DOI:
10.1039/C5RA17226H
(Paper)
RSC Adv., 2015,
5, 78352-78360
Microfluidic synthesis of high-performance monodispersed chitosan microparticles for methyl orange adsorption
Received
25th August 2015
, Accepted 4th September 2015
First published on 4th September 2015
Abstract
High-performance monodispersed chitosan microparticles for methyl orange (MO) adsorption were synthesized on a microfluidic platform coupled with a cross-linking approach. Batch adsorption experiments were carried out to evaluate the capacity and kinetics of the as-prepared microparticles on the adsorption of MO. Due to the advantage of microfluidics that all the manipulations and operations are related to independent droplets, the prepared microparticles are controlled to within a narrow size distribution (CV = 1.86%) while exhibiting uniform high performance (adsorption capacity = 182 mg g−1). Adsorption experiments were carried out under various design and operation conditions. It is found that the adsorption isotherm was well described by the Langmuir model and the adsorption kinetics followed a pseudo-second-order kinetic model. The high performance together with biodegradable feature and low-cost raw material give the microfluidic-synthesized chitosan microparticles a promising potential for future dye effluent treatment.
1. Introduction
Discharge of synthetic dyes in textiles, leather and paper industries currently imposes a major threat on the environment.1 Dyes are stable, recalcitrant, colorant, and sometimes carcinogenic and toxic. Their release into the environment results in serious environmental, aesthetical and health issues.2 With respect to both number and production volume, azo dyes are the largest group of colourants, constituting 70% of all organic dyes produced in the world.3 Due to the existence of the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– chromophore group and the aromatic structure, they are non-biodegradable and resistant to light and oxidizing agents.4,5 Adsorption methods have been proven more advantageous for azo dyestuff treatment as compared to various physical, chemical, photochemical and biological methods, owing to their high efficiency, economic feasibility and simple operation.6–9 Recently, considerable attention has been directed towards low-cost biosorbents due to their sustainable sources and excellent biodegradable nature.10,11 Biological materials including peat, biomass, cellulose and chitosan are commonly used biosorbents to concentrate and remove dyes from solutions.12
N– chromophore group and the aromatic structure, they are non-biodegradable and resistant to light and oxidizing agents.4,5 Adsorption methods have been proven more advantageous for azo dyestuff treatment as compared to various physical, chemical, photochemical and biological methods, owing to their high efficiency, economic feasibility and simple operation.6–9 Recently, considerable attention has been directed towards low-cost biosorbents due to their sustainable sources and excellent biodegradable nature.10,11 Biological materials including peat, biomass, cellulose and chitosan are commonly used biosorbents to concentrate and remove dyes from solutions.12
Chitosan (CS), synthesized from chitin deacetylation, second most abundant polysaccharide after cellulose, has been the subject of several studies due to its relevant physicochemical properties with promising applications in antibacterial coatings and adsorbents. Its high adsorption capability is provided by the presence of several chelating groups including primary and secondary hydroxyl groups as well as highly reactive amino-groups.13 Combined with special characteristics such as biocompatibility, biodegradability and non-toxicity, chitosan can provide a promising and competitive alternative as green adsorbent for dyestuff wastewater treatment.14 Cross-linking treatments are usually needed to stabilize chitosan forming insoluble beads and/or particles with improved (bio)chemical and mechanical resistance.15
Particle adsorption materials possess numerous applications in process equipment such as fixed bed reactors, absorption columns, fluidized beds and cyclone separators for wastewater treatment,16–18 with advantages including cost-effectiveness, simplicity in construction and adaptability to a wide range of operating conditions. In these advanced process designs, microparticles with controlled shapes, compartments, nanostructures and size distributions are required to achieve not only a high adsorption capacity but also improved process efficiency and reliability. Most conventional methods for particle synthesis have limitations to achieve a precise control of size distribution. Therefore, microfluidic-based platform with its intrinsic feature in high degree of product control can provide a novel approach towards the synthesis of advanced emulsion-based templates and/or microparticles.19,20 Microfluidics have already proven useful in applications related to bio-technologies, diagnostics, drug encapsulation and controlled release.21,22 Yet, little or no attempts have been considered to develop microfluidic-synthesised materials for large-scale industrial processes including wastewater treatment, probably due to the high costs and limited production capacity associated with microfluidic methods. Recent breakthroughs in microreactors and their scale-up/integration technologies have nevertheless allowed an additional flexibility and further opportunities for future large-scale application of microfluidic-synthesized materials.21,23 Fundamental research is consequently highly demanded to fill the gap by exploring the possibilities and potential benefits of using microfluidic synthesized materials in large-scale industrial processes.
Few attempts have been reported to date in order to produce chitosan microparticles under microfluidics,24–26 with these investigations mostly limited to the development and control of various chitosan particle structures. No investigations have been conducted to utilize chitosan microparticles in applications related to dye adsorption and/or detailed adsorption characterization studies. In this study, we have developed a simple, reproducible and high performing microfluidics approach combining crosslinking methods towards the design of chitosan microparticles as a model azo dye (MO) adsorption materials. As-prepared particles show a narrow size distribution (CV = 1.86%) while exhibiting high performance (adsorption capacity = 182 mg g−1). The mechanism of adsorption was also clarified and the adsorption isotherm could be well described by the Langmuir model, with adsorption kinetics following a pseudo-second-order kinetic model.
2. Experimental
2.1 Materials
85% deacetylated chitosan (Haidebei Marine Bioengineering Co., Ltd) was dissolved in acetic acid (Jingchun Biochemical Technology Co., Ltd), forming an aqueous solution, which serves as dispersed phase. The solution was stirred overnight and utilised after several filtrations and vacuum degassing. An emulsifier, Span 80, was added as in continuous phase for the stabilization of chitosan droplets. The continuous phase was made from Span 80 dissolved in a mixture of liquid paraffin and petroleum ether with volume ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (all from Jingchun Biochemical Technology Co., Ltd). Glutaraldehyde Saturated Toluene (GST, Jingchun Biochemical Technology Co., Ltd) was used as cross-linking reagent for solidification. All reagents were of analytical grade and used as received. Deionized distilled water was used throughout the experiments.
:5 (all from Jingchun Biochemical Technology Co., Ltd). Glutaraldehyde Saturated Toluene (GST, Jingchun Biochemical Technology Co., Ltd) was used as cross-linking reagent for solidification. All reagents were of analytical grade and used as received. Deionized distilled water was used throughout the experiments.
2.2 Design and fabrication of a microfluidic system
The microfluidic flow-focusing chip (55 mm × 35 mm × 3 mm) was curved on 1 mm PMMA plate with a Laser Microfabrication Machine (Universal VLS2.30). The chip comprises three layers (Fig. 1a and b) which are, from top to bottom, the cover layer (containing three inlet orifices holes), the main layer (containing the flow-focusing junction) and the bottom layer (containing one outlet orifices hole). The channel width at the flow-focusing junction is 500 μm and the broadened channel expands to 2 mm to reduce pressure resistance and facilitate in situ observation (Fig. 1c). The three layers were sealed with hot-pressing bonding, thus forming the enclosed microchannels with a certain degree of strength and minimizing deformation and leakage. Single-channel syringe pumps (Longer LSP01-1A) were used to pump the dispersed phase and a dual-channel syringe pump (Longer LSP01-1B) was used to pump the continuous phase. The pumps were connected to the chip with Teflon tubes. When the emulsion was formed, it was led and collected in a container on a shaker where it will further be solidified upon cross-linking. The overview of the experimental system as well the cross-linking process is shown in Fig. 2.
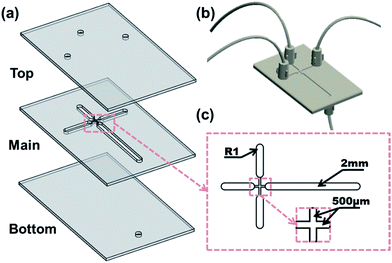 |
| | Fig. 1 Schematic drawing of the structure and dimension of microfluidic chip (a) the chip in expanded view, (b) the chip in assembly view and (c) the partial enlarged view of the flow-focusing junction. | |
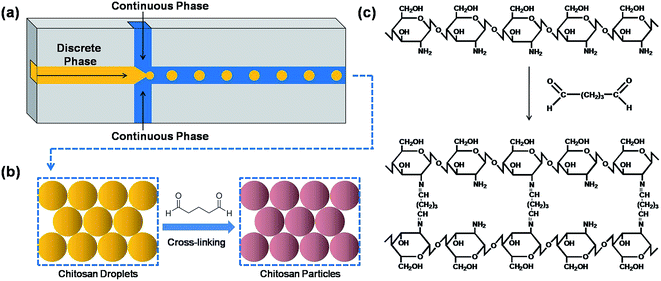 |
| | Fig. 2 Overview of the microfluidic system and cross-linking process: (a) microfluidic system, (b) sketch figure of cross-linking process and (c) schematic representation of the cross-linking process. | |
2.3 Formation of monodispersed chitosan microspheres
To prepare monodispersed chitosan microspheres, an aqueous solution containing 2.0 wt% chitosan and 2.0 wt% acetic acid was injected into the central channel, while 4.0 wt% Span 80 liquid paraffin and petroleum mixed solution was injected into the side channels. When reaching the flow-focusing geometry, the dispersed phase immediately breaks up into droplets by the shear force of the continuous phase as shown in Fig. 3. Chitosan droplets were subsequently solidified under Schiff's base reaction by adding the cross-linking agent (glutaraldehyde). Chitosan microspheres were eventually washed several times with ethanol, acetone and then deionized water to remove the remaining impurities on the particle surface, before drying at 40 °C for 12 h.
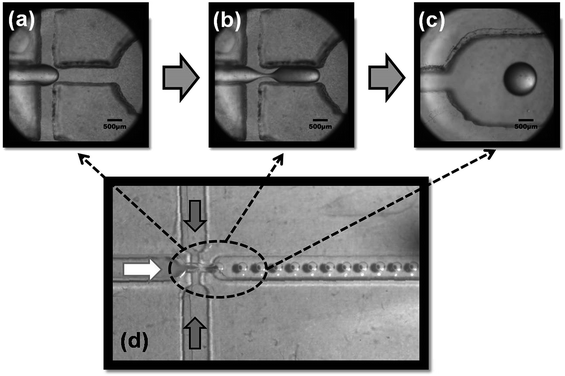 |
| | Fig. 3 Time-serial images of the generation of chitosan micro-emulsions including (a) chitosan solution flow, (b) primary droplet and thinned thread, (c) chitosan droplet and (d) overall representation of the formation of emulsions. | |
2.4 Analysis and characterization of microspheres
The droplets and microspheres were observed with an optical microscope (OLYMPUS SZX7) and on-line high-speed CCD (IDT Motion XtraNX4) with a frequency of 200 images per second. Particle monodispersity was defined using coefficients of variation (CV). 300 particles were measured each time in this experiment. Lower CV values indicate narrower microsphere size distributions. SEM (JEOL JSM-6380LV) was also employed to observe more detailed structures of the particles.| |
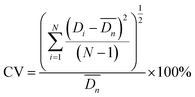 | (1) |
2.5 Batch adsorption experiments
A methyl orange (MO) stock solution (1.0 g L−1) was first prepared and further diluted to desired MO concentrations. For each adsorption experiment, 30 mL of the dye solution with a specified concentration was prepared and a certain amount of chitosan microspheres were added. The adsorption experiments were carried out in a mini-shaker (Mei Xiang Instrument, KR-B, oscillation frequency 800 min−1, and vibration amplitude 8 mm) at constant temperature under continuous shaking. At given time intervals, upper samples were withdrawn by a pipette and centrifuged. The residual concentration was then determined from a constructed calibration curve by measuring the absorbance at 465 nm using UV-Vis (Xin Mao Instrument, UV-7502). It should be noted that the calibration curves under different pH conditions (pH = 2.9, 5.0, 6.8, 9.0, 11.0) were measured and used separately to avoid any influence from the pH variation.
Adsorption properties were measured by varying adsorbent dosage, the initial concentration of MO and pH. Original parameters used for this study included adsorbent dosage 5 mg, 40 mg L−1 initial concentration of MO and pH 6.84 of MO solution (pH value of MO in DI water) at 298 K (room temperature) as initial conditions. These values were carefully chosen after several pilot experiments and acted as conditional control group (CCG). Under pH parameter experiments, solutions were adjusted to a desired value by adding 0.1 mol L−1 NaOH or HCl solution, and pHs were measured with a pH meter (Aolilong, AOLL-01).
The adsorption capacity (qt) and adsorption efficiency (η) was calculated according to the following equations:
| |
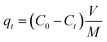 | (2) |
| |
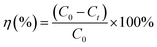 | (3) |
where
C0 is the initial concentration of MO (mg L
−1),
Ct (mg L
−1) is the instant concentration of MO at a predetermined time
t.
V (mL) is the volume of solution and
M (mg) is the adsorbent weight. The time interval was set after the adsorption–desorption equilibrium was reached.
Each experiment is replicated 3 to 8 times and the standard deviation for each group of data is found to be under 2%.
2.6 Desorption experiments
In desorption study, 5 mg chitosan microspheres was added to 30 mL MO solution with initial concentration of 40 mg L−1. After 10 h, the MO-loaded absorbent was collected, quickly rinsed with deionized water, and stirred for 6 h in 30 mL of 2 mol L−1 NaCl and 0.5 mol L−1 NaOH solution. The amount of regeneration MO and the corresponding desorption rate of the material were analyzed and calculated in the same way as that in the adsorption experiments. To test the reusability of the adsorbent, this adsorption/desorption cycle was repeated five times.
3. Results and discussion
3.1 Size and monodispersity control of chitosan microspheres
As-prepared microspheres have been depicted in Fig. 4a, clearly illustrating a remarkable size uniformity and sphericity could be achieved for synthesized chitosan microspheres under microfluidics. Further measurements demonstrated the high monodispersity in as-synthesized materials (CV = 1.86%, Fig. 4b), considerably lower to that of microparticles synthesized using conventional methods including stirring (CV > 10%) and membrane methods (CV ∼ 10%). The monodispersity of the material can provide a high degree of control at the process level, which is significantly important in large-scale industrial applications, particularly for fluidized bed and cyclone separators.27,28 Fig. 4c shows a digital photo of chitosan microspheres in water solution, indicating that particles are insoluble in water and can be homogeneously dispersed in aqueous solutions. SEM images of chitosan microspheres (Fig. 4d) also pointed out a homogeneous and reproducible spherical geometry and surface smoothness of particles. Glutaraldehyde was uniformly available to cross-link the surface of the droplets due to the solubility of toluene in liquid paraffin.
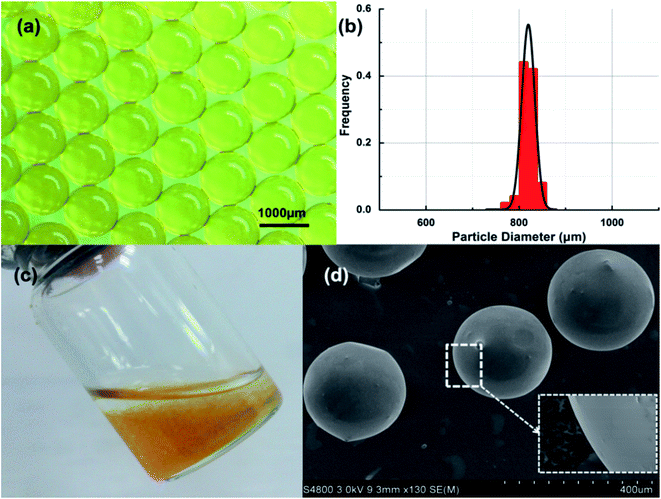 |
| | Fig. 4 (a) Micrograph image of chitosan microspheres, (b) size distribution of chitosan microspheres, (c) photograph of chitosan microspheres and (d) SEM image of chitosan microspheres. | |
The effect of continuous/dispersed phase flow rates on the size control of chitosan microspheres was subsequently studied as shown in Fig. 5. The diameter of chitosan microspheres was found to decrease with an increase in the flow rate of the continuous phase (Qc), while synchronized with the change in flow rate of the dispersed phase (Qd). These findings pointed out that the size of chitosan microspheres can be easily controlled in a wide range (e.g., in our experiment, from 1002 μm to 735 μm).
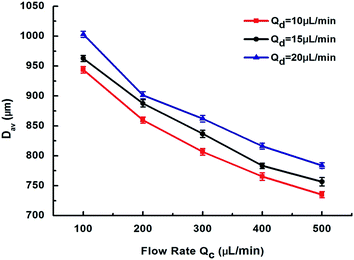 |
| | Fig. 5 Effects of continuous phase flow rate Qc and dispersed phase flow rate Qd on microsphere diameter Dav. | |
3.2 Adsorption experiment studies
3.2.1 Effect of adsorbent dosage. Adsorbent dosage is an important parameter in wastewater treatment processes as this factor determines the capacity of an adsorbent for a given initial concentration of the adsorbate.29 Fig. 6a illustrates the effect of chitosan microspheres dosage on adsorption efficiency of MO solution. MO adsorption efficiency increased from 15 to >95% with a slight increasing of adsorbent dosage (from 1 to 7 mg). Available adsorption sites obviously increase with an increase in the adsorbent dosage, which consequently result in the increase of the amount of adsorbed dye. The adsorption capacity was comparatively reduced as the adsorbent dosage increased. The amount adsorbed per unit mass of MO decreased from 182 mg g−1 to 158 mg g−1 with increasing adsorbent dosage (as shown in Fig. 6b), which can be attributed to the increase of unsaturated adsorption active sites during the adsorption process.30
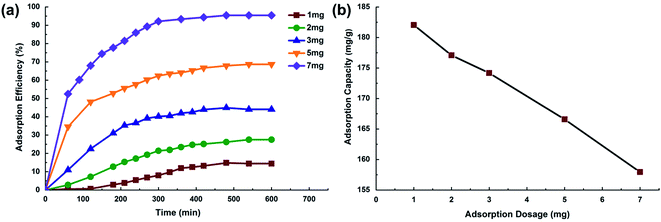 |
| | Fig. 6 (a) Effect of adsorbent dosage on MO removal adsorption efficiency, (b) effect of adsorbent dosage on MO adsorption capacity. | |
3.2.2 Effect of initial MO concentration. The effect of initial MO concentration on adsorption efficiency is depicted in Fig. 7a. A rapid initial adsorption of MO took place within the first 220 min, after which the adsorption slowed down and finally reached equilibrium after 10 h. MO adsorption efficiency tended to decrease from 89 to 49% when increasing the initial dye concentration from 20 to 60 mg L−1. Fig. 7b shows the effect of initial concentration on MO adsorption capacity. The adsorption uptake of MO was improved from 107 mg g−1 to 175 mg g−1, with the increase in initial concentration. The higher performance at more concentrated dye solution can be attributed to an increase of mass gradient between the solution and adsorbent, which acts as a driving force to transfer dye molecules from bulk solution to the particle surface.31
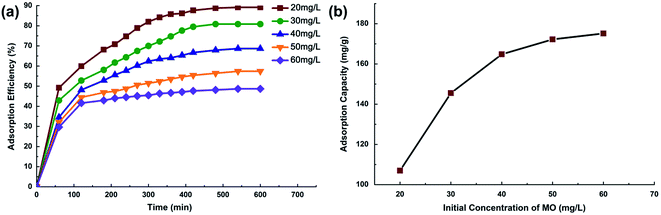 |
| | Fig. 7 (a) Effect of initial concentration on MO removal adsorption efficiency, (b) effect of initial concentration on MO adsorption capacity. | |
3.2.3 Effect of pH. The pH of the dye solution affects the surface charge of the adsorbent, the degree of ionization of the materials, the dissociation of functional groups on the active sites of the adsorbent and eventually structure of the dye molecule.32 The adsorption efficiency of MO at different pH values is plotted in Fig. 8. The pKa value for MO is 3.4. To avoid the pH effect on the accuracy of MO concentration determination, the UV-Vis curves under different pH conditions (pH = 2.9, 5.0, 6.8, 9.0, 11.0) were measured and used separately. We also measured the pH value after the adsorption experiment, and the variations of pH were lower than ±0.1. The result showed that an increase in pH from 3.0 to 11.0 remarkably reduced MO adsorption efficiency from 83 to 11% (as shown in Fig. 8a) while MO adsorption capacity decreased from 201 mg g−1 to 26 mg g−1 (as shown in Fig. 8b). These results indicated that an acid environment can be more favorable for chitosan microparticles functioning as dye adsorbent. Chitosan has a positively charged surface below pH 6.5 (point of zero potential).33 In acidic solution, amino groups (–NH2) present in chitosan can be easily protonated. As a result, the electrostatic interactions between MO anions and positively-charged chitosan surfaces can be enhanced.30,34 Furthermore, the lower adsorption capacity observed under alkaline solutions can be also attributed to the presence of excessive hydroxyl ions, which competed with MO molecules for the adsorption sites.35
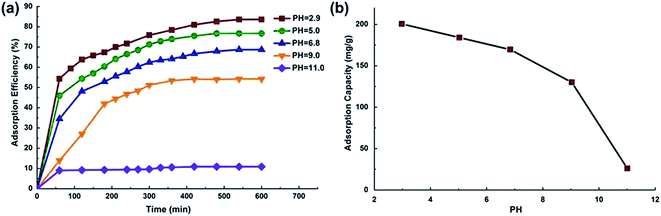 |
| | Fig. 8 (a) Effect of pH on MO removal adsorption efficiency, (b) effect of pH on MO adsorption capacity. | |
3.3 Adsorption isotherms
To describe the adsorption process and investigate adsorption mechanisms, two well-known models (Langmuir and Freundlich isotherms) were selected to correlate chitosan–dye interactions in this study. Langmuir's adsorption model depends on the assumption that intermolecular forces rapidly decrease with distance and consequently predicts the existence of monolayer coverage of the adsorbate at the outer surface of the adsorbent. The isotherm equation further assumes that adsorption takes place at specific homogeneous sites within the adsorbent. Once a dye molecule occupies a site, no further adsorption can take place at that site.36 The Langmuir isotherm model can be represented as:| |
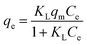 | (4) |
where Ce is the equilibrium concentration (mg L−1); qe is the amount of dye adsorption at equilibrium (mg g−1), qm is for a complete monolayer (mg g−1), which gives the maximum adsorption capacity of adsorbent; and KL is the sorption equilibrium constant (L mg−1).
Comparatively, the Freundlich isotherm model is valid for multilayer adsorption on the surface of the adsorbent where there is a possibility that the amount of dye being adsorbed on the adsorbent surface is not constant at a given concentration.37 The Freundlich isotherm model can be represented as:
where
Ce is the equilibrium concentration (mg L
−1);
qe is the amount of dye adsorption at equilibrium (mg g
−1).
KF and
n are Freundlich constants.
n gives an indication of adsorption intensity and
KF (mg g
−1 (L mg
−1)1/
n) is the adsorption capacity of the adsorbent.
A general consensus has been made by comparing the results among Langmuir, Freundlich and experimental data for the adsorption of MO on chitosan microspheres. The comparisons have been summarized in Fig. 9 and Table 1. The calculations revealed that the equilibrium data showed a perfect agreement with the Langmuir model with R2 = 0.992, as compared to the less fitted Freundlich model (R2 = 0.928). The results suggest that once a MO molecule occupies homogeneous sites within the adsorbent surface, the adsorption is completed and MO monolayers are consequently formed.38 From Langmuir adsorption isotherms, the maximum MO adsorption capacities for chitosan microspheres were calculated to be 182 mg g−1, significantly higher to those reported for similar chitosan-based adsorption materials in literature (ca. 30–70 mg g−1) (Table 2). The superior capacity demonstrated in this study may be attributed to the natural advantage of microfluidics for a high-degree of control in the production of microparticles, which lead to an increasing number of more homogeneous adsorption sites together with an ideal cross-linked surface, with respect to film, flake or irregular shapes present in similar adsorbent systems.
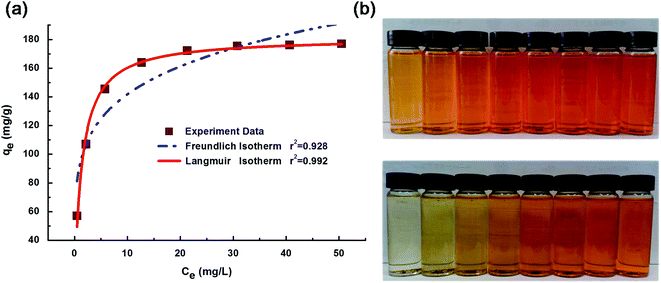 |
| | Fig. 9 (a) Freundlich and Langmuir adsorption isotherms for MO adsorption, and (b) digital photos of dye adsorption by chitosan microspheres at 25 °C (equilibration time = 10 h; from left to right: C0 = 10, 20, 30, 40, 50, 60, 70, 80 mg L−1). | |
Table 1 Isotherm parameters for MO adsorption
| Freundlich constants |
Langmuir constants |
| KF (mg g−1) (L mg−1)1/n |
n |
R2 |
KL (L g−1) |
qm (mg g−1) |
R2 |
| 92.569 |
5.395 |
0.928 |
0.754 |
181.462 |
0.992 |
Table 2 Comparison of the adsorption capacity of chitosan microspheres for the removal of MO with other chitosan based adsorbents
| Adsorbent |
qm (mg g−1) |
pH |
Ref. |
| Maghemite/chitosan (films) |
29 |
6.0 |
39 |
| Chitosan/alumina composite (flake) |
33 |
6.0 |
40 |
| γ-Fe2O3/SiO2/CS composite (irregular shape) |
34 |
4.7 |
6 |
| γ-Fe2O3/MWCNTs/chitosan (powder) |
60–66 |
6.5 |
41 |
| Monodispersed chitosan microparticles by this study |
182 |
6.8 |
This work |
3.4 Adsorption kinetics
Kinetics models are generally employed to correlate adsorption rates and equilibrium time. Through adsorption kinetics, the mechanism for MO uptake rate and the residence time for the adsorption process can be determined. Two of the most widely used kinetic models, i.e. Lagergren-first-order model, and pseudo-second-order model were used to investigate the adsorption kinetic behavior of MO onto chitosan microspheres.
Lagergren first-order kinetics can be generally expressed as:
| |
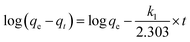 | (6) |
where
qe and
qt are the amounts of adsorbed MO at equilibrium and at time
t, respectively,
k1 is the rate constant. The value of adsorption capacity, rate constant and correlation coefficient (
R2) are determined from the plot log(
qe −
qt)
versus t.
The pseudo-second-order kinetics model is expressed using the following equation:
| |
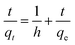 | (7) |
where
k2 is rate constant for pseudo-second-order model and
h =
k2qe2 is the initial adsorption rate of MO. The slope and intercept of the plot
t/
qt versus t denotes the rate constant and adsorption capacity, respectively.
Kinetic parameters were determined from linear plots of log(qe − qt) versus t for the Lagergren-first-order model, and t/qt versus t for the pseudo-second-order model (shown in Fig. 10a and b). All experimental data obtained was in better agreement with pseudo-second-order kinetics in terms of higher correlation coefficient values (R2) as shown in Fig. 10. In addition, experimental qe values (qe,exp) also exhibited a better fit with calculated values (qe2,cal) obtained from the pseudo-second-order kinetic model. Both results suggested that the adsorption behavior of MO onto chitosan microspheres follows pseudo-second-order kinetics (R2 > 0.99) rather than first-order, indicating that the chemisorptions step is the rate-limiting step of the adsorption mechanism, with mass transfers in solution being less relevant to overall adsorption rates.31 Similar kinetic results were found for MO on various adsorbents previously reported by other researchers.6,41
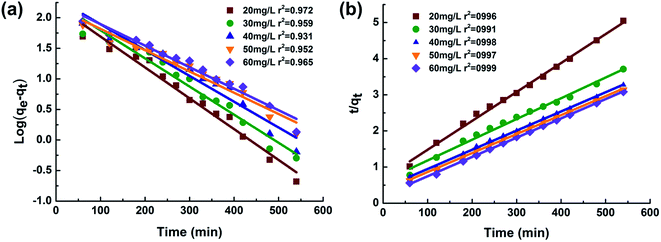 |
| | Fig. 10 Kinetic models for MO adsorption for different initial MO concentrations: (a) Lagergren-first-order model and (b) pseudo-second-order model. | |
3.5 Material regeneration capability
We have also studied the material regeneration capability to provide indications for possible future solutions for the spent sorbent. Fig. 11 shows MO recovery performance by potting the adsorption rate (%) after successive adsorption/desorption cycles. A mixture of NaCl and NaOH was used as desorption agent in each batch. Our material demonstrated a considerable regenerative property, as around 85% of the adsorbed dye can be desorbed, indicating same percentage of adsorption capacity being recovered after each cycle. A part of the MO dye (about 15–16%) remained adsorbed on the surface of adsorbent leading to the reduced adsorption capacity of the particle material.42 In addition, the adsorption capacity of the adsorbent show a slight decrease (<10%) after 5 adsorption–desorption cycles. Therefore, further improvement of the material regeneration capacity is highly desired by better material design.
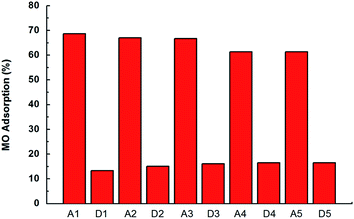 |
| | Fig. 11 MO adsorption (%) for five adsorption (Ai)/desorption (Di) cycles. Adsorbent dosage 5 mg, initial concentration of MO was 40 mg L−1 and pH 6.8. | |
In addition to that, more solutions for the treatment of spent sorbent can be proposed. Firstly, because of the low cost nature of the raw material, the sorbent material is possible to be disposed (e.g. burned or naturally degraded) after use, as long as the total production cost of the material can be kept under acceptable level. Secondly, with the advantage of microfluidic platform, more advanced materials, such as core–shell particles integrating adsorption–photocatalysis functions, can be designed and fabricated for in situ degradation of the dye and other organics. These researches are currently undergoing in our lab.
4. Conclusions
Microfluidics has been successfully utilized to design high performing, reproducible and highly monodispersed chitosan microparticles with unprecedented adsorption properties for dyes. Aiming to develop advanced and efficient materials for dye wastewater treatment, chitosan microparticles synthesized under microfluidics integrating crosslinking methods exhibited an excellent monodispersity (CV = 1.86%) together with a high adsorption capacity (182 mg g−1) for MO adsorption. This demonstrated benefits of using microfluidic synthesized materials in dyestuff wastewater treatment opens up a new direction for future efforts in both material synthesis and alternative industrial process development for large-scale applications of advanced micro-materials.
Acknowledgements
This work is supported by The National Basic Research Program of China (973 Program, 2014CB748500), National Natural Science Foundation of China (51406057) and Research Fund for the Doctoral Program of Higher Education of China (20130074120019).
References
- M. Arshadi, F. SalimiVahid, J. Salvacion and M. Soleymanzadeh, RSC Adv., 2014, 4, 16005–16017 RSC.
- G. Moussavi and M. Mahmoudi, J. Hazard. Mater., 2009, 168, 806–812 CrossRef CAS PubMed.
- C. M. Carliell, S. J. Barclay, C. Shaw, A. D. Wheatley and C. A. Buckley, Environ. Technol., 1998, 19, 1133–1137 CrossRef CAS PubMed.
- T. S. Anirudhan, P. G. Radhakrishnan and K. Vijayan, Sep. Sci. Technol., 2013, 48, 947–959 CrossRef CAS PubMed.
- W. Cheah, S. Hosseini, M. A. Khan, T. G. Chuah and T. S. Y. Choong, Chem. Eng. J., 2013, 215, 747–754 CrossRef PubMed.
- H. Y. Zhu, R. Jiang, Y. Q. Fu, J. H. Jiang, L. Xiao and G. M. Zeng, Appl. Surf. Sci., 2011, 258, 1337–1344 CrossRef CAS PubMed.
- S. Sengupta and R. Mondal, J. Mater. Chem. A, 2014, 2, 16373–16377 CAS.
- M. Z. Iqbal and A. A. Abdala, RSC Adv., 2013, 3, 24455–24464 RSC.
- S. Ray, M. Takafuji and H. Ihara, RSC Adv., 2013, 3, 23664–23672 RSC.
- W. S. Wan Ngah and M. A. K. M. Hanafiah, Bioresour. Technol., 2008, 99, 3935–3948 CrossRef CAS PubMed.
- M. P. Elizalde-González, Trends Chem. Eng., 2006, 10, 55–66 Search PubMed.
- G. Crini, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef CAS PubMed.
- I. M. N. Vold, K. M. Vårum, E. Guibal and O. Smidsrød, Carbohydr. Polym., 2003, 54, 471–477 CrossRef CAS PubMed.
- M. N. V. Ravi Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef.
- J. Ma, C. Liu, R. Li and J. Wang, Bio-Med. Mater. Eng., 2012, 22, 129–135 Search PubMed.
- A. A. Ahmad and B. H. Hameed, J. Hazard. Mater., 2010, 175, 298–303 CrossRef CAS PubMed.
- I. Ali, Sep. Purif. Rev., 2014, 43, 175–205 CrossRef CAS PubMed.
- M. Colic, W. Morse and J. D. Miller, Int. J. Environ. Pollut., 2007, 30, 296–312 CrossRef CAS.
- J. H. Kim, T. Y. Jeon, T. M. Choi, T. S. Shim, S.-H. Kim and S.-M. Yang, Langmuir, 2014, 30, 1473–1488 CrossRef CAS PubMed.
- W. Wang, M.-J. Zhang and L.-Y. Chu, Acc. Chem. Res., 2014, 47, 373–384 CrossRef CAS PubMed.
- A. van den Berg, H. G. Craighead and P. Yang, Chem. Soc. Rev., 2010, 39, 899–900 RSC.
- W. J. Duncanson, T. Lin, A. R. Abate, S. Seiffert, R. K. Shah and D. A. Weitz, Lab Chip, 2012, 12, 2135–2145 RSC.
- M. B. Romanowsky, A. R. Abate, A. Rotem, C. Holtze and D. A. Weitz, Lab Chip, 2012, 12, 802–807 RSC.
- C.-H. Yang, K.-S. Huang, P.-W. Lin and Y.-C. Lin, Sens. Actuators, B, 2007, 124, 510–516 CrossRef CAS PubMed.
- L. Liu, J.-P. Yang, X.-J. Ju, R. Xie, Y.-M. Liu, W. Wang, J.-J. Zhang, C. H. Niu and L.-Y. Chu, Soft Matter, 2011, 7, 4821–4827 RSC.
- J. H. Xu, H. Zhao, W. J. Lan and G. S. Luo, Adv. Healthcare Mater., 2012, 1, 106–111 CrossRef CAS PubMed.
- G. Wan, G. Sun, X. Xue and M. Shi, Powder Technol., 2008, 183, 94–104 CrossRef CAS PubMed.
- Z. Ji, Z. Xiong, X. Wu, H. Chen and H. Wu, Powder Technol., 2009, 191, 254–259 CrossRef CAS PubMed.
- Y. Bulut and H. Aydın, Desalination, 2006, 194, 259–267 CrossRef CAS PubMed.
- H.-Y. Zhu, R. Jiang, L. Xiao and W. Li, J. Hazard. Mater., 2010, 179, 251–257 CrossRef CAS PubMed.
- F.-C. Wu, R.-L. Tseng and R.-S. Juang, Water Res., 2001, 35, 613–618 CrossRef CAS.
- G. Crini, H. N. Peindy, F. Gimbert and C. Robert, Sep. Purif. Technol., 2007, 53, 97–110 CrossRef CAS PubMed.
- N. Sakkayawong, P. Thiravetyan and W. Nakbanpote, J. Colloid Interface Sci., 2005, 286, 36–42 CrossRef CAS PubMed.
- P. Senthil Kumar, S. Ramalingam, C. Senthamarai, M. Niranjanaa, P. Vijayalakshmi and S. Sivanesan, Desalination, 2010, 261, 52–60 CrossRef CAS PubMed.
- Y. Yao, H. Bing, X. Feifei and C. Xiaofeng, Chem. Eng. J., 2011, 170, 82–89 CrossRef CAS PubMed.
- Y. C. Wong, Y. S. Szeto, W. H. Cheung and G. McKay, Langmuir, 2003, 19, 7888–7894 CrossRef CAS.
- W. Cheah, S. Hosseini, M. A. Khan, T. G. Chuah and T. S. Y. Choong, Chem. Eng. J., 2013, 215–216, 747–754 CrossRef CAS PubMed.
- B. Tanhaei, A. Ayati, M. Lahtinen and M. Sillanpää, Chem. Eng. J., 2015, 259, 1–10 CrossRef CAS PubMed.
- R. Jayakumar, H. Nagahama, T. Furuike and H. Tamura, Int. J. Biol. Macromol., 2008, 42, 335–339 CrossRef CAS PubMed.
- J. Zhang, Q. Zhou and L. Ou, J. Chem. Eng. Data, 2012, 57, 412–419 CrossRef CAS.
- H. Y. Zhu, R. Jiang, L. Xiao and G. M. Zeng, Bioresour. Technol., 2010, 101, 5063–5069 CrossRef CAS PubMed.
- Q. Li, H. Su and T. Tan, Biochem. Eng. J., 2008, 38, 212–218 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence















