
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combination of Ru(II) complexes and light: new frontiers in cancer therapy
Cristina
Mari†
a,
Vanessa
Pierroz†
ab,
Stefano
Ferrari
b and
Gilles
Gasser
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: gilles.gasser@chem.uzh.ch; Web: http://www.gassergroup.com Fax: +41 44 635 6803; Tel: +41 44 635 4630
bInstitute of Molecular Cancer Research, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
First published on 13th January 2015
Abstract
The synergistic action of light, oxygen and a photosensitizer (PS) has found applications for decades in medicine under the name of photodynamic therapy (PDT) for the treatment of skin diseases and, more recently, for the treatment of cancer. However, of the thirteen PSs currently approved for the treatment of cancer over more than 10 countries, only two contain a metal ion. This fact is rather surprising considering that nowadays around 50% of conventional chemotherapies involve the use of cisplatin and other platinum-containing drugs. In this perspective article, we review the opportunities brought by the use of Ru(II) complexes as PSs in PDT. In addition, we also present the recent achievements in the application of Ru(II) complexes in photoactivated chemotherapy (PACT). In this strategy, the presence of oxygen is not required to achieve cell toxicity. This is of significance since tumors are generally hypoxic. Importantly, this perspective article focuses particularly on the Ru(II) complexes for which an in vitro biological evaluation has been performed and the mechanism of action (partially) unveiled.
 (From left to right:) Gilles Gasser, Vanessa Pierroz, Cristina Mari and Stefano Ferrari | Cristina Mari (third from left) was born in Milan (Italy) where she grew up and completed her education. She obtained a Master’s in Chemistry at the University of Milan in 2010, with a one year project on the functionalization of peptide nucleic acids with peptides or a rhenium dye. In 2011, she moved to the group of Prof. Gilles Gasser at the University of Zurich (Switzerland), where she is heading towards the end of her PhD. Her current research focuses on the investigation of new DNA intercalating photoactivatable ruthenium complexes as anticancer drug candidates. |
Vanessa Pierroz (second from left) was born and completed her whole education in Switzerland. After completion of her matriculation examination in Saint-Maurice in 2006, she studied biology at the University of Lausanne (Switzerland). After obtaining her Bachelor’s degree in biology in 2009, she undertook a Master’s in medical biology at the same institution. During these studies, she first completed a pre-Master thesis, under the supervision of Dr Benjamin Boutrel at the Centre of Psychiatric Neurosciences, where she investigated the interindividual vulnerability to develop alcohol abuse in rats. Afterwards, she joined the laboratories of Dr Pascal Schneider at the Department of Biochemistry to perform her Master’s thesis, investigating the proteolytic maturation of Tumor Necrosis Factors (TNF) family members. She obtained her Master’s degree in 2011. She then moved to Zurich to start her PhD thesis in the groups of Prof. Gilles Gasser and PD Dr Stefano Ferrari, where she is studying the photo-activation of metallodrugs in living cells. |
Stefano Ferrari (fourth from left) was born in Trento (Italy) and completed his undergraduate studies in Biology at the Universities of Padua (Italy) and Dundee (Scotland). He received his PhD in Biochemistry from the University of Padua in 1988. After a post-doctoral stage with Prof. George Thomas at the Friedrich Miescher Institute in Basel (Switzerland), he took up a Junior Group Leader position at the Institute of Experimental Cancer Research of the University of Freiburg (Germany) and subsequently a Program-Team Head position at the Department of Oncology of Novartis, Basel (Switzerland). Since 2001 he is a lecturer at the Institute of Molecular Cancer Research of the University of Zurich. Over the years, Stefano's studies have addressed the role of signaling in cell growth and proliferation as well as in the control of cell cycle transitions. |
Gilles Gasser (first from left) was born, raised and educated in Switzerland. He received his PhD thesis from the University of Neuchâtel in 2004 after working in the group of Prof. Helen Stoeckli-Evans. After post-doctoral stays at Monash University (Australia) with Prof. Leone Spiccia and at the Ruhr-University Bochum (Germany) with Prof. Nils Metzler-Nolte, Gilles started his independent career at the Department of Chemistry of the University of Zurich (Switzerland) first as a Swiss National Science Foundation (SNSF) Ambizione fellow and then, since 2011, as a SNSF Assistant Professor. Over his career, Gilles has been the recipient of several awards including a SNSF fellowship for prospective researchers, an Alexander von Humboldt fellowship (2007) and the Werner Prize from the Swiss Chemical Society (2015). Gilles's group is currently working on several topics which all involve the use of metal complexes in a medicinal or chemical biology context. |
Introduction
The biological activity of ruthenium (Ru) compounds has been known for decades.1–3 Two Ru complexes are currently in phase II clinical trials (NAMI-A and KP1339) as anticancer drug candidates and a third one, RAPTA-C, is progressing towards clinical trials (see Fig. 1 for the structures of these compounds).4–10 The increasing interest in the biological behavior of Ru compounds is due to their appealing physico-chemical properties. Among others, such complexes can have different geometries (e.g. tetrahedral or octahedral) allowing for the design of compounds with a specific cellular target (e.g. proteins). Hence, the rigid and well-defined spatial arrangement of a series of Ru complexes has enabled the preparation of highly potent and selective enzyme inhibitors. The group of Meggers has notably demonstrated such a concept with kinase inhibitors.2,11,12 Other attractive features of Ru complexes include their generally lower systemic toxicity compared to platinum complexes and their higher cellular uptake, thanks to the specific transport of ruthenium inside cells by transferrin.13 Of utmost importance, ruthenium complexes can easily be obtained in two oxidation states (II and III) and are prone to ligand exchange. Such properties have been found to play a pivotal role in the mode of action of both NAMI-A and KP1339.14 Ru(III) complexes are thus prodrugs – meaning that the compound which is administered to the patient is not the active species. Ru(III) complexes are reduced into a more active Ru(II) form when localized in an hypoxic environment, which is a property characteristic of tumors.6 This phenomenon is normally referred to as “activation by reduction” and was also exploited for the in situ activation of Pt-based anticancer drug candidates, like satraplatin.15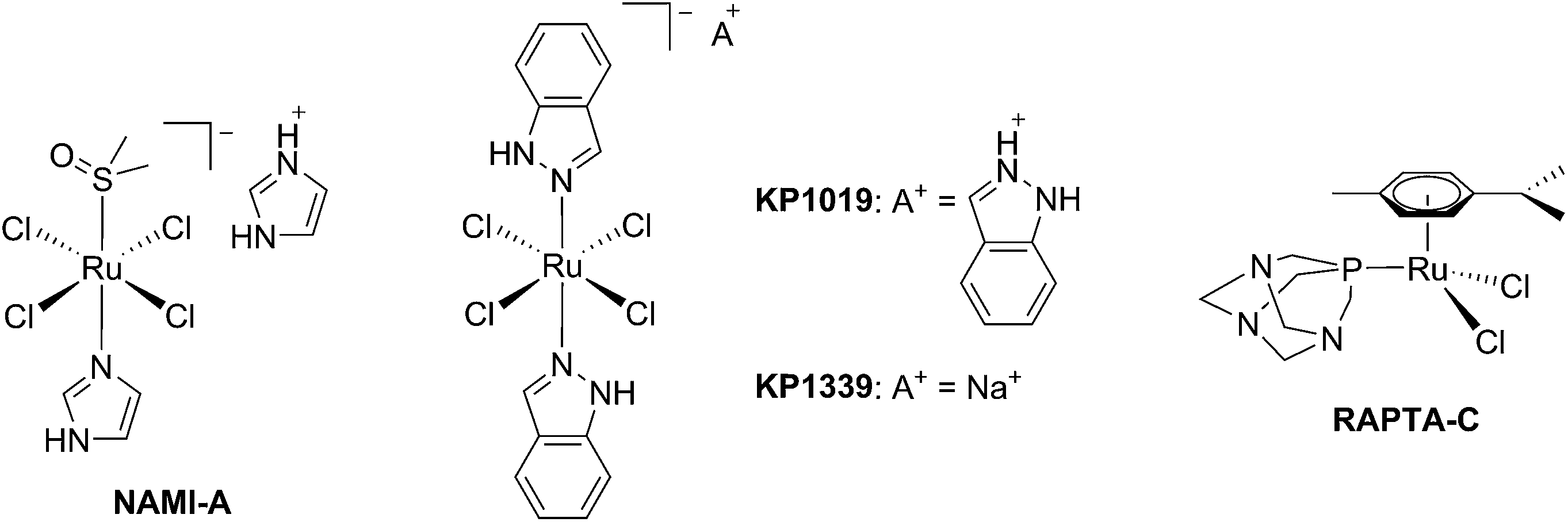 | ||
| Fig. 1 Structures of NAMI-A, KP1339, KP1019 and RAPTA-C. | ||
Nowadays, the use of the “prodrug approach” is very appealing to reduce the systemic toxicity of a drug candidate.16 In order to activate the prodrugs, two different kinds of stimuli can be employed, namely an internal stimulus (reducing cellular conditions, hypoxia, enzymatic reactions, etc.) or an external stimulus (magnetic field, temperature, light, etc.). The first approach, however, presents a significant disadvantage, in that it completely relies on intracellular parameters. In other words, once the prodrug is injected into the patient, physicians have no more control over the fate of the compound. On the contrary, this is exactly the kind of control that can be achieved using an external stimulus. The latter indeed provides complete spatial and temporal control over the generation of the toxic molecule. As of today, the most commonly applied technique to induce the formation of active species is via light irradiation.15,17,18
The light-mediated activation of prodrugs in the field of anticancer research can be generally divided into two categories: photodynamic therapy (PDT) and photoactivated chemotherapy (PACT). PDT relies mainly on the generation of the toxic reactive oxygen species (ROS) singlet oxygen (1O2). On the other hand, PACT exploits different mechanisms to induce cell death such as ligand ejection, DNA crosslinking and caging approaches. In this perspective article, we intend to give an overview of recent progress in the application of ruthenium complexes in both PDT and PACT, focusing particularly on those compounds for which an in vitro evaluation of the biological activity has been performed and the mechanism of action (partially) unveiled. Notably, these topics have been partially reviewed in the past but an article covering all subjects is, to the best of our knowledge, currently missing.17–26
Ruthenium complexes as photosensitizers in PDT
Photodynamic therapy is an approved medical technique, which is applied in dermatology for the treatment of several diseases such as acne or psoriasis and in ophthalmology for age-related macular degeneration. Since relatively recently, this technique has been used for the treatment of some types of cancer. For example, Photofrin® (Fig. 2), the only FDA-approved PDT drug, is employed to treat esophageal and non-small cell lung cancers. In the UK, on the other hand, there are several photoactive agents which are clinically approved (i.e. Foscan®, Fig. 2) to treat a wide range of cancer types, from skin to internal organs.27,28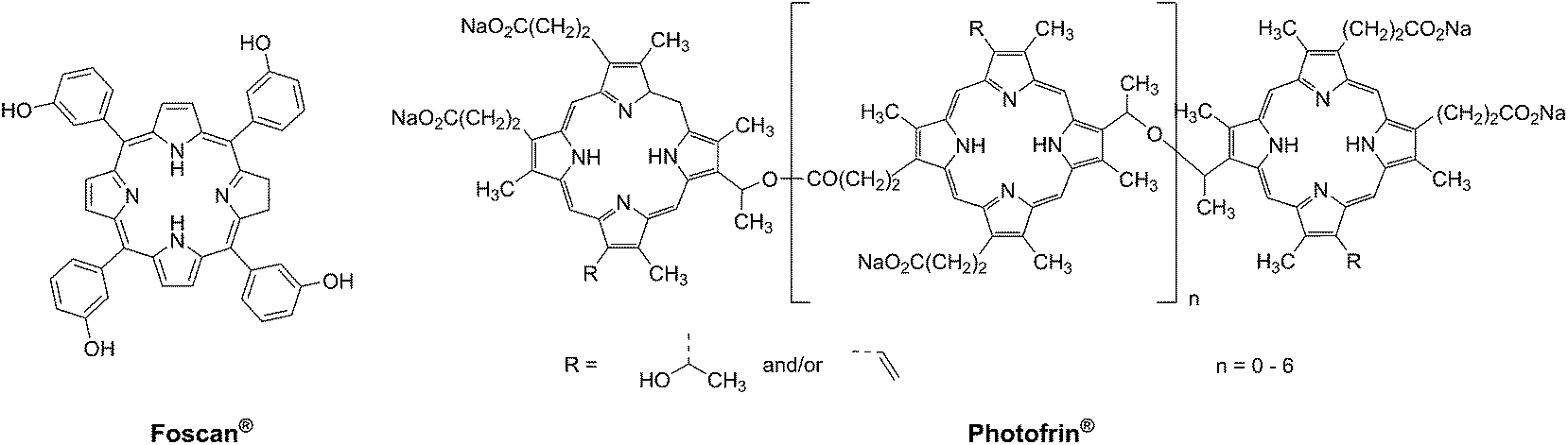 | ||
| Fig. 2 Structures of porphyrin-based approved PDT agents. | ||
More specifically, PDT relies on the synergistic activity of an ideally non-toxic molecule called a photosensitizer (PS), light and molecular oxygen. The PS is administrated to the patient either locally or systemically. Upon light irradiation at a wavelength in its range of absorption, the PS is able to reach its singlet excited state 1PS* (Fig. 3). Very importantly, the PS must then undergo an intersystem crossing (ISC) so that the excited state has a triplet character (3PS*). At this point, PDT relies on two different mechanisms called Type I and Type II. A Type I reaction consists of an electron or proton transfer from the triplet excited state of the PS to the surrounding biological substrates (or the other way around). This leads to the formation of radicals that can further interact with molecular oxygen to form ROS such as superoxide, hydroxyl radicals or peroxides. At the same time, an energy transfer from the triplet excited state of the PS to molecular oxygen in its ground triplet state (3O2) can occur (a Type II reaction). In this case, singlet oxygen (1O2) is generated. 1O2 is a very reactive form of oxygen with an estimated half life of 40 ns in a biological environment.27 Consequently, it will rapidly react just with the surrounding biomolecules, generating topical cellular damage that can ultimately lead to cell death. PSs which are nowadays applied in clinics mainly rely on the Type II mechanism of action.29
 | ||
| Fig. 3 Mechanisms of action of PDT. | ||
PDT is a very appealing medical technique due to its intrinsic selectivity. The toxic species are generated just at the site of light irradiation, with complete spatial and temporal control. Furthermore, due to the very fast reactivity of 1O2, damage is limited to the irradiated areas. The outcomes of PDT treatment depend on the performance of the PS but also on other very important factors (e.g. the light component, the in vivo dosimetry or the oxygen tension). To be clinically applicable, a PS should, among other requirements, (i) localize mainly (ideally only) in cancer cells; (ii) should be non-toxic in the absence of light, while displaying strong phototoxicity. This behavior is normally described by the so-called phototoxic index (PI), defined for a compound as the ratio of its IC50 in the dark to its IC50 upon light irradiation. Finally, the PS (iii) should be excited in the red or near-IR region of the spectrum (>600 nm). This last requirement is very important to avoid cytotoxicity deriving from high energy light irradiation. In addition, the use of long wavelength light allows for a deeper penetration through the human tissues.21,30
The great majority of PSs that are currently applied in clinics are based on a cyclic tetrapyrrolic scaffold. The photophysical and biological characteristics of porphyrins, phthalocyanines and chlorins match the requirements for a PDT agent relatively well. On the other hand, their performances are also limited by important side-effects. As an example, treatment with Photofrin® results in light sensitivity for several weeks due to slow clearance of the drug from the body.31 As a consequence, an important effort has been undertaken to improve the performances of the current PSs following two approaches: the modification of a conventional porphyrin-based PS or the optimization of entirely new systems that can outperform porphyrins in their PDT activity. In this specific section, we present a description of the influence of the insertion of ruthenium fragments into porphyrin-based PSs, focusing our attention on the works that report on the biological behavior of these new systems. Furthermore, we present the recent achievements in the use of ruthenium polypyridyl complexes as novel PSs in the innovative attempt to move away from the traditional porphyrin-as-PS paradigm.
Ruthenium-containing porphyrin PSs
The derivatization of the porphyrin core with metal complexes is an appealing opportunity to improve the activity of a PS. This functionalization was exploited for the first time fifteen years ago by Brunner and coworkers.32,33 They synthesized hematoporphyrin–platinum conjugates to combine the strong anticancer activity of platinum-based drugs with the phototoxic effect of porphyrins. The metal derivatization of a porphyrin core can enhance the intrinsic properties of a PS by modifying its physico-chemical characteristics. For example, the metal fragment can change the lipophilicity of the PS, increase its water solubility or improve its cellular uptake. As mentioned above, ruthenium complexes display very promising biological behavior. Consequently, several research groups have recently evaluated the possibility of introducing Ru(II) moieties on the periphery of porphyrins. For instance, Therrien et al. synthesized a wide range of Ru-modified porphyrin systems and studied their biological performances.34 More specifically, they appended a number of Ru-arene fragments to the meso-4′-tetrapyridylporphyrin scaffold to evaluate the influence of the different aromatic moieties (1a–e, Fig. 4, top). All the compounds were found to induce 60–80% mortality in human Me300 melanoma cells at a 10 μM concentration, using light at 652 nm with a dose of 5 J cm−2. The photoactivity of the metal-functionalized systems was found to be independent of the nature of the arene. This flexibility can give access to the use of arenes which are derivatized with targeting agents or chemotherapeutic compounds. Fig. 5, which shows the phototoxicity evaluation of the compounds synthesized by Therrien et al., demonstrates that the improved behavior of their systems required the presence of the Ru fragment, since the Rh analog 3 was not internalized by cells and was therefore not toxic. In addition, the Os derivative 2 exerted just a weak phototoxic effect (see Fig. 4 for the structures of the latter compounds).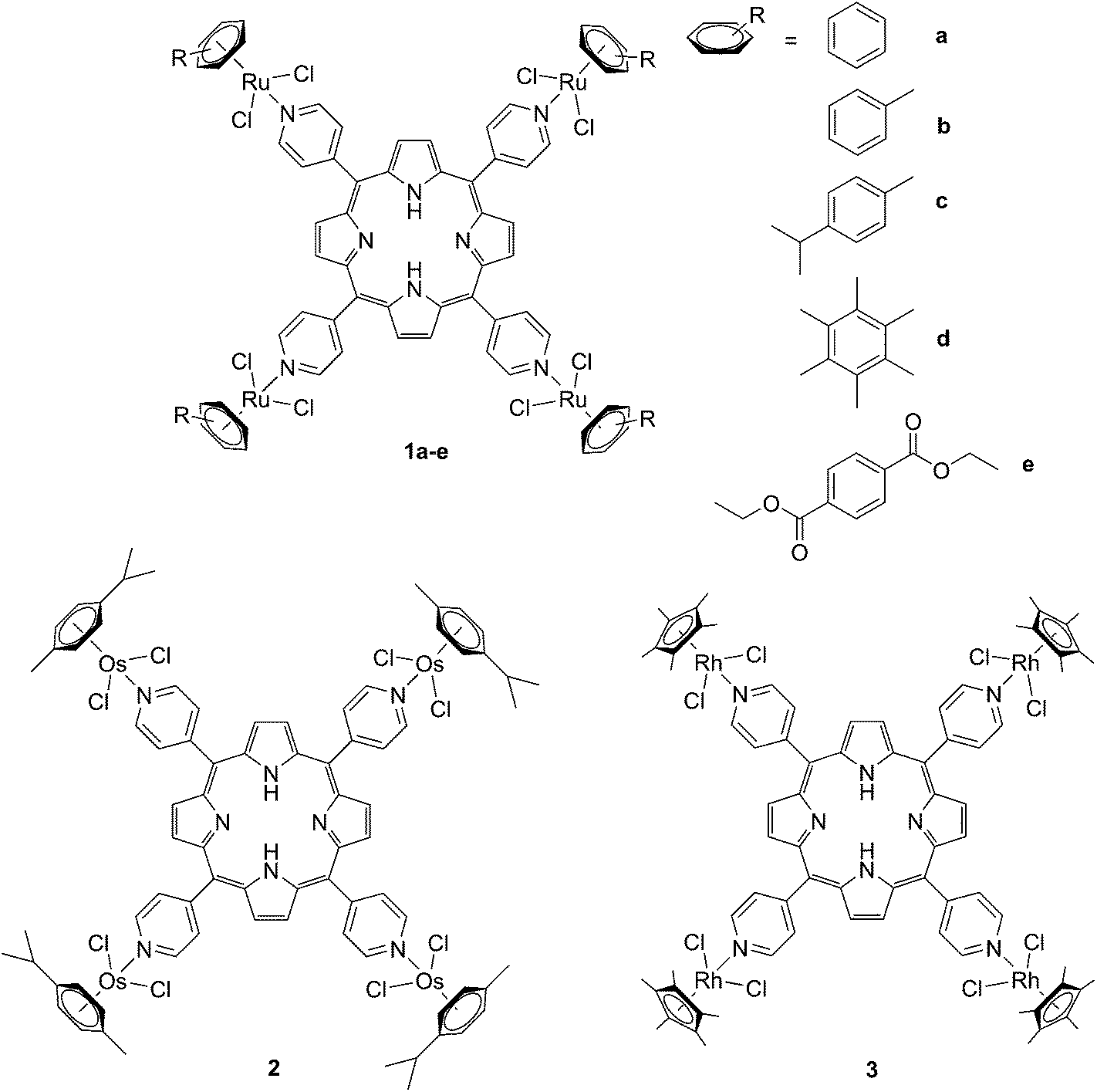 | ||
| Fig. 4 Structures of Ru–porphyrin conjugates (top, 1a–e), and Os and Rh analogs (bottom, 2 and 3).34 | ||
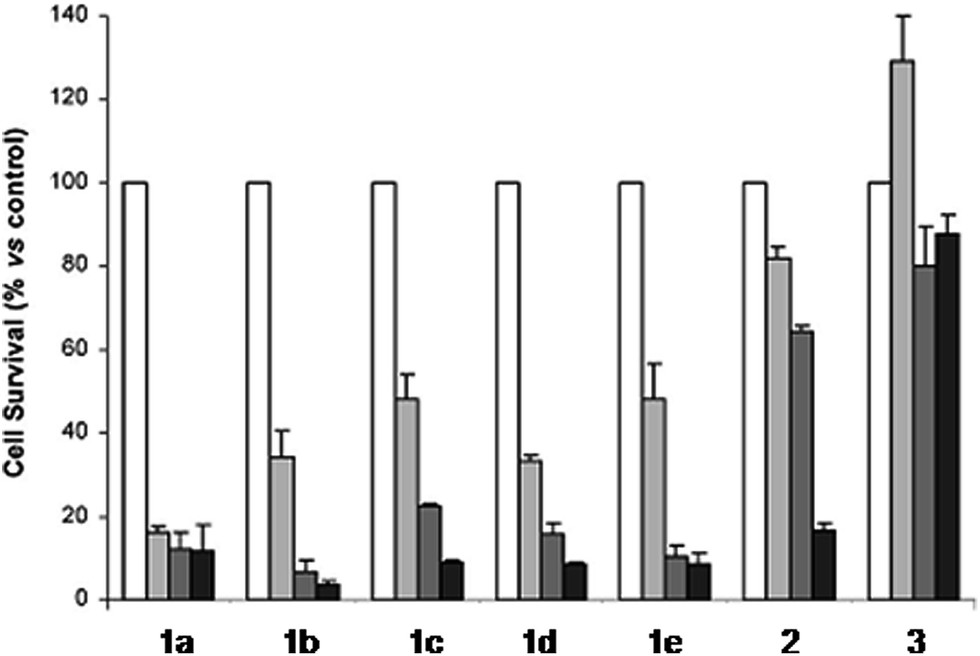 | ||
| Fig. 5 Phototoxicity evaluation of compounds 1a–e, 2 and 3 on Me300 melanoma cells. Cells were incubated with 10 μM of the compounds, incubated for 24 h, then irradiated at 652 nm with 0 J cm−2 (white bar), 5 J cm−2 (light grey bar), 15 J cm−2 (dark grey bar) or 30 J cm−2 (black bar) light doses. Adapted with permission from ref. 34. Copyright 2008 American Chemical Society. | ||
The same authors also studied the influence of tetra- vs. mono-metallic derivatization (4a-b/6a–bvs.5a–b/7a–b, Fig. 6), as well as the nature of the pyridylporphyrin isomers, by comparing 4′-pyridylporphyrin or 3′-pyridylporphyrin derivatives (4a–b/5a–bvs.6a–b/7a–b, Fig. 6).35 Several conclusions could be drawn from this small structure–activity relationship (SAR) study. For example, the type of pyridylporphyrin isomer was shown to play a major role in the observed activity, since the 3′-pyridyl substituted compounds showed a greater phototoxic effect than the 4′-pyridyl analogs. On the other hand, the number of Ru atoms or the arene derivatization seemed to have less influence on the biological activity.
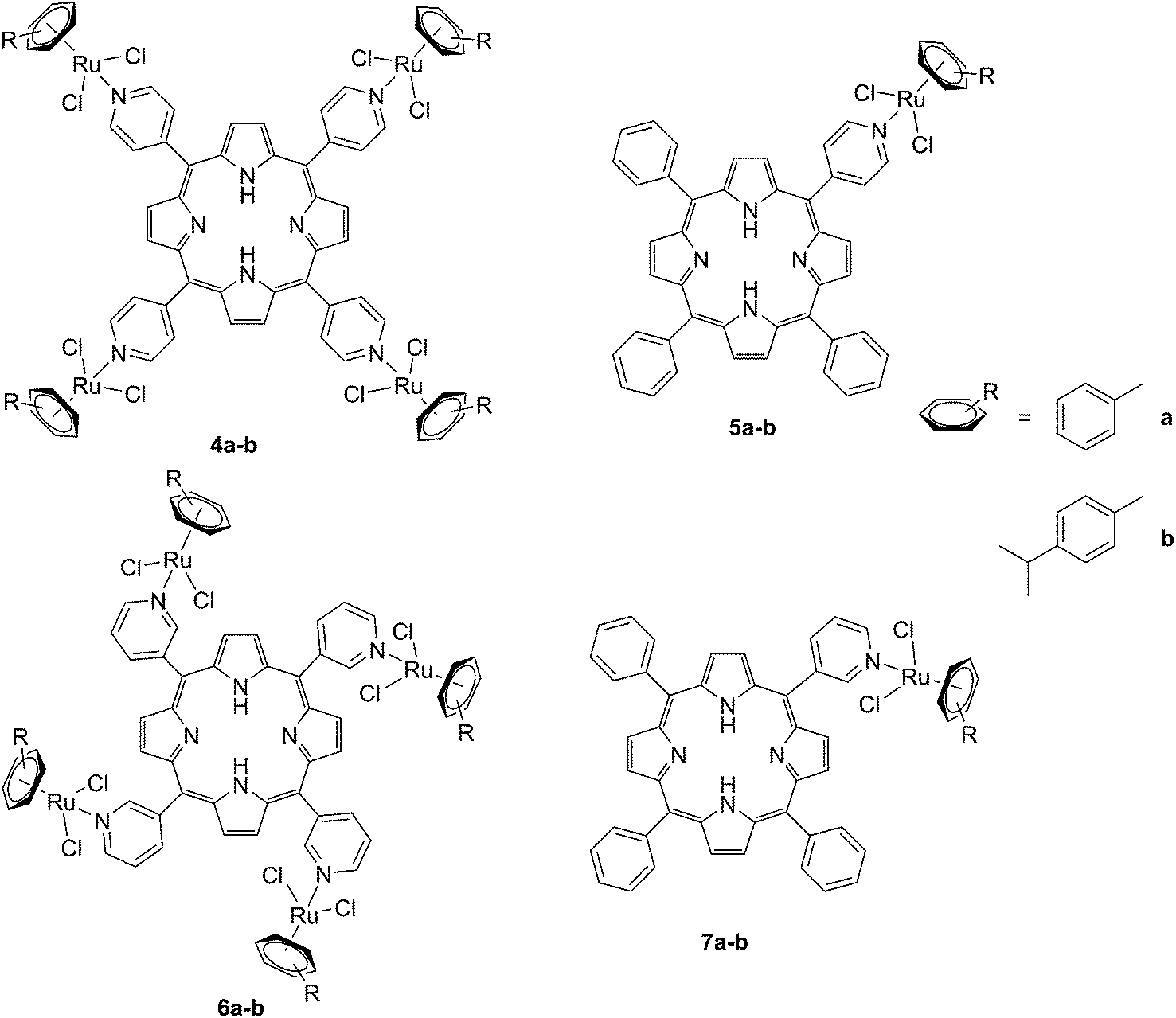 | ||
| Fig. 6 Structures of the Ru–porphyrin conjugates evaluated in the SAR study by Schmitt et al.35 | ||
In more detail, upon the 652 nm light irradiation of human Me300 melanoma cells, a LD50 of 5 μM was reached with a light dose of 0.5 J cm−2 for compounds 6a and 6b and with a light dose of 2.5 J cm−2 for 7a and 7b. For the 4-pyridyl derivatives, 5 or even 10 J cm−2 were necessary to achieve the same potency. This difference in biological activity was explained by luminescence microscopy studies, where 4a (more hydrophobic) was shown to form aggregates inside the cytoplasm (Fig. 7A), although the authors did not discuss further about accumulation in a specific organelle. This aggregation could lead to a quenching of the ROS production. On the contrary, compound 6a was shown to be evenly distributed in the cytoplasm, where it could exert its phototoxic activity (Fig. 7B).
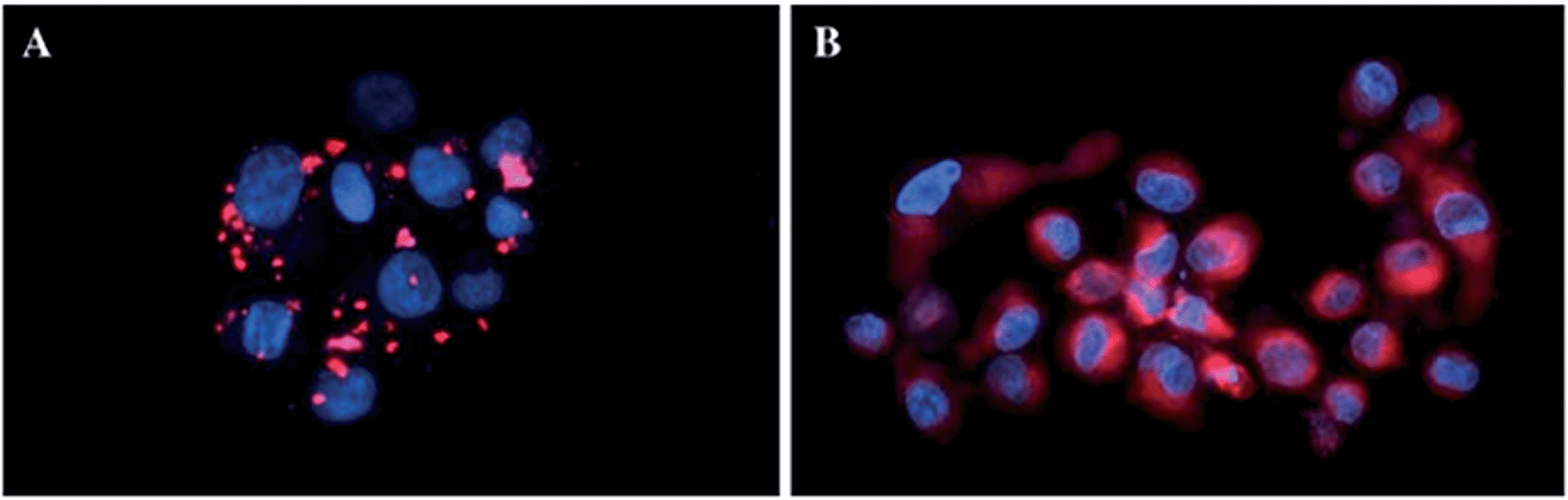 | ||
| Fig. 7 Fluorescence microscopy images of human Me300 melanoma cells incubated for 24 h with 5 μM of 4a (A) and 6a (B), displaying red luminescence. The blue luminescence in the nuclei derives from DAPI co-staining. With kind permission from Springer Science and Business Media.35 | ||
The two best compounds in this study, namely [Ru(η6-p-iPrC6H4Me)(PMP)Cl2] (PMP = 5-(3-pyridyl)-10,15,20-triphenylporphyrin) and [Ru4(η6-p-iPrC6H4Me)4(PTP)Cl8] (PTP = 5,10,15,20-tetra(3-pyridyl)porphyrin) (6b and 7b) were evaluated in vivo on nude mice xenografted with human head and neck carcinoma KB cells.36 Since PDT is a synergistic cooperation of different components (PS, light and O2), the evaluation of its in vivo efficacy depends on the combination of a complex system of parameters, which reciprocally affect each other. As a consequence, the authors determined that crucial factors to be optimized during in vivo studies were not just the concentration of the drug, but also the interval between PS administration and light treatment (the drug-light interval, DLI), light fluence and the fluence rate.36 They therefore adopted a statistical approach to find the combination of parameters that would yield the best therapeutic outcomes, thereby reducing as much as possible the number of required experiments. The study showed that, if PS concentration and light fluence were not crucial parameters, a long DLI and the use of the tetranuclear species led to statistically significant tumor growth stabilization up to at least 30 days.
Since the study on these systems highlighted that the number of ruthenium modifications is correlated with an increase in phototoxicity, the authors synthesized two cationic octanuclear metalla-cubes 8 and 9 (Fig. 8). These compounds, thanks to their higher ruthenium content, showed better activities when compared to their tetranuclear analogs.37 An LD50 of 1 μM was reached upon irradiation with 652 nm light and a 2–7 J cm−2 light dose for both compounds, whereas for the tetranuclear analogs, a light dose of 5–10 J cm−2 at the same wavelength resulted in a LD50 of 5 μM.
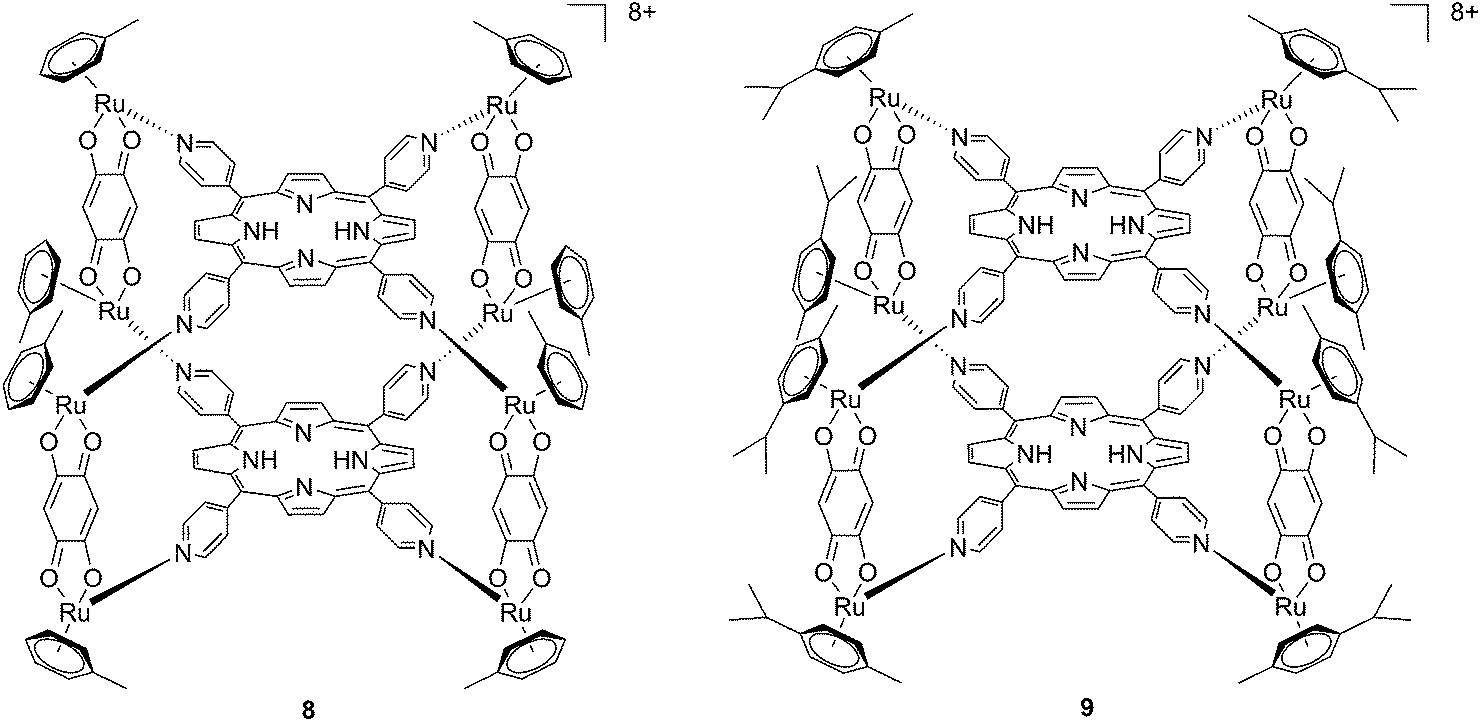 | ||
| Fig. 8 Polynuclear metalla-cubes 8 and 9 synthesized by Therrien to increase phototoxicity.37 | ||
Another interesting approach used by this group for the combination of Ru complexes and PDT is the application of Ru-cages as carriers for porphyrin photosensitizers inside cancer cells. The authors developed the two cages presented in Fig. 9, namely hexa- (10) and octanuclear (11), which were characterized by different mechanisms of release.38 In the case of 10, the cage must be disrupted to allow the release of the PS, whereas for 11, the PS can diffuse through the sides of the cage. As a consequence of this difference, 11 was found to be 10 times more photoactive than 10. The authors obtained phototoxicity in the submicromolar range and a PI of about 20 for 11 on cervical cancer HeLa cells, upon irradiation at 455 nm with an impressively weak light dose (0.2 J cm−2). This result demonstrated the release of the porphyrin after cellular internalization, as was also shown by luminescence microscopy (Fig. 10). In these pictures, it is possible to notice the red luminescence from the free PS and the blue emission originating from the empty cage. This also indicates that the two systems are localizing in different cellular compartments after release. Furthermore, the internalization of the porphyrin in both cages resulted in a hypochromic effect on the porphyrin. This means that when the PS is trapped, its emission is dramatically reduced and consequently also the phototoxic effect. This phenomenon leads to a safe delivering agent that does not display undesired phototoxicity outside of cells.
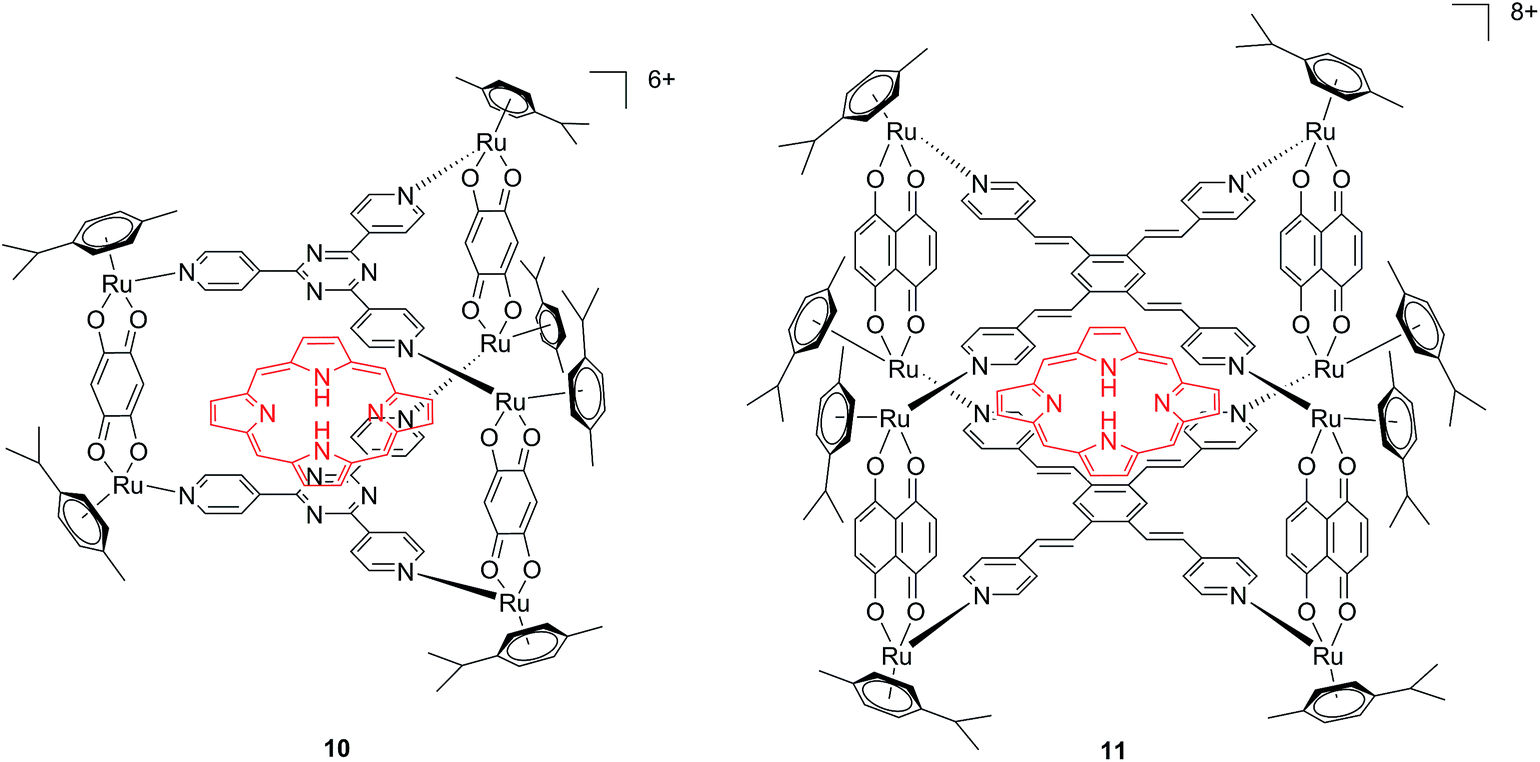 | ||
| Fig. 9 Ruthenium cages 10 and 11 applied as carriers of a porphyrin PS inside cancer cells.38 | ||
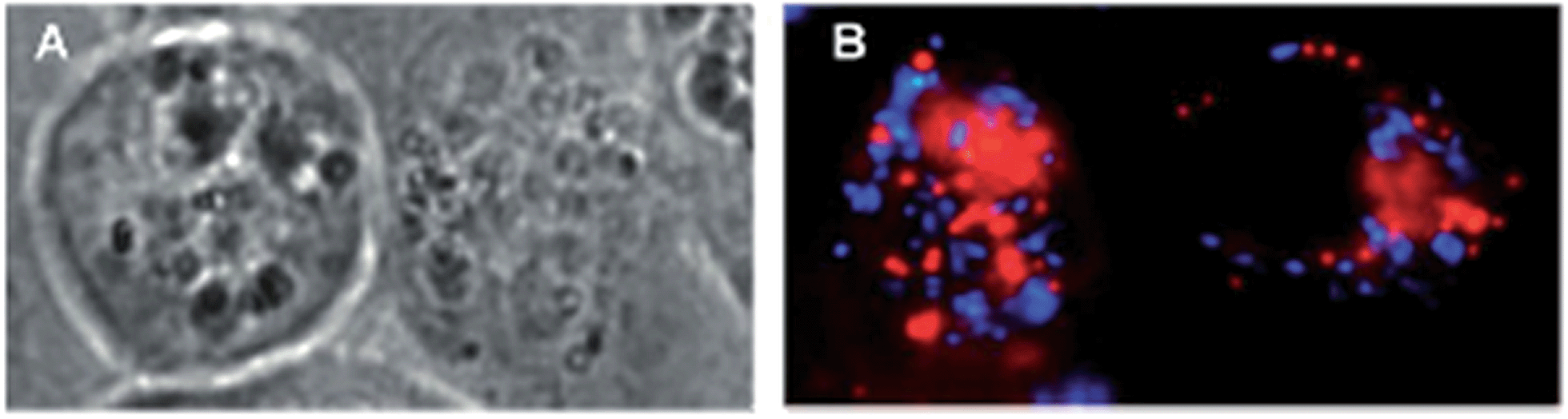 | ||
| Fig. 10 Fluorescence microscopy of HeLa cells incubated with 11 (2 μM, 2 h): (A) white light and (B) fluorescence. Reprinted with permission from ref. 38. Copyright 2012 American Chemical Society. | ||
With the same idea in mind, namely to obtain a synergistic biological effect owing to the conjugation of porphyrin and ruthenium fragments, Alessio and coworkers synthesized a library of compounds where meso-tetraphenylporphyrin or meso-4′-tetrapyridylporphyrin cores were modified on their peripheries with Ru complexes.39 The authors then selected five cationic species for biological evaluation.40 The most active compounds 13 and 14 (Fig. 11) contain four ruthenium moieties and their coordination sphere is a slight modification of the [Ru([9]aneS3)(en)Cl]+ complex (12, Fig. 11, top left, [9]aneS3 = 1,4,7-trithiacyclononane, en = ethylenediamine), which was already shown by the same group to be characterized by a strong cytotoxicity.41,42
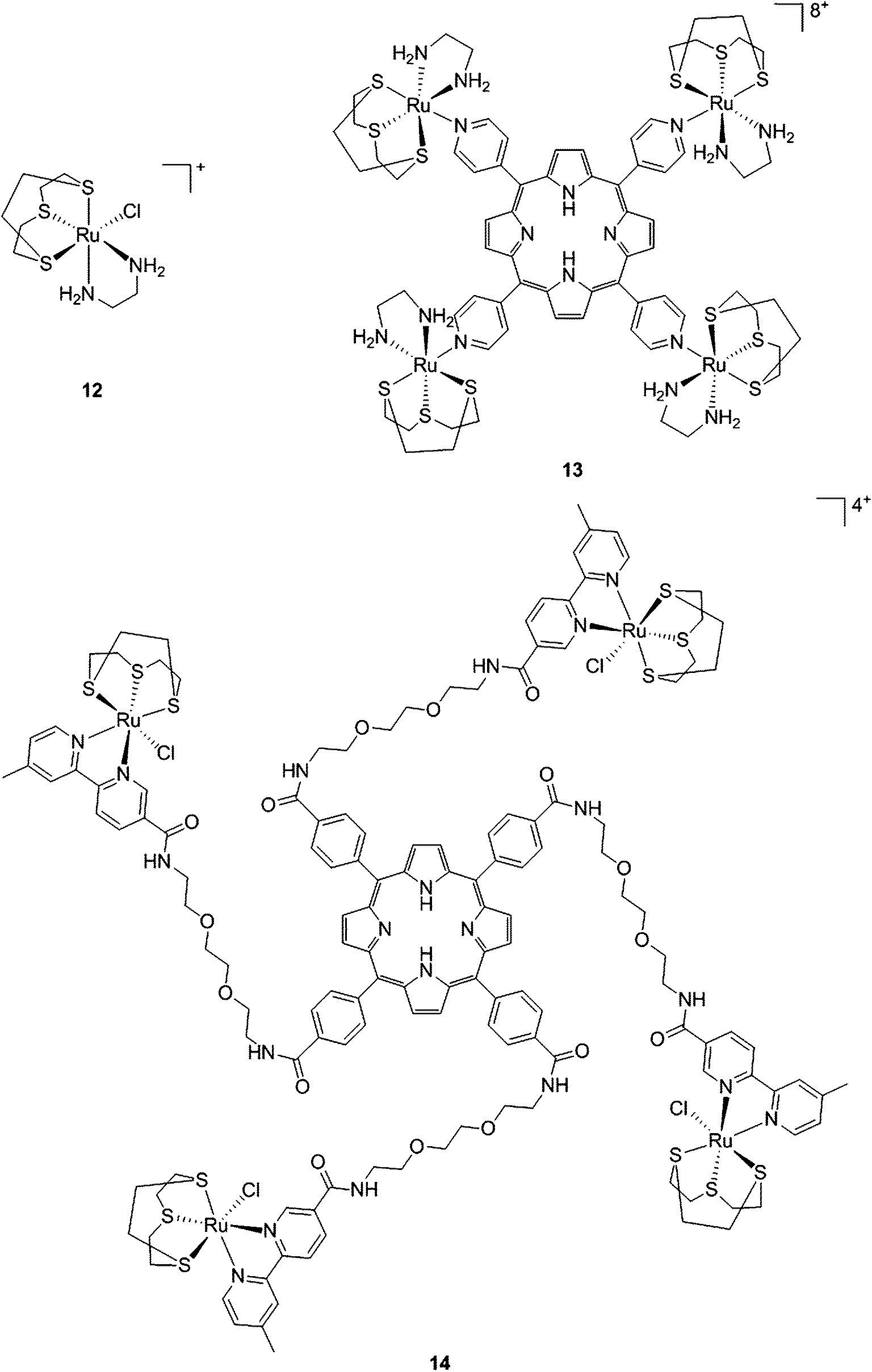 | ||
| Fig. 11 Structures of Ru([9]aneS3)(en)Cl]+ (top, left) and of the ruthenium-derivatized porphyrin systems 13 and 14.40 | ||
As expected, the ruthenium fragments strongly improved the physicochemical behavior of the porphyrin core. This resulted in a clear increase in cytotoxicity of the compounds, most likely, as speculated by the authors, due to higher cellular accumulation. Furthermore, the potency of the systems in human breast cancer cells MDA-MB-231 was improved by one order of magnitude upon exposure to 5 J cm−2 of 590–700 nm light, thus reaching the nanomolar range. Following the same strategy, Swavey et al. explored a range of possible modifications of porphyrins to improve their activity and selectivity.43 In particular, they introduced a Ru(bipy)2 moiety (bipy = 2,2′-bipyridine) with a labile Cl ligand to obtain additional DNA binding and light-induced DNA cleavage. Two pentafluoroaryl groups, which are known to increase the excited state lifetime of a PS, were also linked to the porphyrin, to give compound 15 (Fig. 12, left). The authors obtained a very strong affinity for DNA and consequent photocleavage of plasmid supercoiled DNA. Furthermore, they demonstrated that the compound exerted a higher phototoxicity on melanoma cells when compared to normal skin fibroblast cells.
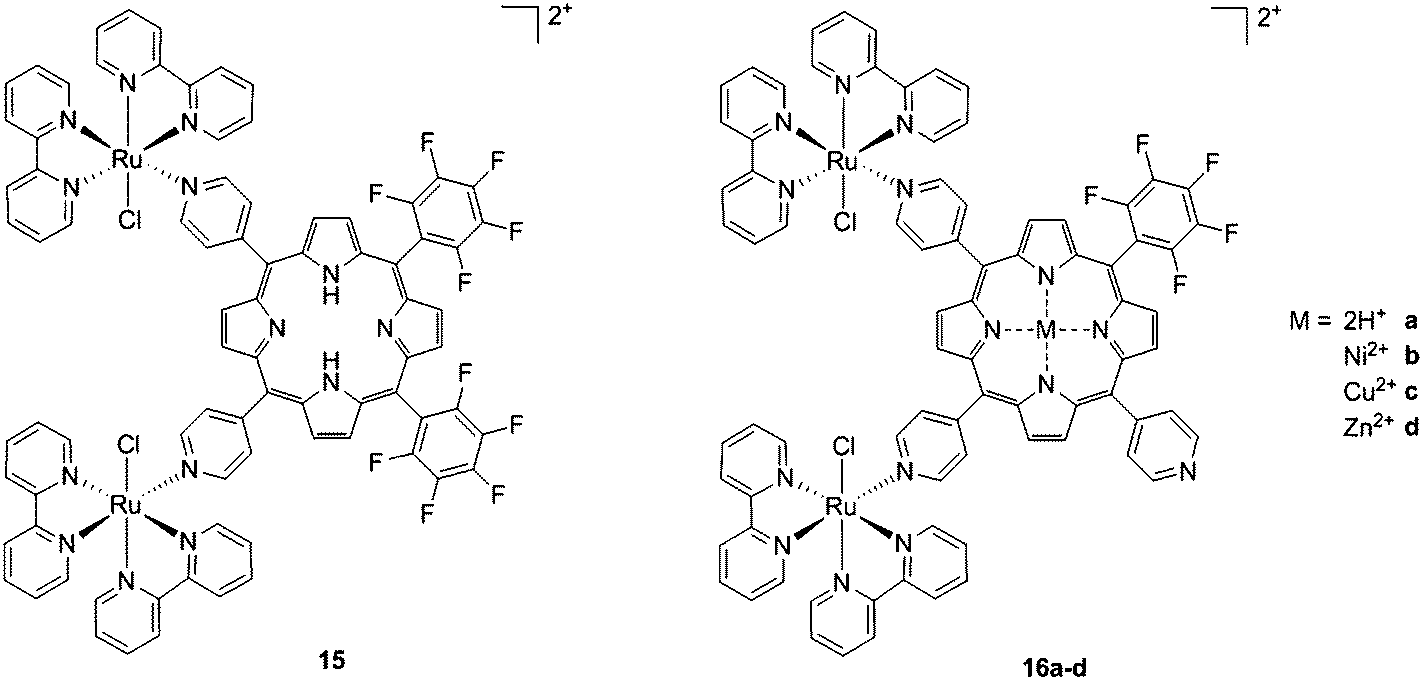 | ||
| Fig. 12 Porphyrin with pentafluoroaryl and Ru(bipy)2Cl fragments to give 15 (left) and Ru–porphyrin conjugates containing different metals in the ring (16a–d, right).43,44 | ||
To improve the efficacy and the selectivity of their system, the same authors removed one pentafluoroaryl group and evaluated the effect of the insertion of a metal into the porphyrin ring (16a–d, Fig. 12, right).44 Upon coordination of a metal ion in the porphyrin, the photophysical properties of the system undergo an important change due to the metal perturbing the energy levels of the free ligand. For instance, it was noticed that the complexation of Zn(II) increases the lifetime of the excited state of the porphyrin.19 In this work, they demonstrated that all three metal-coordinated systems were able to nick plasmid DNA upon induction with light, with the Zn(II) system 16d also generating double strand breaks. In cellular studies, Ni(II) and Cu(II)–porphyrins were inactive as photosensitizers. On the other hand, the Zn(II) system at a concentration of 5 μM induced cell death very efficiently on a melanoma cell line upon white light irradiation (Fig. 13, bottom). Interestingly, the same treatment did not show any efficacy on normal skin fibroblast cells (Fig. 13, top), providing indications of a very selective system.
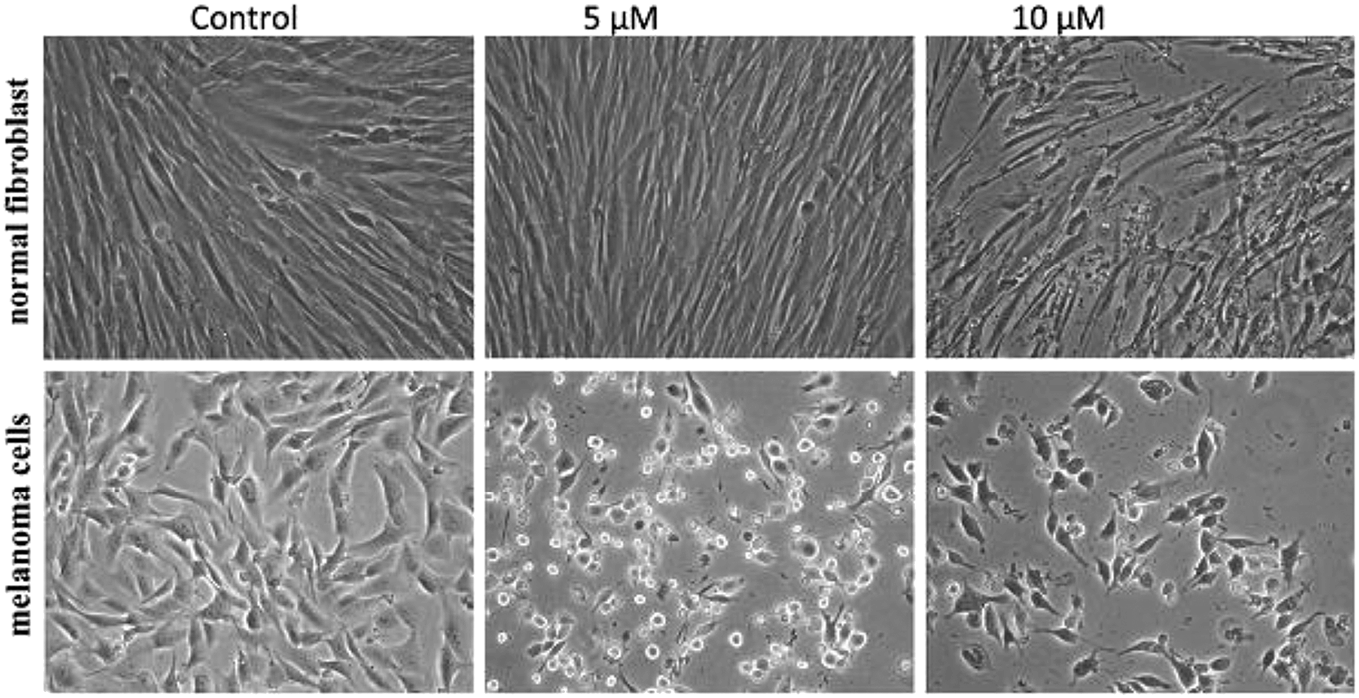 | ||
| Fig. 13 Phase contrast microscopy images of cells irradiated with a 60 W tungsten lamp for 30 min. Normal fibroblast cells (top) and melanoma cells (bottom) without 16d (control) and in the presence of 5 and 10 μM concentrations of 16d. Reproduced from ref. 44 with permission from The Royal Society of Chemistry. | ||
Of utmost interest, the authors performed in vivo studies with compound 16d on Drosophila melanogaster to assess its general toxicity in the dark as well as biodistribution.45 The compound was found to be harmless for the larvae and during their development. Cellular localization studies were also performed by feeding the larvae with the compound. Confocal microscopy revealed that the molecule was able to accumulate in the cytosol, but also in the nuclei at higher concentration. This suggests that the compound is not readily metabolized.
Another interesting class of compounds includes the coordinatively saturated ruthenium polypyridyl complexes. These compounds are known to be kinetically inert and substitutionally stable. Therefore, they do not have a labile ligand that can covalently bind DNA. Nevertheless, it was shown that, with the use of appropriate ligands such as dipyrido[3,2-a:2′,3′-c]phenazine (dppz) or tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine (tpph), these complexes can interact very strongly with double-stranded DNA via intercalation or groove binding. Thanks to these interesting characteristics, these compounds were extensively studied as DNA intercalating probes46,47 or as cytotoxic agents.48–51 Furthermore, it was demonstrated that these compounds are also able to produce 1O2 (see next paragraph for more information on this topic). To exploit this property, Wong and co-workers conjugated a [Ru(bipy)2phen]2+ (phen = 1,10-phenanthroline) moiety to a porphyrin core via three different linkers on the phen (Fig. 14) and evaluated the biochemical behavior of the resulting systems 17a–c.52 The ruthenium conjugation was also introduced here to improve the two-photons absorption (TPA) characteristics of the compounds. As a consequence, by virtue of the simultaneous absorption of two photons, the molecule can be excited at 800 nm, a more tissue penetrating and less harmful wavelength. Therefore, this interesting characteristic allows for the development of bifunctional PDT and tumor imaging agents.
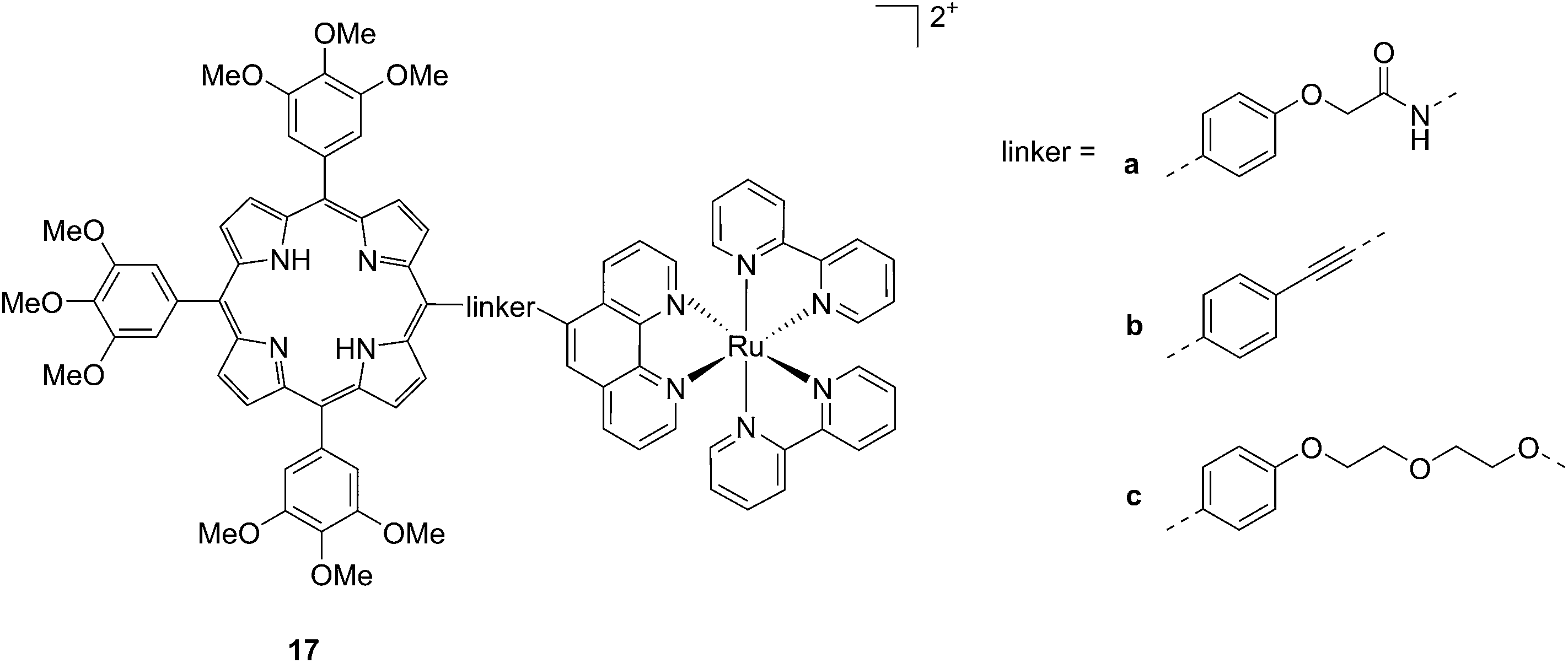 | ||
| Fig. 14 Structures of Ru–porphyrin conjugates 17a–c, with three different bridging linkers.52 | ||
Interestingly, the authors could achieve a different cellular localization based on the type of linker used to connect the porphyrin core to the ruthenium moiety. This difference allowed for studying the effects of PDT in different cellular compartments. Compounds 17a and 17b were characterized by the best cellular uptake, as demonstrated by flow cytometry analysis. Comparably, they also displayed the best phototoxic behavior with a toxicity of 118 and 175 μM on HeLa cells in the dark and LD50 of 1 μM upon yellow light irradiation with doses of 6.5 and 2.0 J cm−2, respectively. Compound 17b also showed its activity as a TPA-PDT agent, causing cell shrinkage upon irradiation at 850 nm. The compound, which localized in the mitochondria before light exposure, was found to relocate in the nuclei after light irradiation. The authors therefore assumed that 17b induced light-mediated damage to mitochondria, from which it is then released. Once in the cytosol, the compound can damage the nuclear membrane and cause cell death. Interestingly, they also showed that the presence of the Zn atom in their conjugates had a detrimental effect on the emission quantum yields of the systems in DMSO, going from values of 1.93–5.3% for the free base compounds to <1% when Zn(II) was inserted in the porphyrin ring. The authors considered this difference in the photophysical behavior to be related to an energy transfer from the Soret band of Zn–porphyrins to the ruthenium fragment.
Ruthenium complexes as PSs
As discussed above, porphyrins certainly have good characteristics as PSs due to their intrinsic physico-chemical properties. On the other hand, the PSs available on the market still display a number of drawbacks such as their low solubility in biological media, lack of selective cancer accumulation and the frequently encountered photosensitivity in patients undergoing PDT treatments. Over the last few years, several research groups have explored the possibility to move away from tetrapyrrolic systems, studying the potential of metal complexes as PSs themselves. The application of ruthenium complexes as PSs is a reasonable approach due to their tunable photophysics and the aforementioned advantages for biological applications (see Introduction). As an example of this approach, our group synthesized six [Ru(bipy)2dppz]2+ complexes 18a–f with different functional groups on the dppz ligand (Fig. 15).53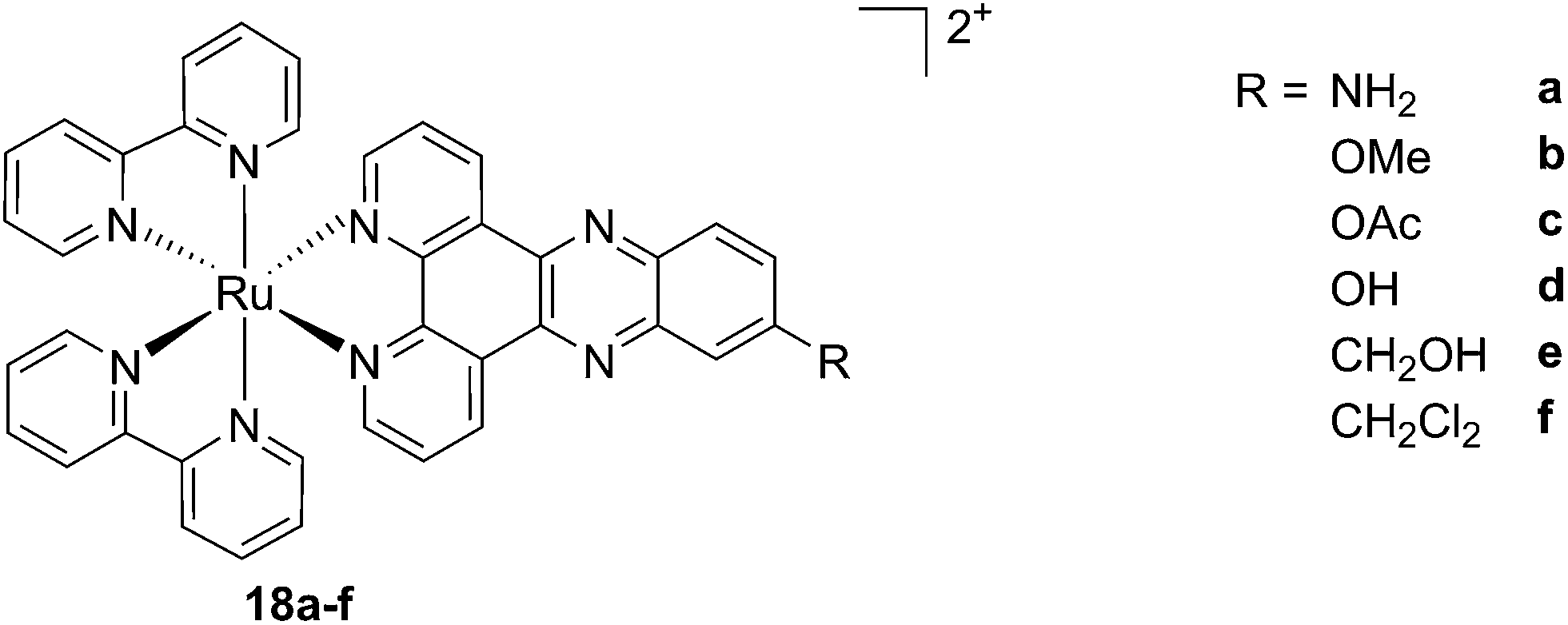 | ||
| Fig. 15 Structures of the six different DNA intercalating Ru complexes 18a–f.53 | ||
As highlighted before, the presence of the dppz intercalative ligand was meant to increase the affinity of the compounds for DNA, so that a targeted delivery of singlet oxygen to the genetic material can be achieved. All Ru complexes were found to be non-toxic (up to 100 μM) to both normal fetal lung fibroblast cells (MRC-5) and cervical cancer HeLa cells in the dark. Nevertheless, the amino- and methoxy-substituted Ru complexes showed impressive photoactivities. When HeLa cells were irradiated with a light dose of 9.27 J cm−2 at 420 nm, IC50 values in the low micromolar range were obtained for 18a and 18b. An impressive PI of 43 for the latter and even >150 for the former were obtained. Cellular distribution studies were performed on both compounds by means of confocal microscopy and high-resolution continuum source atomic absorption spectrometry (HR-CS AAS) and the results are reported in Fig. 16. These techniques indicated a very good cellular uptake of both compounds. Furthermore, HR-CS AAS analysis confirmed the nuclear localization for both complexes after 4 h incubation, allowing for target delivery of 1O2 to DNA. Compounds 18a and 18b also showed good efficiency in generating strand breaks of supercoiled plasmid DNA upon light irradiation. This feature strongly suggested the involvement of DNA in the mechanism of phototoxicity. Further studies are ongoing to investigate the interaction of 18b with DNA, and the exact mechanism of cell death engendered by light activation.
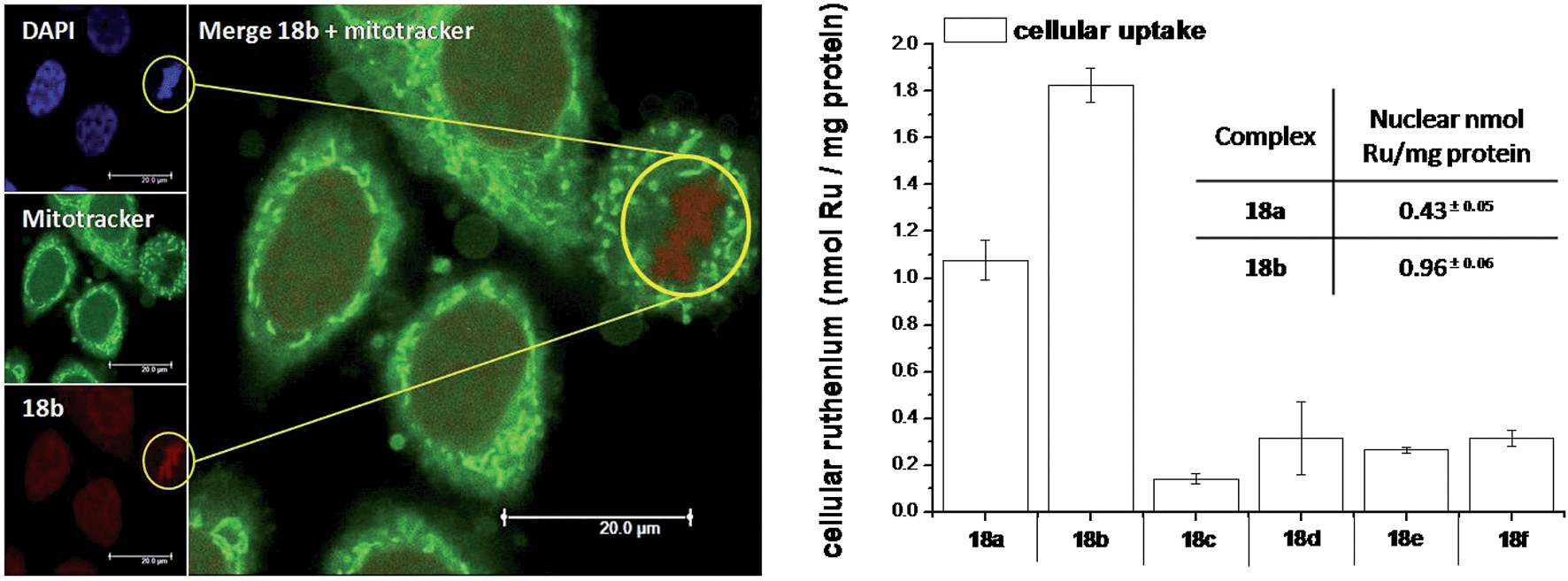 | ||
| Fig. 16 Left: Confocal microscopy images of HeLa cells treated for 2 h with 100 μM of complex 18b (excitation at 488 nm, emission above 600 nm, bottom left) and stained with DAPI (nuclear staining, top left) and with Mitotracker green (mitochondrial staining, middle left); in the yellow circle a representative example of the different localization of 18b and Mitotracker green is found (picture on the right). Right: Cellular uptake into HeLa cells treated for 4 h with 20 μM solutions of the complexes 18a–f. Results are expressed as the mean ± error of independent experiments. In the inset: nuclear uptake for complexes 18a and 18b. Reproduced with permission from ref. 53. © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
With the similar goal of targeting and photocleaving DNA, Brewer et al. studied mono-metallic or supramolecular complexes of Ru, Pt, Rh and their abilities to interact with DNA upon light irradiation in depth (see also the PACT section below). In particular, they demonstrated the ability of three [(TL)2Ru(dpp)]2+ compounds (dpp = 2,3-bis(2-pyridyl)pyrazine, with TL = bipy, phen or Ph2phen = 4,7-diphenyl-1,10-phenanthroline) to efficiently photocleave supercoiled pUC18 plasmid DNA upon irradiation at λ = 450 nm thanks to the formation of 1O2.54 However, the biological activity of compounds of the type [(TL)2Ru(dpp)]2+ in cells was not evaluated. Turro and coworkers are also very active in the field of light-activated ruthenium complexes. They synthesized and characterized many compounds and studied their photophysics, and light-mediated interactions with DNA and proteins due to the formation of singlet oxygen,55,56 or to other mechanisms (see also the PACT section below). To further highlight the mode of action of these photoactivated compounds, these researchers investigated their light-induced effects on DNA and proteins in fibroblasts.57 The two complexes, [Ru(tpy)(pydppn)]2+ (19) and [Ru(pydppn)2]2+ (20) reported in Fig. 17, with tpy = [2,2′;6′,2′′]-terpyridine and pydppn = 3-(pyrid-2′-yl)-4,5,9,16-tetraaza-dibenzo[a,c]naphthacene,56 displayed very long lifetimes of the excited states (20–24 μs), thanks to the pydppn ligand, which allows for singlet oxygen generation with an efficiency of almost 100%.
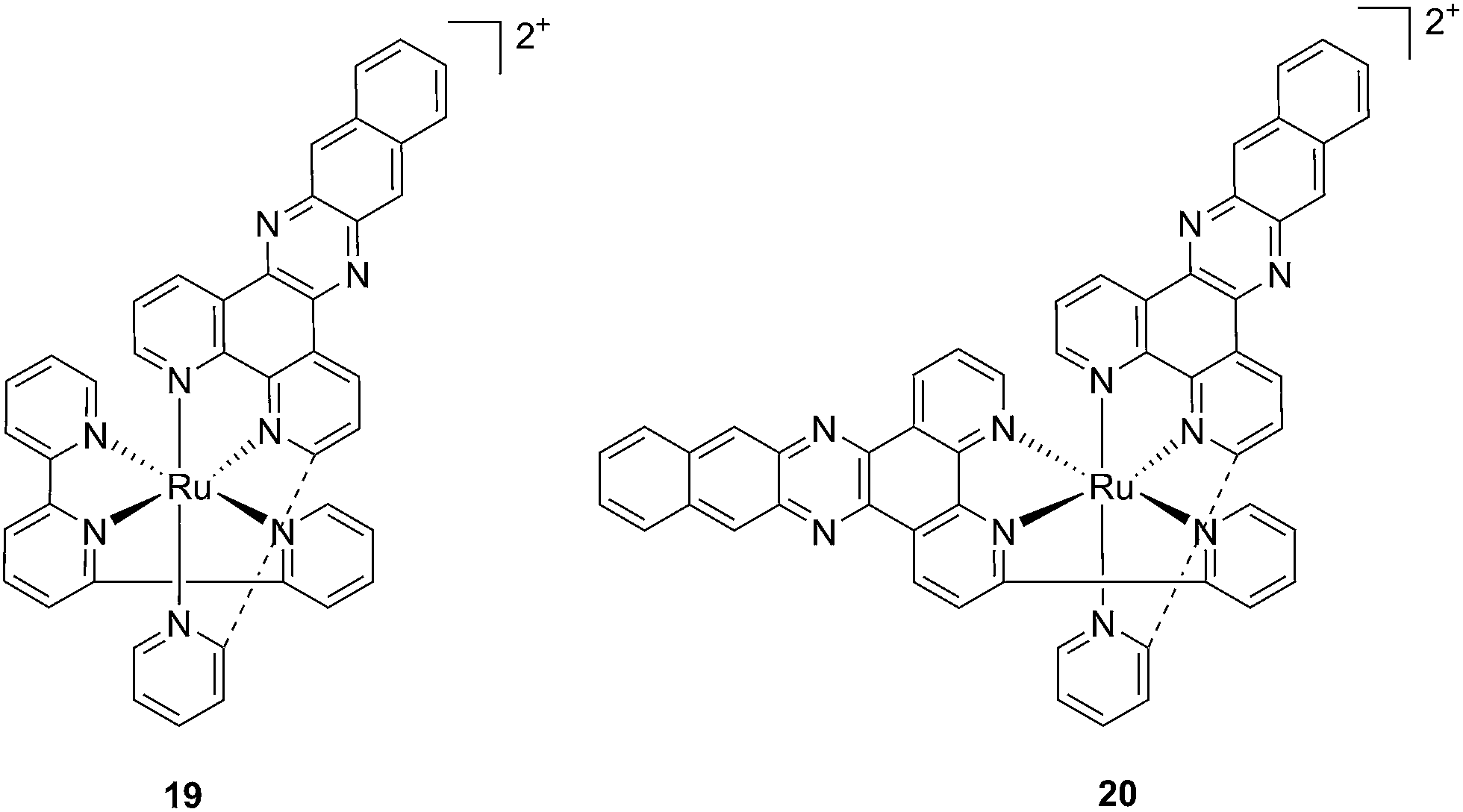 | ||
| Fig. 17 Structures of the Ru complexes 19 and 20 bearing the tridentate pydppn ligand, which confers very long excited state lifetimes.57 | ||
The authors were then able to demonstrate that 19 and, to a lesser extent, 20 induced photodynamic damage to the tumor suppressor p53 and the DNA polymerase processivity factor PCNA (proliferating cell nuclear antigen), both of them being key components of DNA maintenance and repair pathways. Upon light irradiation of cells and cell lysates (3.15 J cm−2 of visible light), the compounds induced covalent crosslinking of the protein subunits, the formation of DNA–protein adducts and, as a consequence, the inhibition of DNA replication. p53 crosslinking was previously demonstrated to correlate with the formation of singlet oxygen,58 and the work of Turro and colleagues57 demonstrated a strong reduction in the efficiency of p53 photodamage by the presence of sodium azide, a known singlet oxygen quencher. In addition, protein–DNA crosslinking was demonstrated to depend on singlet oxygen-mediated formation of 8-oxo-7,8-dihydroguanine and its further reaction with amino groups in the protein. Also in this case though, the evaluation of the phototoxic profile of the compounds on cells was not explored.
Ruthenium polypyridyl complexes also have an excellent record of performance in the field of dye-sensitized solar cells (DSSCs)59 due to their absorption in the visible range and very long lifetimes. Interestingly, and as previously noted, these characteristics are also of extreme importance in the field of PDT. Consequently, our group decided to explore the photodynamic behavior of two derivatives of ruthenium complexes bearing a benzenedithiol (21) and a tridentate polypyridyl ligand (22), respectively (Fig. 18), which were previously employed in the field of DSSCs.60
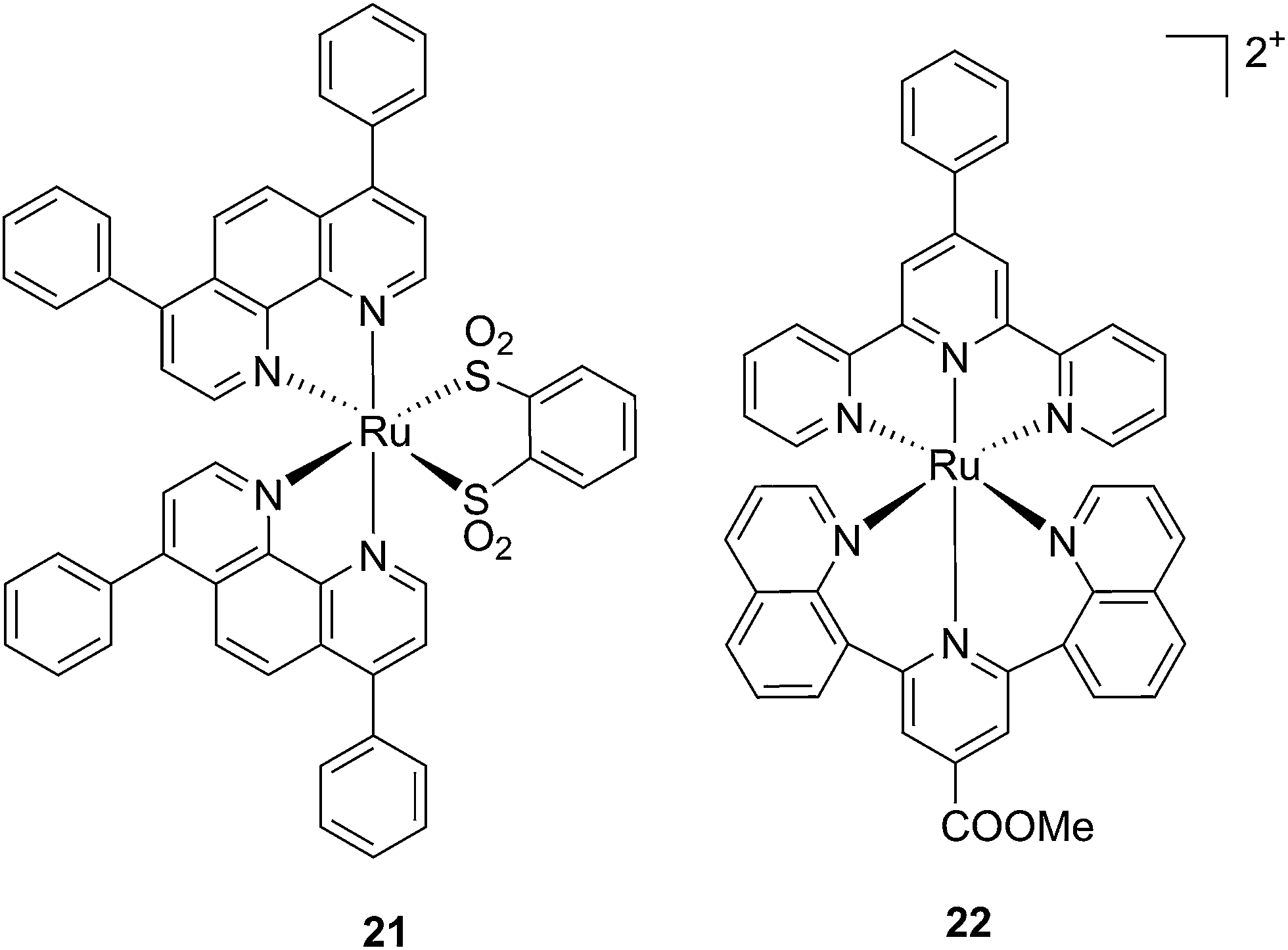 | ||
| Fig. 18 Structures of the ruthenium complexes 21 and 22 which have PDT and aPDT activity.60 | ||
Both compounds were characterized by moderate uptake by HeLa cells, as indicated by inductively coupled plasma mass spectrometry (ICP-MS) analysis performed after 4 h incubation. 21 accumulated preferentially in mitochondria (67% of the entire Ru uptake) as also confirmed by fluorescence confocal microscopy (Fig. 19). 22, on the other hand, was shown to target the nuclei, where 50% of the total Ru that entered cells was localized.
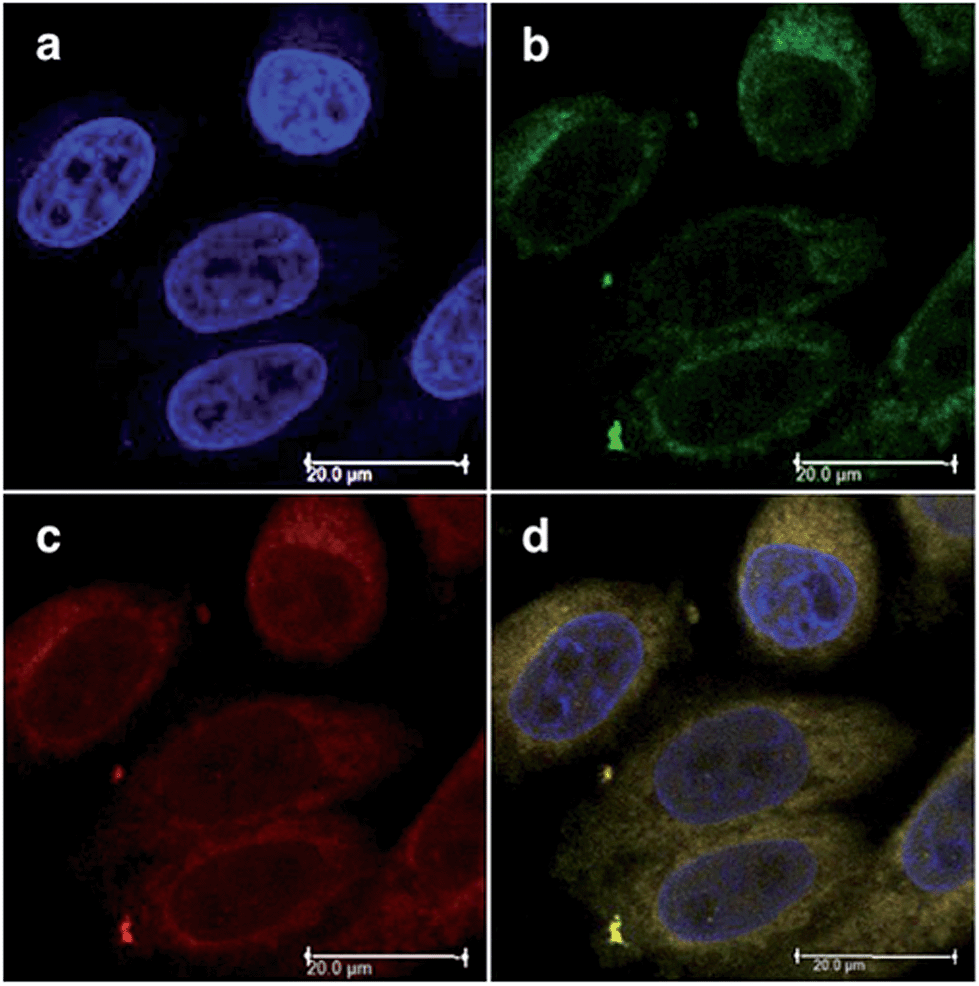 | ||
| Fig. 19 Fluorescence confocal microscopy images of HeLa cells incubated with 40 μM of 21 for 4 h: (a) DAPI staining, (b) Mitotracker green FM staining, (c) visualization of 21 by excitation at 405 nm, (d) overlay of a–c. Reprinted with permission from ref. 60. Copyright 2014 American Chemical Society. | ||
Phototoxicity was evaluated on HeLa cells. 21 was found to be most active upon irradiation at 420 nm with 6.95 J cm−2. Its PI was equal to 80, with an IC50 of 620 nM upon light irradiation. It is important to notice that although the uptake of 21 was not as high as those reported for similar complexes, the amount of compound present in cells was sufficient to produce a strong phototoxic effect. On the contrary, 22 displayed a lower phototoxicity against HeLa with an IC50 of 25.3 μM under the same irradiation conditions. Of utmost interest, the compounds were also evaluated for their potential activity as PSs in antibacterial PDT (aPDT). The use of PDT to kill bacteria was recently exploited to overcome the problematic occurrence of resistance to available antibiotics. This is essentially due to the fact that a resistance mechanism is far more difficult to develop for bacteria since PDT does not have a specific target but can affect the entire cell. The antibacterial activities of 21 and 22 were tested on the Gram-(−) Staphylococcus aureus and on the Gram-(+) Escherichia coli. Surprisingly, 22 was active against both strains, with a reduction of >6 log10 of the viability of the S. aureus and >4 log10 of that of E. coli at a concentration of 50 μM and with a dose of 8 J cm−2 of light at 420 nm. Under the same conditions, 21 displayed the same activity towards S. aureus, while being completely non-toxic towards E. coli. The very good performance of 22 is particularly promising considering that it is reported that normally Gram-(−) bacteria are less sensitive to PDT treatment.
In the last few years, Glazer and coworkers have thoroughly investigated the application of Ru polypyridyl complexes as PACT agents (see also PACT section). However, they also recently performed an in-depth biological characterization of two potential PDT agents. In particular, they evaluated [Ru(Ph2phen)3]2+ (23) and [Ru(Ph2phen-SO3)3]4− (24) (Fig. 20), which are known dyes for solar cells or biological staining, but which were never investigated as PDT agents.61 The two compounds have very similar structures but extremely different physical properties, mainly due to their different charges, namely +2 for 23 and −4 for 24. This, along with the subsequent difference in hydrophilicity of the two molecules, was expected to induce distinct cellular responses. Nonetheless, both molecules were found to be able to produce singlet oxygen when photo-irradiated.
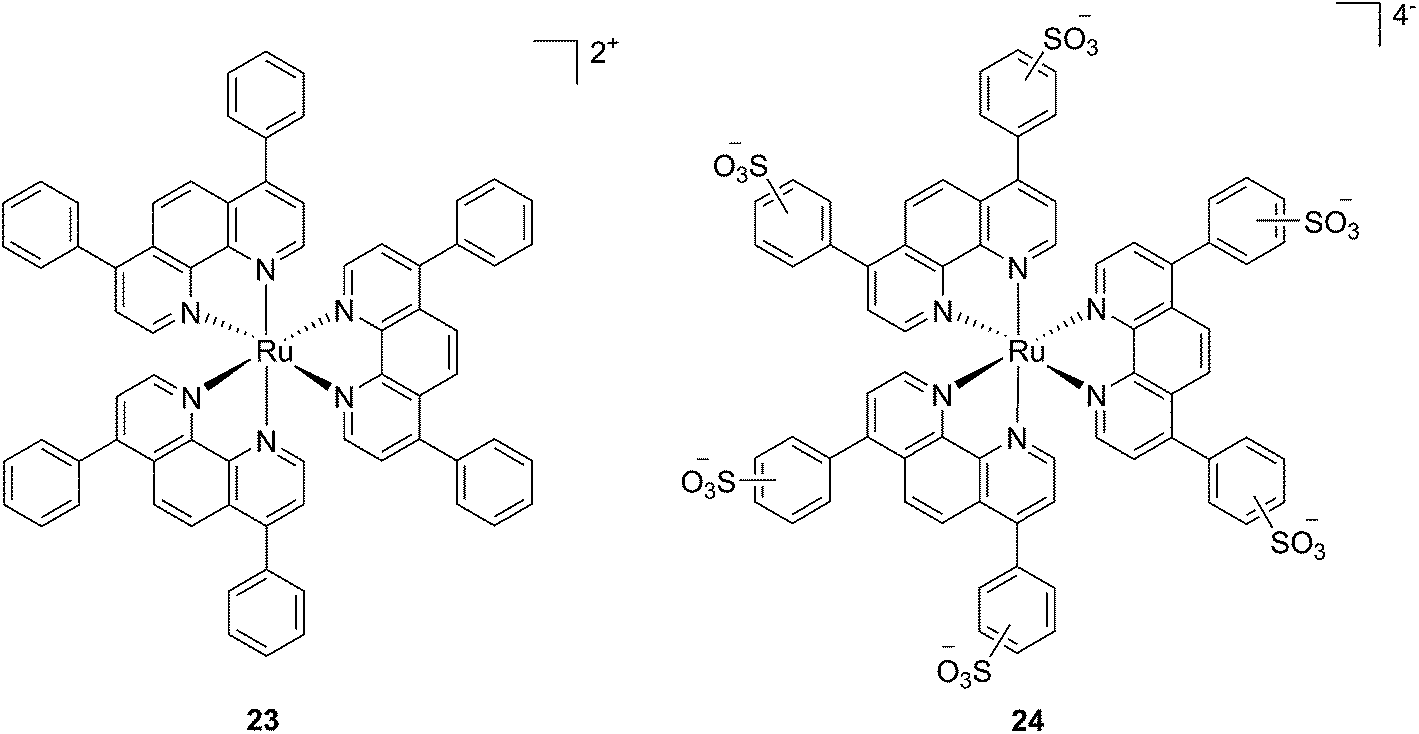 | ||
| Fig. 20 Structures of the Ph2phen complexes 23 and 24 with different charges investigated by Glazer and co-workers.61 | ||
Toxicity experiments were performed on three different cell lines (A549 human non-small lung cancer cells, HL60 human promyelocytic leukemia cells and Jurkat human T lymphoblastoid cells) in the dark and upon irradiation with 7 J cm−2 of >400 nm light. 23 showed a very good cytotoxic effect on all cell lines studied. Irradiation brought a further increment in potency, with IC50 values ranging from 0.075 μM to 0.35 μM, depending on the cell lines employed. However, the PI was just around 10–20. Surprisingly, 24 appeared to be non-toxic in the dark (up to 300 μM) on all cell lines studied. Nevertheless, irradiation induced strong toxicity with IC50 values in the low micromolar range, resulting in a larger therapeutic window compared to 23. The compounds also displayed a different subcellular localization, with 23 accumulating in mitochondria and lysosomes and 24 displaying a non-specific accumulation in the cytoplasm (Fig. 21). Interestingly, mitochondrial uptake of 23 was proposed by the authors as the cause of toxicity in the dark. Upon light irradiation, 23 relocalized from mitochondria and lysosomes to the nucleus. This phenomenon was explained by the authors as the consequence of damage to the nuclear membrane induced by 23 upon light irradiation. On the other hand, when cells incubated with 24 were irradiated, the compound was mainly observed in lysosomes, suggesting that no damage occurred to the nuclear membrane in this case.
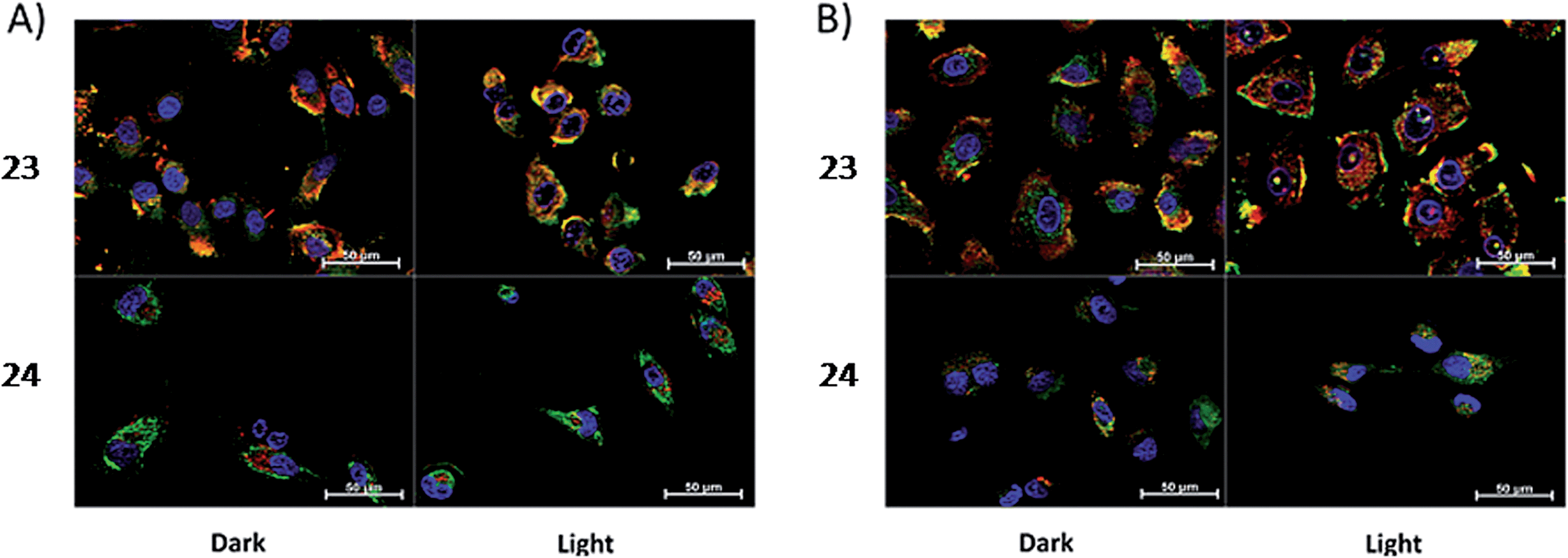 | ||
| Fig. 21 ApoTome microscopy showing subcellular localization of 23 and 24 at 8 h. Co-localization of 23 and 24 in mitochondria or lysosomes is indicated by the apparent yellow emission. (A) Mitotracker green FM was used to image mitochondria. (B) Lysotracker green DND-26 was used to image lysosomes. Red color denotes intrinsic emission of 23 and 24, whereas blue color denotes Hoechst staining of the nucleus. The yellow color results from overlap of the red emission from the ruthenium complexes and green emission of the organelle-specific dyes, indicating co-localization. Compound 23 localizes in both the mitochondria and the lysosomes, while 24 was not predominantly found in either organelle. Reprinted with permission from ref. 61. Copyright 2014 American Chemical Society. | ||
Investigation of the mechanism of cell death using distinct assays and read-outs revealed a role for light-induced apoptotic pathways in the case of 24. On the other hand, initial necrotic cell death in the dark, followed by a combination of necrotic and apoptotic pathways, was observed for 23 upon light irradiation.
While one of the main problems of PDT is its reliance on oxygen, which is often present at low concentrations in the tumor environment (hypoxic conditions), the application of metal complexes as PSs also has its drawbacks, which are due to the need for light at a high energy (blue or green) for the excitation of the PS. McFarland and co-workers addressed both issues by taking advantage of the possibility to fine tune the photophysical characteristics of coordination compounds. More specifically, by modifying the structures of the ligands coordinated to the metal centre, the authors developed Ru polypyridyl PSs characterized by a triplet intraligand (3IL) excited state with remarkably long lifetimes. Oxygen was reported to be able to quench this excited state even when present at very low concentrations (3.5%). Furthermore, the strong photosensitizing ability of this excited state allowed PDT effects to be achieved in the red and NIR regions where compounds have marginal absorptions (ε values in the order of 10 M−1 cm−1). The first series of compounds bearing a pyrenylethynyl moiety on the phenathroline ligand was strongly effective on the cell line Malme-3M, a malignant melanoma lung metastasis.62 Melanoma cells are able to grow at very low oxygen concentrations and have a remarkable ability to resist the outburst of ROS.63 Nevertheless, compound 25 (Fig. 22, left) could induce cell death in a melanoma cell line, with a toxicity increase of two orders of magnitude upon irradiation with white light at 7 J cm−2. In these conditions, EC50 went from 62 μM in the dark to 200 nM upon irradiation.
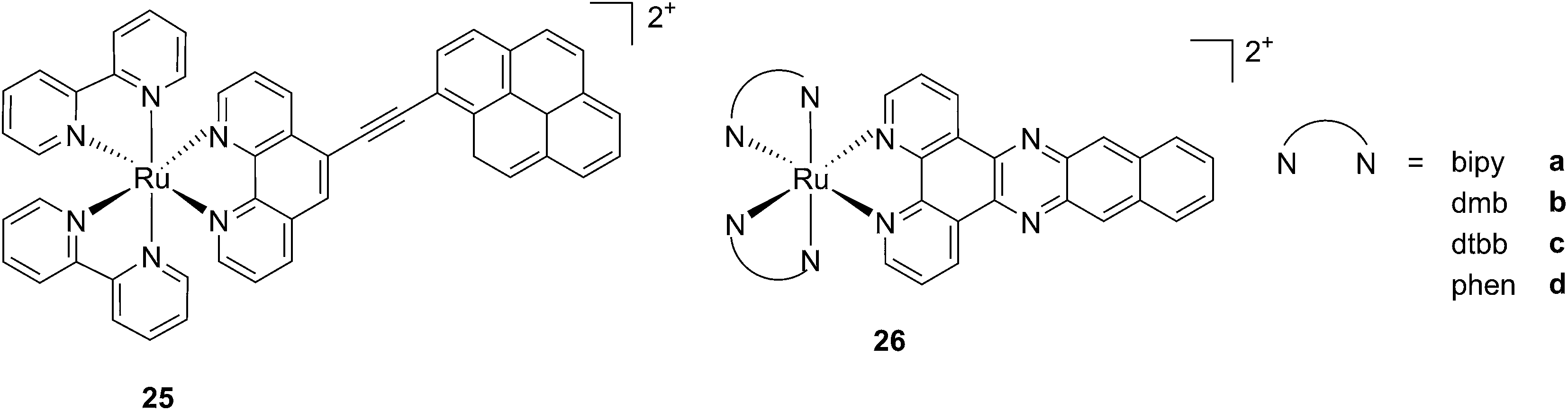 | ||
| Fig. 22 Structures of ruthenium complexes 25 and 26 studied by McFarland, characterized by 3IL excited states.62,64 | ||
A second class of compounds studied by the same group contained the extensively conjugated benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine ligand (dppn, Fig. 22, right).64 The authors could exploit the 3IL excited state of these compounds with very long lifetimes to obtain a remarkable PDT effect. Impressively, EC50 values in the low micromolar range were obtained upon irradiation with 100 J cm−2 light at 625 nm, where the compounds have marginal absorption. This efficacy demonstrated that it is possible to achieve good photoactivity with compounds that mainly absorb in the blue-green region of the light spectrum. Furthermore, the same authors developed a system where the Ru polypyridyl complexes are connected to polythiophene chains of variable lengths (Fig. 23). This conjugation gave access to a low-lying 3IL excited state and to a strong non covalent DNA association.65 Gel electrophoresis experiments were performed on the complexes to elucidate the interaction with plasmid DNA. These analyses suggested that compounds bearing more than one thiophene unit are able to induce light-mediated damages to plasmid DNA via an oxygen-independent pathway. This was indicated by the fact that compound 27c was still able to induce single strand breaks when the experiment was performed under argon atmosphere. Therefore, the authors speculated that these thiophene conjugates could act via photoinitiated Type II reaction in the case of high oxygen tension. On the contrary, under low oxygen concentration, the compounds induced damage to DNA via a Type I pathway. This behavior was already observed for this class of compounds in the photoinactivation of bacteria.66 A PS with the ability to act via a dual Type I/II photosensitization could allow for the treatment of hypoxic tissues, broadening the spectra of applicability of PDT.
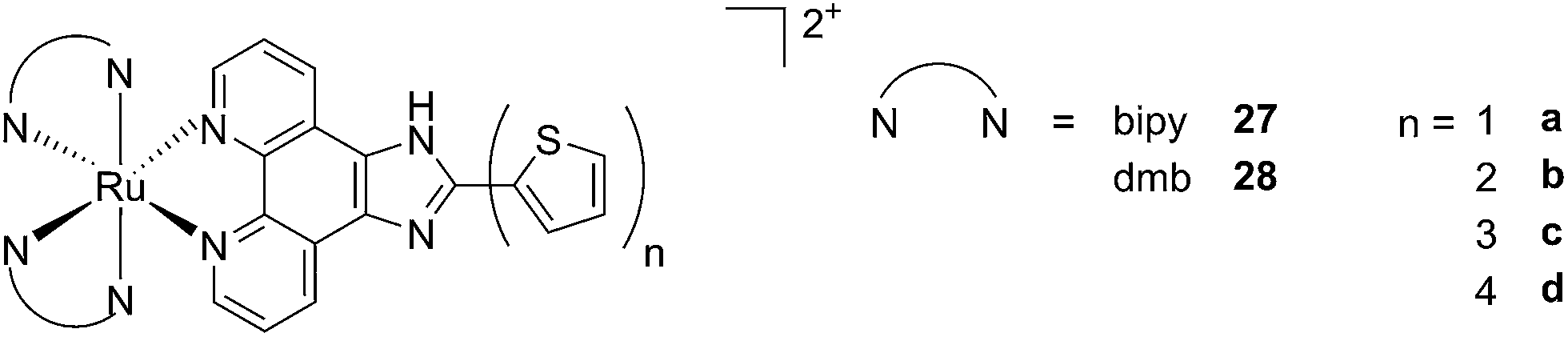 | ||
| Fig. 23 Structures of Ru polypyridyl complexes conjugated with different polythiophene moieties to achieve dual Type I/II photosensitization.65 | ||
The in vitro PDT effect of these compounds was found to be directly proportional to the polythiophene chain length, with a PI of > 200 when 4 thiophene units were present in the complex. The two best compounds 27c and 28c, bearing three thiophene units, were also tested in vivo on mice, which were inoculated with colon carcinoma cells (CT26.WT). In animals treated with compound 28c (53 mg kg−1), administration of 525 nm continuous wave light (192 J cm−2) resulted in complete tumor regression, with no recurrence up to 52 days after the end of the treatment. These compounds are currently under optimization for clinical phase I trials.65
A very elegant approach to effectively increase the selectivity of PDT treatment is the so-called CALI (chromophore-assisted light inactivation). This technique is based on the functionalization of a modest protein inhibitor with a PS, allowing for strong enhancement of the inhibitory properties through photo-triggered 1O2 generation in close proximity to the active site (see the mechanism in Fig. 24). The Kodadek group recently explored this technique using a [Ru(bipy)3]2+ derivative,67 demonstrating the feasibility of this technique on both membrane and intracellular proteins.
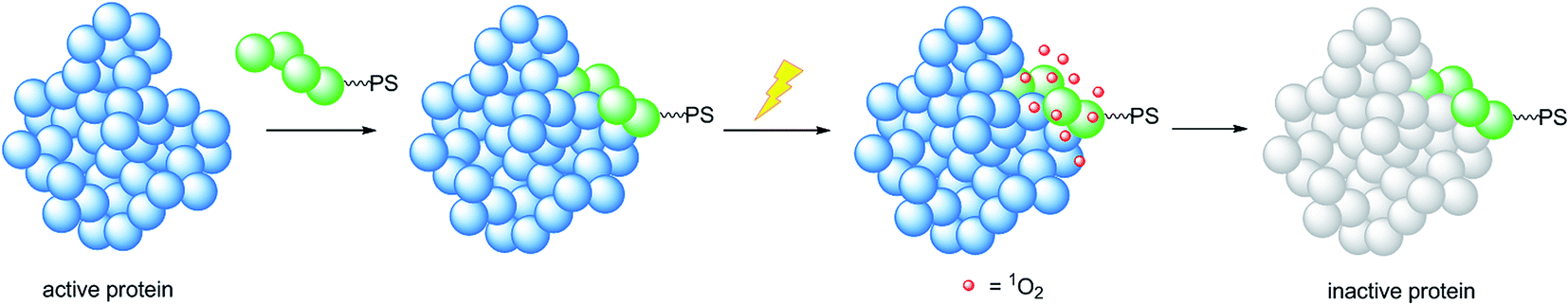 | ||
| Fig. 24 Mechanism of the CALI strategy to inhibit enzymes, applied by the Kodadek group. | ||
Furthermore, the authors showed selective inhibition of RBBP9 serine hydrolase, which is implicated in pancreatic cancer, in protein-enriched cell lysate.68 The limitation of this approach is related to the choice of PS, since [Ru(bipy)3]2+ derivatives do not allow efficient photosensitization due to the short wavelengths required for excitation. A careful optimization of the system could provide a very useful tool for future targeted PDT applications.
Ruthenium complexes in PACT
As mentioned above, PDT relies on the presence of oxygen to induce cell death. However, most tumors are hypoxic in their internal core,69 limiting the efficacy of PDT. Hence, increasing efforts are devoted to the optimization of novel photo-activation strategies that do not rely on an oxygen-dependent mechanism, but which would still allow for spatial and temporal control of the toxicity engendered to cells. Strategies of this type are normally referred to as photoactivated chemotherapy. In this section of the review, we describe the recent efforts in the use of ruthenium complexes for PACT. We have divided this section into two main parts depending on the photo-activation strategy employed. In the first part, we will focus on ruthenium-based DNA photobinders acting (1) in a cisplatin-like mode of action resulting in DNA helix distortion; (2) via intercalation yielding DNA cleavage; and (3) via conjugated oligodeoxyribonucleotides (ODNs) to allow for gene silencing. In the second part, we will discuss photo-activated release approaches involving Ru(II) complexes. In this part, we will first introduce the use of Ru(II) complexes as caging agents for the selective release of bioactive molecules upon light activation. We will then present a parallel approach consisting of the photorelease of cytotoxic Ru(II) complexes rendered inactive upon caging.Photo-activated Ru complexes targeting DNA
Cancer cells differ from their original healthy precursor cells by their ability, inter alia, to continuously proliferate.70 This feature, conferred by mutations in tumor suppressor genes or by the altered expression/activity of proto-oncogenes, implies continuous activation of DNA replication, which is not the case in healthy cells, which rather display the ability to enter quiescence after a certain number of cell divisions. This hallmark of cancer has been extensively exploited to selectively target cancer cells by means of chemotherapeutic drugs, inhibiting components of the DNA replication/transcription machineries, such as topoisomerase I (e.g. camptothecin)71 or covalently binding to DNA (e.g. cisplatin)72. In this section, we will introduce photo-triggered strategies designed to target DNA.In contrast to square planar Pt(II) compounds, Ru(II) complexes offer octahedral conformations. It was shown that complexes with distorted octahedral geometry can undergo ligand dissociation after photo-irradiation,82,83 which is followed by the formation of an aqua complex that can bind to DNA in a manner similar to cisplatin. To exploit this concept, Glazer and coworkers recently investigated the potential use of methyl substituents on one polypyridyl ligand to obtain highly distorted geometries.84 To this end, an unstrained [Ru(bipy)2phen]2+ (29) and two methylated derivatives of [Ru(bipy)2(2,2′-bypiridyl)]2+ (30) and [Ru(bipy)2(dipirido[3,2-f:2′,3′-h]-quinoxaline)]2+ (31) were synthesized (see Fig. 25 for structures). As expected, after >450 nm light irradiation using a 200 W projector, the authors could monitor the photoejection of the methylated ligand of 30 and 31, with half-lives (t1/2) of 2 and 60 min, respectively. Since photoejection of the latter ligand resulted in the formation of a similar aqua species to cisplatin, the authors naturally explored the activity of the photo-product on biologically relevant molecules. In the presence of plasmid DNA pUC19, only irradiated (>450 nm, 200 W, 1 h) products showed DNA damage. Complex 29 produced DNA photo-cleavage, 30 showed only DNA photobinding whilst 31 combined both properties. To verify if the DNA damage observed in vitro would reflect in decreased viability in cancer cells in culture, the authors treated HL60 leukemia and A549 lung cancer cells for 12 h with the complexes in the dark prior to >450 nm irradiation for 3 min at 410 W, followed by a further 72 h of incubation (see Table 1 for complete IC50 values). As a control, they used aminolevulinic acid (ALA) which is a clinically available PS. Complexes 30 and 31 showed no toxicity in the dark with IC50 values of > 100 μM. However, a strong effect after irradiation on both cells with IC50 on HL60 cells of 1.6 and 2.6 μM, respectively, and of 1.1 and 1.2 μM, respectively, on A549 cells, was observed. In order to efficiently mimic the three-dimensional tumor environment, the authors also assessed the photo-toxicity of the compounds on A549 spheroids. As reported for the monolayer cell culture, complex 30 was confirmed to be efficient with an IC50 of 21 μM upon light irradiation, a value that corresponds to twice the potency of cisplatin on the same spheroids. Worthy of note, glutathione (GSH), responsible for cisplatin inhibition in cells, had no deleterious effect on DNA binding or cleavage efficiency nor on the toxicity of the ruthenium complexes 30 and 31.
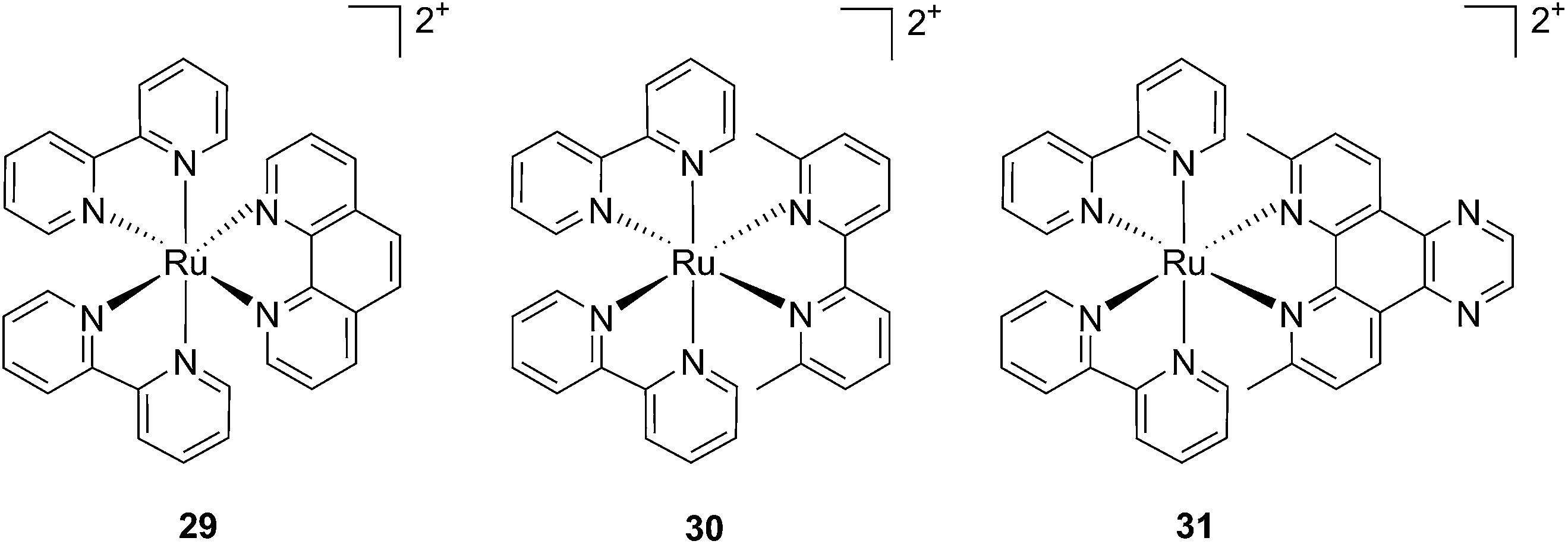 | ||
| Fig. 25 Structures of the strained Ru complexes 30 and 31 that undergo ligand photoejection and the inert control 29.84 | ||
| IC50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | Light | Dark | PI | |||||
| HL60 | A549 | A549 spheroid | HL60 | A549 | A549 spheroid | HL60 | A549 | |
| a n. d. = not determined. ALA = aminolevulinic acid. | ||||||||
| Cisplatin | 3.1 ± 0.2 | 3.4 ± 0.6 | n. d. | 3.1 ±0.1 | 3.5 ± 0.6 | 42 ± 3.6 | 1 | 1 |
| 29 | 8.1 ± 1.9 | 40 ± 4 | >300 | 240 ± 9 | 250 ± 5 | >300 | 3 | 6.3 |
| 30 | 1.6 ± 0.2 | 1.1 ± 0.3 | 21.3 ± 2.3 | >300 | 150 ± 7 | >300 | >188 | 136 |
| 31 | 2.6 ± 1.0 | 1.2 ± 0.1 | 64.6 ± 4.7 | 108 ± 1.9 | 250 ± 5 | >300 | 42 | 208 |
| ALA | 16.2 ± 3.2 | 21 ± 3.5 | >300 | >300 | 87.8 ± 5.5 | >300 | >18 | 4.2 |
More recently, Glazer et al. applied the same methylation strategy to a novel strained Ru(bipy)2 complex bearing a 2,3-dihydro-1,4-dioxino[2,3-f]-1,10-phenanthroline (dop) (32) ligand.85 The methylated form of 32 is 2,3-dihydro-1,4-dioxino[2,3-f]-2,9-dimethyl-1,10-phenanthroline (dmdop) (33, Fig. 26). To further increase the straining, the authors also synthesized a [Ru(dmphen)2(dop)]2+ (dmphen = 2,9-dimethyl-1,10-phenanthroline) (34, Fig. 26). As anticipated by the authors, after irradiation at >400 nm with a 200 W projector at a distance of 12 inches from the cuvette, both methylated analogs showed photoejection. The process was found to be 10-fold faster for complex 34 (t1/2 = 4 min) than for 33 (t1/2 = 42 min). The authors further analyzed photo-induced DNA damage. Upon the same irradiation settings, complex 32 created single strand breaks (SSBs) in pUC19, likely via1O2 production. In comparison, complex 34 showed covalent binding while 33 showed a combination of both mechanisms. Regarding cell photo-toxicity, complex 34 exerted the highest toxicity against leukemia cells HL60 with a PI of >1880. The IC50 was 300 μM in the dark while the value was 0.16 μM after 12 h incubation, 3 minutes irradiation at >400 nm with 410 W projector and 72 h recovery. This impressive PI was explained by the fact that 34 binds and distorts DNA, whereas the mechanism of action of complex 33 is characterized by a dual mode of action including SSBs formation via1O2 production and DNA distortion, which possibly lowers its distortion efficiency.
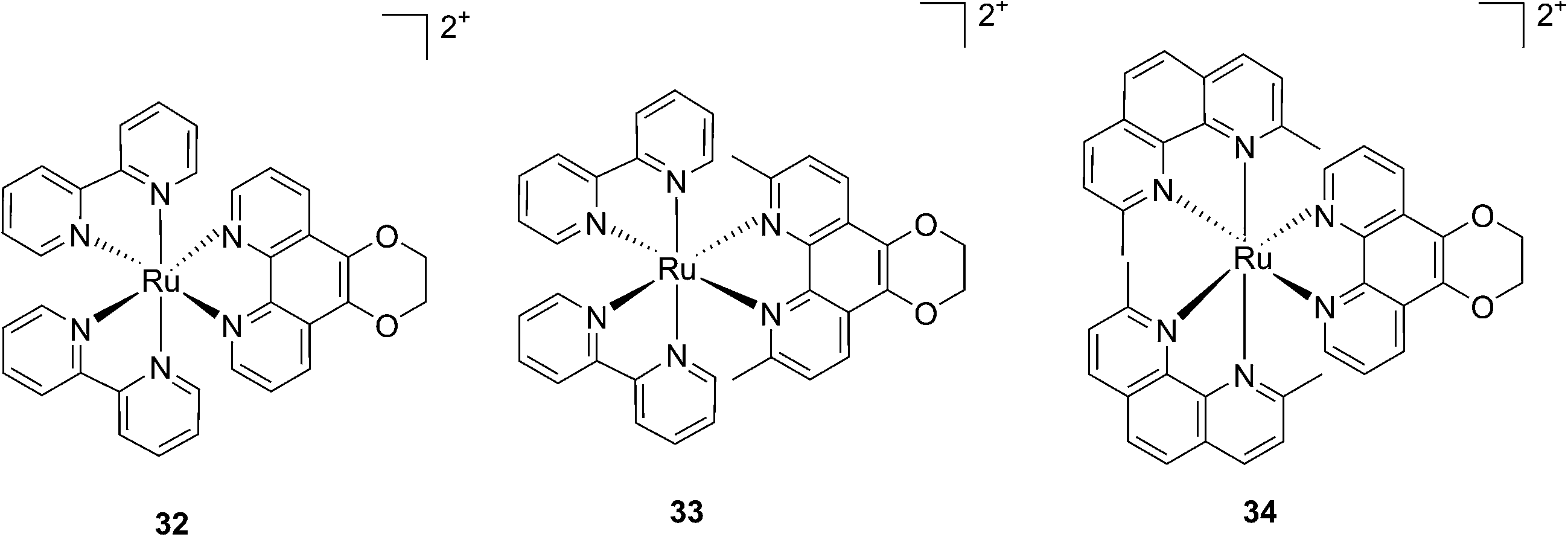 | ||
| Fig. 26 Structures of the photostable control compound 32 and the strained 33 and 34 that undergo ligand photoejection.85 | ||
Following a similar distortion strategy, the same group studied the coordination of biquinoline (biq) to ruthenium-based complexes. Such a ligand would act as a potent geometry distorter, and at the same time, improve the light activation process by pushing the absorption maximum to higher wavelengths. Indeed, the resulting strained Ru(II) 2,2′-biquinoline complexes (see Fig. 27 for structures) were shown to be active in the PDT therapeutic window. 35, which bears one biq, and 36, which contains two biq ligands, can absorb light up to 700 and 800 nm, respectively.86 Both complexes induced decreased electrophoretic migration of the DNA plasmid pUC19 only upon illumination (samples were placed at 12 inches from a 200 W lamp equipped with either blue, green, red or near-IR cut-off filters and irradiated for 1 h or 3 h). Since the appearance of open circular or linear DNA was not observed, the mechanism involved clearly coincides with photobinding. Maximal activity has been observed with blue light irradiation, which is consistent with the absorption profile of the Ru(II) complex. Cytotoxicity against HL60 leukemia cells followed the same light-dose/-wavelength profile as DNA photo-cleavage. After 12 h incubation followed by 7 J cm−2 light irradiation and 72 h recovery, a cell viability assay revealed that complex 36 had the best photo-toxicity profile with IC50 values of between 2.3 and 5.1 μM among the different wavelengths used, compared to 47.3 μM in the dark (see Table 2 for IC50 values). The interesting PIs (blue: 19.7, IR: 9.2) obtained for complex 36 hold great promise for this type of Ru(II) complex, which can be activated with red light or even with near-IR wavelengths. Interestingly, a complex similar to compound 36 but bearing a 2-phenylpyridine (phpy) instead of the phen, had different photophysical properties. Indeed, Dunbar, Turro and coworkers found that the latter complex had enhanced phototoxicity against HeLa cells.87 Nevertheless, its mode of action remains unclear, since no ligand dissociation was observed. Moreover, the short lifetime of the compound rules out any singlet oxygen mechanism.88
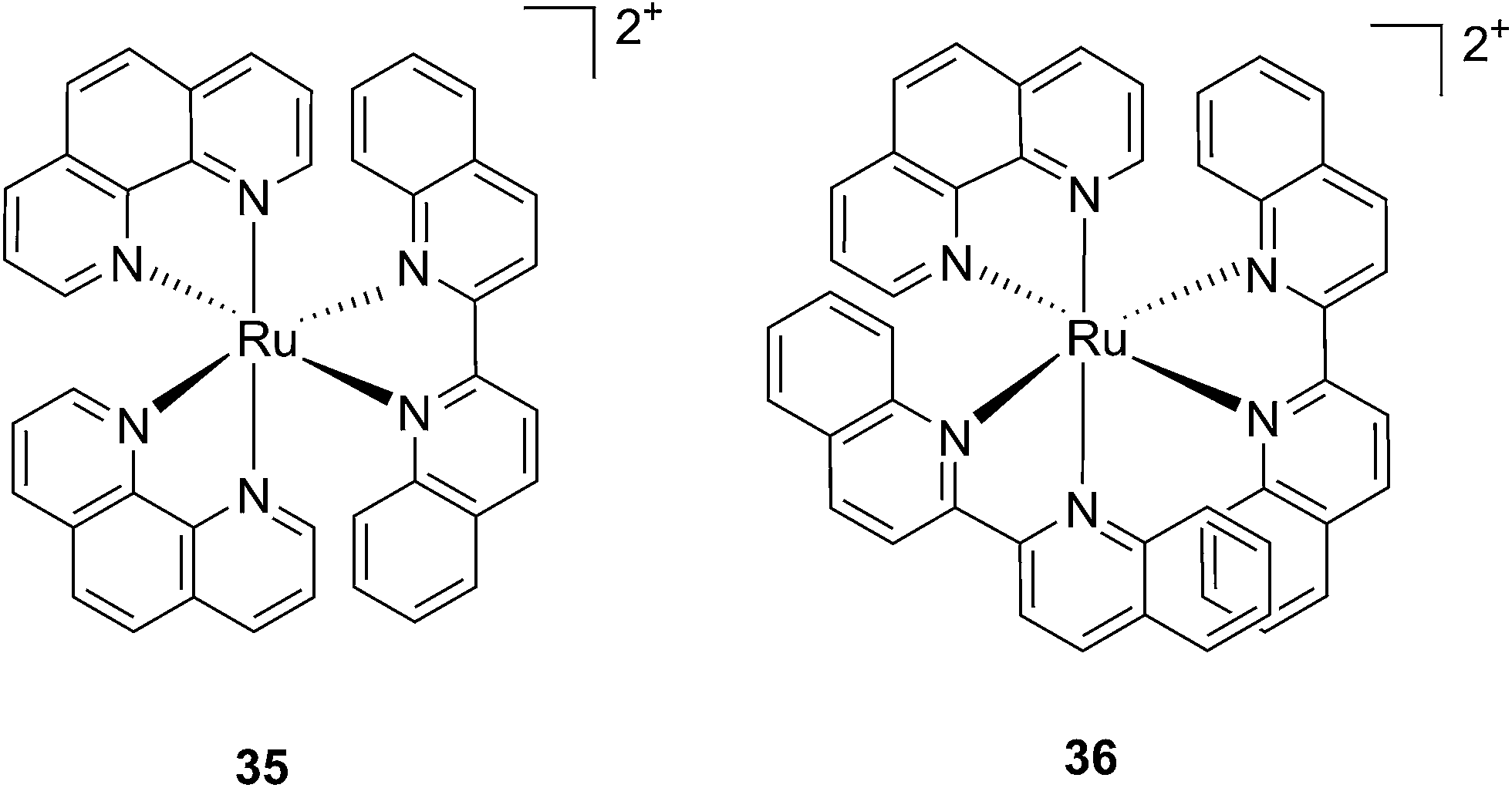 | ||
| Fig. 27 Structures of the two Ru(II) complexes 35 and 36 with the biq ligands that are exchanged upon irradiation.86 | ||
| IC50 (μM) | PI | ||||||
|---|---|---|---|---|---|---|---|
| Compounds | Dark | Blue (3 min) | Red (3 min) | Red (6 min) | IR (25 min) | Blue | IR |
| a n. d.= not determined. | |||||||
| 35 | 52.5 | 1.2 | 13.8 | 7.6 | 15.8 | 43.8 | 3.32 |
| 36 | 47.3 | 2.4 | 4.5 | 2.3 | 5.1 | 19.2 | 9.2 |
| Cisplatin | 3.1 | 3.1 | n. d. | n. d. | n. d. | 1 | |
Another manner to shift the MLCT absorption of a Ru(II) complex to the red (PDT window) is by insertion of a cyclometallating ligand.89 For this purpose, Turro and coworkers focused on the ligand phpy. More specifically, they investigated the photo-induced ligand release of cis-[Ru(phpy)(phen)(CH3CN)2]2+ (37, Fig. 28), a complex known to decrease tumor growth in mice,90 but whose photo-induced ligand release potential had never been evaluated. First, they observed that 3 min of irradiation at 690 nm were sufficient to eject one CH3CN, while 30 min were needed to release the second acetonitrile ligand. They could also observe an enhancement of cytotoxicity upon light irradiation (100 s irradiation at 690 nm, 5 J cm−2). As expected, the compound displayed a potent cytotoxicity on human advanced ovarian epithelial cancer cells (OVCAR-5) in the dark, with an IC50 of 1 μM (15 h incubation followed by rinsing and then by 24 h recovery). Upon light irradiation, the IC50 reached 70 nM, a 14-fold increase compared to dark conditions. According to agarose gel shift assay, this increase in toxicity upon light illumination is due to photobinding of the complex to DNA. Of note, the authors could demonstrate that GSH enhanced the photo-dissociation process, but still further analyses are needed to fully understand its role.91
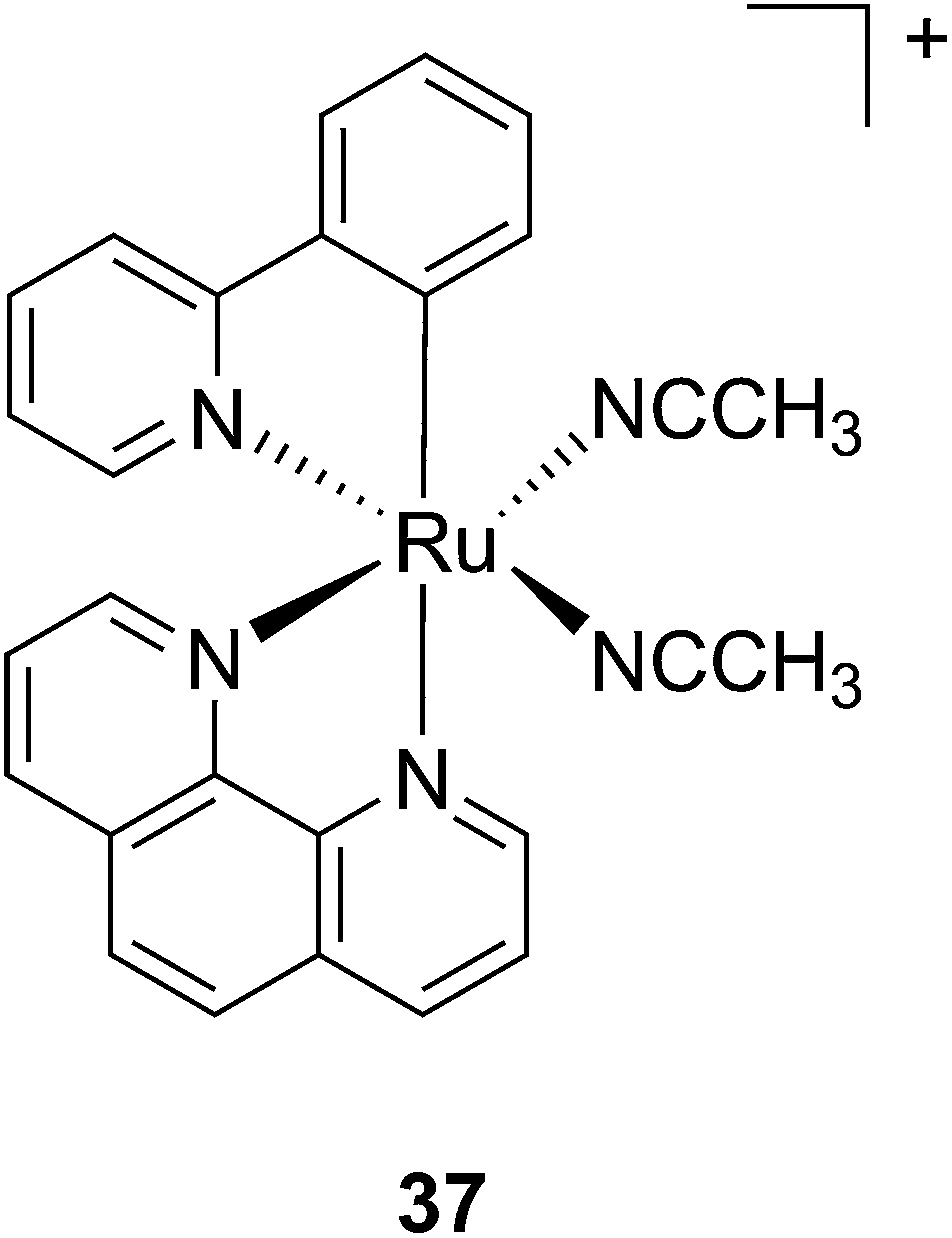 | ||
| Fig. 28 Structure of the Ru(II) complex 37 with labile CH3CN ligand.91 | ||
Although light-activated prodrugs offer an already high temporal and spatial selectivity per se, they still suffer from the recurrent problem of photosensitivity. Despite the fact that Ru complexes may be mimicking Fe uptake and thereby accumulate in cancer cells overexpressing transferrin,92,93 and assuming an intravenous administration, the light-activatable prodrugs presented above are supposed to be transported everywhere in the body without special selectivity for cancer cells. This implies that surrounding healthy tissues are not free from deleterious effects. To tackle this important drawback, several groups have envisaged coupling a targeting moiety to light-activatable prodrugs. In this perspective, Marchán et al. coupled two different receptor-binding peptides, which are known to target receptors overexpressed on the membrane of some cancer cells, to a photoactivated Ru(II) arene complex (Fig. 29).94 Since tumor endothelial cells overexpress two types of integrins, ανβ3 and ανβ5, the authors attached the specific targeting peptide sequence Arg-Gly-Asp (RGD). On the other hand, they decided to target the somatostatin receptor sst2, which is located at the membranes of malignant cells in supernumerary copies, using the peptide octreotide, a somatostatin agonist. The authors reported interesting and promising in vitro studies. First, they showed that conjugation to the peptides did not affect the photo-activation process, since the reactive aqua species was formed after pyridine ligand loss upon visible light irradiation (420 nm lamps). Second, they reported that DNA binding of the Ru-conjugates was not compromised by the presence of the peptides. When pre-irradiated (8 h) peptide-conjugated Ru(II) complexes 38-octreotide or 38-RGD were incubated overnight with the 9-ethylguanine, monofunctional adducts were formed. The same was true for the incubation with a short oligonucleotide sequence (5′dCATGGCT), as shown in Fig. 29, left branch. The formation of monofunctional adducts 41a and 41b was due to the release of the pyridyl ligand 39-octreotide or 39-RGD (depending on the compound used) upon irradiation, followed by formation of the aqua species [Ru(η6-p-cymene)(bpm)(H2O)]2+ (bpm = 2,2′-bipyrimidine) 40. This intermediate then reacted with one guanine present in the sequence, yielding the isomers 41a and 41b. On the other hand, when the solution containing the Ru–peptide conjugates and the oligonucleotide was intensively irradiated (9 J cm−2, 9 h irradiation), the bifunctional adduct 41c appeared, due to consequent arene release (see Fig. 29, right branch). Encouragingly, the authors demonstrated the specificity of the ruthenium complex for guanine over other potential biological ligands present in octreotide such as histidine or methionine, since no interactions between the ruthenium complex and these amino acids were observed. Nevertheless, these promising results and targeted strategy still need to be verified in cell-based assays.
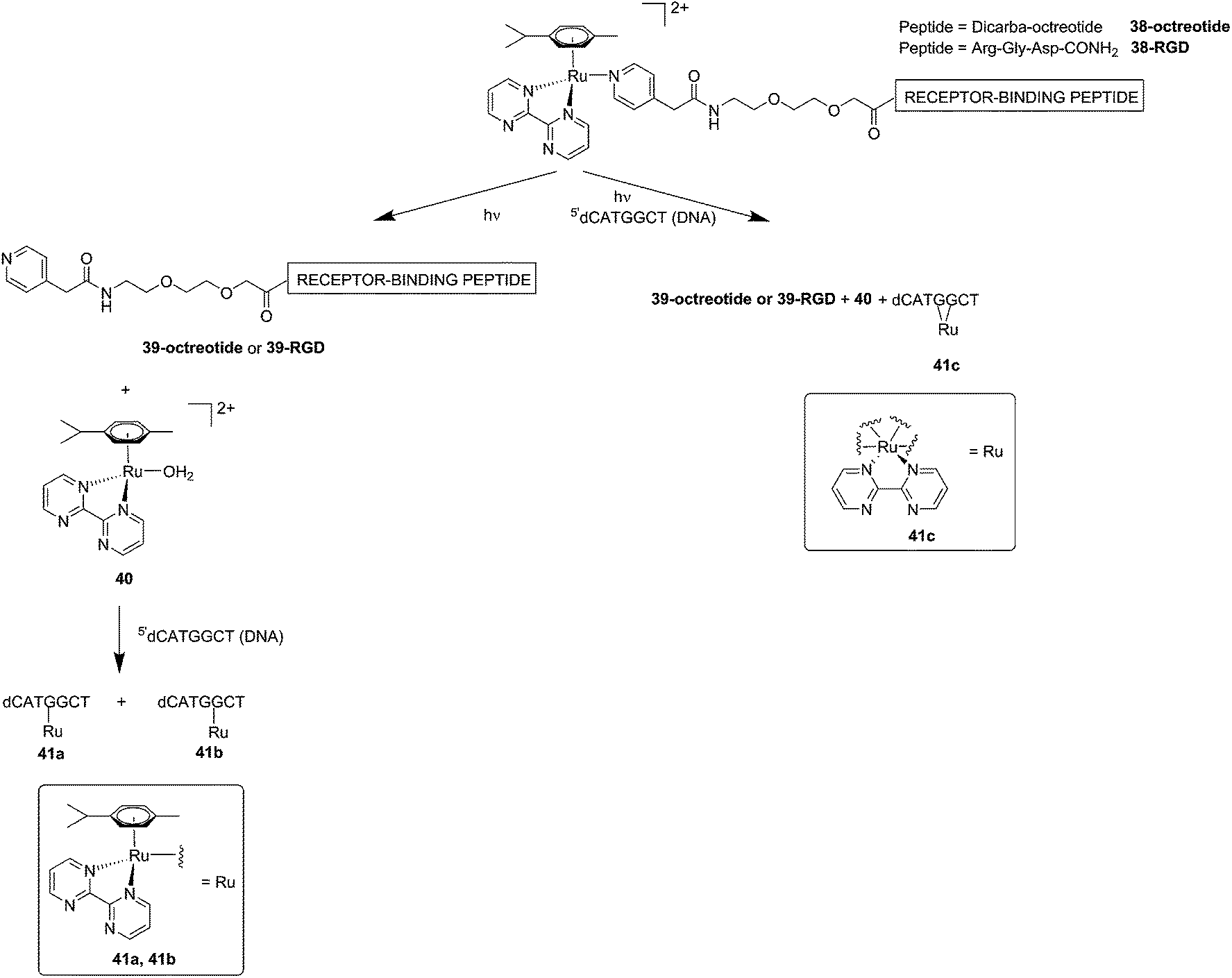 | ||
| Fig. 29 Schematic representation of the photo-dissociation process. Left branch: pre-irradiated Ru–peptide conjugates followed by addition of the oligonucleotide. Right branch: irradiation of a mixture of peptide-conjugated Ru(II) complex and the oligonucleotide.94 | ||
 | ||
| Fig. 30 Structures of [Ru(TAP)2PHEHAT]2+42 studied for DNA intercalation and nicking by de Feyter and [Ru(η6-p-cymene)(dpb)(py)]2+43 synthesized by Wang, characterized by high wavelength absorption and ligand release.95,96 | ||
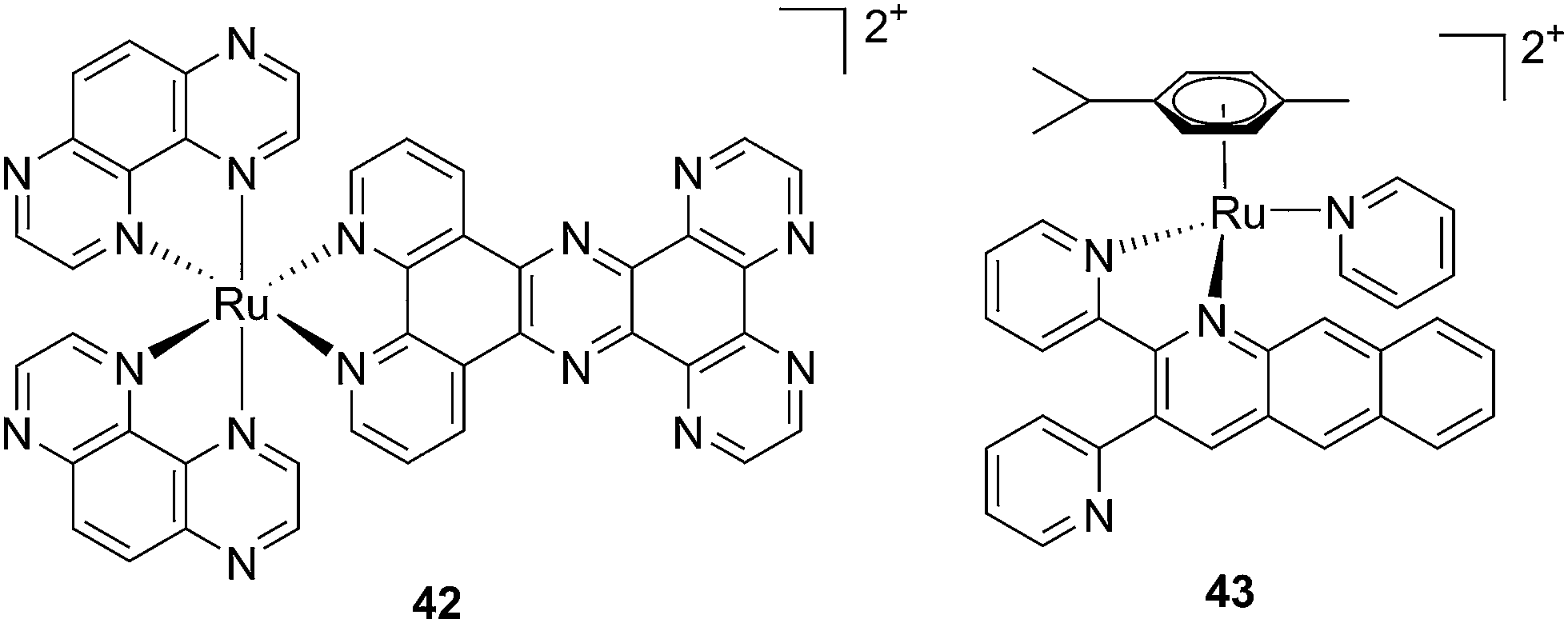
Recently, Wang et al. succeeded to shift the absorption of a Ru complex to longer wavelengths in order to obtain ligand photorelease in the PDT window. At the same time, the authors achieved an increase of the lifetime of the excited state, which facilitates 1O2 production, thus combining PDT and PACT mechanisms. The authors undertook the latter by introduction of a 2,3-bis(2-pyridyl)-benzoquinoxaline (dpb) ligand into the structure of the Ru(II) complex, whose delocalized π system was able to shift 1MLCT absorption to lower energy.96 The authors observed the formation of several photoproducts when [Ru(η6-p-cymene)(dpb)(py)]2+ (py = pyridine) (43) (Fig. 30) was irradiated with visible light (>400 nm) with the moieties (py) and (dpb) released in a 3.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Moreover, the authors showed that the complex was a modest 1O2 generator with a 1O2 quantum yield of 0.25 in CH3CN. Further investigation of the direct effects on DNA revealed the photo-binding and photo-cleaving ability of the complex. These behaviors were subsequently examined in cell-based assays. After 4 h incubation in A549 cells, followed by 1 h irradiation at >400 nm and an additional incubation of 20 h in the dark, the complex showed enhanced toxicity after light irradiation, although with a moderate PI of about 7 (IC50 in the dark: 27.6 μM, upon light exposure: 4.0 μM). Brewer and co-workers also aimed to tune the absorption of the Ru(II) complexes to lower energy wavelengths and to not be, at the same time, dependent on the presence of oxygen to achieve cell killing. To this end, they designed a mixed-metal supramolecular complex [{(bipy)2Ru(dpp)}2RhCl2]5+ (44, Fig. 31) containing two Ru(II) centers to absorb visible light and one Rh(III) atom.97 Complexes containing Rh(III) have previously been shown to photo-cleave DNA.98 By agarose gel shift assays, the authors were able to characterize the structural requirements for the photo-cleavage process. First, they could show that the presence of rhodium in the molecule is needed for the DNA cleavage to occur. Indeed, when DNA was irradiated for 10 min at >475 nm, no cleavage was observed in the presence of the analog compounds lacking Rh(III), both in the presence and absence of oxygen. Second, they could demonstrate that the process followed metal-to-metal charge transfer (MMCT) and not the ordinary MLCT process. To show this, the authors compared the DNA photo-cleavage efficiency of two analogs with inaccessible Rh(dσ*) and Ir(dσ*) orbitals, namely [{(bipy)2Ru(bpm)}2RhCl2]5+ and [{(bipy)2Ru(dpp)}2IrCl2]5+ (45 and 46 in Fig. 31). As expected, both analogs failed to cleave DNA. The same group then tested the photo-triggered impact of their Ru–Rh mixed-metal complex on African green monkey kidney epithelial cells (vero cells) replication.99 The authors demonstrated that when cells were pretreated with 44 and exposed to >460 nm light, limited growth was observed up to 12 μM (conditions: 48 h incubation with compounds, then removal of the medium and 4 min irradiation at >460 nm, followed by 48 h recovery). At higher concentrations, cell death was observed. Interestingly, cell death was not observed with cells pretreated with the osmium analog complex 47, shown in Fig. 31. This last observation highlighted the fundamental role of the Ru atoms in 44 in triggering cell death.
:1 ratio. Moreover, the authors showed that the complex was a modest 1O2 generator with a 1O2 quantum yield of 0.25 in CH3CN. Further investigation of the direct effects on DNA revealed the photo-binding and photo-cleaving ability of the complex. These behaviors were subsequently examined in cell-based assays. After 4 h incubation in A549 cells, followed by 1 h irradiation at >400 nm and an additional incubation of 20 h in the dark, the complex showed enhanced toxicity after light irradiation, although with a moderate PI of about 7 (IC50 in the dark: 27.6 μM, upon light exposure: 4.0 μM). Brewer and co-workers also aimed to tune the absorption of the Ru(II) complexes to lower energy wavelengths and to not be, at the same time, dependent on the presence of oxygen to achieve cell killing. To this end, they designed a mixed-metal supramolecular complex [{(bipy)2Ru(dpp)}2RhCl2]5+ (44, Fig. 31) containing two Ru(II) centers to absorb visible light and one Rh(III) atom.97 Complexes containing Rh(III) have previously been shown to photo-cleave DNA.98 By agarose gel shift assays, the authors were able to characterize the structural requirements for the photo-cleavage process. First, they could show that the presence of rhodium in the molecule is needed for the DNA cleavage to occur. Indeed, when DNA was irradiated for 10 min at >475 nm, no cleavage was observed in the presence of the analog compounds lacking Rh(III), both in the presence and absence of oxygen. Second, they could demonstrate that the process followed metal-to-metal charge transfer (MMCT) and not the ordinary MLCT process. To show this, the authors compared the DNA photo-cleavage efficiency of two analogs with inaccessible Rh(dσ*) and Ir(dσ*) orbitals, namely [{(bipy)2Ru(bpm)}2RhCl2]5+ and [{(bipy)2Ru(dpp)}2IrCl2]5+ (45 and 46 in Fig. 31). As expected, both analogs failed to cleave DNA. The same group then tested the photo-triggered impact of their Ru–Rh mixed-metal complex on African green monkey kidney epithelial cells (vero cells) replication.99 The authors demonstrated that when cells were pretreated with 44 and exposed to >460 nm light, limited growth was observed up to 12 μM (conditions: 48 h incubation with compounds, then removal of the medium and 4 min irradiation at >460 nm, followed by 48 h recovery). At higher concentrations, cell death was observed. Interestingly, cell death was not observed with cells pretreated with the osmium analog complex 47, shown in Fig. 31. This last observation highlighted the fundamental role of the Ru atoms in 44 in triggering cell death.
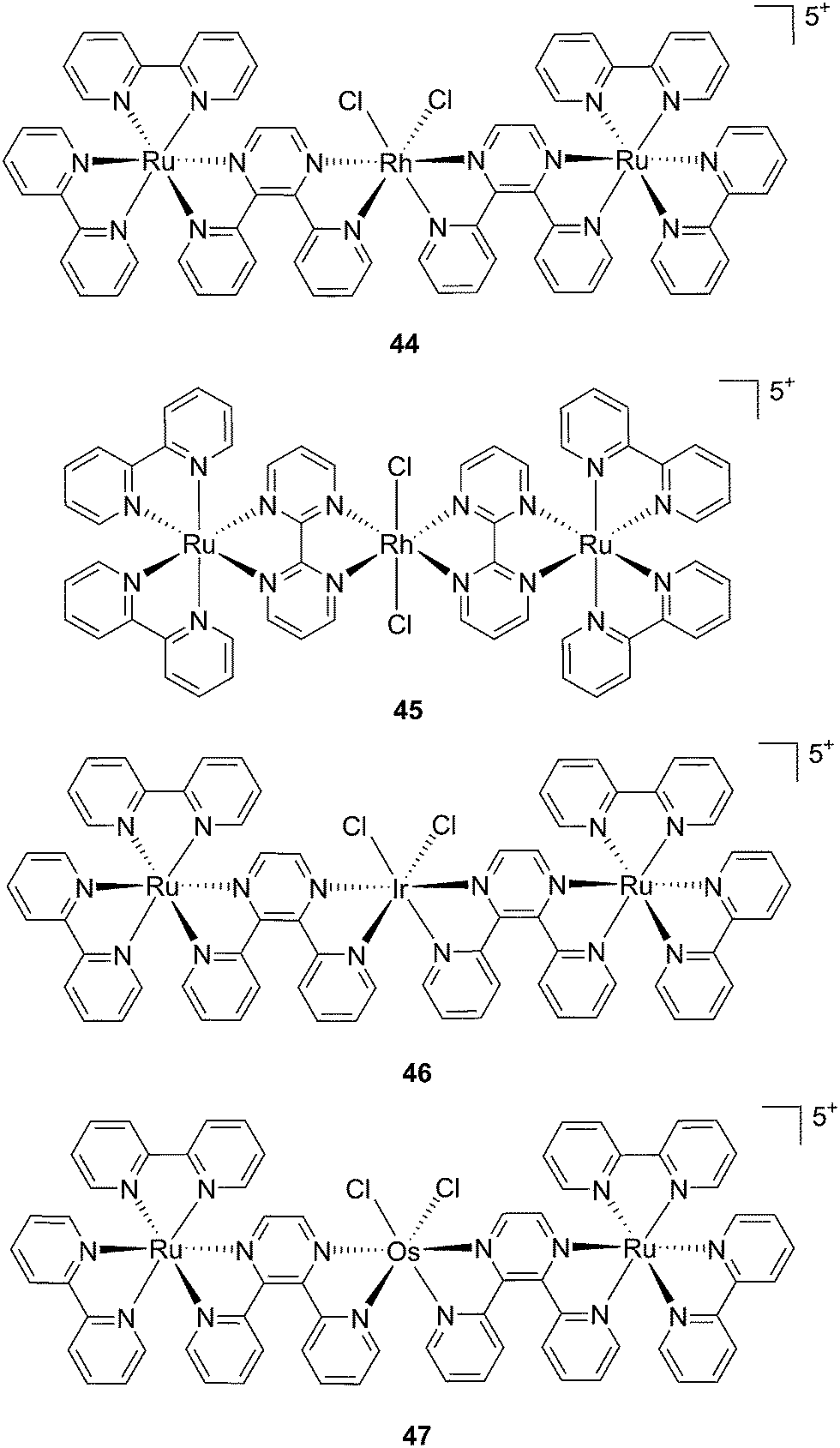 | ||
| Fig. 31 Structures of the mixed-metal supramolecular complexes; (44) [{(bipy)2Ru(dpp)}2RhCl2]5+, (45) [{(bipy)2Ru(bpm)}2RhCl2]5+, (46) [{(bipy)2Ru(dpp)}2IrCl2]5+, and (47) [{(bipy)2Ru(dpp)}2OsCl2]5+.97,99 | ||
Gene silencing (ODN strategy)
Another attractive approach to target DNA is the use of oligodeoxyribonucleotides (ODNs) to inhibit gene expression. ODNs can act on different kinds of targets, namely double-stranded DNA by triple helix formation (antigene strategy) or mRNA (antisense strategy). However, these strategies suffer from the low stability of ODNs, their ineffective delivery into cells and from the low affinity of the ODNs for the target sequence. To overcome these drawbacks, chemically modified ODNs have been investigated with different moieties, including ruthenium complexes. In the last few years, the group of Kirsch-De Mesmaeker focused its attention on the detection of nucleic acids using metal complexes.100 As an example, they employed highly photo-reactive Ru complexes to irreversibly crosslink ODNs to a DNA target sequence. A photoinduced electron transfer (PET) takes place between a Ru complex and the guanine present in close vicinity in the complementary DNA sequence. This results in covalent binding between the Ru complex and the guanine, forming a crosslink (see Fig. 32). The authors first examined the different geometric factors influencing adduct formation using [Ru(TAP)2dip]2+ (dip = 4,7-diphenylphenanthroline) (48, Fig. 33). They demonstrated that guanines at the 3′ side of the complementary strand, compared to the ruthenium complex anchoring position, are more favorable for the recombination of radicals formed by PET (irradiation settings: 1 h at 4 °C with a mercury/xenon lamp (200 W) using a filter (2M KNO3 solution)).101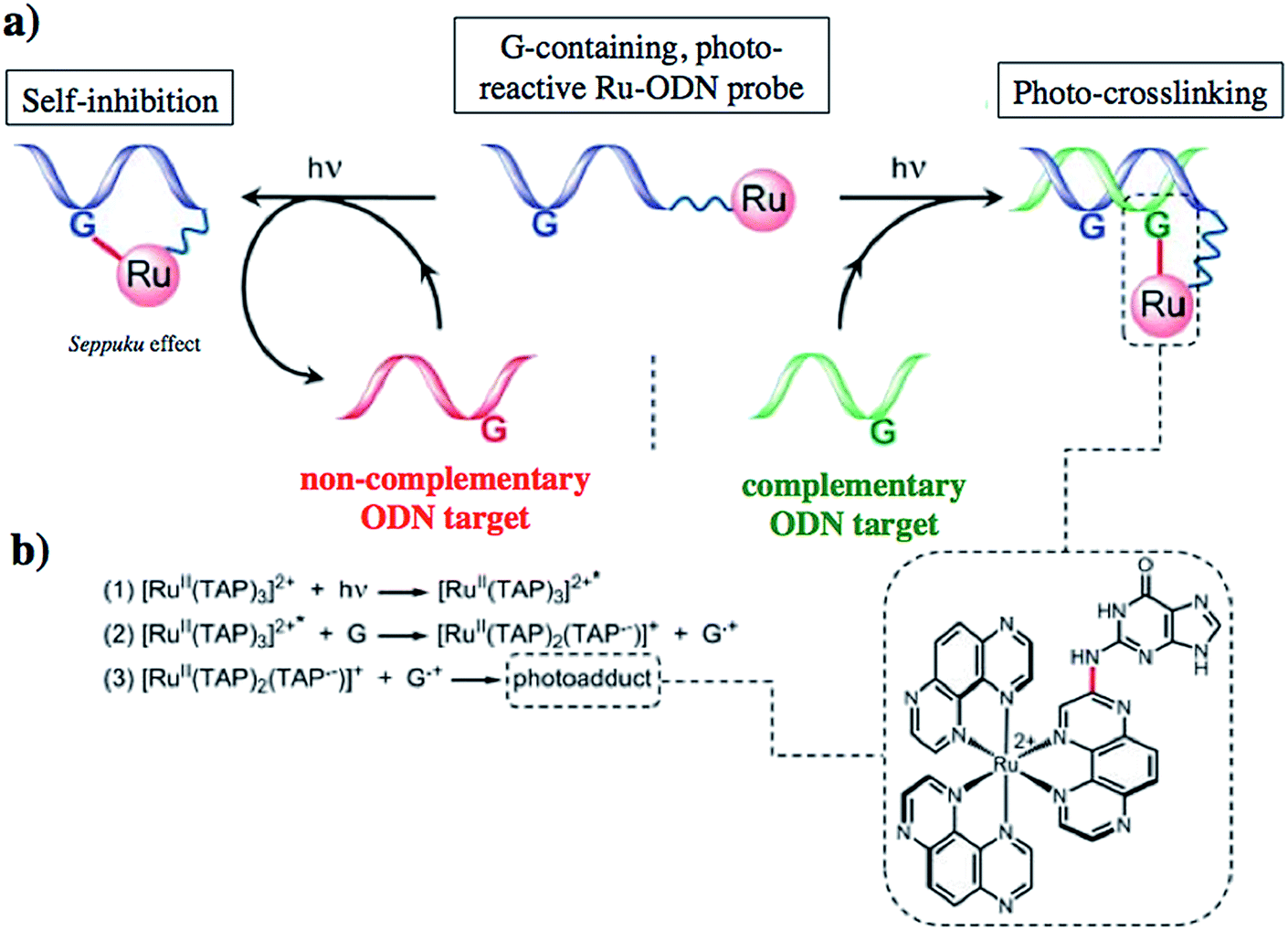 | ||
| Fig. 32 (a) Schematic representation of the Ru–ODN strategy, (b) explanation of the adduct formation. Adapted from ref. 102. | ||
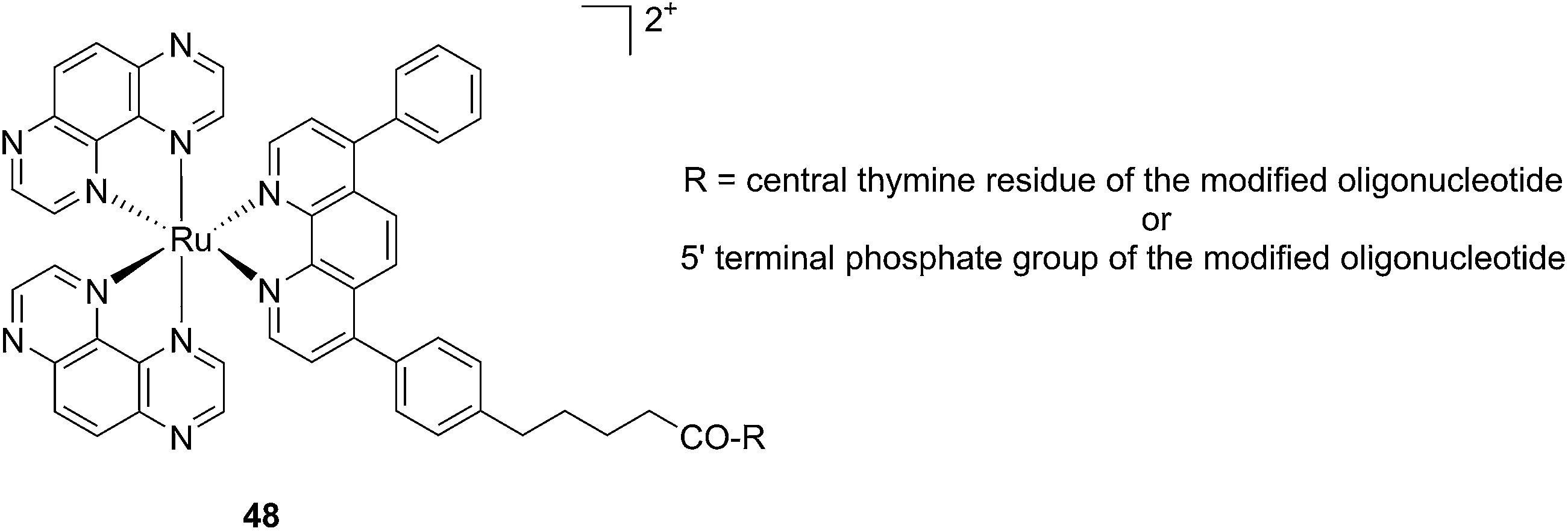 | ||
| Fig. 33 Structure of the [Ru(TAP)2dip]2+ conjugated to the ODN sequences.101 | ||
In a follow-up study, the same group explored the importance of the anchoring position of the Ru complex with respect to the ODN sequence. To do so, the photo-reactive polyazaaromatic complex [Ru(TAP)2(dppz)]2+ was coupled to the ODN sequence either via the dppz (alias Ru(D) (49) in Fig. 34) or the TAP moiety (alias Ru(T) (50) in Fig. 34). Adduct formation efficiencies and DNA interactions were evaluated. The authors found that both versions of anchored Ru complexes had the same DNA photo-ligation efficiency upon light irradiation (442 nm, 50 mW, 60 min) but, interestingly, they interacted with DNA in a different fashion. Whilst Ru(T) interacts by intercalation with the dppz ligand, Ru(D) interacts via TAP without intercalation. On one hand, dppz intercalation places the ruthenium center in direct contact with the guanine, favoring the PET process and back electron transfer. However, this geometrical conformation secludes the reduced TAP˙− and oxidized G+˙, thus reducing the photo-crosslinking efficiency. On the other hand, the TAP interaction puts TAP˙− and oxidized guanine in an optimal orientation for the photo-crosslinking reaction.102
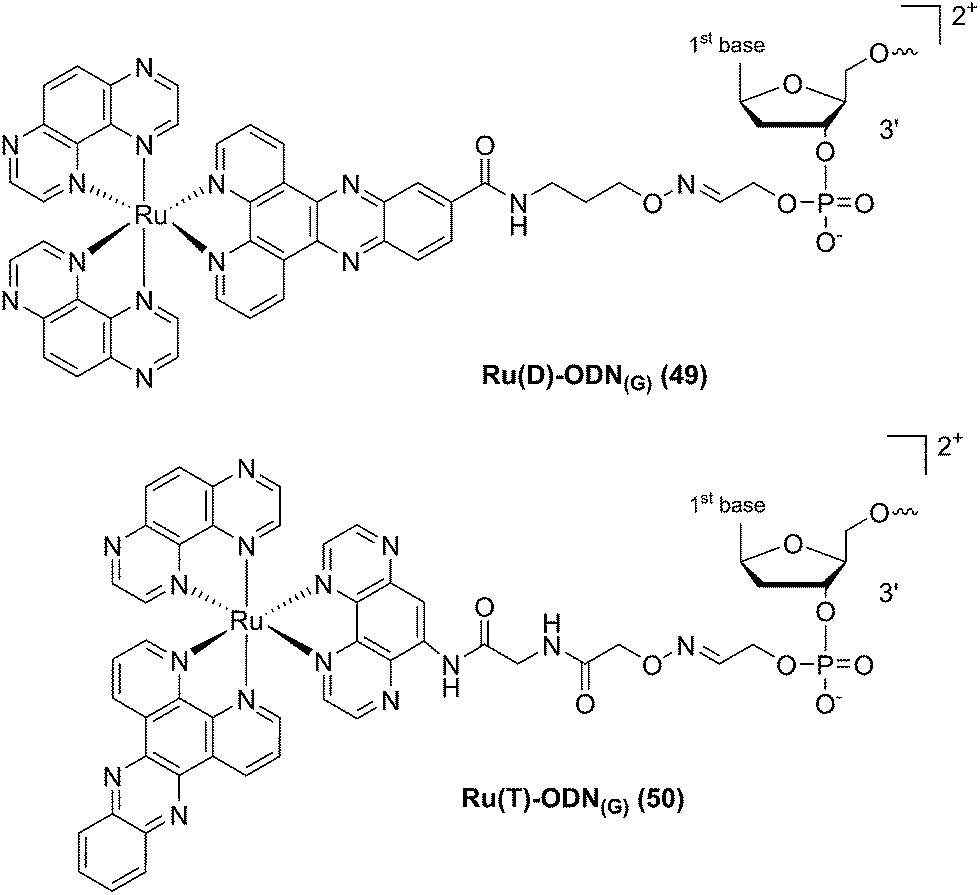 | ||
| Fig. 34 Structures of the Ru(II) complexes with an ODN sequence coupled either on the dppz (49) or on the TAP (50) moiety.102 | ||
This Ru–ODN strategy was examined in cell-based assays.103 In their study, Delvenne and co-authors coupled the complementary sequence of the oncogene E6, stimulated after HPV16 infection and responsible for the silencing of p53, to a polyazaaromatic complex [Ru(TAP)2(phen)]2+ (51, Fig. 35). With this tool in hand, they tested the efficiency of the conjugate for impairing HPV16 positive cervical cancer cell (SiHa) proliferation upon visible illumination. They could demonstrate an irreversible crosslink between the target and the Ru-conjugated probe. The authors could not only show reduced cell proliferation (45–50% growth inhibition on SiHa cells, 24 h post illumination at 380–480 nm for 2 h 30), but also a restored amount of p53, which is the principal target of E6, as well as a reduced E6 protein level. The use of photo-reactive Ru complexes for conjugation to ODN sequences offers, in light of the studies presented above, an attractive tool in the field of gene silencing. On one hand, the presence of a metal complex can enhance cell delivery of the Ru–ODN due to the positive charge brought by the Ru complex. On the other hand, the affinity of the probe for the target is improved since an irreversible crosslink is induced upon light irradiation. Moreover, undesired non-specific interactions are avoided if the DNA target sequence is not present, since the complex is capable of auto-inhibition (the ‘seppuku effect’), thus eliminating any collateral inhibition or crosslink (see Fig. 32).
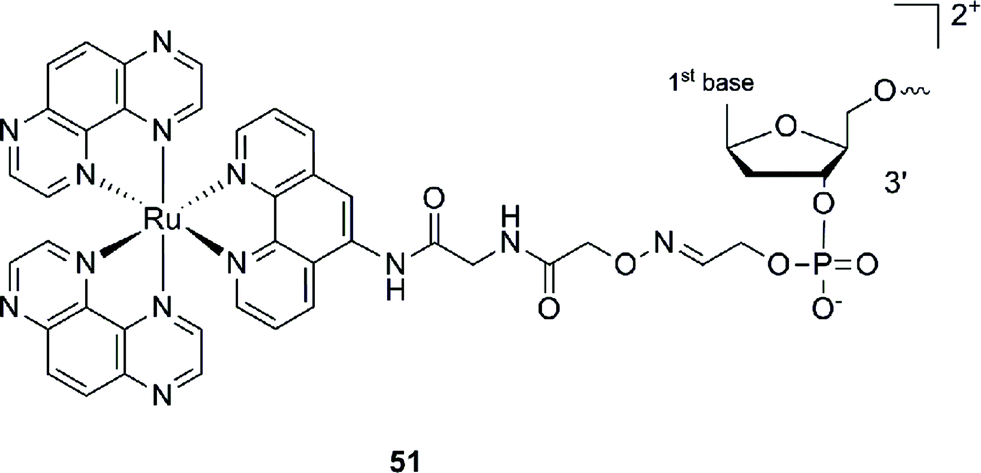 | ||
| Fig. 35 Structure of [Ru(TAP)2(phen)]2+.103 | ||
Photo-release strategy
Modification of the activity of a compound can be achieved by masking the functional groups involved in the toxicity with a cleavable moiety, acting as a cage. The idea of using light-responsive cages to deliver biologically active compounds into living cells or organisms is extremely appealing, offering control over the cytotoxicity and improved cellular uptake thanks to the modulation of the lipophilicity or the insertion of a charge. However, to date, there are only a few examples reported in the literature. Those are described below. | ||
| Fig. 36 Structure of the Ru(II) caged neuroactive 4-AP.104 | ||
Kodanko and collaborators, in turn, considered the latter study and the opportunity offered by Ru complexes to act as an effective photo-caging group for nitriles. They synthesized a Ru(II)tris(2-pyridylmethyl)amine complex 53, functionalized with two molecules of a known cathepsin K inhibitor 54 containing a nitrile group (Cbz-Leu-NHCH2CN) (Fig. 37).112 In healthy tissue, cathepsin K is a proteinase secreted by osteoclasts to degrade bones. It was shown to be expressed by breast cancer metastasized to bones as well. The authors were able to demonstrate the inhibition of the enzymatic activity of cathepsin K upon light activation (365 nm for 15 min) even if only one molecule of the inhibitor was released (IC50 values of 5.6 μM in the dark vs. 63 nM upon light irradiation).
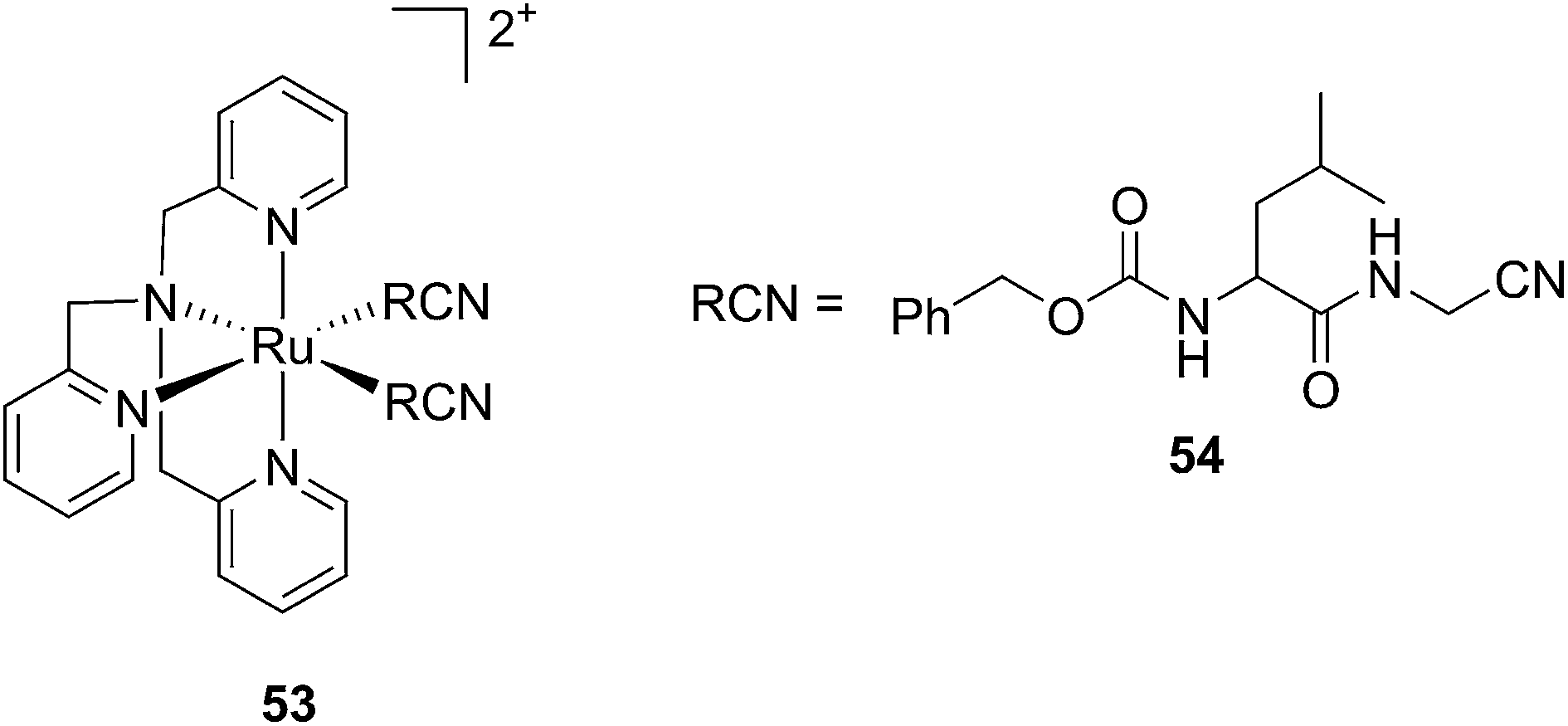 | ||
| Fig. 37 Structures of caged cathepsin K inhibitor (Cbz-Leu-NHCH2CN) (RCN).112 | ||
Driven by these promising in vitro results, the same authors chose a different Ru cage and a second cathepsin K inhibitor 55, yielding the compounds 56 and 57 reported in Fig. 38.113
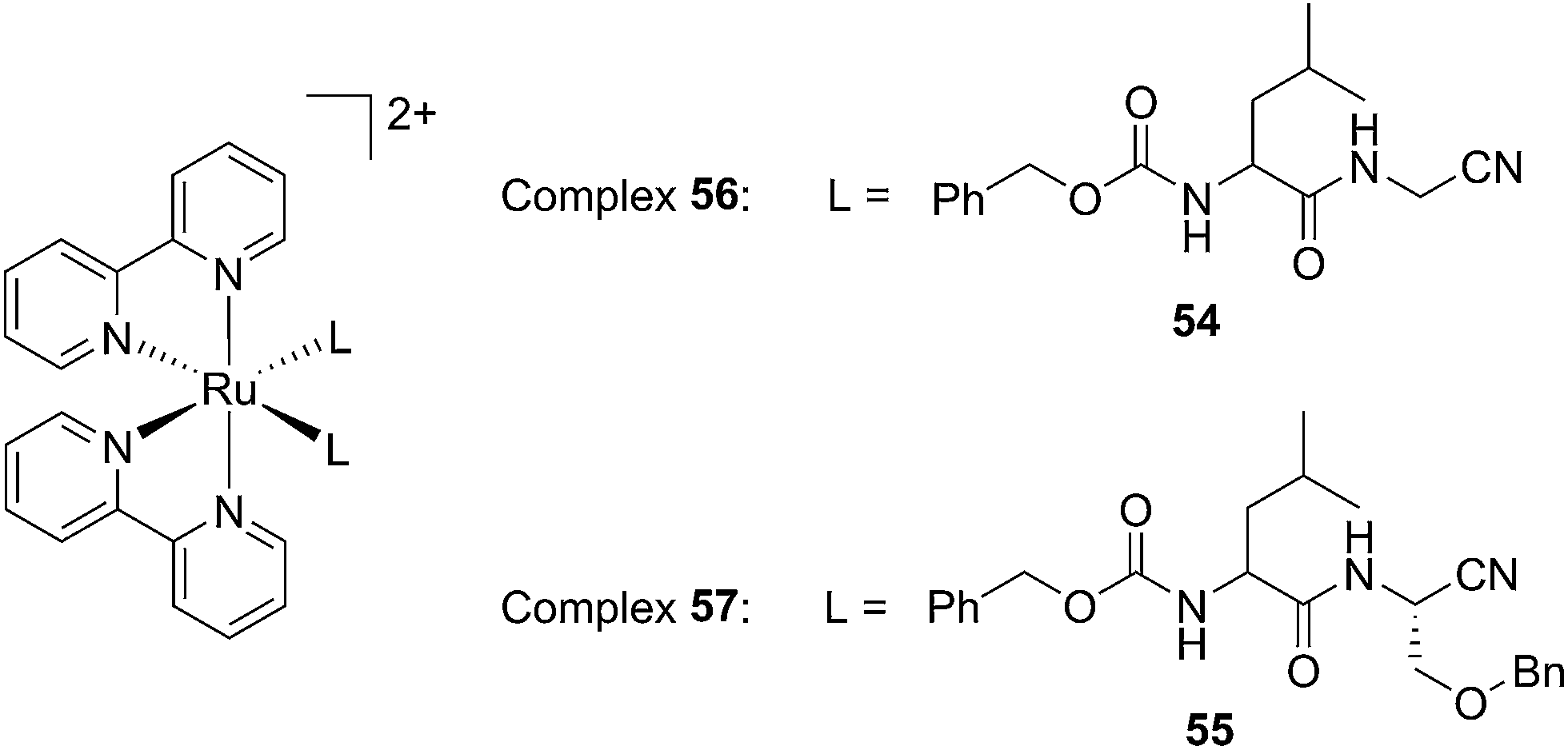 | ||
| Fig. 38 Structures of the Ru(II) complexes containing cathepsin K inhibitors.113 | ||
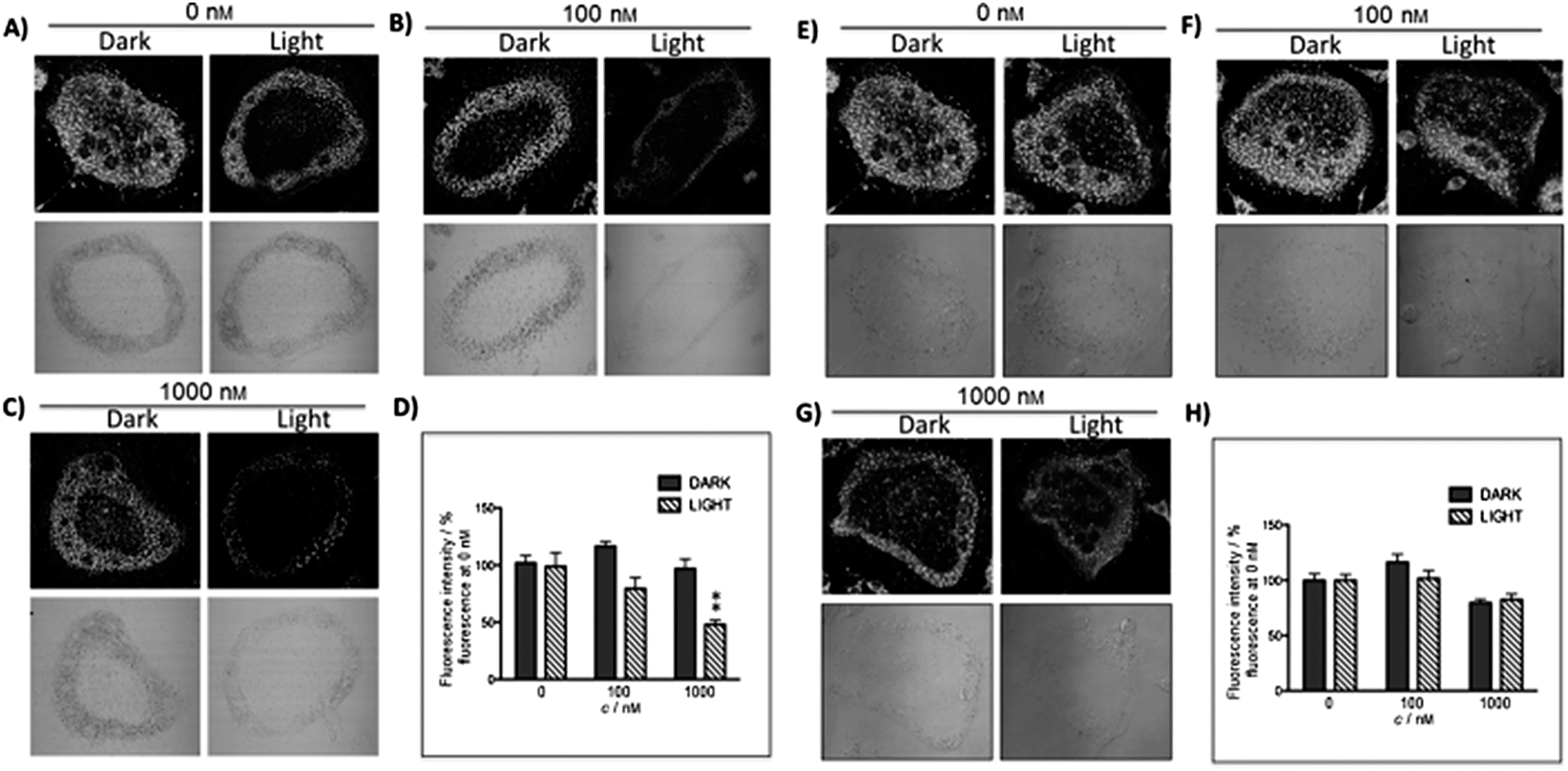
The new inhibitor 55 was chosen because of its better inhibition potency than 54 (reported IC50: 35 and 9 nM for 54 and 55, respectively). Moreover, using the Ru(bipy)2 fragment as a cage, they could demonstrate that both nitrile ligands could be photoreleased. However, complex 57 required a longer exposure time to release the inhibitor (up to 60 min of irradiation) than complex 56 (15 min). Using the same experimental conditions (tungsten lamp, 250 W, >395 nm, H2O filter), 56 and 57 showed significantly enhanced inhibition activities compared to the parent inhibitors 54 and 55 (IC50 values are 36 nM and 28 nM, respectively). Complex 56 showed a dark to light IC50 improvement from 560 nM to 16 nM (PI = 35). The photo-activated inhibition of cathepsin K is twice as effective as the parent inhibitor alone, correlating with the two molecules of inhibitor released. In the case of complex 57, the dark to light IC50 enhancement was from 2.2 μM to 25 nM (PI = 88). The light-triggered inhibition is in good agreement with the slow release rate of the second inhibitor molecule, reaching a similar value to the free parent molecule 55. In order to verify that none of the drugs or photoproducts were toxic in cells, the authors tested the viability of Bone Marrow Macrophages (BMM) and PC3 cells after 30 min incubation with the complexes, followed by a dark environment or 15 min irradiation for 56 or 40 min for 57 and 24 h additional incubation. They were able to confirm that no toxicity was found in murine BMM or PC3 cells up to 10 μM for complex 56 and up to 1 μM for 57. Since 56 showed the most promising features, the authors evaluated its enzymatic inhibition ability in a cell-based assay. Enzymatic activity in osteoclasts decreased by 25% and 50% when treated with 100 nM and 1 μM, respectively, either with 54 or with the photo-activated 56 (see the enzymatic inhibition studies in mouse osteoclasts in Fig. 39 for 54 and Fig. 40 for 56). These findings suggest that the photo-released enzyme inhibitors can play a potent role in the treatment of diseases where increased enzymatic activity is observed, sparing normal activity in surrounding tissues. These studies widen the perspective on the use of Ru(II) complexes as caging groups for the release of a large variety of biomolecules, from the pioneering Ru(II)(bipy)2 fragment as a neurotransmitter releaser, to nitrile-based protease inhibitors.
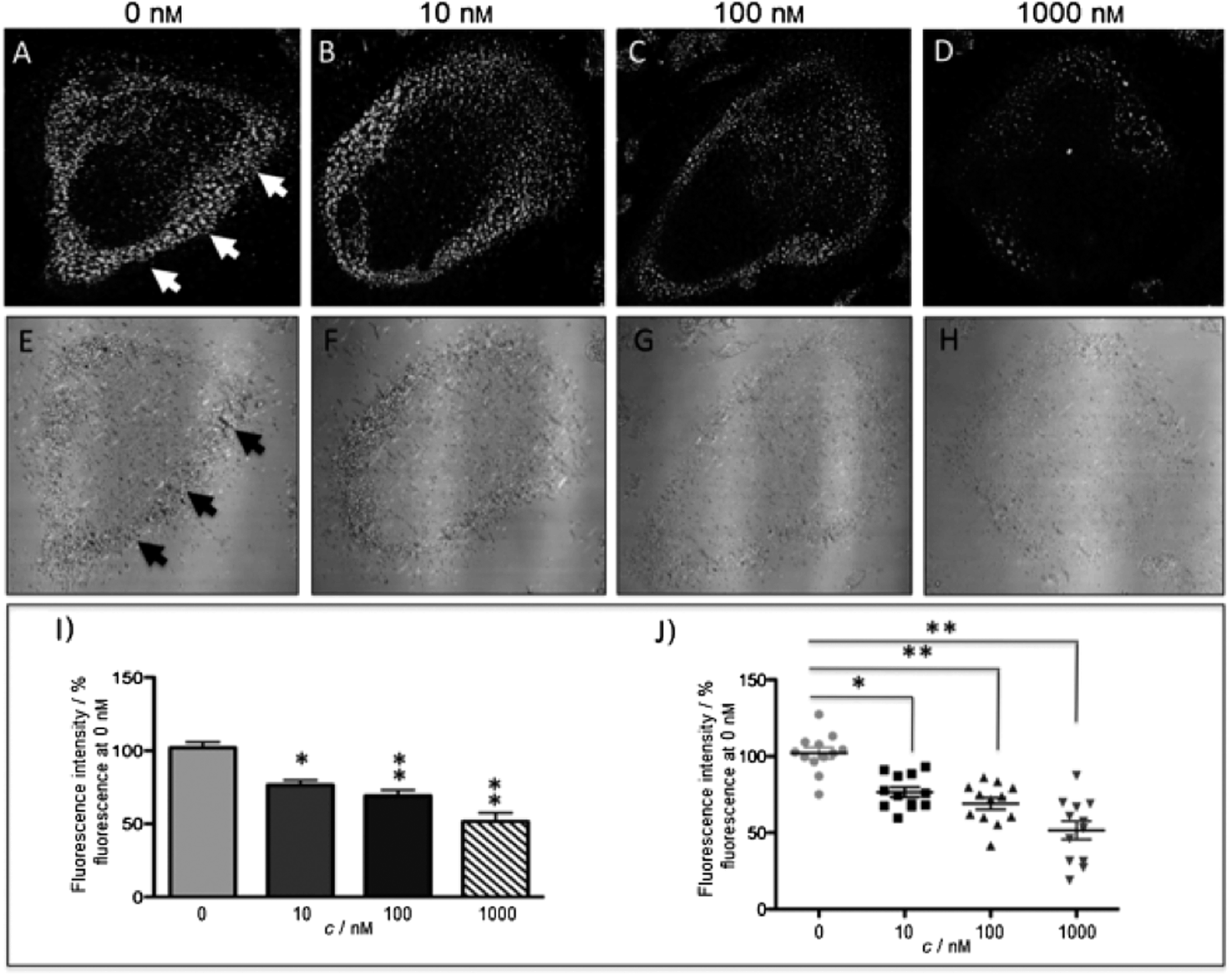 | ||
| Fig. 39 Confocal microscopy images of mouse osteoclast cells treated with 54. Cells were pre-incubated with 54 (10–1000 nm) for 30 min at 37 °C in the presence of cathepsin B inhibitor CA074Me (1 mm). Cells were treated with the cathepsin K substrate Z-LR-4MbNA (0.25 mm) and nitrosalicylaldehyde (1.0 mm, a precipitating agent), leading to the release of 4MbNA (green fluorescent precipitate indicative of cathepsin activity, arrows). Cells were fixed and imaged with a confocal laser scanning microscope (Zeiss LSM 780) using a 40× oil immersion lens. For each of the conditions at least six images of individual osteoclast cells were acquired, and fluorescence intensity per osteoclast area was measured and quantified using ImageJ software (NIH). The intensity of green fluorescence is a direct measure of the quantity of hydrolyzed and precipitated substrate (A–D), also visible on DIC images (E–H). The quantified data are shown as column (I) and dot (J) plots; *p < 0.05, **p < 0.001. Results are representative of at least three experiments. Reproduced with permission from ref. 113. © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
 | ||
| Fig. 40 Confocal microscopy images of mouse osteoclast cells treated with the ruthenium-caged inhibitor 56 (A–D) or cis-[Ru(bpy)2(MeCN)2](PF6)2 (E–H). Cells were pre-incubated with either complex (0–1000 nm) for 30 min at 37 °C in the presence of cathepsin B inhibitor CA074Me (1 mm), then exposed to dark (no irradiation) or light (irradiation at 250 W, 395–750 nm) conditions for 15 min. Cells were treated with the cathepsin K substrate Z-LR-4MbNA (0.25 mm) and nitrosalicylaldehyde (1.0 mm, a precipitating agent), leading to the release of 4MbNA (green fluorescent precipitate indicative of cathepsin activity). Cells were fixed and imaged with a confocal laser scanning microscope (Zeiss LSM 780) using a 40× oil immersion lens. For each of the conditions at least six images of individual osteoclast cells were acquired, and fluorescence intensity per osteoclast area was measured and quantified using ImageJ (NIH) software as described for Fig. 39 above; **p < 0.001. Results are representative of at least three experiments. Reproduced with permission from ref. 113. © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Kodanko and colleagues applied the Ru(bipy)2 fragment as a cage. However, compared to this, the Ru(II)tpy fragment offers a lower energy 1MLCT absorption, fitting within the PDT window (600–850 nm). Turro et al. took this opportunity and designed a Ru(II)tpy complex able to induce the release of 5-cyanouracil (5CNU), a known pyrimidine catabolism inhibitor, upon irradiation with visible light (>400 nm). Since it was shown that the bis-aqua Ru derivative can bind DNA, the following [Ru(tpy)(5CNU)3]2+ complex (59, Fig. 41) can potentially be used as a dual-action therapeutic agent.114
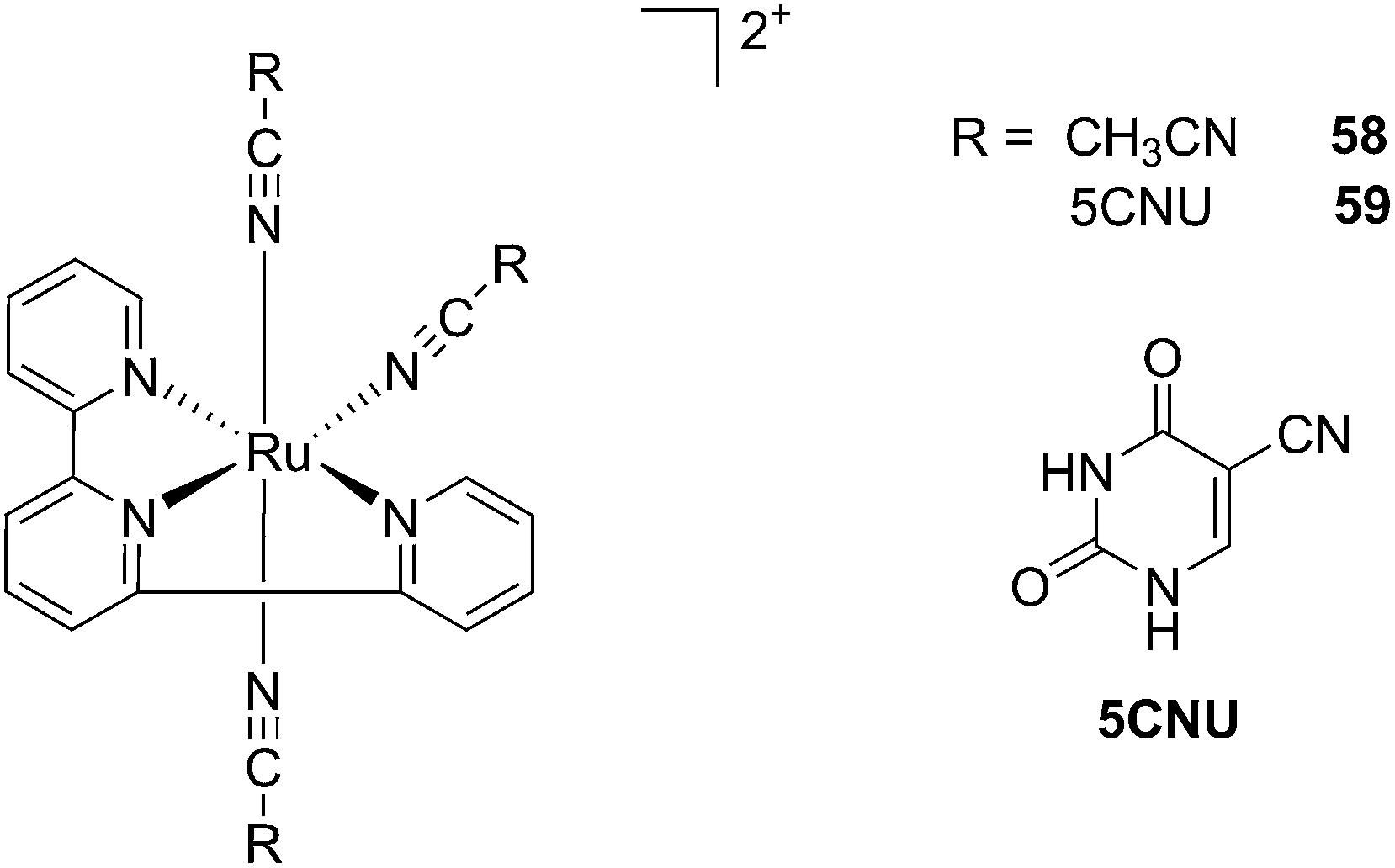 | ||
| Fig. 41 Structures of the Ru-inhibitor complexes synthesized by Turro.114 | ||
Indeed, the authors demonstrated that similarly to its analog complex bearing (CH3CN)3 (58, Fig. 41), the complex efficiently released the two axial ligands when irradiated with visible light (150 W Xe lamp housed in a Milliarc compact arc lamp housing), concomitantly producing the bis-aqua species. Only the latter photoproduct was then able to bind to DNA as observed by a reduction in the plasmid pUC19 mobility, when complexes 58 and 59 were irradiated for 5 or 15 min, respectively. However, extending these observations to cellular studies was revealed to be more challenging. Indeed, when human cervical cancer HeLa cells were treated with 100 μM of the Ru complexes for 2 h in the dark, followed by 1 h light irradiation, only complex 59 was shown to be capable of generating damage (no damage was observed in non-irradiated cells for 58 and 59). Moreover, cells treated with 100 μM of free 5CNU showed the same extent of damage as for complex 59 upon light irradiation, coinciding with only one molecule of 5CNU being released. The latter observation was confirmed when the LC50 value of irradiated 59 matched the one of free 5CNU (156 and 151 μM, respectively). In both cases, the mono-aqua photoproduct formed was not able to bind DNA. Accordingly, no decreased mobility was observed in agarose gel shift assay. This was also confirmed by the absence of cytotoxicity upon irradiation in the case of 58 or increased toxicity for 59.
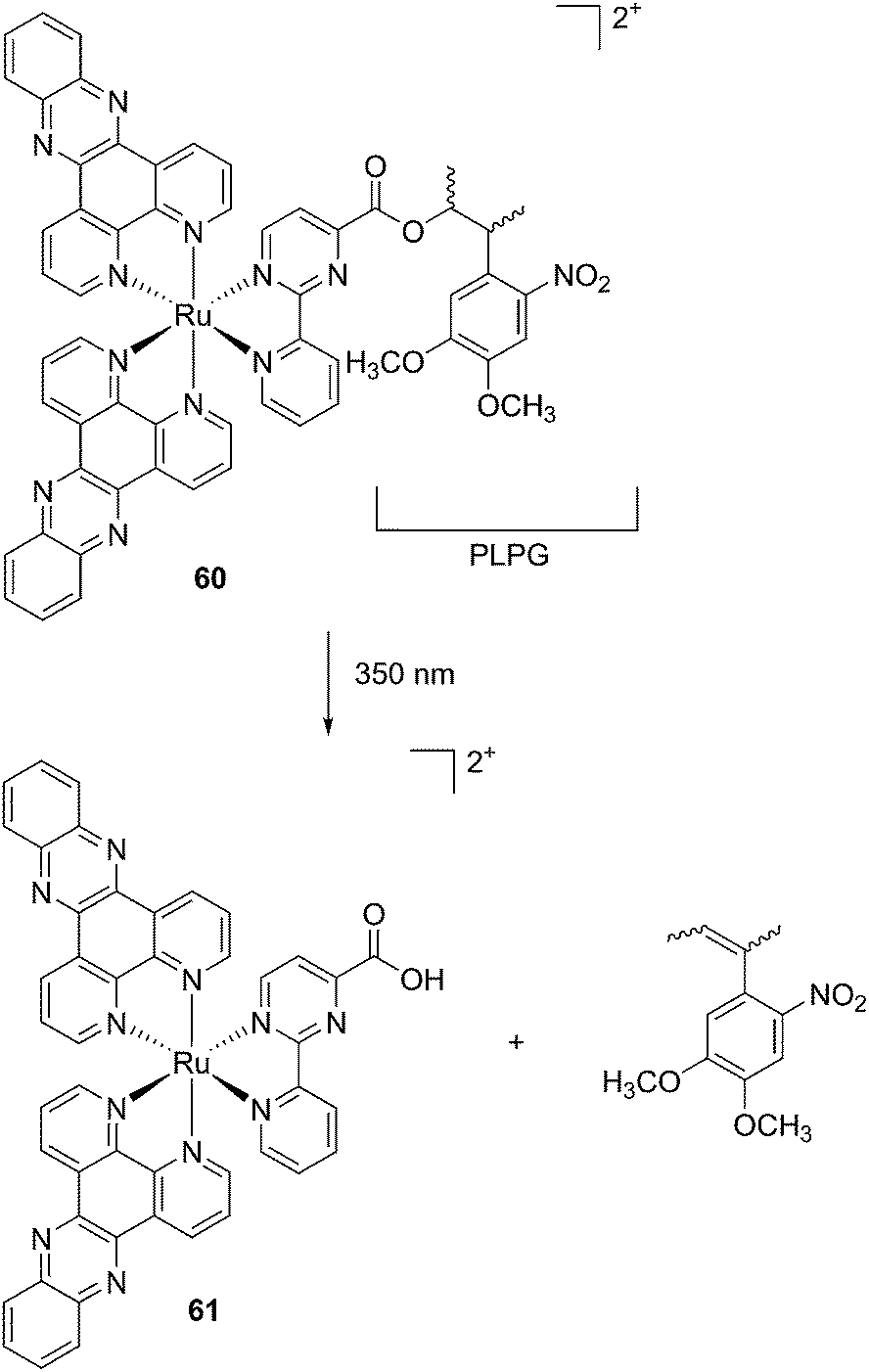 | ||
| Fig. 42 Structures of the caged Ru(II) complex 60 and of the toxic photoproduct 61 which is released.51 | ||
Conclusions
As shown in this perspective article, Ru(II) complexes offer several opportunities as light-activated drug candidates. Although very promising in vitro results have been achieved so far, the lack of in vivo studies (to the best of our knowledge, there have only been three reported so far) undoubtedly does not allow for a full assessment of the suitability of such compounds in a clinical context. However, we are confident that more such studies will be reported in the near future, shedding light on the full potential of these compounds.Abbreviations
| [9]aneS3 | 1,4,7-Trithiacyclononane |
| 3IL | Triplet intraligand |
| aPDT | Antibacterial PDT |
| ALA | Aminolevulinic acid |
| bipy | 2,2′-Bipyridine |
| biq | 2,2′-Biquinoline |
| bpm | 2,2′-Bipyrimidine |
| CALI | Chromophore-assisted light inactivation |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DLI | Drug to light interval |
| dmb | 4,4′-Di-methyl-2,2′-bipyridine |
| dmdop | 2,3-Dihydro-1,4-dioxino[2,3-f]-2,9-dimethyl-1,10-phenanthroline |
| dmphen | 2,9-Dimethyl-1,10-phenanthroline |
| dop | 2,3-Dihydro-1,4-dioxino[2,3-f]-1,10-phenanthroline |
| dpb | 2,3-Bis(2-pyridyl)-benzoquinoxaline |
| dpp | 2,3-Bis(2-pyridyl)pyrazine |
| dppn | Benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine |
| dppz | Dipyrido[3,2-a:2′,3′-c]phenazine |
| DSSCs | Dye-sensitized solar cells |
| dtbb | 4,4′-Di-t-butyl-2,2′-bipyridine |
| en | Ethylenediamine |
| GSH | Glutathione |
| HR-CS AAS | High-resolution continuum source atomic absorption spectrometry |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| IPPBA | 3-(1H-Imidazo[4,5-f][1,10]phenanthrolin-2yl)phenylboronic acid |
| ISC | Intersystem crossing |
| MLCT | Metal to ligand charge transfer |
| MMCT | Metal to metal charge transfer |
| ODNs | Oligodeoxyribonucleotides |
| PACT | Photoactivated chemotherapy |
| PCNA | Proliferating cell nuclear antigen |
| PDT | Photodynamic therapy |
| PET | Photoinduced electron transfer |
| Ph2phen | 4,7-Diphenyl-1,10-phenanthroline |
| PHEHAT | 1,10-Phenanthrolino[5,6-b]1,4,5,8,9,12-hexaazatriphenylene |
| phen | 1,10-Phenanthroline |
| phpy | 2-Phenylpyrydine |
| PI | Phototoxic index |
| PMP | 5-(3-Pyridyl)-10,15,20-triphenylporphyrin |
| PS | Photosensitizer |
| PTP | 5,10,15,20-Tetra(3-pyridyl)porphyrin |
| py | Pyridine |
| pydppn | 3-(Pyrid-2′-yl)-4,5,9,16-tetraaza-dibenzo[a,c]naphthacene |
| ROS | Reactive oxygen species |
| SAR | Structure activity relationship |
| SSBs | Single strand breaks |
| TAP | 1,4,5,8-Tetraazaphenanthrene |
| TPA | Two photon absorption |
| tpph | Tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′]phenazine |
| tpy | [2,2′;6′,2′′]-Terpyridine |
Acknowledgements
This work was financially supported by the Swiss National Science Foundation (SNSF Professorship PP00P2_133568 to G.G.), the University of Zurich (G.G.), the Stiftung für Wissenschatliche Forschung of the University of Zurich (G.G. and S.F.), the Stiftung zur Krebsbekämpfung (S.F), the Huggenberger-Bischoff Stiftung (S.F) and the Hartmann-Müller Stiftung (S.F.).References
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- N. L. Kilah and E. Meggers, Aust. J. Chem., 2012, 65, 1325–1332 CrossRef CAS.
- L. Salassa, Eur. J. Inorg. Chem., 2011, 4931–4947 CrossRef CAS.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. DiGiovine, G. Pezzoni, L. Sartor and S. Garbisa, Clin. Cancer Res., 2003, 9, 1898–1905 CAS.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- N. R. Dickson, S. F. Jones and H. A. Burris, J. Clin. Oncol., 2011, 29 Search PubMed , suppl., abstr 2607.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Muller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- J. Maksimoska, L. Feng, K. Harms, C. Yi, J. Kissil, R. Marmorstein and E. Meggers, J. Am. Chem. Soc., 2008, 130, 15764–15765 CrossRef CAS PubMed.
- M. Doerr and E. Meggers, Curr. Opin. Chem. Biol., 2014, 19, 76–81 CrossRef CAS PubMed.
- F. Kratz, M. Hartmann, B. Keppler and L. Messori, J. Biol. Chem., 1994, 269, 2581–2588 CAS.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS PubMed.
- P. J. Bednarski, F. S. Mackay and P. J. Sadler, Anti-Cancer Agents Med. Chem., 2007, 7, 75–93 CrossRef CAS.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 1451–1467 CrossRef CAS.
- N. A. Smith and P. J. Sadler, Philos. Trans. R. Soc., A, 2013, 371, 20120519 CrossRef PubMed.
- O. J. Stacey and S. J. A. Pope, RSC Adv., 2013, 3, 25550–25564 RSC.
- N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690–10701 RSC.
- S. L. H. Higgins and K. J. Brewer, Angew. Chem., Int. Ed. Engl., 2012, 51, 11420–11422 CrossRef CAS PubMed.
- S. Swavey, Ruthenium Complexes as Photosensitizers: New Possibilities in Photodynamic Therapy, Nova Science Publishers, Inc., New York, 2011 Search PubMed.
- G. S. Smith and B. Therrien, Dalton Trans., 2011, 10793–10800 RSC.
- T. Joshi and G. Gasser, Synlett, 2014 DOI:10.1055/s-0034-1379426.
- J. Ghesquiere, S. Le Gac, L. Marcelis, C. Moucheron and A. Kirsch-De Mesmaeker, Curr. Top. Med. Chem., 2012, 12, 185–196 CrossRef CAS.
- E. C. Glazer, Isr. J. Chem., 2013, 53, 391–400 CrossRef CAS.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2004, 1, 279–293 CrossRef CAS.
- Z. Huang, Technol. Cancer Res. Treat., 2005, 4, 283–293 CrossRef CAS PubMed.
- K. Plaetzer, B. Krammer, J. Berlanda, F. Berr and T. Kiesslich, Laser. Med. Sci., 2009, 24, 259–268 CrossRef CAS PubMed.
- S. Bonnet, Comments Inorg. Chem., 2014 DOI:10.1080/02603594.02602014.02979286.
- R. K. Pandey and C. K. Herman, Chem. Ind., 1998, 18, 739–743 Search PubMed.
- C. Lottner, K.-C. Bart, G. Bernhardt and H. Brunner, J. Med. Chem., 2002, 45, 2064–2078 CrossRef CAS PubMed.
- C. Lottner, K.-C. Bart, G. Bernhardt and H. Brunner, J. Med. Chem., 2002, 45, 2079–2089 CrossRef CAS PubMed.
- F. Schmitt, P. Govindaswamy, G. Süss-Fink, W. H. Ang, P. J. Dyson, L. Juillerat-Jeanneret and B. Therrien, J. Med. Chem., 2008, 51, 1811–1816 CrossRef CAS PubMed.
- F. Schmitt, P. Govindaswamy, O. Zava, G. Süss-Fink, L. Juillerat-Jeanneret and B. Therrien, J. Biol. Inorg. Chem., 2009, 14, 101–109 CrossRef CAS PubMed.
- M. Pernot, T. Bastogne, N. P. E. Barry, B. Therrien, G. Koellensperger, S. Hann, V. Reshetov and M. Barberi-Heyob, J. Photochem. Photobiol., B, 2012, 117, 80–89 CrossRef CAS PubMed.
- F. Schmitt, N. P. E. Barry, L. Juillerat-Jeanneret and B. Therrien, Bioorg. Med. Chem. Lett., 2012, 22, 178–180 CrossRef CAS PubMed.
- F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed.
- T. Gianferrara, I. Bratsos, E. Iengo, B. Milani, A. Ostric, C. Spagnul, E. Zangrando and E. Alessio, Dalton Trans., 2009, 10742–10756 RSC.
- T. Gianferrara, A. Bergamo, I. Bratsos, B. Milani, C. Spagnul, G. Sava and E. Alessio, J. Med. Chem., 2010, 53, 4678–4690 CrossRef CAS PubMed.
- B. Serli, E. Zangrando, T. Gianferrara, C. Scolaro, P. J. Dyson, A. Bergamo and E. Alessio, Eur. J. Inorg. Chem., 2005, 3423–3434 CrossRef CAS.
- I. Bratsos, S. Jedner, A. Bergamo, G. Sava, T. Gianferrara, E. Zangrando and E. Alessio, J. Inorg. Biochem., 2008, 102, 1120–1133 CrossRef CAS PubMed.
- S. Rani-Beeram, K. Meyer, A. McCrate, Y. Hong, M. Nielsen and S. Swavey, Inorg. Chem., 2008, 47, 11278–11283 CrossRef CAS PubMed.
- P. Sweigert, Z. Xu, Y. Hong and S. Swavey, Dalton Trans., 2012, 5201–5208 RSC.
- J. Yoho, C. Stroh, S. Swavey and M. Kango-Singh, Genesis, 2014, 52, 309–314 CrossRef CAS PubMed.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC.
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed.
- U. Schatzschneider, J. Niesel, I. Ott, R. Gust, H. Alborzinia and S. Wölfl, ChemMedChem, 2008, 3, 1104–1109 CrossRef CAS PubMed.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef CAS PubMed.
- T. Joshi, V. Pierroz, S. Ferrari and G. Gasser, ChemMedChem, 2014, 9, 1419–1427 CrossRef CAS PubMed.
- T. Joshi, V. Pierroz, C. Mari, L. Gemperle, S. Ferrari and G. Gasser, Angew. Chem., Int. Ed. Engl., 2014, 53, 2960–2963 CrossRef CAS PubMed.
- J. X. Zhang, J. W. Zhou, C. F. Chan, T. C. K. Lau, D. W. J. Kwong, H. L. Tam, N. K. Mak, K. L. Wong and W. K. Wong, Bioconjugate Chem., 2012, 23, 1623–1638 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem.–Eur. J., 2014, 20, 14421–14436 CrossRef CAS PubMed.
- M. T. Mongelli, J. Heinecke, S. Mayfield, B. Okyere, B. S. J. Winkel and K. J. Brewer, J. Inorg. Biochem., 2006, 100, 1983–1987 CrossRef CAS PubMed.
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 2426–2428 RSC.
- Y. Liu, R. Hammitt, D. A. Lutterman, L. E. Joyce, R. P. Thummel and C. Turro, Inorg. Chem., 2009, 48, 375–385 CrossRef CAS PubMed.
- R. Zhao, R. Hammitt, R. P. Thummel, Y. Liu, C. Turro and R. M. Snapka, Dalton Trans., 2009, 10926–10931 RSC.
- S. I. Bae, R. Zhao and R. M. Snapka, Biochem. Pharmacol., 2008, 76, 1653–1668 CrossRef CAS PubMed.
- G. C. Vougioukalakis, A. I. Philippopoulos, T. Stergiopoulos and P. Falaras, Coord. Chem. Rev., 2011, 255, 2602–2621 CrossRef CAS PubMed.
- A. Frei, R. Rubbiani, S. Tubafard, O. Blacque, P. Anstaett, A. Felgenträger, T. Maisch, L. Spiccia and G. Gasser, J. Med. Chem., 2014, 57, 7280–7292 CrossRef CAS PubMed.
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370–10377 CrossRef CAS PubMed.
- R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161–17175 CrossRef CAS PubMed.
- B. Zbytek, J. A. Carlson, J. Granese, J. Ross, M. Mihm and A. Slominski, Expert Rev. Dermatol., 2008, 3, 569–585 CrossRef CAS PubMed.
- H. Yin, M. Stephenson, J. Gibson, E. Sampson, G. Shi, T. Sainuddin, S. Monro and S. A. McFarland, Inorg. Chem., 2014, 53, 4548–4559 CrossRef CAS PubMed.
- G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Coord. Chem. Rev., 2014, 282–283, 127–138 Search PubMed.
- Y. Arenas, S. Monro, G. Shi, A. Mandel, S. McFarland and L. Lilge, Photodiagn. Photodyn. Ther., 2013, 10, 615–625 CrossRef CAS PubMed.
- J. Lee, D. G. Udugamasooriya, H.-S. Lim and T. Kodadek, Nat. Chem. Biol., 2010, 6, 258–260 CrossRef CAS PubMed.
- X. Liu, M. Dix, A. E. Speers, D. A. Bachovchin, A. M. Zuhl, B. F. Cravatt and T. J. Kodadek, ChemBioChem, 2012, 13, 2082–2093 CrossRef CAS PubMed.
- A. L. Harris, Nat. Rev. Cancer, 2002, 2, 38–47 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- Y. H. Hsiang, R. Hertzberg, S. Hecht and L. F. Liu, J Biol Chem, 1985, 260, 14873–14878 CAS.
- B. Lippert, Cisplatin, Chemistry and Biochemistry of a Leading Anticancer Drug, Verlag Helvetica Chimica Acta: Zürich, Wiley_VCH, Weinheim, Germany, 1999, pp. 111–134 Search PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- K. M. Comess, J. N. Burstyn, J. M. Essigmann and S. J. Lippard, Biochemistry, 1992, 31, 3975–3990 CrossRef CAS.
- Y. Corda, M. F. Anin, M. Leng and D. Job, Biochemistry, 1992, 31, 1904–1908 CrossRef CAS.
- J. M. Rosenberg and P. H. Sato, Mol. Pharmacol., 1993, 43, 491–497 CAS.
- A. J. Deans and S. C. West, Nat. Rev. Cancer, 2011, 11, 467–480 CrossRef CAS PubMed.
- F. S. Mackay, J. A. Woods, H. Moseley, J. Ferguson, A. Dawson, S. Parsons and P. J. Sadler, Chem.–Eur. J., 2006, 12, 3155–3161 CrossRef CAS PubMed.
- F. S. Mackay, J. A. Woods, P. Heringová, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed.
- A. F. Westendorf, J. A. Woods, K. Korpis, N. J. Farrer, L. Salassa, K. Robinson, V. Appleyard, K. Murray, R. Grünert, A. M. Thompson, P. J. Sadler and P. J. Bednarski, Mol. Cancer Ther., 2012, 11, 1894–1904 CrossRef CAS PubMed.
- Y. Zhao, J. A. Woods, N. J. Farrer, K. S. Robinson, J. Pracharova, J. Kasparkova, O. Novakova, H. Li, L. Salassa, A. M. Pizarro, G. J. Clarkson, L. Song, V. Brabec and P. J. Sadler, Chem.–Eur. J., 2013, 19, 9578–9591 CrossRef CAS PubMed.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS.
- P. C. Ford, Coord. Chem. Rev., 1982, 44, 61–82 CrossRef CAS.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- A. N. Hidayatullah, E. Wachter, D. K. Heidary, S. Parkin and E. C. Glazer, Inorg. Chem., 2014, 53, 10030–10032 CrossRef CAS PubMed.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC.
- B. Peña, A. David, C. Pavani, M. S. Baptista, J.-P. Pellois, C. Turro and K. R. Dunbar, Organometallics, 2014, 33, 1100–1103 CrossRef.
- B. A. Albani, B. Peña, K. R. Dunbar and C. Turro, Photochem. Photobiol. Sci., 2014, 13, 272–280 CAS.
- P. G. Bomben, K. C. D. Robson, P. A. Sedach and C. P. Berlinguette, Inorg. Chem., 2009, 48, 9631–9643 CrossRef CAS PubMed.
- C. Gaiddon, P. Jeannequin, P. Bischoff, M. Pfeffer, C. Sirlin and J. P. Loeffler, J. Pharmacol. Exp. Ther., 2005, 315, 1403–1411 CrossRef CAS PubMed.
- A. M. Palmer, B. Peña, R. B. Sears, O. Chen, M. El Ojaimi, R. P. Thummel, K. R. Dunbar and C. Turro, Philos. Trans. R. Soc., A, 2013, 371, 20120135 CrossRef PubMed.
- W. Guo, W. Zheng, Q. Luo, X. Li, Y. Zhao, S. Xiong and F. Wang, Inorg. Chem., 2013, 52, 5328–5338 CrossRef CAS PubMed.
- M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46–51 RSC.
- F. Barragán, P. López-Senín, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchán, J. Am. Chem. Soc., 2011, 133, 14098–14108 CrossRef PubMed.
- W. Vanderlinden, M. Blunt, C. C. David, C. Moucheron, A. Kirsch-De Mesmaeker and S. De Feyter, J. Am. Chem. Soc., 2012, 134, 10214–10221 CrossRef CAS PubMed.
- Y. Chen, W. Lei, G. Jiang, Y. Hou, C. Li, B. Zhang, Q. Zhou and X. Wang, Dalton Trans., 2014, 15375–15384 RSC.
- S. Swavey and K. J. Brewer, Inorg. Chem., 2002, 41, 6196–6198 CrossRef CAS PubMed.
- A. Sitlani, E. C. Long, A. M. Pyle and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 2303–2312 CrossRef CAS.
- A. A. Holder, D. F. Zigler, M. T. Tarrago-Trani, B. Storrie and K. J. Brewer, Inorg. Chem., 2007, 46, 4760–4762 CrossRef CAS PubMed.
- L. Marcélis, W. Vanderlinden and A. Kirsch - De Mesmaeker, in Inorganic Chemical Biology, ed. G. Gasser, Wiley-VCH Verlag GmbH & Co. KGaA, 2014 Search PubMed.
- O. Lentzen, J.-F. Constant, E. Defrancq, M. Prévost, S. Schumm, C. Moucheron, P. Dumy and A. Kirsch-De Mesmaeker, ChemBioChem, 2003, 4, 195–202 CrossRef CAS PubMed.
- S. Le Gac, M. Foucart, P. Gerbaux, E. Defrancq, C. Moucheron and A. Kirsch-De Mesmaeker, Dalton Trans., 2010, 9672–9683 RSC.
- A. Reschner, S. Bontems, S. Le Gac, J. Lambermont, L. Marcélis, E. Defrancq, P. Hubert, C. Moucheron, A. Kirsch-De Mesmaeker, M. Raes, J. Piette and P. Delvenne, Gene Ther., 2013, 20, 435–443 CrossRef CAS PubMed.
- L. Zayat, C. Calero, P. Alborés, L. Baraldo and R. Etchenique, J. Am. Chem. Soc., 2003, 25, 882–883 CrossRef PubMed.
- L. Zayat, M. G. Noval, J. Campi, C. I. Calero, D. J. Calvo and R. Etchenique, ChemBioChem, 2007, 8, 2035–2038 CrossRef CAS PubMed.
- V. Lopes-dos-Santos, J. Campi, O. Filevich, S. Ribeiro and R. Etchenique, Braz. J. Med. Biol. Res., 2011, 44, 688–693 CAS.
- O. Filevich and R. Etchenique, Photochem. Photobiol. Sci., 2013, 12, 1565–1570 CAS.
- E. Fino, R. Araya, D. S. Peterka, M. Salierno, R. Etchenique and R. Yuste, Front. Neural Circuits, 2009, 3, 1–9 Search PubMed.
- M. Salierno, E. Marceca, D. S. Peterka, R. Yuste and R. Etchenique, J. Inorg. Chem., 2010, 104, 418–422 CAS.
- O. Filevich, M. Salierno and R. Etchenique, J. Inorg. Biochem., 2010, 104, 1248–1251 CrossRef CAS PubMed.
- R. Araya, V. Andino-Pavlovsky, R. Yuste and R. Etchenique, ACS Chem. Neurosci., 2013, 4, 1163–1167 CrossRef CAS PubMed.
- R. Sharma, J. D. Knoll, P. D. Martin, I. Podgorski, C. Turro and J. J. Kodanko, Inorg. Chem., 2014, 53, 3272–3274 CrossRef CAS PubMed.
- T. Respondek, R. Sharma, M. K. Herroon, R. N. Garner, J. D. Knoll, E. Cueny, C. Turro, I. Podgorski and J. J. Kodanko, ChemMedChem, 2014, 9, 1306–1315 CrossRef CAS PubMed.
- M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274–11282 CrossRef CAS PubMed.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed. Engl., 2006, 45, 4900–4921 CrossRef CAS PubMed.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef CAS PubMed.
Footnote |
| † These authors have contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2015 |