
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereocontrolled protein surface recognition using chiral oligoamide proteomimetic foldamers†
Valeria
Azzarito
ab,
Jennifer A.
Miles
ab,
Julie
Fisher
a,
Thomas A.
Edwards
bc,
Stuart L.
Warriner
ab and
Andrew J.
Wilson
*ab
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: A.J.Wilson@leeds.ac.uk
bAstbury Centre for Structural and Molecular Biology, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
cSchool of Molecular and Cellular Biology, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
First published on 30th January 2015
Abstract
The development of foldamers capable of selective molecular recognition of solvent exposed protein surfaces represents an outstanding challenge in supramolecular chemical biology. Here we introduce an oligoamide foldamer with well-defined conformation that bears all the hallmarks of an information rich oligomer. Specifically, the foldamer recognizes its target protein hDM2 leading to inhibition of its protein–protein interaction with p53 in a manner that depends upon the composition, spatial projection and stereochemistry of functional groups appended to the scaffold. Most significantly, selective inhibition of p53/hDM2 can be achieved against four other targets and the selectivity for p53/hDM2 inhibition versus Mcl-1/NOXA-B inhibition is critically dependent upon the stereochemistry of the helix mimetic.
Introduction
The ability to understand and manipulate biological function using molecules programmed with specific and selective molecular recognition properties is a challenging but crucial objective. In its ultimate embodiment, the ability to design and synthesize molecules that effectively mimic the structure and function of protein secondary and tertiary structure1–5 using abiotic repeat backbones would begin to answer the question: “is the proteinogenic code for 3D structure and recognition limited to polymers of α-amino acids?” A well-defined “code” has been elaborated for DNA recognition that makes use of polyamides assembled from a limited set of monomer building blocks,6 but an equivalent code remains elusive for protein surface recognition. There are some notable examples of supramolecular binding motifs for protein surfaces that have been developed to target particular residues;7,8 however, these are not sufficiently information rich to achieve selective recognition when challenged with similar targets. Non-natural scaffolds are hence required that can reproduce the protein-binding specificity and selectivity of the natural-ligands upon which they are based9 and which are amenable to predictable sequence based optimization.10–12 In this context we and others have been developing inhibitors of α-helix mediated13–15 protein–protein interactions (PPIs).16,17 Although inhibitors of helix mediated PPIs have been identified through conventional drug discovery methods18,19 with the specific objective of advancing therapeutics discovery/development, a different approach is needed to realize the goals of sequence based design. Two generic approaches have been proposed: (i) by using an oligomer which reproduces the local topography of the helical fold13 and (ii) by employing a small molecule scaffold termed a “proteomimetic”20 to orient functionality in a manner that reproduces the spatial and angular positioning of key side chains presented by the helix donor.13 For the former, the development of conformationally constrained peptides has shown promising advantages for functionality21 and therapeutic potential,22 whilst studies on β-peptides have illustrated that mixed α/β-sequences10,11,23,24 or wholly β-sequences,25 can effectively inhibit PPIs. Although such studies illustrate the requirement for correct spatial relationship of hot-spot26 side-chains, the approach relies on reproducing the helical fold as closely as possible to achieve recognition. In contrast, the proteomimetic approach seeks to identify modular sequences of reduced complexity that are orthogonal to the natural sequence in terms of topology but compatible in terms of recognition complementarity. Proteomimetics20 have been identified that are selective for their targets in biophysical assays27–29 depending on the composition of variable functionality added to the scaffold, alongside reports illustrating targeting in cellulo30 and in vivo.31 Despite these promising results, the fundamental parameters determining selective recognition by proteomimetics remain poorly defined. The purpose of this study was to determine whether the spatial and angular projection of key side chains can interplay in reduced complexity proteomimetic systems to provide recognition motifs that exhibit the hallmarks of an information rich system capable of programmed or coded interaction with proteins. We do so using an oligoamide proteomimetic20 scaffold assembled using robust solid-phase synthesis that is capable of reproducing the vectoral presentation of side chains projecting from the i, i + 4 and i + 7 positions of an α-helix. As a model protein we selected hDM2 which is a well characterized32 helix binding protein. Our goal was not to add to the plethora of optimized small molecules already identified and in clinical development for this target. Instead, our goal was to understand the rules that govern molecular recognition. Our biophysical analyses, reveal that recognition of hDM2 – and therefore inhibition of the p53/hDM2 interaction – is dependent upon the side chain composition and spacing between monomer units and that the selectivity of inhibition of p53/hDM2 versus Mcl-1/NOXA-B33 can be switched by varying the stereochemistry of the central monomer in the mimetic – an observation that illustrates for the first time the importance of stereochemistry for α-helix mimetic foldamers. This study suggests these scaffolds show all the hallmarks required of an informational oligomer34 and show promise for the further construction of recognition codes which extend beyond oligomers of α-amino acids.Results
Design of a new hybrid α-helix mimetic and preliminary in silico studies
A key premise upon which helix mimetics are typically designed is that they should be based on a relatively rigid scaffold and reproduce exactly the distances and angular relationship between key side chains from the native α-helical template upon which they are based.35 However, experimental36 and computational investigations37,38 have shown that such scaffolds normally adopt a range of conformations, and only some of these are able to mimic the required pharmacophore. The design of an effective helix mimetic ought, hence, to be refined to consider the conformational plasticity of the mimetic itself alongside the dynamic nature of the target protein and thus the potential for induced fit between ligand and protein to occur.39 Indeed whether rigid conformational control is actually desirable for effective mimicry remains to be established.Building on our studies of homo-oligobenzamide α-helix mimetics,36,40,41 we designed a new mimetic with distinct H-bonding capabilities and stereoelectronic restraints. The backbone was varied by substitution of the middle aryl unit with an α-amino acid to generate a ‘hybrid’ α-helix mimetic (Fig. 1a) designed to mimic the i, i + 4 and i + 7 side-chains of an α-helix. Although heterofoldamers42 containing both aromatic and aliphatic amino acid building blocks are known,43,44 the scaffold described in this work has not previously been described. As a helix donor to mimic, we selected p53 which forms a PPI with hDM2 as an acceptor; the Phe19, Trp23 and Leu26 of p53 are known hot-spot residues which should be mimicked (Fig. 1b).32 This PPI represents a classic model system for therapeutics discovery45 and test system for novel helix mimetic scaffolds.25,27,46–50 Appendage of functional groups that effectively mimic hot-spot residues to the scaffold would be expected to result in hDM2 recognition and inhibition of the p53/hDM2 PPI (Fig. 1c). It was envisaged that this new scaffold could adopt relatively well defined conformations through intramolecular hydrogen bonding at the top and bottom of the sequence, but that the α-amino acid may allow access to multiple conformers of similar energy and thus a wider range of pharmacophores.
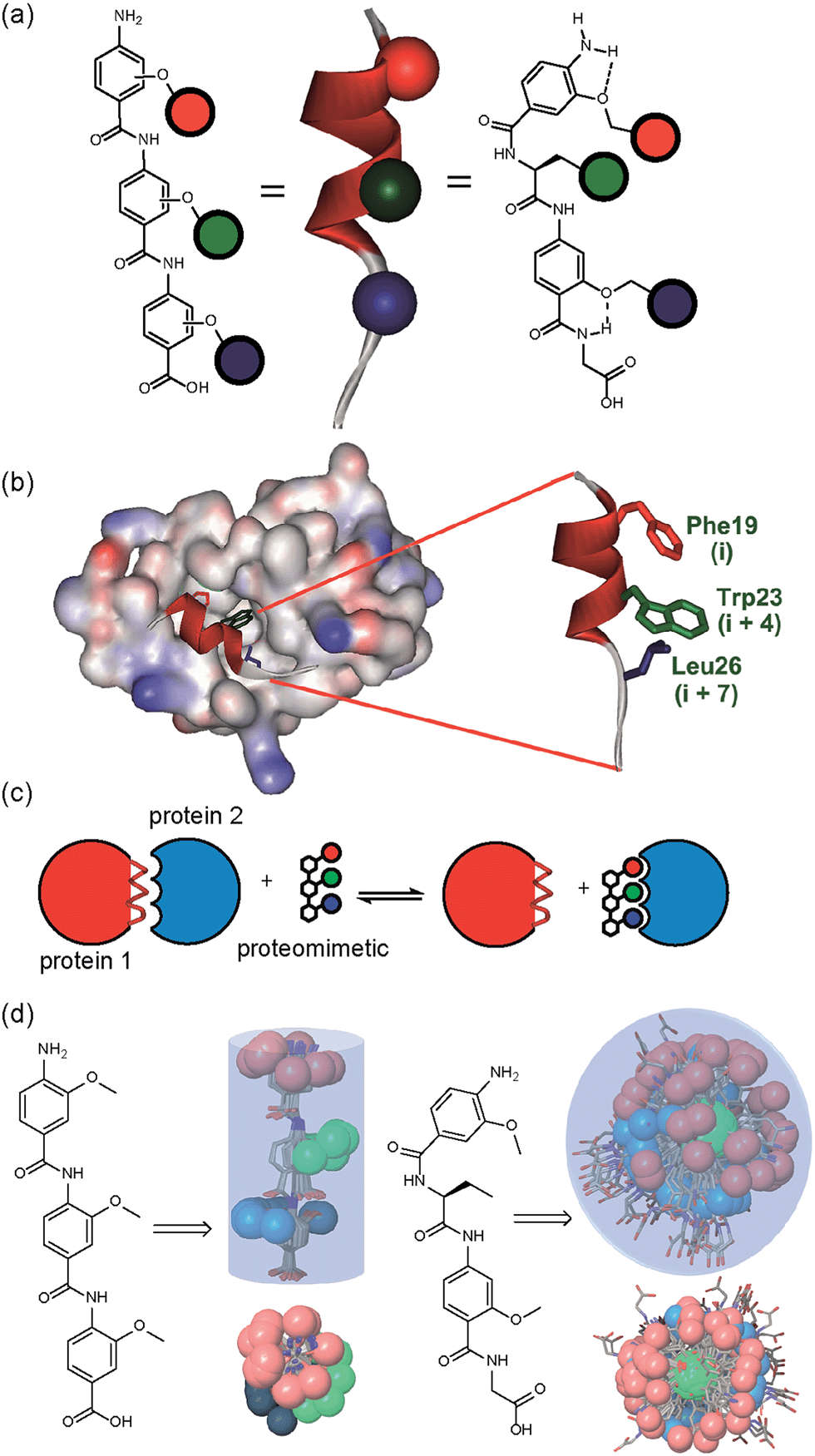 | ||
| Fig. 1 (a) Schematic representing the design of a hybrid α-helix mimetic (right) and its comparison with an O-alkylated benzamide mimetic (left) and an α-helix (middle). (b) p53/hDM2 PPI with expansion on the native p53 peptide (PDB ID:1YCR). (c) Schematic illustrating the process of PPI inhibition with a proteomimetic. (d) Investigation of the accessible conformational space (shown as a shaded 3D object) highlighting the orientation of the side chains (shown in CPK format): side (top left) and top (bottom left) view of a 3-O-alkylated trimer; side (top right) and top (bottom right) view of a hybrid mimetic (i, i + 4 and i + 7 side chains together with residues that mimic them are shown in red, green and blue respectively). | ||
To assess the conformational plasticity further, molecular modelling was used to perform a qualitative comparison of a 3-O-alkylated trimethyl benzamide and a methyl functionalised hybrid mimetic (Fig. 1d). Methyl side chains were chosen to exclude side chain rotations and focus only on the inherent flexibility of the backbone. The structures were minimised by performing a full Monte Carlo search using the software Macromodel® with the MMFFs method. The set of conformers within 1.5 kJ mol−1 of the minimum were superimposed without further manipulation. As shown in Fig. 1d, the 3-O-alkylated oligobenzamide trimer presented different combinations of anti and syn orientations51 of the side chains but the accessible conformational space was restricted by the rigidity of the scaffold and by intramolecular hydrogen bonding such that variation arises only through rotation about each of the Ar–CO axes. In contrast, for the hybrid mimetic a difference of a few degrees in a bond torsion angle can result in diverse side chain orientations. Within the limitations of this qualitative analysis, the study revealed that this new scaffold could therefore mimic a wider range of pharmacophores, suggesting that a greater number of conformers for effective mimicry can be sampled and the possibility for induced-fit binding to occur.
Synthesis of hybrid α-helix mimetics and conformational analyses
In designing the new α-helix mimetic scaffold, a solid phase synthesis (SPS) strategy was desired to allow rapid access to large numbers of compounds/libraries. The synthesis (Scheme 1) uses the well-established Fmoc (9-fluorenylmethyloxycarbonyl) strategy. Hybrid α-helix mimetics were built using an automated microwave assisted CEM Liberty® peptide synthesiser with Fmoc–Gly preloaded Wang resin as solid support. After resin deprotection with piperidine, protected monomers were coupled using HATU ((1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate)). The successful use of HATU as a coupling reagent is particularly noteworthy, as standard peptide synthesis coupling reagents have been previously shown to be ineffective to couple poorly nucleophilic aminobenzoic acid building blocks.52,53 With a synthetic route in hand, a small library of compounds (1–17, Fig. S2–144†) was synthesized, including parent mimetic 1 presenting R1 = iPr, R3 = Bn and L-Phe as the central amino acid. Five control molecules were also prepared (Fig. 2a): three dimeric versions of the hybrid scaffold (2–4), a hybrid with an unfunctionalised p-aminobenzoic acid top unit (5, syntheses shown in Schemes S3 and S4, ESI†) and finally a control with the central aromatic residue replaced with histidine 6 R1 = iPr, R2 = L-His, R3 = Bn. Histidine was chosen as a control rather than alanine, because it is sterically comparable to phenylalanine but is less hydrophobic.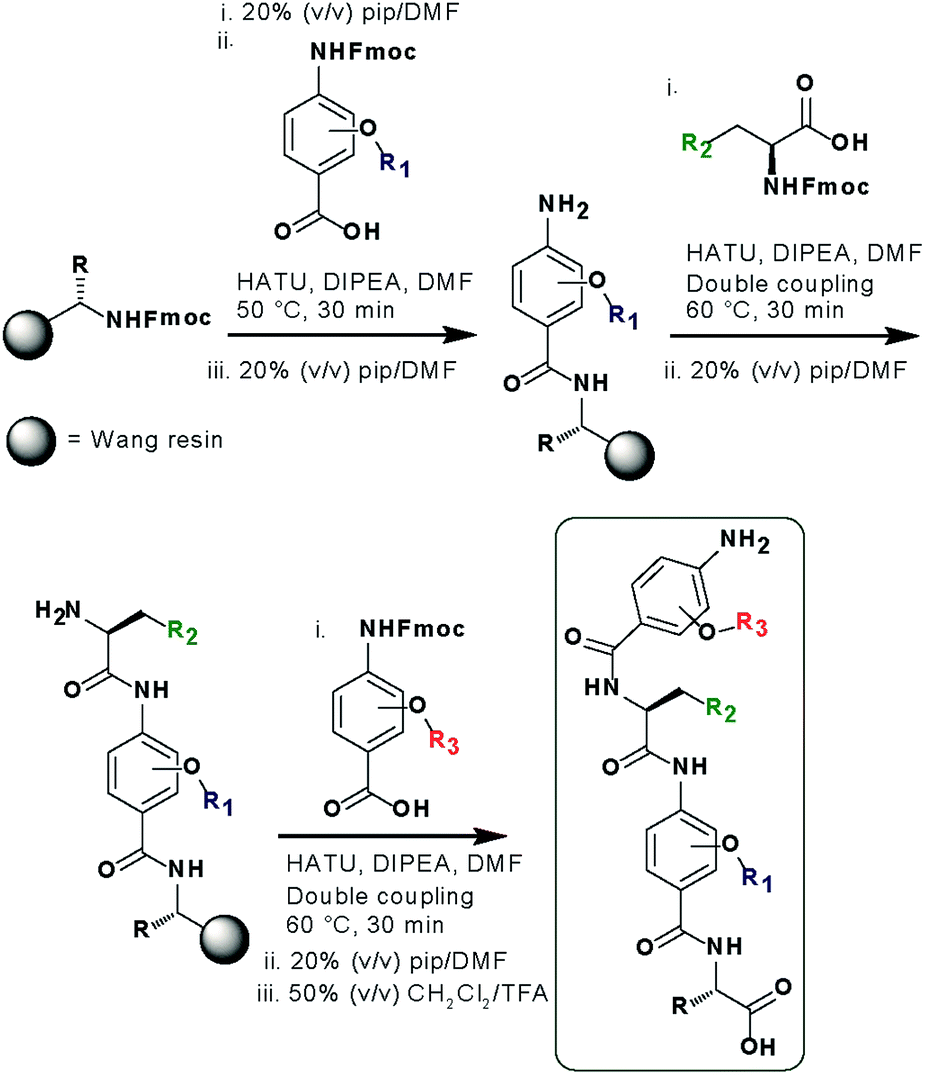 | ||
| Scheme 1 SPS synthesis of hybrid α-helix mimetics. | ||
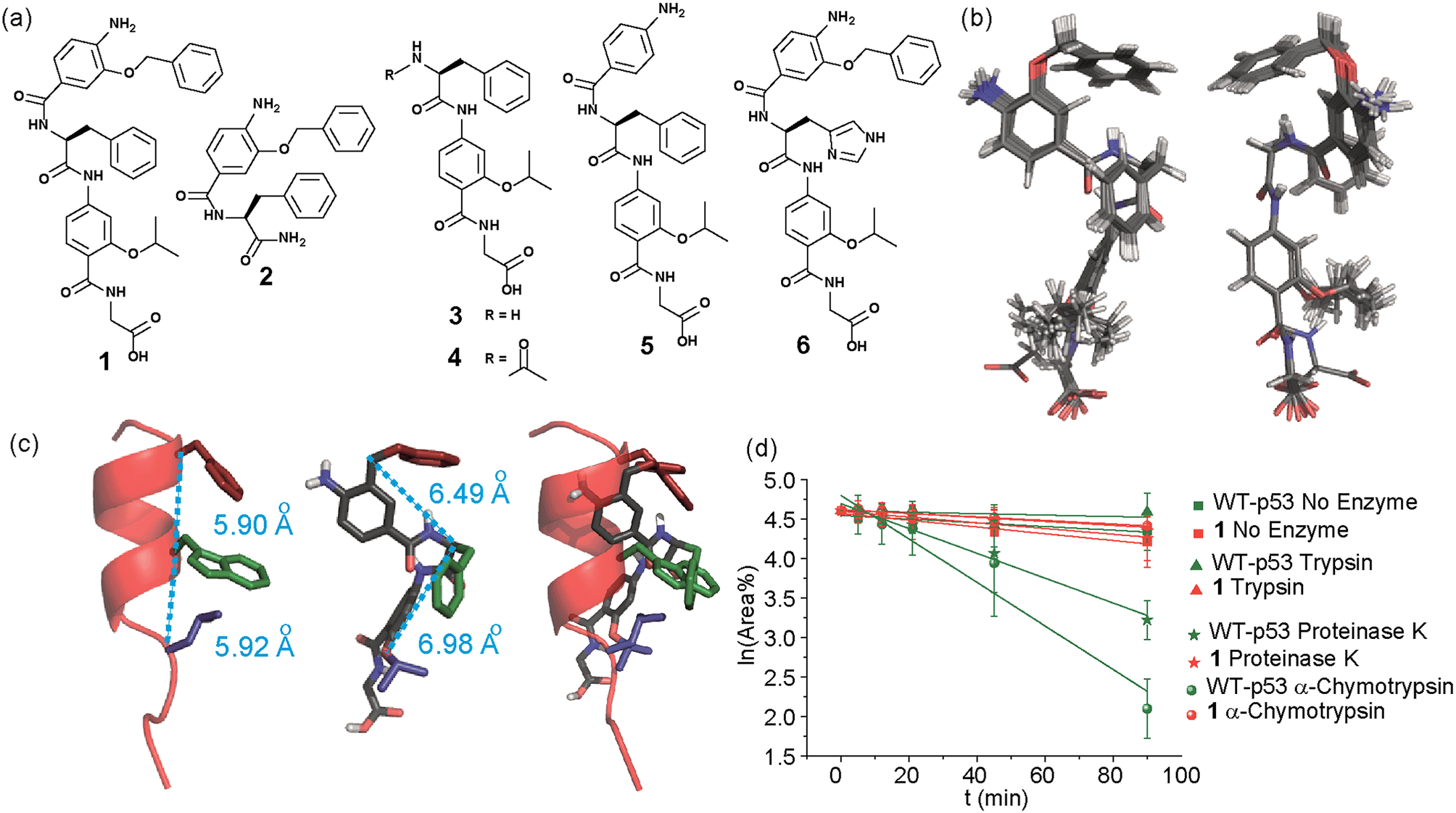 | ||
Fig. 2 (a) Structures of a model hybrid mimetic 1 and five control derivatives 2–6. (b) NMR derived structure of compound 1 (left and right represent the view from opposite faces). (c) Overlay of the NMR structure of mimetic 1 and p53 transactivation domain illustrating good matching of side chains between mimetic and helix together with distances between key positions (left; p53 helix, middle; mimetic 1, right; overlay). (d) Kinetics of degradation from proteolytic studies performed on hybrid 1 (red) and WT-p53 (green) treated with no enzyme (square), trypsin (triangle), α-chymotrypsin (sphere) and proteinase K (star) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10000 enzyme/substrate ratio. :10000 enzyme/substrate ratio. | ||
Solution-state analyses were conducted on the model hybrid mimetic 1 to elucidate its conformational properties. The 2D 1H–1H NOESY experiment (Fig. S145 and 146†) showed nOe correlations between the amide protons of the top two units and the ArCH resonances in the ortho position of the adjacent monomer units, indicating free rotation around the Ar–CO and NH–Ar axes. Furthermore, nOe correlations between the NH and the Hα proton of the isopropyl side chain, together with the absence of cross peaks to the ArCH resonances in the ortho and meta positions of the adjacent monomer unit indicated restricted rotation around this Ar–CO axis and intramolecular S(6) H-bonding with the oxygen of the adjacent isopropyl moiety. Intramolecular H-bonding was further confirmed by dilution and variable temperature (VT) NMR studies (see ESI, Fig. S147 and 148†). The fully assigned spectra were used to produce a model of the minimum energy structure of helix mimetic 1. Assigned ROESY cross-peaks were integrated using SPARKY 3.111 (ref. 54) and volumes converted to distances with reference to the fixed and known distance between the H5 and H6 protons of residue 4 (the N-terminal amino benzoic acid). These distances were then used within CNS-solve55 to generate a set of low energy structures, following a simulated annealing process, all satisfying the distance constraints, in a similar manner to that adopted recently for a similar sized peptide.56 Of particular note are strong, inter-residue rOes (indicative of short inter-proton separations) between 4-H2 and 3-Phe–NH, 4-H2 and 3-Phe–Hβ. The data indicate the presence of one major conformer (although it is noteworthy that additional nOe's (and rOe's) were indicative of a second conformer, for which there was insufficient data to obtain a structure). The structure obtained (Fig. 2b) illustrates the minimum energy structure – as can be seen, all the side chains are presented on one face of the molecule as is required for effective mimicry of the helix. Thus, the NMR data would appear to indicate that the conformational landscape of mimetic 1 is more well-defined than is indicated by the modelling, although the two methods are not necessarily comparable. Furthermore, this does not preclude the mimetic from readily adopting different conformations in the presence of a target protein. Nonetheless, structure alignment with the p53 transactivation domain, as shown in Fig. 3b (and also Fig. S162 and 163†), further indicates the compound is capable of effective α-helix mimicry (Fig. 2c); each side chain of the mimetic can adopt an orientation that overlays well with the Phe19, Trp23 and Leu26 side chains of p53.
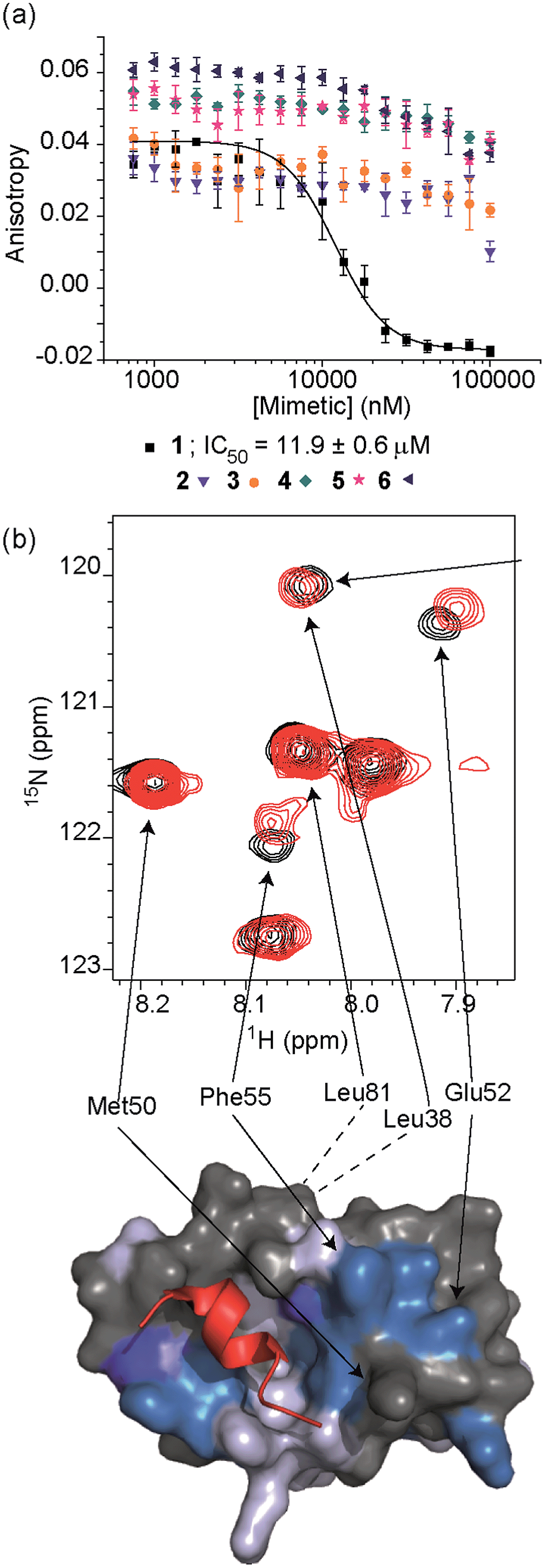 | ||
| Fig. 3 (a) Dose–response curves of hybrids 1 (black), 2 (purple), 3 (orange), 4 (dark cyan), 5 (magenta) and 6 (dark blue) in a FA competition assay against the p53/hDM2 PPI (errors bars correspond to three experimental replicates). (b) Top; partial 1H–15N HSQC spectrum of the 15N-labelled 125 μM solution of hDM2 (black) overlaid with the HSQC spectrum of hDM2 (125 μM) in complex with a 200 μM solution of 1 (red), highlighting shift changes (dark blue; large shift, light blue; medium shift, grey; no shift, white unassigned), of pertinent residues (600 MHz, 100 mM sodium phosphate buffer pH 7.3, 2.5% glycerol, 1 mM DTT, 6% DMSO, 25 °C), bottom; chemical shift perturbation mapping onto the crystal structure of p53/hDM2 (PDB ID:1YCR). | ||
We also performed proteolysis studies on mimetic 1 to ascertain to what extent introduction of an α-amino acid in the middle of the hybrid sequence could render the mimetics susceptible to enzymatic degradation. The wild type (WT) p53 helix was also used for direct comparison. α-Chymotrypsin and proteinase K were chosen for this analysis, as these enzymes preferentially cleave amide bonds adjacent to aromatic functionalities. Trypsin was used as a control, as this protease cleaves amide bonds adjacent to arginine or lysine residues and therefore should not degrade either substrate. The mimetic 1 and WT-p53 were treated with each enzyme (see ESI for further details and Fig. S149 and 150†) and degradation was followed by analytical HPLC. The data obtained were then analysed to extract kinetic values (ESI†). As shown in Fig. 2d, the hybrid mimetic displayed complete resistance to all three proteases.
Selective inhibition of the p53/hDM2 PPI
To test the potential of the proteomimetic scaffold to act as a functional α-helix mimetic, hybrid 1 and controls 2–6 were tested in a p53/hDM2 fluorescence anisotropy (FA) competition assay (Fig. 3a and Table 1). Hybrid 1 displayed low micromolar inhibition of this PPI (IC50 of 11.9 ± 0.6 μM) whereas, Nutlin-3a – a well known and potent p53/hDM2 inhibitor,57 displayed an IC50 of 0.53 ± 0.02 μM in this assay (see ESI Fig. S155†). The value compares well with a previously described 3-O-alkylated aromatic oligomer we described previously (R1 = Bn, R2 = 2-Bn, R3 = iPr; IC50 of 5.1 ± 0.4 μM).40 Although mimetic 1 was shown to be a weaker inhibitor than Nutlin-3a, it represents an impressive starting point for a 1st generation compound and augurs well for further potency enhancement in future studies. The data also demonstrated that the entirety of the sequence was required to mimic the key ‘hot spot’ residues and therefore achieve inhibition. In this regard, the inactivity of hybrid 2 indicated that the ‘bottom’ unit plays a significant role in binding. Furthermore, absence of activity for hybrids 3 and 4 proved that the ‘top’ monomer was also essential for activity and the lack of inhibition of hybrid 5 revealed that this unit needs to be functionalised with an interacting side chain in order to achieve molecular recognition and retain binding affinity. Finally, substitution of the central L-phenylalanine residue with a more hydrophilic residue (hybrid 6) – L-histidine – abrogated inhibition demonstrating the importance of the composition of side chains.| Compound | p53/hDM2 | Mcl-1/NOXA-B |
|---|---|---|
| Nutlin-3 | 0.53 ± 0.02 μM | |
| 1 | 11.9 ± 0.6 μM | >100 μM |
| 2 | No inhibition | No inhibition |
| 3 | No inhibition | No inhibition |
| 4 | No inhibition | No inhibition |
| 5 | No inhibition | No inhibition |
| 6 | No inhibition | No inhibition |
| 9 | 21.2 ± 2.5 μM | >100 μM |
| 12 | 59.7 ± 31.9 μM | >100 μM |
| 15 | 9.2 ± 0.4 μM | 27.1 ± 1.1 μM |
| 16 | 11.5 ± 0.3 μM | 24.1 ± 1.4 μM |
| 17 | 25.2 ± 1.4 μM | >100 μM |
The binding mode of the hybrid mimetic 1 was further investigated via1H–15N HSQC studies. HSQC spectra were acquired for either the 15N labelled apo form of the protein (125 μM protein 100 mM sodium phosphate buffer at pH 7.3, 2.5% glycerol, 1 mM DTT, 5% DMSO, 25 °C) or the protein in complex with a 200 μM solution of hybrid 1 after overnight incubation; distinct shifts were induced upon addition of 1. Once mapped onto the crystal structure of p53/hDM2 (PDB ID:1YCR; Fig. 2f),32 the study showed that changes were induced that were comparable to those observed previously for WT-p53 peptide.41 Shifts of residues around the helix binding cleft such as Phe55 and His73 (not shown in the expansion shown in Fig. 3b), which are located at opposite edges of the hydrophobic cleft, support the notion that the mimetic binds in the peptide binding site. Notably, shift changes at both ends of the hDM2 cleft also supported the hypothesis that the hybrid adopts an extended conformation. We also tested control compound 3 in the 1H–15N HSQC experiment (see ESI Fig. S164 and 165†): only minor shifts in the HSQC spectrum were observed supporting further the notion of a direct and specific interaction of 1 with hDM2.
We then tested the compounds against four further protein–protein interactions using FA competition assays (see ESI†) to ascertain the extent to which mimetic 1 acts as a selective inhibitor. We selected these targets having developed assays on each in prior work.40,58–60 No inhibition of Bcl-xL/BAK,61 HIF-1α/p30062 or eIF4E/eIF4G63 (see ESI Fig. S159–161†) was observed whilst only weak inhibition of Mcl-1/NOXA-B33 interaction (>100 μM, Table 1) was observed (see below). Proteins of the Bcl-2 family have a central role in the regulation of apoptosis64 and have attracted attention as targets for molecular therapeutics.19 Since BH3-only pro apoptotic proteins of this family mediate PPIs through three or four key residues of an α-helix placed along one face, we envisaged that hybrid α-helix mimetics might also bind to their antiapoptotic partner, particularly given that p53 itself has been shown to interact with both Mcl-1 and Bcl-xL.65 Thus the absence of strong inhibition for these targets is particularly noteworthy.
The role of side-chain spacing
The results obtained for hybrids 1–6 suggested that the compositional properties of these mimetics play a key role for effective recognition of the target protein. To investigate the role of spacing between interacting side chains, we designed and synthesised hybrids 7–14 including a combination of 2-O, 3-O and N-alkylated monomers (Fig. 4a–c and Table 1 for key compounds). Since the alkylation topography is different for each of these building blocks, spacing between side chain residues is different and should impact upon inhibitor potency if this is an essential feature for inhibition of the PPI. This role of side chain spacing has not been effectively explored in previous studies on helix mimetics.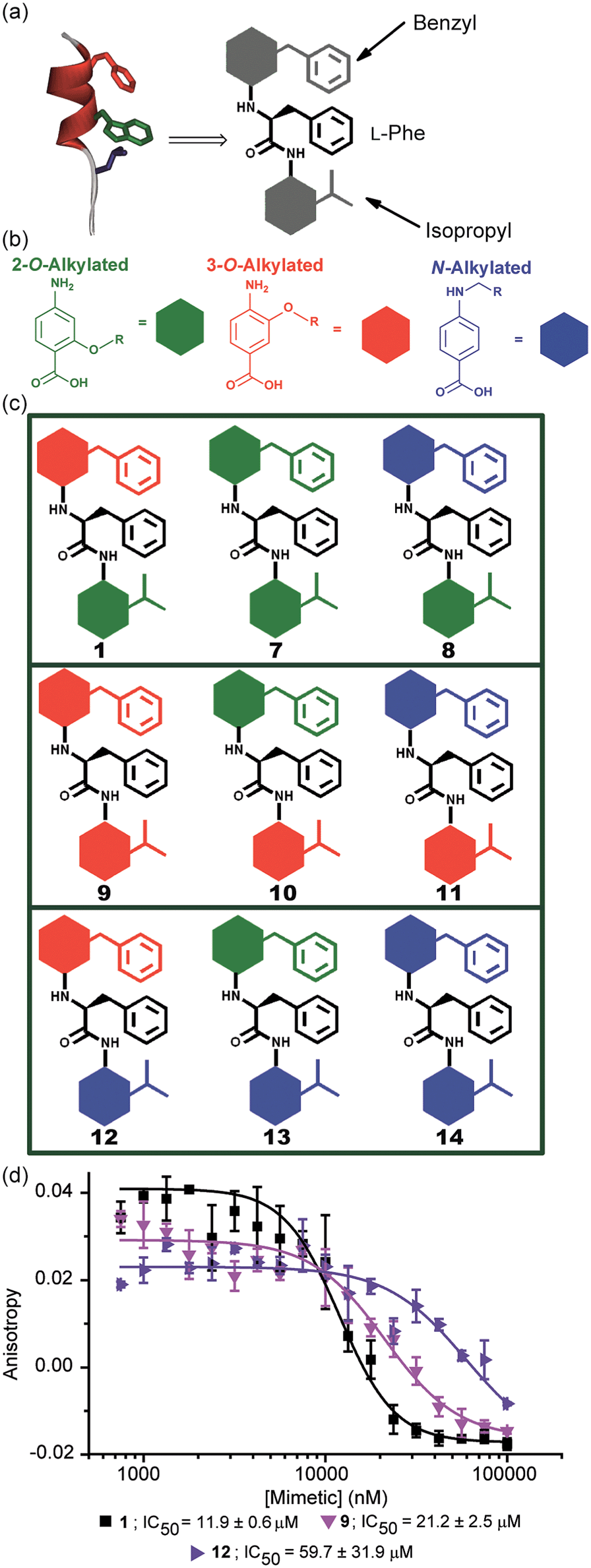 | ||
| Fig. 4 (a) Cartoon depicting the design of a library of hybrid mimetics for sequence-dependent SAR studies. (b) 2-O (green), 3-O (red) and N-alkylated (blue) aminobenzoic acid building blocks; (c) cartoon representation of the library of hybrid mimetics for sequence-dependent SAR studies; (d) dose–response curves of hybrids 1 (black), 9 (magenta) and 12 (violet) in a FA competition assay against the p53/hDM2 PPI. | ||
The mimetics were divided into three families (2-O, 3-O and an N-alkylated series, named after the first monomeric unit at the bottom of the sequence), and were tested in the p53/hDM2 FA competition assay. Full competition curves for the whole library are shown in Fig. S152–154 (ESI†) and revealed a similar trend of activity within each family. Fig. 4d shows the results for hybrids 1, 9 and 12, which are representative of each series. Mimetics containing a 2-O alkylated building block at the bottom position (6.98 Å between O and α position) displayed better inhibitory activity than the corresponding 3-O-alkylated hybrids (4.50 Å between O and α position, see Fig. S178†) and five fold increased potency than hybrids of the N-alkylated series (3.10 Å between N and α position, see Fig. S178†). The result demonstrates that the side chain spacing has a significant effect on the binding affinity, as a preferential sequence was identified. Thus, mimetic 1 with the largest distance between the R1 and R2 side chains (mimicking the i + 7 and i + 4 positions of the helix) is optimal for recognition of hDM2 and inhibition of the p53/hDM2 interaction in this instance. The result is significant; despite having a different degree of backbone curvature, the previously reported regioisomeric 2-O and 3-O-alkylated oligobenzamide mimetics were shown to have comparable potency for inhibition of the p53/hDM2 interaction36 because free rotation of multiple bonds within a repeating structure permit similar vectoral presentation of hot-spot mimicking residues. Here, for the hybrid mimetic 1 – which might be thought of as more flexible due to greater variation in the accessible conformational space of the scaffold – the irregular backbone allows the spatial relationship to be varied for only two of the three residues in the mimetic at a time and so the distance between these residues must differ. As the hDM2 cleft is hydrophobic in nature, the preliminary Structure Activity Relationship (SAR) data for 1–6 might simply reflect an increase in non-specific hydrophobic interactions, however the fact that inhibitory potency varies for the series 1, 9 and 12 critically illustrates that in addition to the compositional complementarity of the helix mimetic and protein-surface, there must also be some shape complementarity. Finally, the series 1 and 7–14 was also tested in the Mcl-1/NOXA-B competition assay. All these mimetics showed little inhibitory activity against this PPI (Table 1 and full competition curves shown in Fig. S156–158†) indicating a good level of selectivity towards recognition of the hDM2 cleft is retained.
Stereodependent inhibitory behaviour of α-helix mimetics
Whilst several helix mimetics incorporate stereogenic centres in the backbone or chiral appendages,29,48,50,66–70 a role for stereochemistry in molecular recognition has not been demonstrated. The use of an α-amino acid as the central monomer of the mimetic allows access to enantiomeric helix mimetics and thus permits the role of stereochemistry on inhibitory activity to be probed. The chirality of many small molecules is pivotal for effective binding to their target and represents a hallmark of specific and selective molecular recognition. We therefore synthesised the enantiomer of 1i.e.15 with a D-Phe residue (Fig. 5a). Disappointingly, there was little difference in the inhibitory potencies of 1 and 15 against the p53/hDM2 interaction (Fig. 5b and Table 1). In our preliminary selectivity studies (above) we had observed weak inhibition of the Mcl-1/NOXA-B interaction with compound 1, however the compound was not sufficiently potent to obtain a full competition curve in this assay. We therefore attempted to confirm that the role of stereochemistry was not important in this series of helix mimetics by studying the inhibition of the Mcl-1/NOXA-B interaction with 15. Significantly, 15 was observed to act as an inhibitor of the Mcl-1/NOXA-B interaction being around one order of magnitude more potent than the L-Phe variant 1 (Fig. 5b and Table 1). It is also noteworthy that the Mcl-1/NOXA-B interaction involves a significantly longer helix than does p53/hDM2 (20 versus 9 residues) hence the result with 15 demonstrates that the scaffold may be useful for recognition of longer helix binding clefts. We performed 1H–15N HSQC perturbation shift studies to investigate this behaviour. The HSQC of the complex 15/hDM2 (see ESI, Fig. S166 and 167†) was consistent with the perturbation shifts obtained with hybrid 1. Shown in Fig. 5c is the hDM2 structure with shifts mapped onto the surface – as can be seen these are similar in nature for both 1 and 15 emphasising the absence of any difference in inhibitory potency. 1H–15N HSQC analyses of both hybrids in complex with Mcl-1 (see Fig. S168–171†), indicated different behaviour for 1 and 15 (Fig. 5d). For compound 15, a significantly higher number of resonances exhibit a larger shift or disappear completely from the 1H–15N HSQC spectrum of Mcl-1 than for compound 1. This disappearance of resonances is significant as it indicates slower exchange and therefore higher affinity binding. Again, we observed only minor shifts in the 1H–15N HSQC spectrum upon addition of the non-binding control compound 3 (Fig. S172 and 173†).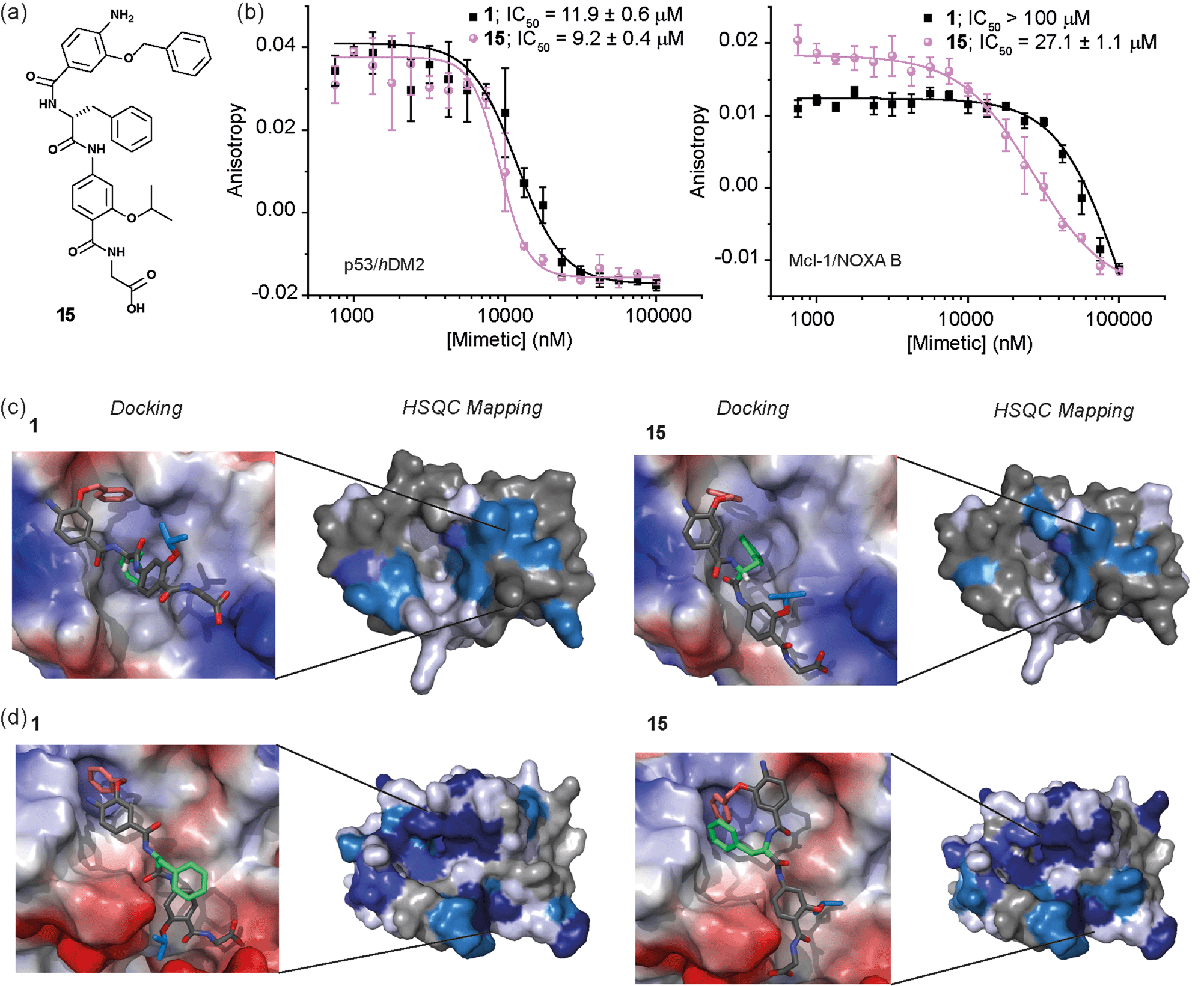 | ||
| Fig. 5 (a) Chemical structure of the D-Phe functionalised hybrid mimetic 15. (b) Dose–response curves of L-Phe (1) and D-Phe (15) hybrid mimetics in FA competition assays against p53/hDM2 (left) and Mcl-1/NOXA-B (right). (c) Docking and 1H–15N HSQC perturbation shift studies (dark blue; large shift/peak disappears, light blue; medium shift, grey; no shift, white unassigned) with hDM2 (PDB ID:1YCR): docked hybrid 1 with protein surface 3D representation and chemical shift mapping (left), docked hybrid 15 with protein surface 3D representation and chemical shift mapping (right). (d) Docking and 1H–15N HSQC perturbation shift studies with Mcl-1 (PDB ID:2JM6): docked hybrid 1 with protein surface 3D representation and chemical shift mapping (left), docked hybrid 15 with protein surface 3D representation and chemical shift mapping (right). Spectra recorded at 600 MHz, in 20 mM HEPES pH 7.0, 50 mM NaCl, 0.5 mM DTT, 2.5% glycerol, 5% DMSO at 25 °C. | ||
To provide a molecular hypothesis for this behaviour, we also performed docking studies. The structures of hybrids 1 and 15 were minimised by performing a full Monte Carlo search using the software Macromodel® with the MMFFs method. The set of structures within 1.5 kJ mol−1 from the lowest energy conformation was initially docked with the crystal structure of hDM2 (PDB ID:1YCR) using the software Glide®. Of all the poses generated from hybrids 1 and 15, 77% and 69% respectively assumed conformations which were binding in the hDM2 cleft. Representative examples are shown in Fig. 5c. The docking results indicate that the mimetics are indeed capable of acting as structural mimics of the α-helix and suggest why there is no discrimination between the enantiomers 1 and 15, employing L-Phe and D-Phe respectively; clearly both are capable of adopting conformations where the top residue and the amino acid can occupy the Phe19 and Trp23 pockets respectively. Docking experiments were also performed with the crystal structure of Mcl-1 (PDB ID:2JM6). Only 3% of the poses generated from L-hybrid 1 assumed conformations which were binding in the Mcl-1 cleft. Most of the structures instead adopt conformations where the backbone of the two bottom residues binds to the cleft thus inducing the side chains to engage in interactions with amino acids outside the NOXA-B binding pocket. A representative example is shown in Fig. 5d. Within the constraints of the method (limitations of the force fields, assumptions regarding conformational restrictions and media for simulation), this analysis indicates that the side chains of this hybrid are not matched to the NOXA-B sequence and that this molecule does not act as a good mimetic of the NOXA-B helix. On the other hand, of the poses generated from hybrid 15, 62% assumed conformations binding into the Mcl-1 cleft (Fig. 5d). The D-hybrid mimetic binds in the Mcl-1 cleft with all three residues through hydrophobic contacts (between the benzyl side chain and Phe251, between the middle Phe residue and Phe209/Ala208 and between the isopropyl side chain and Val246). These interactions suggest good matching between the position of the side chains of this mimetic to the NOXA-B sequence representing a possible explanation for the enantioselective recognition of the mimetic 15 over 1 by the Mcl-1 protein. Why Mcl-1 discriminates between 1 and 15 more effectively than does hDM2 is unclear at this stage, however the helix binding clefts differ in terms of shape and composition – how the mimetic is able to exploit its conformational landscape to target the respective proteins will be the focus of future studies.
The role of stereochemistry was further highlighted using hybrids 16 and 17, which presented a tryptophan in lieu of the phenylalanine to mimic the natural ‘hot-spot’ residue of p53 (Fig. 6a). These compounds were synthesized in the expectation that incorporation of the native helix side chain within the hybrid mimetic might enhance binding to hDM2, however this was not observed, perhaps indicating that the scaffold is not yet optimal for perfect helix mimicry and that it needs further refinement in future studies. The biophysical analyses with both hDM2 and Mcl-1 however, reveal additional binding properties that depend on mimetic chirality. Firstly, as for 1 and 15, the protein selectivity is influenced by mimetics stereochemistry (Fig. 6b); for 17 the preference for hDM2 over Mcl-1 is four-fold; where for the Phe derivatives 1 and 15 the D-derivative bound to Mcl-1, for Trp derivatives 16 and 17 the opposite trend i.e.L-Trp residue is preferred. The reason for a difference in enantiopreference is unclear at this stage. Another pivotal feature, not as clearly observed for the Phe series, was shown by this series as each of the proteins exhibit a preference for one of the enantiomers. In this instance, hDM2 displays a 2 fold preference for the L-Trp hybrid 16 over 17, whereas for Mcl-1 the preferences is at least four-fold. Once again docking studies supported the experimental trend for inhibition (see Fig. S174–177†).
 | ||
| Fig. 6 (a) Chemical structure of the Trp-functionalised hybrid mimetics 16 and 17. (b) Dose–response curves of hybrid mimetics in FA competition assays against p53/hDM2 (top) and Mcl-1/NOXA-B (bottom). | ||
Conclusions
We have described the design, synthesis and characterization of a new oligoamide proteomimetic scaffold. In order for the scaffold to act as a ligand for protein surface recognition, we illustrate that the side-chains mimicking the α-helical template upon which they are based must be positioned appropriately to effectively reproduce the spatial and angular projection of the i, i + 4 and i + 7 side-chains of a canonical α-helix and present appropriate composition to recapitulate these hot-spot residues. The stereochemistry of the central monomer of our hybrids was found to play a significant role in the inhibition of target PPIs. In particular, as would be required for an information rich oligomer exhibiting defined molecular recognition, both hDM2 and Mcl-1 differentiate between enantiomers of the same helix mimetic. Finally, the initial mimetic 1 was observed to be a selective inhibitor of the p53/hDM2 interaction when compared with 4 other PPIs. However, we were able to show that its protein selectivity could be modulated simply by varying the central chiral unit in the mimetic, thus leading to stereodependent selectivity or dual inhibition of p53/hDM2 and Mcl-1/NOXA-B. A definitive explanation for the difference in stereodependent recognition and protein selectivity requires further study and, similarly, the binding affinity of the mimetic might be further optimised in future work. However, the primary goal of the current study, was to demonstrate that, when appropriately functionalized, the scaffold developed herein exhibits all the hallmarks expected of an information rich oligomer and therefore is well-suited for future sequence based design. Specifically, the combination of stereodependent recognition behaviour, married with correct side chain spacing as observed in this work is unprecedented for helix mimetics. Of consequence for future studies is the observation that this can be achieved using a scaffold that has considerable flexibility in terms of its conformational properties, which contrasts with the expectation that a helix mimetic should employ a rigid scaffold. As our results clearly illustrate, the scope for induced fit binding and conformational selection should also be accounted for. Finally, this new scaffold represents the most simple helix mimetic described to date in terms of its synthesis (achieved by SPS) and functional group tolerance, and hence will allow access to large libraries of helix mimetics48,71,72 facilitating high-throughput studies of protein-surface recognition.Acknowledgements
This work was supported by the European Research Council [ERC-StG-240324] and The Wellcome Trust [104920/Z/14/Z]. We would like to thank George Burslem and Hannah Kyle for assistance with modelling and testing of compound 1 in FA assays and Anna Barnard for useful discussions. The authors thank Arnout Kalverda and Gary Thompson for assistance with NMR experiments.Notes and references
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- Foldamers: Structure, Properties, and Applications, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, 2007 Search PubMed.
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- L. Delaurière, Z. Dong, K. Laxmi-Reddy, F. Godde, J.-J. Toulmé and I. Huc, Angew. Chem., Int. Ed., 2012, 51, 473–477 CrossRef PubMed.
- C. Mayer, M. M. Müller, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 6978–6981 CrossRef CAS PubMed.
- P. B. Dervan and B. S. Edelson, Curr. Opin. Struct. Biol., 2003, 13, 284–299 CrossRef CAS PubMed.
- R. i. E. McGovern, H. Fernandes, A. R. Khan, N. P. Power and P. B. Crowley, Nat. Chem., 2012, 4, 527–533 CrossRef CAS PubMed.
- D. Bier, R. Rose, K. Bravo-Rodriguez, M. Bartel, J. M. Ramirez-Anguita, S. Dutt, C. Wilch, F.-G. Klärner, E. Sanchez-Garcia, T. Schrader and C. Ottmann, Nat. Chem., 2013, 5, 234–239 CrossRef CAS PubMed.
- L.-G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Chem. Rev., 2014, 114, 4695–4748 CrossRef CAS PubMed.
- W. S. Horne, M. D. Boersma, M. A. Windsor and S. H. Gellman, Angew. Chem., Int. Ed., 2008, 47, 2853–2856 CrossRef CAS PubMed.
- M. D. Boersma, H. S. Haase, K. J. Peterson-Kaufman, E. F. Lee, O. B. Clarke, P. M. Colman, B. J. Smith, W. S. Horne, W. D. Fairlie and S. H. Gellman, J. Am. Chem. Soc., 2011, 134, 315–323 CrossRef PubMed.
- B. J. Smith, E. F. Lee, J. W. Checco, M. Evangelista, S. H. Gellman and W. D. Fairlie, ChemBioChem, 2013, 14, 1564–1572 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- T. Edwards and A. Wilson, Amino Acids, 2011, 41, 743–754 CrossRef CAS PubMed.
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS PubMed.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS PubMed.
- O. Keskin, A. Gursoy, B. Ma and R. Nussinov, Chem. Rev., 2008, 108, 1225–1244 CrossRef CAS PubMed.
- I. Ray-Coquard, J.-Y. Blay, A. Italiano, A. Le Cesne, N. Penel, J. Zhi, F. Heil, R. Rueger, B. Graves, M. Ding, D. Geho, S. A. Middleton, L. T. Vassilev, G. L. Nichols and B. N. Bui, Lancet Oncol., 2012, 13, 1133–1140 CrossRef CAS PubMed.
- W. H. Wilson, O. A. O'Connor, M. S. Czuczman, A. S. LaCasce, J. F. Gerecitano, J. P. Leonard, A. Tulpule, K. Dunleavy, H. Xiong, Y.-L. Chiu, Y. Cui, T. Busman, S. W. Elmore, S. H. Rosenberg, A. P. Krivoshik, S. H. Enschede and R. A. Humerickhouse, Lancet Oncol., 2010, 11, 1149–1159 CrossRef CAS PubMed.
- B. P. Orner, J. T. Ernst and A. D. Hamilton, J. Am. Chem. Soc., 2001, 123, 5382–5383 CrossRef CAS PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed.
- J. L. LaBelle, S. G. Katz, G. H. Bird, E. Gavathiotis, M. L. Stewart, C. Lawrence, J. K. Fisher, M. Godes, K. Pitter, A. L. Kung and L. D. Walensky, J. Clin. Invest., 2012, 122, 2018–2031 CAS.
- E. F. Lee, B. J. Smith, W. S. Horne, K. N. Mayer, M. Evangelista, P. M. Colman, S. H. Gellman and W. D. Fairlie, ChemBioChem, 2011, 12, 2025–2032 CrossRef CAS PubMed.
- R. W. Cheloha, A. Maeda, T. Dean, T. J. Gardella and S. H. Gellman, Nat. Biotechnol., 2014, 32, 653–655 CrossRef CAS PubMed.
- E. A. Harker and A. Schepartz, ChemBioChem, 2009, 10, 990–993 CrossRef CAS PubMed.
- T. Clackson and J. A. Wells, Science, 1995, 267, 383–386 CrossRef CAS PubMed.
- H. Yin, G.-I. Lee, H. S. Park, G. A. Payne, J. M. Rodriguez, S. M. Sebti and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 2704–2707 CrossRef CAS PubMed.
- T. Shahian, G. M. Lee, A. Lazic, L. A. Arnold, P. Velusamy, C. M. Roels, R. K. Guy and C. S. Craik, Nat. Chem. Biol., 2009, 5, 640–646 CrossRef CAS PubMed.
- B. B. Lao, K. Drew, D. A. Guarracino, T. F. Brewer, D. W. Heindel, R. Bonneau and P. S. Arora, J. Am. Chem. Soc., 2014, 136, 7877–7888 CrossRef CAS PubMed.
- A. Kazi, J. Sun, K. Doi, S.-S. Sung, Y. Takahashi, H. Yin, J. M. Rodriguez, J. Becerril, N. Berndt, A. D. Hamilton, H.-G. Wang and S. M. Sebti, J. Biol. Chem., 2011, 286, 9382–9392 CrossRef CAS PubMed.
- S. Kushal, B. B. Lao, L. K. Henchey, R. Dubey, H. Mesallati, N. J. Traaseth, B. Z. Olenyuk and P. S. Arora, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15602–15607 CrossRef CAS PubMed.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS PubMed.
- C. L. Day, C. Smits, F. C. Fan, E. F. Lee, W. D. Fairlie and M. G. Hinds, J. Mol. Biol., 2008, 380, 958–971 CrossRef CAS PubMed.
- G. K. Mittapalli, K. R. Reddy, H. Xiong, O. Munoz, B. Han, F. De Riccardis, R. Krishnamurthy and A. Eschenmoser, Angew. Chem., Int. Ed., 2007, 46, 2470–2477 CrossRef CAS PubMed.
- I. Saraogi and A. D. Hamilton, Biochem. Soc. Trans., 2008, 36, 1414–1417 CrossRef CAS PubMed.
- V. Azzarito, P. Prabhakaran, A. I. Bartlett, N. S. Murphy, M. J. Hardie, C. A. Kilner, T. A. Edwards, S. L. Warriner and A. J. Wilson, Org. Biomol. Chem., 2012, 10, 6469–6472 CAS.
- D. Xin, E. Ko, L. M. Perez, T. R. Ioerger and K. Burgess, Org. Biomol. Chem., 2013, 11, 7789–7801 CAS.
- E. Ko, A. Raghuraman, L. M. Perez, T. R. Ioerger and K. Burgess, J. Am. Chem. Soc., 2012, 135, 167–173 CrossRef PubMed.
- J. M. Rogers, A. Steward and J. Clarke, J. Am. Chem. Soc., 2013, 135, 1415–1422 CrossRef CAS PubMed.
- J. P. Plante, T. Burnley, B. Malkova, M. E. Webb, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Commun., 2009, 5091–5093 RSC.
- F. Campbell, J. P. Plante, T. A. Edwards, S. L. Warriner and A. J. Wilson, Org. Biomol. Chem., 2010, 8, 2344–2351 CAS.
- A. Roy, P. Prabhakaran, P. K. Baruah and G. J. Sanjayan, Chem. Commun., 2011, 47, 11593–11611 RSC.
- M. Kudo, V. Maurizot, B. Kauffmann, A. Tanatani and I. Huc, J. Am. Chem. Soc., 2013, 135, 9628–9631 CrossRef CAS PubMed.
- S. S. Kale, S. M. Kunjir, R. L. Gawade, V. G. Puranik, P. R. Rajamohanan and G. J. Sanjayan, Chem. Commun., 2014, 50, 2886–2888 RSC.
- K. K. Hoe, C. S. Verma and D. P. Lane, Nat. Rev. Drug Discovery, 2014, 13, 217–236 CrossRef PubMed.
- F. Lu, S.-W. Chi, D.-H. Kim, K.-H. Han, I. D. Kuntz and R. K. Guy, J. Comb. Chem., 2006, 8, 315–325 CrossRef CAS PubMed.
- R. Fasan, R. L. A. Dias, K. Moehle, O. Zerbe, D. Obrecht, P. R. E. Mittl, M. G. Grütter and J. A. Robinson, ChemBioChem, 2006, 7, 515–526 CrossRef CAS PubMed.
- A. Shaginian, L. Whitby, S. Hong, I. Hwang, B. Farooqi, M. Searcey, J. Chen, P. Vogt and D. Boger, J. Am. Chem. Soc., 2009, 131, 5564–5572 CrossRef CAS PubMed.
- R. Hayashi, D. Wang, T. Hara, J. A. Iera, S. R. Durell and D. H. Appella, Bioorg. Med. Chem., 2009, 17, 7884–7893 CrossRef CAS PubMed.
- Z. Z. Brown, K. Akula, A. Arzumanyan, J. Alleva, M. Jackson, E. Bichenkov, J. B. Sheffield, M. A. Feitelson and C. E. Schafmeister, PLoS One, 2012, 7, e45948 CAS.
- P. Prabhakaran, V. Azzarito, T. Jacobs, M. J. Hardie, C. A. Kilner, T. A. Edwards, S. L. Warriner and A. J. Wilson, Tetrahedron, 2012, 68, 4485–4491 CrossRef CAS.
- N. Murphy, P. Prabhakaran, V. Azzarito, J. Plante, M. Hardie, C. Kilner, S. Warriner and A. Wilson, Chem.–Eur. J., 2013, 19, 5546–5550 CrossRef CAS PubMed.
- K. Long, T. Edwards and A. Wilson, Bioorg. Med. Chem., 2013, 21, 4034–4040 CrossRef CAS PubMed.
- T. D. Goddard and D. G. Kneller, Sparky 3, University of California, San Fransisco, USA, 2006 Search PubMed.
- A. T. Brünger, P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J.-S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson and G. L. Warren, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 905–921 CrossRef.
- P. A. Lichtor and S. J. Miller, J. Am. Chem. Soc., 2014, 136, 5301–5308 CrossRef CAS PubMed.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS PubMed.
- G. M. Burslem, H. F. Kyle, A. L. Breeze, T. A. Edwards, A. Nelson, S. L. Warriner and A. J. Wilson, ChemBioChem, 2014, 15, 1083–1087 CrossRef CAS PubMed.
- A. Barnard, K. Long, D. J. Yeo, J. A. Miles, V. Azzarito, G. M. Burslem, P. Prabhakaran, T. A. Edwards and A. J. Wilson, Org. Biomol. Chem., 2014, 12, 6794–6799 CAS.
- D. J. Yeo, S. L. Warriner and A. J. Wilson, Chem. Commun., 2013, 49, 9131–9133 RSC.
- M. Sattler, H. Liang, D. Nettesheim, R. P. Meadows, J. E. Harlan, M. Eberstadt, H. S. Yoon, S. B. Shuker, B. S. Chang, A. J. Minn, C. B. Thompson and S. W. Fesik, Science, 1997, 275, 983–986 CrossRef CAS PubMed.
- S. A. Dames, M. Martinez-Yamout, R. N. De Guzman, H. J. Dyson and P. E. Wright, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5271–5276 CrossRef CAS PubMed.
- C. J. Brown, C. S. Verma, M. D. Walkinshaw and D. P. Lane, Cell Cycle, 2009, 8, 1905–1911 CrossRef CAS PubMed.
- J. M. Adams and S. Cory, Oncogene, 2007, 26, 1324–1337 CrossRef CAS PubMed.
- H. Yao, S. Mi, W. Gong, J. Lin, N. Xu, S. Perrett, B. Xia, J. Wang and Y. Feng, Biochemistry, 2013, 52, 6324–6334 CrossRef CAS PubMed.
- H. Yin, G.-i. Lee, K. A. Sedey, J. M. Rodriguez, H.-G. Wang, S. M. Sebti and A. D. Hamilton, J. Am. Chem. Soc., 2005, 127, 5463–5468 CrossRef CAS PubMed.
- I. C. Kim and A. D. Hamilton, Org. Lett., 2006, 8, 1751–1754 CrossRef CAS PubMed.
- S. M. Biros, L. Moisan, E. Mann, A. Carella, D. Zhai, J. C. Reed and J. Rebek Jr, Bioorg. Med. Chem. Lett., 2007, 17, 4641–4645 CrossRef CAS PubMed.
- P. Restorp and J. Rebek Jr, Bioorg. Med. Chem. Lett., 2008, 18, 5909–5911 CrossRef CAS PubMed.
- P. Maity and B. König, Org. Lett., 2008, 10, 1473–1476 CrossRef CAS PubMed.
- J. H. Lee, Q. Zhang, S. Jo, S. C. Chai, M. Oh, W. Im, H. Lu and H.-S. Lim, J. Am. Chem. Soc., 2010, 133, 676–679 CrossRef PubMed.
- M. Oh, J. H. Lee, W. Wang, H. S. Lee, W. S. Lee, C. Burlak, W. Im, Q. Q. Hoang and H.-S. Lim, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11007–11012 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03559c |
| This journal is © The Royal Society of Chemistry 2015 |