxide powder. This apparent decomposition of bromonitromethane in aqueous base has been previously reported.31 Reducing the equivalents of bromonitromethane from 5 to 1.5 (Table 1, entries 1 and 2) appeared to mitigate this direct reaction by lowering the concentration of bromonitromethane. However, incomplete conversion remained correlated to the formation of an orange-brown residue on the solid cesium hydroxide, a material that resulted from bromonitromethane decomposition.32 Alternative bases were identified that formed little or no residue, but these bases failed to provide significant conversion (Table 1, entries 3, 4, 5 and 7). CsOH·H2O and KOH (Table 1, entries 2 and 6) remained the only reagents with sufficient reactivity, and these provided promising conversion coupled with moderate enantioselection (62–72% ee). The products were generally formed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, but this was not of concern as the bromine-substituted sp3 carbon becomes an amide sp2-hybridized carbon in the UmAS step.
:1 dr, but this was not of concern as the bromine-substituted sp3 carbon becomes an amide sp2-hybridized carbon in the UmAS step.
|
|
||||
|---|---|---|---|---|
| Entrya | Base | BrCH2NO2 (equiv.) | Conversionb (%) | eec (%) |
| a All reactions were conducted using sulfone (1 equiv.), 12 mol% catalyst, base (1.3 equiv.) and bromonitromethane (1.5 equiv.) in toluene (0.3 M) for 96 h. b Measured by 1H NMR relative to an internal standard (CH2Br2). c Enantiomeric excesses determined by chiral HPLC using an OD-H column (Chiral Technologies). d Isolated yield. | ||||
| 1 | CsOH·H2O | 5 | 0 | — |
| 2 | CsOH·H2O | 1.5 | 79 (47)d | 72/72 |
| 3 | K2CO3 | 1.5 | 15 | 59/43 |
| 4 | Na2CO3 | 1.5 | 3 | — |
| 5 | CaCO3 | 1.5 | 0 | — |
| 6 | KOH | 1.5 | 74 (41)d | 62/68 |
| 7 | Cs2CO3 | 1.5 | 0 | — |
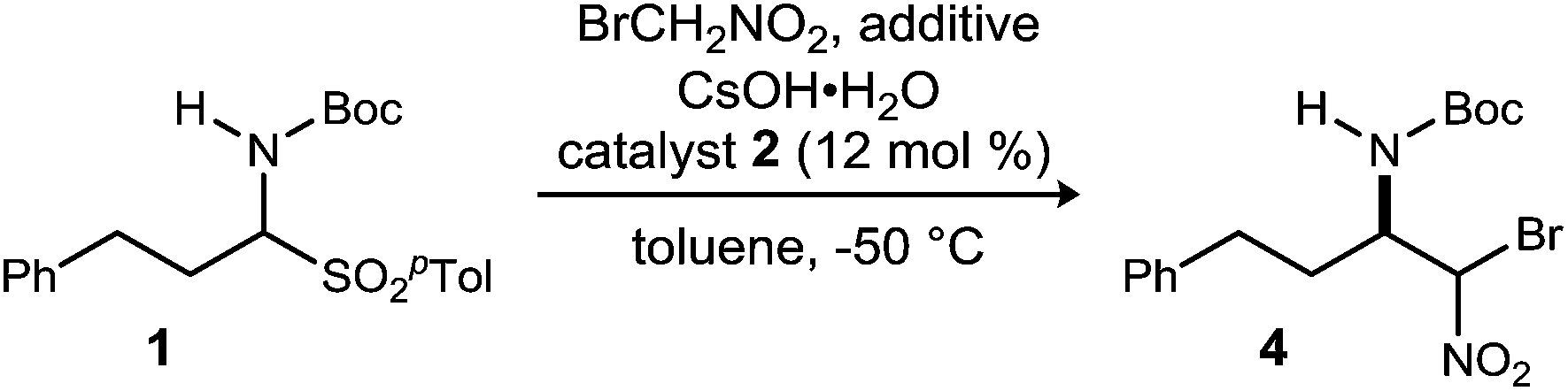
Further exploration of the contrasting behavior of nitromethane and bromonitromethane with cesium hydroxide led to the addition of nitromethane to the reaction mixture in an effort to improve conversion.23 The introduction of 1.5 equivalents of nitromethane resulted in near full conversion and an increase in yield (47–62%) and enantioselection (72/72 to 81/78% ee) (Table 2, cf. entries 1 and 2). The corresponding nitromethane adduct accounted for the mass balance of product, an odd observation considering the significant difference in acidity between bromonitromethane and nitromethane.33 Further increase in the equivalents of nitromethane resulted, predictably, in diminishing yield of 4 due to increased formation of the nitromethane adduct (3). However, this led to the unexpected observation that enantioselection increased up to 95/94% ee with increasing equivalents of nitromethane (Table 2, entries 2–5). In order to attenuate the reactivity of the nitroalkane additive while maintaining the apparent benefit of its presence, a series of increasingly hindered nitroalkanes were examined (Table 2, entries 5, 7 and 8). Nitroalkane additives with additional steric bulk resulted in an increased yield, and a decreased amount of its addition product was noted as anticipated. Use of 10 equivalents of nitroethane provided the desired adduct in 59% yield and 96/96% ee (Table 2, entry 6). 2-Nitropropane (Table 2, entry 7) provided similar yield but significantly lower enantioselection, suggesting a more limited interaction with the solid base. The potential for nitrobenzene to act as a non-acidic additive was examined, and although improvement of enantioselection was clear, significant decomposition of bromonitromethane was again noted. Our observations overall suggest that the acidity of the nitroalkane is related to its ability to temper the reactivity of the solid base, but the nitro functionality operates in a distinct manner to positively influence enantioselectivity. Attempts to use other additives, including phenol and sodium acetate, resulted in poor conversion (<20%).
|
|
|||
|---|---|---|---|
| Entrya | Additive (equiv.) | Yieldb (%) | eec (%) |
| a All reactions were conducted using sulfone (1 equiv.), 12 mol% catalyst, CsOH·H2O (1.3 equiv.) and bromonitromethane (1.5 equiv.) in toluene (0.3 M). b Isolated yield. c Enantiomeric excesses determined by chiral HPLC using an OD-H column (Chiral Technologies). | |||
| 1 | None | 47 | 72/72 |
| 2 | MeNO2 (1.5) | 62 | 81/78 |
| 3 | MeNO2 (3) | 56 | 88/88 |
| 4 | MeNO2 (5) | 30 | 89/90 |
| 5 | MeNO2 (10) | 27 | 95/94 |
| 6 | EtNO2 (5) | 50 | 92/92 |
| 7 | EtNO 2 (10) | 59 | 96/96 |
| 8 | iPrNO2 (10) | 53 | 87/86 |
| 9 | C6H5NO2 | 36 | 92/92 |
The dependence of enantioselection on the equivalents of nitroalkane employed was intriguing, and the trend can be documented for the addition of nitromethane as well, under similar conditions (Scheme 2). Increasing the equivalents of nitromethane from 1 to 5 resulted in an increase in enantioselection from 75% to 93% ee. Further increasing the amount to 10 equivalents did not improve the enantioselection observed. The enhancement of enantioselection with increasing nitromethane equivalents might also suggest an equilibrium between bound/unbound nitroalkane. Unfortunately, we were unable to better characterize the nature of this interaction.
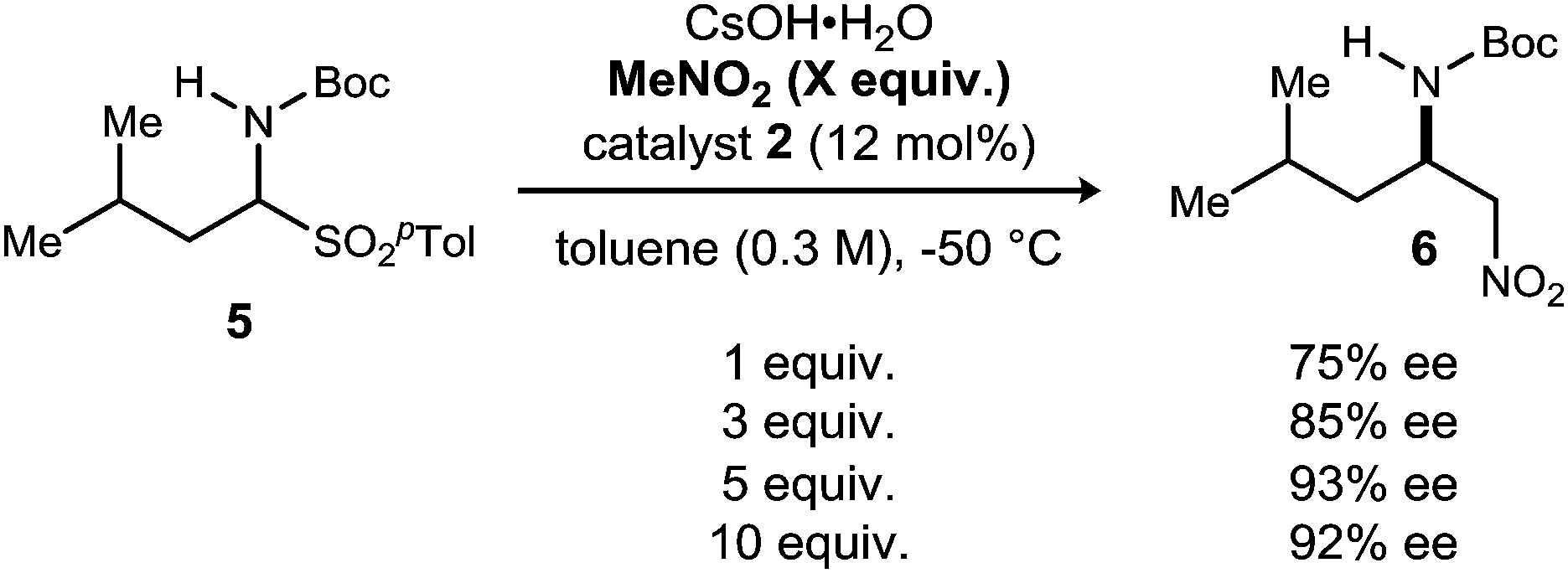 | ||
| Scheme 2 Enantioselectivity dependence on nitromethane equivalents. | ||
A combination of 10 equivalents of nitroethane and 1.5 equivalents of bromonitromethane using catalyst 2 and CsOH·H2O as the base were chosen as optimal conditions to examine a variety of α-amido sulfones (Table 3). Selectivity is provided as a ratio to emphasize the stereospecific nature of UmAS; enantiomeric ratios of nitroalkane donors should translate to diastereomeric ratios for the amide products. Straight carbon chain substrates provided the corresponding α-bromonitroalkane donors for Nle, Nva, and Adod (Table 3, entries 6–8) with high er and moderate yields. Substrates with branching β to the imine carbon (Table 3, entries 2, 3, 9 and 11) became precursors for Leu, Npg, Cha, and Phe, furnishing the corresponding products with high er and moderate yield, with one exception (Phe: Table 3, entry 11, 38% yield). Branching α to the imine resulted in diminished yield and enantioselection. The isopropyl derivative (Table 3, entry 4) leading to Val, and the cyclopropyl derivative leading to cyclopropyl glycine (Table 3, entry 15) gave diminished enantioselection down to 5:1 and 8:1 er, respectively. A cyclohexyl substituent (Table 3, entry 5) leading to the Chg donor resulted in 16:1 er, albeit with poor yield (36%). The catalyzed aza-Henry was tolerant of unsaturation in the imine substituent, maintaining moderate yield and high enantioselection. Alkenyl side chains (Table 3, entries 12 and 14) provided the desired α-bromonitroalkanes in 13:1 and 16:1 er, respectively. The terminal alkyne substrate (Table 3, entry 10) leading to Hpg was provided in 42% yield with 15:1 er. The inclusion of an electron-poor trifluoromethyl side chain (Table 3, entry 13) resulted in 20:1 er, while an electron-rich side chain (Table 3, entry 16) for Et-Hse resulted in diminished enantioselectivity at 7:1 er. The intent of this study was to outline the tolerance of the enantioselective addition to substituent variations; the adducts were often crystalline solids for which fractional recrystallization could deliver enantiopure material (vide infra).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entryab | R | Name | Yieldc (%) | erd | dre | Yieldc (%) | ||
| a All reactions were conducted using sulfone (1 equiv., 0.3 M in toluene), 12 mol% catalyst, CsOH·H2O (1.3 equiv.), nitroethane (10 equiv.) and bromonitromethane (1.5 equiv.) at −50 °C. b All reactions were conducted using bromonitroalkane (1 equiv.), H2O (5 equiv.), (S)-α-Me-benzylamine (1.2 equiv.), K2CO3 (3 equiv.) and NIS (0.1 equiv.) in DME (0.2 M) under an O2 atmosphere at 0 °C. c Isolated yield. d Determined by HPLC using a chiral stationary phase and reported as an average of diastereomers. e Determined by HPLC using AD-H column (Chiral Technologies). | ||||||||
| 1 | CH2CH2Ph | Homophenylalanine (Hph) | 4 | 59 | 45:1 |
64:1 |
22 | 61 |
| 2 | iBu | Leucine (Leu) | 7 | 57 | 15:1 |
16:1 |
23 | 58 |
| 3 | (CH3)3CCH2 | Neopentylglycine (Npg) | 8 | 60 | 37:1 |
35:1 |
24 | 58 |
| 4 | iPr | Valine (Val) | 9 | 44 | 5:1 |
5:1 |
25 | 73 |
| 5 | Cy | Cyclohexylglycine (Chg) | 10 | 36 | 16:1 |
14:1 |
26 | 56 |
| 6 | n Bu | Norleucine (Nle) | 11 | 54 | 49:1 |
41:1 |
27 | 52 |
| 7 | n Pr | Norvaline (Nva) | 12 | 52 | 14:1 |
25:1 |
28 | 69 |
| 8 | C10H21 | 2-Amino-dodecanoic acid (Adod) | 13 | 51 | 22:1 |
29:1 |
29 | 65 |
| 9 | (C6H11)CH2 | Cyclohexylalanine (Cha) | 14 | 53 | 46:1 |
71:1 |
30 | 53 |
| 10 | HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2CH2 CCH2CH2 |
Homopropargylglycine (Hpg) | 15 | 42 | 15:1 |
45:1 |
31 | 48 |
| 11 | PhCH2 | Phenylalanine (Phe) | 16 | 38 | 14:1 |
14:1 |
32 | 69 |
| 12 | H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH2 CHCH2CH2 |
Homoallylglycine (Hag) | 17 | 57 | 13:1 |
20:1 |
33 | 42 |
| 13 | CF3CH2 | 2-Amino-4-trifluorobutyric acid (Atb) | 18 | 46 | 20:1 |
35:1 |
34 | 38 |
| 14 |
cis-CH3CH2CHCH(CH2)2 |
— | 19 | 57 | 16:1 |
17:1 |
35 | 38 |
| 15 | c-C3H5 | Cyclopropylglycine (Δpg) | 20 | 50 | 8:1 |
8:1 |
36 | 49 |
| 16 | EtOCH2CH2 | Ethyl homoserine (Et-Hse) | 21 | 37 | 7:1 |
8:1 |
37 | 61 |
Details regarding the subsequent umpolung amide synthesis using substoichiometric NIS are also included in Table 3.34 The chiral enantioenriched β-amino-α-bromonitroalkanes were coupled to enantioenriched (S)-α-methylbenzylamine (99% ee) to demonstrate that the diastereomers are homochiral at the β-carbon, and that selectivity translates with complete fidelity to the amide product, as expected based on the UmAS mechanism.16,17 All α-bromonitroalkanes coupled without event including those containing unsaturation that is potentially reactive toward NIS. The resulting α-amino amides were isolated in diastereomeric ratios greater than or equal to their corresponding enantiomeric ratios with few exceptions.
The goal of this work was to develop a general approach to D-amino amide homologation with aliphatic side chains, while improving access to their peptides. In many of the cases in Table 3, there is no existing homologative procedure for the α-amino amide preparation that relies on enantioselective synthesis as an alternative to α-amino acid salt resolution or fermentation. In the case of α-aminododecanoic acid (Adod), however, two preparations of the enantiopure α-amino acid donor have been described and are outlined in Fig. 2.35 Beginning from protected α-amino malonate 39, a sequence of alkylation–hydrolysis–decarboxylation leads to N-Boc-Adod in racemic form. A chlorinated derivative (40) has been resolved using an enzyme-catalyzed kinetic resolution. An alternative to resolution involves a chiral auxiliary, which leads to N-Boc-D-Adod in enantiopure form in two steps from 41. By comparison, N-Boc-D-Adod donor is prepared in enantiopure form in only two steps and a recrystallization from undecanal (38).
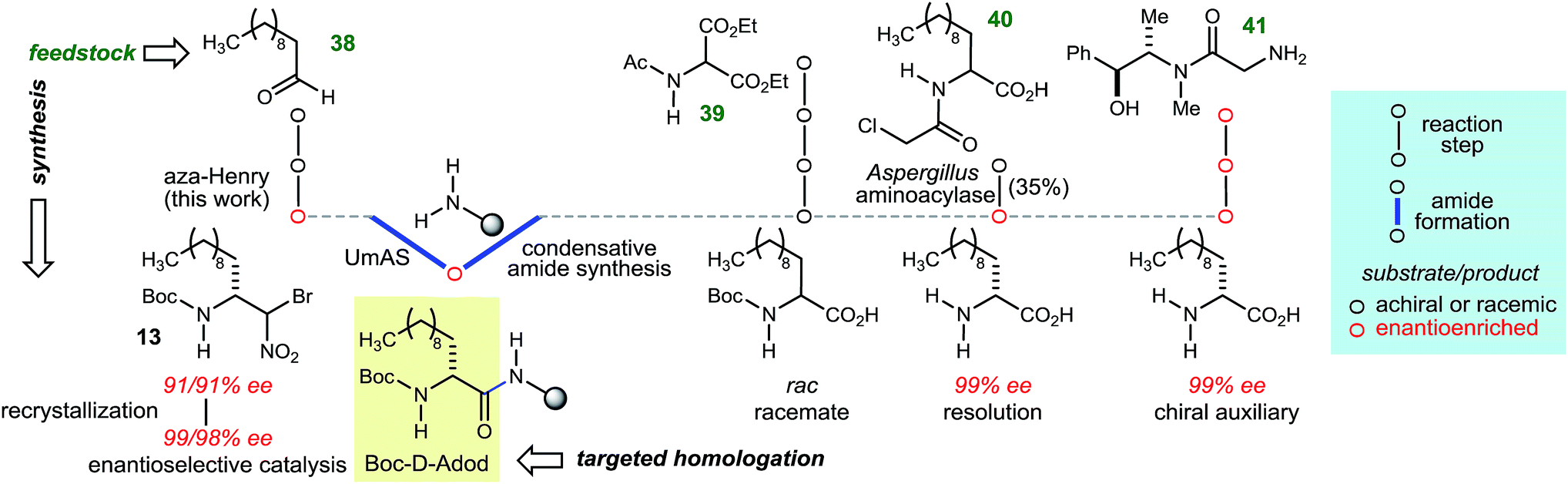 | ||
| Fig. 2 Comparative analysis of Boc-D-Adod homologation using stereoselective synthesis. | ||
In order to illustrate the fully modular construction of each aliphatic D-amino acid and an iterative assembly, the synthesis of tripeptide 46 containing three noncanonical amino acids, neopentylglycine (Npg), homophenylalanine (Hph) and 2-amino-dodecanoic acid (Adod) was targeted (Scheme 3). α-Bromonitroalkane 8 was synthesized on gram-scale while maintaining yield (55%) and enantioselection (90% ee). Subsequent UmAS coupling on gram-scale provided the desired amide 24 in 57% yield in 20:1 dr. Boc-deprotection with HCl·dioxane proceeded uneventfully in 97% yield. Amine 43 was submitted to UmAS conditions with D-homophenylalanine donor 4 to afford dipeptide 44 in 47% yield. At this point, the minor diastereomer(s) were undetectable after standard flash column chromatography. Boc-deprotection provided free amine 45 in 94% yield, and a final UmAS coupling with D-Adod donor 13 provided tripeptide 46 in 51% yield.
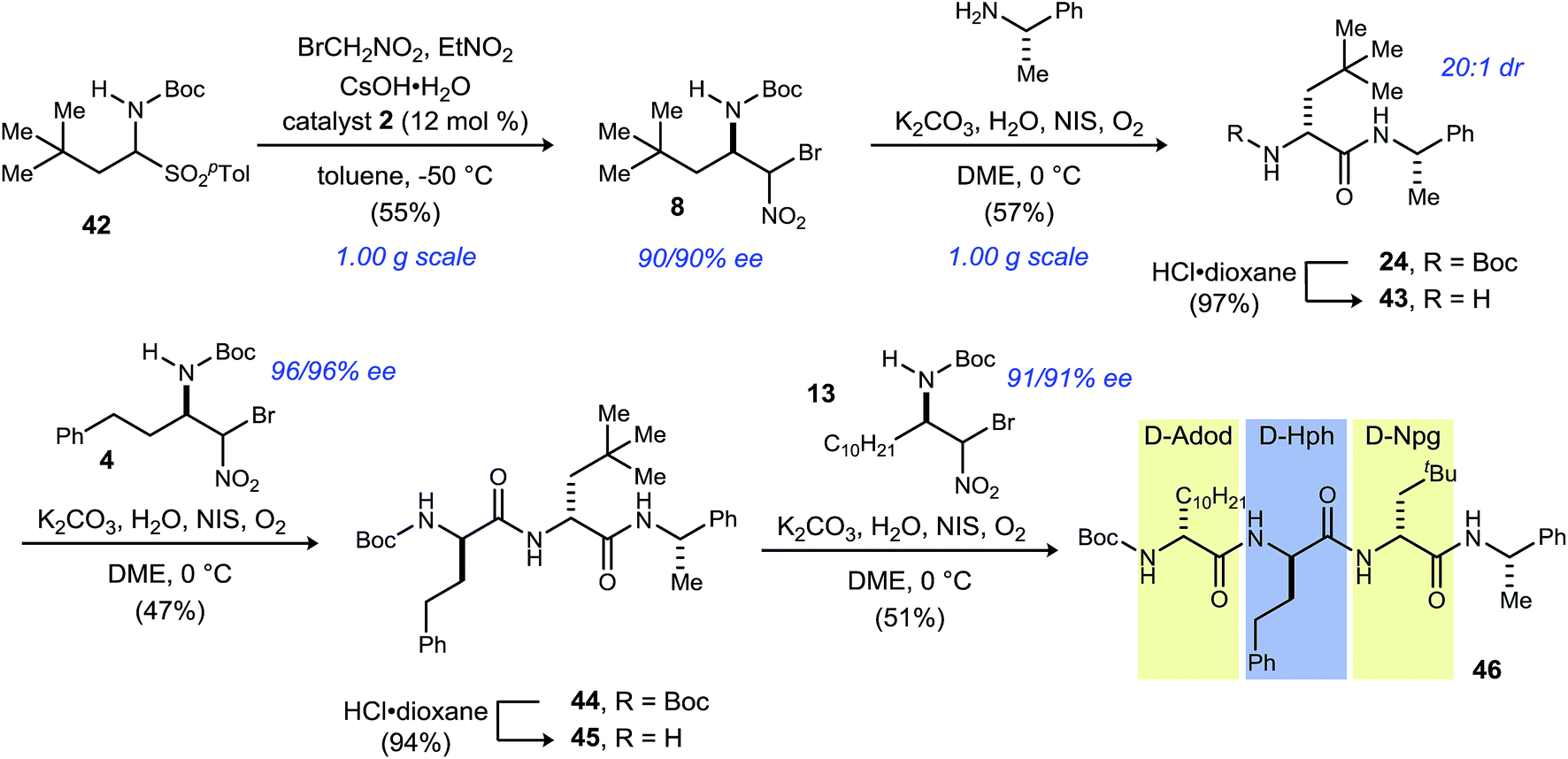 | ||
| Scheme 3 Iterative tripeptide synthesis. | ||
Conclusion
In three steps from commercially available, inexpensive aldehydes, peptides containing one or more D-amino amides can be readily prepared using a combination of a cinchona alkaloid-catalyzed aza-Henry and UmAS chemistry. In order to achieve this, the rapid decomposition of bromonitromethane with cesium hydroxide was circumvented by effectively tempering the reactivity of the solid base with a less acidic nitroalkane. This solution exhibited the added benefit of increasing enantioselection, albeit for reasons yet unclear. Overall, the catalytic enantioselective synthesis of peptides containing aliphatic D-amino acids is both possible and efficient (3 steps), and derived from aldehyde feedstock that is commercially diverse and inexpensive.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM 063557).Notes and references
- T. M. Woodruff, T. V. Arumugam, I. A. Shiels, R. C. Reid, D. P. Fairlie and S. M. Taylor, J. Immunol., 2003, 171, 5514–5520 CrossRef CAS.
- C. Lin, A. D. Kwong and R. B. Perni, Infect. Disord.: Drug Targets, 2006, 6, 3–16 CrossRef CAS.
- K. Shiosaki, A. S. Tasker, G. M. Sullivan, B. K. Sorensen, T. W. von Geldern, J. R. Wu-Wong, C. A. Marselle and T. J. Opgenorth, J. Med. Chem., 1993, 36, 468–478 CrossRef CAS.
- A. R. J. Anas, T. Kisugi, T. Umezawa, F. Matsuda, M. R. Campitelli, R. J. Quinn and T. Okino, J. Nat. Prod., 2012, 75, 1546–1552 CrossRef CAS PubMed.
- P. Vlieghe, V. Lisowski, J. Martinez and M. Khrestchatisky, Drug Discovery Today, 2010, 15, 40–56 CrossRef CAS PubMed.
- C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584–4671 CrossRef PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- W. S. Knowles, M. J. Sabacky and B. D. Vineyard, J. Chem. Soc., Chem. Commun., 1972, 10–11 RSC.
- M. J. Burk, J. E. Feaster, W. A. Nugent and R. L. Harlow, J. Am. Chem. Soc., 1993, 115, 10125–10138 CrossRef CAS.
- M. J. O'Donnell, W. D. Bennett and S. Wu, J. Am. Chem. Soc., 1989, 111, 2353–2355 CrossRef.
- B. Lygo and P. G. Wainwright, Tetrahedron Lett., 1997, 38, 8595–8598 CrossRef CAS.
- E. J. Corey, F. Xu and M. C. Noe, J. Am. Chem. Soc., 1997, 119, 12414–12415 CrossRef CAS.
- T. Li, S. Zhou, J. Wang, J. L. Acena, V. A. Soloshonok and H. Liu, Chem. Commun., 2015, 51, 1624–1626 RSC.
- T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 1999, 121, 6519–6520 CrossRef CAS; K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef PubMed.
- S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed.
- M. S. Iyer, K. M. Gigstad, N. D. Namdev and M. Lipton, J. Am. Chem. Soc., 1996, 118, 4910–4911 CrossRef CAS.
- M. S. Sigman, P. Vachal and E. N. Jacobsen, Angew. Chem., Int. Ed., 2000, 39, 1279–1281 CrossRef CAS.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed.
- J. P. Shackleford, B. Shen and J. N. Johnston, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 44–46 CrossRef CAS PubMed.
- M. W. Leighty, B. Shen and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 15233–15236 CrossRef CAS PubMed.
- D. A. Evans, D. Seidel, M. Rueping, H. W. Lam, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2003, 125, 12692–12693 CrossRef CAS PubMed.
- G. Blay, L. R. Domingo, V. Hernández-Olmos and J. R. Pedro, Chem. –Eur. J., 2008, 14, 4725–4730 CrossRef CAS PubMed; G. Blay, V. Hernandez-Olmos and J. R. Pedro, Chem. Commun., 2008, 4840–4842 RSC.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed; M. C. Dobish, F. Villalta, M. R. Waterman, G. I. Lepesheva and J. N. Johnston, Org. Lett., 2012, 14, 6322–6325 CrossRef PubMed; D. M. Makley and J. N. Johnston, Org. Lett., 2014, 16, 3146–3149 CrossRef PubMed.
- A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887–2939 CrossRef CAS PubMed.
- C. Palomo, M. Oiarbide, A. Laso and R. López, J. Am. Chem. Soc., 2005, 127, 17622–17623 CrossRef CAS PubMed; E. Gomez-Bengoa, A. Linden, R. López, I. Muúgica-Mendiola, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2008, 130, 7955–7966 CrossRef PubMed; L. S. Aitken, N. R. Arezki, A. Dell'Isola and A. J. A. Cobb, Synthesis, 2013, 2627–2648 Search PubMed.
- J. C. Anderson, G. P. Howell, R. M. Lawrence and C. S. Wilson, J. Org. Chem., 2005, 70, 5665–5670 CrossRef CAS PubMed.
- B. M. Trost and D. W. Lupton, Org. Lett., 2007, 9, 2023–2026 CrossRef CAS PubMed.
- M. T. Robak, M. Trincado and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 15110–15111 CrossRef CAS PubMed.
- S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed.
- A. Monleón, Synlett, 2013, 529–530 Search PubMed.
- B. C. Challis and T. I. Yousaf, J. Chem. Soc., Perkin Trans. 2, 1991, 283–286 RSC.
- See Supporting Information for additional experiments that characterize and support an adverse reaction specifically between cesium hydroxide and bromonitromethane.
- Although bromonitromethane is significantly more acidic than nitromethane, competition by the latter was consistently observed when using these PTC conditions. F. G. Bordwell, J. E. Bartmess and J. A. Hautala, J. Org. Chem., 1978, 43, 3107–3113 CrossRef CAS.
- K. E. Schwieter, B. Shen, J. P. Shackleford, M. W. Leighty and J. N. Johnston, Org. Lett., 2014, 16, 4714–4717 CrossRef CAS PubMed.
- P. González-Bulnes, A. González-Roura, D. Canals, A. Delgado, J. Casas and A. Llebaria, Bioorg. Med. Chem., 2010, 18, 8549–8555 CrossRef CAS PubMed; Y. Endo, S. Takehana, M. Ohno, P. E. Driedger, S. Stabel, M. Y. Mizutani, N. Tomioka, A. Itai and K. Shudo, J. Med. Chem., 1998, 41, 1476–1496 CrossRef PubMed; M. Cox, R. H. Prager and C. E. Svensson, Aust. J. Chem., 2003, 56, 887–896 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc00064e |
| This journal is © The Royal Society of Chemistry 2015 |