
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge-transfer excited states in phosphorescent organo-transition metal compounds: a difficult case for time dependent density functional theory?†
Thomas A.
Niehaus
*a,
Thomas
Hofbeck
b and
Hartmut
Yersin
*b
aInstitut für Theoretische Physik, Universität Regensburg, 93040 Regensburg, Germany. E-mail: thomas.niehaus@ur.de
bInstitut für Physikalische Chemie, Universität Regensburg, 93040 Regensburg, Germany. E-mail: hartmut.yersin@ur.de
First published on 10th July 2015
Abstract
Light emitting organo-transition metal complexes have found widespread use in the past. The computational modelling of such compounds is often based on time-dependent density functional theory (TDDFT), which enjoys popularity due to its numerical efficiency and simple black-box character. It is well known, however, that TDDFT notoriously underestimates energies of charge-transfer excited states which are prominent in phosphorescent metal–organic compounds. In this study, we investigate whether TDDFT is providing a reliable description of the electronic properties in these systems. To this end, we compute 0–0 triplet state energies for a series of 17 pseudo-square planar platinum(II) and pseudo-octahedral iridium(III) complexes that are known to feature quite different localization characteristics ranging from ligand-centered (LC) to metal-to-ligand charge transfer (MLCT) transitions. The calculations are performed with conventional semi-local and hybrid functionals as well as with optimally tuned range-separated functionals that were recently shown to overcome the charge transfer problem in TDDFT. We compare our results against low temperature experimental data and propose a criterion to classify excited states based on wave function localization. In addition, singlet absorption energies and singlet–triplet splittings are evaluated for a subset of the compounds and are also validated against experimental data. Our results indicate that for the investigated complexes charge-transfer is much less pronounced than previously believed.
1. Introduction
Phosphorescent organo-transition metal complexes play a key role in several modern optoelectronic applications, such as organic light emitting diodes (OLEDs),1–11 light emitting electrochemical cells,12 optical sensors,13–15 photo-catalysts,16,17 as well as cell imaging systems.15,18 For OLED triplet emitters, for example, spin–orbit coupling (SOC) induced by the central heavy metal atom leads to efficient intersystem crossing, fast radiative decay from the lowest triplet state (T1) and thus to efficient harvesting of electrically generated electron–hole pairs.1,4,11 Organo-transition metal compounds are also used in dye-sensitized solar cells,19–22 here the long wavelength part of the solar spectrum can be utilized by SOC allowed singlet–triplet excitations.23 Therefore, one of the crucial quantities for a rational design of all these applications is the transition energy (ΔET1 = E(T1) − E(S0)) from the ground to the excited state (or vice versa). Important is also the energy difference between excited singlet and triplet states that are spin–orbit coupled. A small value of this singlet–triplet splitting favors large radiative rates, which for example minimize the roll-off of OLED efficiency due to saturation.24In the past, pseudo-square planar platinum(II) and pseudo-octahedral iridium(III) complexes have shown to be excellent emitter materials for phosphorescent OLEDs and were synthesized with a variety of organic ligands.1,4,25–29 In order to obtain suitable candidate materials for future applications, a reliable computational screening procedure covering a large number of ligands and combinations thereof would be highly rewarding. As a numerically efficient quantum chemical method that provides useful accuracy for the key quantity ΔET1, time dependent density functional theory30–32 (TDDFT) has become the major tool in this regard over the last years. With recent relativistic extensions, spin–orbit interaction can be accounted for and enables the computation of triplet zero-field splittings and radiative rates for phosphorescence.33,34 These parameters can then directly be compared to experimental data. As reviewed by Escudero and Jacquemin,35 also the deactivation pathways of excited phosphors have been the subject of TDDFT simulations recently.
Although TDDFT frequently provides excited state energies with fairly systematic errors of 0.2–0.4 eV, there are also several well documented failures of the theory.36 One of these, the severe underestimation of charge-transfer excited state energies, seems to undermine the potential use of TDDFT in the field of organo-transition metal photophysics and photochemistry. This is because low lying excited states of these compounds are often characterized as MLCT states, based on experimental observations28,37 and also on more sophisticated correlated quantum chemical methods.38 This shortcoming of TDDFT is well understood39–41 by now and we summarize the main arguments below for the later discussion.
Let us assume the system of interest is a closed shell molecule and the excited state is dominated by a transition from the occupied orbital i into the unoccupied orbital a. The difference between the Kohn–Sham orbital energies associated with this transition is called ΔEia. In this case, the TDDFT eigenvalue problem that leads to the singlet (ΔES) and triplet (ΔET) excitation energies may be drastically simplified:42,43
| ΔES/T = ΔEia + KS/T | (1) |
| KS = 2(ia|ia) + (ia|f↑↑xc + f↑↓xc|ia) − b(ii|aa) | (2) |
| KT = (ia|f↑↑xc − f↑↓xc|ia) − b(ii|aa). | (3) |
Here (ia|ia) denotes a two-electron integral in the Mulliken notation44 and 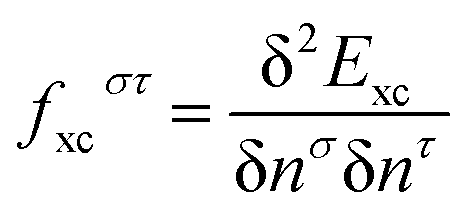 is the functional derivative of the DFT exchange–correlation (xc) energy with respect to the density for spin-up (n↑) and spin-down (n↓) electrons (σ, τ ∈ {↑, ↓} are spin indices). Eqn (1)–(3) are valid both for functionals that depend on the density only (termed local in the following) or depend on the density and its gradient (i.e., semi-local functionals in the generalized-gradient approximation (GGA)). They hold as well for hybrid functionals that incorporate a fraction b of Hartree–Fock (HF) exchange. In the limit fxcστ = 0 and b = 1, the equations above reduce to the TD-HF case. For a general charge-transfer excitation, the overlap of orbitals i and a is small, such that the correction terms KS/T can be neglected for local and semi-local functionals (b = 0). The singlet–triplet splitting therefore tends to zero and excitation energies reduce to the Kohn–Sham gap. The latter differs strongly from the quasiparticle gap, defined as the difference of ionization potential (IP) and electron affinity (EA).45,46 This is because the electrons in occupied and virtual orbitals experience the same local potential in DFT. This needs to be compared with HF theory, where an additional electron in a virtual orbital feels the potential of all N electrons. Because of the exact cancellation of self-interaction, an electron in the occupied set feels the potential of N − 1 electrons in contrast. The orbital energy differences ΔEia for hybrid functionals are therefore much larger as differences from pure functionals and provide reasonable approximations for the quasiparticle gap. In fact, the excitation energy for a transition from donor (D) and acceptor (A) molecules separated by a large distance R is given by ΔE = IPD − EAA − 1/R. In this limit the last term of eqn (2) simplifies to −b/R, which shows that hybrid functionals account at least partially for the electron–hole stabilization which is completely absent for currently available LDA/GGA functionals.‡ Recently, range-separated functionals that exhibit 100% HF exchange at large electron–electron distances gained a lot of popularity.48–53 These functionals are designed to suppress the self-interaction error and provide good approximations to quasiparticle gaps.54 They were also shown to solve the erratic description of long range charge-transfer excitations.55
is the functional derivative of the DFT exchange–correlation (xc) energy with respect to the density for spin-up (n↑) and spin-down (n↓) electrons (σ, τ ∈ {↑, ↓} are spin indices). Eqn (1)–(3) are valid both for functionals that depend on the density only (termed local in the following) or depend on the density and its gradient (i.e., semi-local functionals in the generalized-gradient approximation (GGA)). They hold as well for hybrid functionals that incorporate a fraction b of Hartree–Fock (HF) exchange. In the limit fxcστ = 0 and b = 1, the equations above reduce to the TD-HF case. For a general charge-transfer excitation, the overlap of orbitals i and a is small, such that the correction terms KS/T can be neglected for local and semi-local functionals (b = 0). The singlet–triplet splitting therefore tends to zero and excitation energies reduce to the Kohn–Sham gap. The latter differs strongly from the quasiparticle gap, defined as the difference of ionization potential (IP) and electron affinity (EA).45,46 This is because the electrons in occupied and virtual orbitals experience the same local potential in DFT. This needs to be compared with HF theory, where an additional electron in a virtual orbital feels the potential of all N electrons. Because of the exact cancellation of self-interaction, an electron in the occupied set feels the potential of N − 1 electrons in contrast. The orbital energy differences ΔEia for hybrid functionals are therefore much larger as differences from pure functionals and provide reasonable approximations for the quasiparticle gap. In fact, the excitation energy for a transition from donor (D) and acceptor (A) molecules separated by a large distance R is given by ΔE = IPD − EAA − 1/R. In this limit the last term of eqn (2) simplifies to −b/R, which shows that hybrid functionals account at least partially for the electron–hole stabilization which is completely absent for currently available LDA/GGA functionals.‡ Recently, range-separated functionals that exhibit 100% HF exchange at large electron–electron distances gained a lot of popularity.48–53 These functionals are designed to suppress the self-interaction error and provide good approximations to quasiparticle gaps.54 They were also shown to solve the erratic description of long range charge-transfer excitations.55
Given the discussion above and the fact that the excited states of phosphorescent organo-transition metal complexes have partial MLCT character, it surprising that several previous case studies show very good agreement with the experiment using simple hybrid functionals like B3LYP.56–63 Admittedly, the charge-transfer is not long range inter-molecular but intra-molecular in these systems, but questions remain how general these findings are. This information is of great importance for the rational design and computational screening of OLED emitter materials in order to assess the expected accuracy of a given calculation. In this investigation, we therefore analyze the performance of TDDFT for a comprehensive set of 17 cyclometallated platinum(II) and iridium(III) complexes with high phosphorescence quantum yields. Instead of discussing vertical absorption energies, we compute 0–0 emission energies including zero-point energy corrections to make direct contact with the experimentally relevant quantities. For a subset of the compounds, also the S1 energy and the singlet–triplet splittings (ΔEST = ΔES1 − ΔET1) are determined. Through a comparison of results for GGA, hybrid and range-separated exchange–correlation functionals, we finally attempt to answer the question, whether metal-to-ligand excitations pose significant problems for modern TDDFT.
2. Experimental
Organo-transition metal compounds with Pt(II) and Ir(III) ion centers have been studied extensively in recent years to provide a deeper background, especially, for the understanding of emission properties,1,2,27,28,37,64–73 which are dominantly governed by the lowest excited triplet states and which crucially determine the performance of most opto-electronic devices, in particular of OLEDs. For example, by application of methods of high-resolution spectroscopy, it was possible to determine the 0–0 energies of the substrates I, II, and III of the lowest T1 states and the corresponding transition rates with respect to the electronic ground state S0.1,27,28,37,64,65,67–74 These investigations were particular successful for compounds doped at low concentrations of about 10−5 mol L−1 into adequate crystalline matrices, so-called Shpolskii matrices. In most cases alkane matrices and, in particular, octane were used. The resulting environment around the emitter complexes is relatively rigid and thus, geometry changes upon transitions between the lowest excited triplet state and the ground state are largely prevented. This is displayed in the vibrational satellite structures of the highly resolved emission spectra measured at low temperature. For example, for compound 4 doped into n-octane the Huang–Rhys parameter for the most prominent progression is even smaller than S = 0.1.27,64,75The corresponding studies were usually carried out down to T = 1.3 K and techniques of site-selective emission and excitation spectroscopy under variation of temperature and magnetic fields as well as time-resolving methods were applied. The compounds to which it is referred in this study1,27,28,37,64,65,67–75 are summarized in Fig. 1. Table 1 displays the experimental 0–0 substrate averaged transition energies of the T1 state. Moreover, we give the matrix used and the classification as worked out for the respective T1 state.
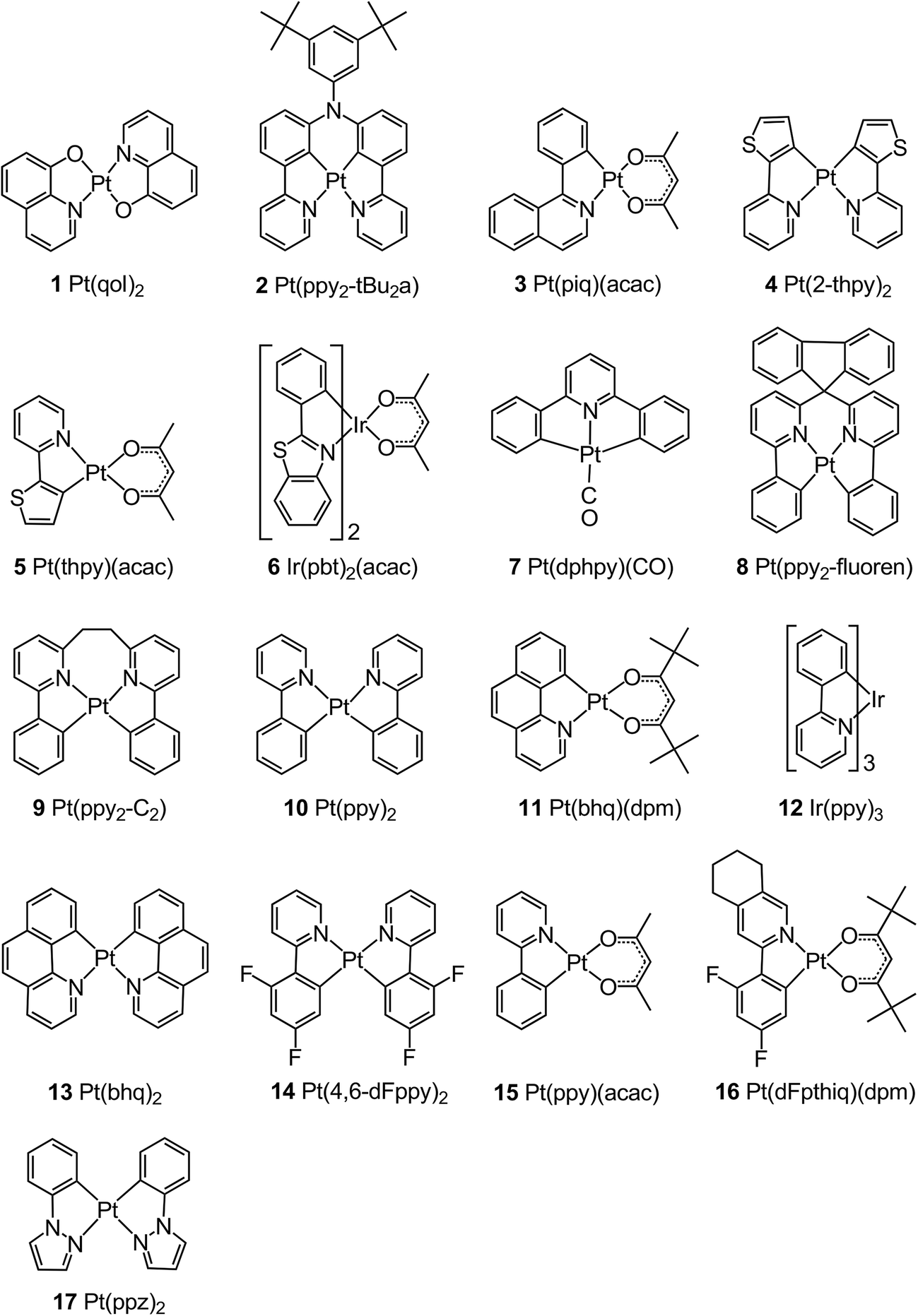 | ||
| Fig. 1 Chemical structures of compounds discussed. Complexes 11 and 13 are occasionally also abbreviated as Pt(bzq)(dpm) and Pt(bzq)2, respectively. | ||
 (6-311G*) for different exchange–correlation functionals compared to experiment. All energies are given in eV. A characterization of the dominant single particle excitation together with the percental participation of the metal atom according to a Löwdin population analysis is given in the third column. Values in parentheses are for the PBE/6-311G* and B3LYP/6-311G* functional, respectively. Metal participations are only shown beyond the threshold of 8% for PBE and 9% for B3LYP. For the LC-PBE functional, the optimal range separation parameter γ [a0−1] is given in parentheses. Experimental
(6-311G*) for different exchange–correlation functionals compared to experiment. All energies are given in eV. A characterization of the dominant single particle excitation together with the percental participation of the metal atom according to a Löwdin population analysis is given in the third column. Values in parentheses are for the PBE/6-311G* and B3LYP/6-311G* functional, respectively. Metal participations are only shown beyond the threshold of 8% for PBE and 9% for B3LYP. For the LC-PBE functional, the optimal range separation parameter γ [a0−1] is given in parentheses. Experimental  values have been measured in a n-octane matrix, apart from 12 (CH2Cl2) and 13 (n-decane). References to the original articles are given in square brackets. The last two columns provide a classification of the excited states based on experimental and theoretical data, respectively, according to the criteria described in the main text
values have been measured in a n-octane matrix, apart from 12 (CH2Cl2) and 13 (n-decane). References to the original articles are given in square brackets. The last two columns provide a classification of the excited states based on experimental and theoretical data, respectively, according to the criteria described in the main text
| # | Compound | S0 → T1 transition | PBE | B3LYP | LC-PBE (γ) | Exp. | Class [exp.] | Class [theo.] |
|---|---|---|---|---|---|---|---|---|
| 1 | Pt(qol)2 | 5d (25%/18%) + πqol → π*qol | 1.48 | 1.85 | 1.99 (0.21) | 1.91 (ref. 65) | 3LC | 3LC |
| 2 | Pt(ppy2-tBu2a) | 5d (25%/19%) + πppy → π*ppy | 1.47 | 1.94 | 1.97 (0.16) | 1.99 (ref. 37 and 98) | 3LC/MLCT | 3LC |
| 3 | Pt(piq)(acac) | 5d (40%/33%) + πpic → π*pic | 1.90 | 2.07 | 2.18 (0.20) | 2.09 (ref. 99) | 3LC | 3LC/MLCT |
| 4 | Pt(2-thpy)2 | 5d (47%/34%) + π2-thpy → π*2-thpy | 1.88 | 2.11 | 2.20 (0.29) | 2.13 (ref. 64) | 3LC/MLCT | 3LC/MLCT |
| 5 | Pt(thpy)(acac) | 5d (34%/26%) + πthpy → 5d* (9%/9%) + π*thpy | 2.12 | 2.19 | 2.06 (0.40) | 2.23 (ref. 71) | 3LC/MLCT | 3LC |
| 6 | Ir(pbt)2(acac) | 5d (53%/46%) + πpbt → π*pbt | 1.93 | 2.29 | 2.28 (0.16) | 2.26 (ref. 37) | 3MLCT | 3MLCT/LC |
| 7 | Pt(dphpy)(CO) | 5d (27%/18%) + πdphpy → 6p* (11%/13%) + π*dphpy + π*CO | 2.06 | 2.32 | 2.46 (0.21) | 2.38 (ref. 37) | 3LC/MLCT | 3LC |
| 8 | Pt(ppy2-fluoren) | 5d (55%/45%) + πppy → π*ppy | 2.05 | 2.42 | 2.45 (0.18) | 2.41 (ref. 37 and 98) | 3MLCT/LC | 3MLCT/LC |
| 9 | Pt(ppy2-C2) | 5d (59%/48%) + πppy → π*ppy | 2.05 | 2.48 | 2.66 (0.27) | 2.42 (ref. 37 and 98) | 3MLCT/LC | 3MLCT/LC |
| 10 | Pt(ppy)2 | 5d (61%/50%) + πppy → π*ppy | 2.05 | 2.48 | 2.67 (0.29) | 2.43 (ref. 37) | 3MLCT/LC | 3MLCT/LC |
| 11 | Pt(bhq)(dpm) | 5d (33%/26%) + πbhq → π*bhq | 2.02 | 2.40 | 2.28 (0.36) | 2.44 (ref. 37 and 73) | 3LC | 3LC/MLCT |
| 12 | Ir(ppy)3 | 5d (60%/52%) + πppy → π*ppy | 2.05 | 2.53 | 2.47 (0.16) | 2.45 (ref. 70) | 3MLCT | 3MLCT |
| 13 | Pt(bhq)2 | 5d (56%/42%) + πbhq → π*bhq | 1.99 | 2.46 | 2.55 (0.28) | 2.51 (ref. 37) | 3MLCT/LC | 3MLCT/LC |
| 14 | Pt(4,6-dFppy)2 | 5d (55%/42%) + π4,6-dFppy → π*4,6-dFppy | 2.20 | 2.57 | 2.72 (0.26) | 2.56 (ref. 67) | 3MLCT/LC | 3MLCT/LC |
| 15 | Pt(ppy)(acac) | 5d (41%/35%) + πppy + πacac → π*ppy | 2.29 | 2.57 | 2.43 (0.41) | 2.56 (ref. 4 and 73) | 3LC/MLCT | 3LC/MLCT |
| 16 | Pt(dFpthiq)(dpm) | 5d (33%/31%) + πdFpthiq + πdpm → π*dFpthiq | 2.48 | 2.69 | 2.80 (0.21) | 2.71 (ref. 37) | 3LC/MLCT | 3LC/MLCT |
| 17 | Pt(ppz)2 | 5d (57%/35%) + πppz → 6p* (8%/10%) + π*ppz | 2.66 | 2.95 | 3.11 (0.28) | 2.85 (ref. 100) | 3LC/MLCT | 3LC/MLCT |
The classification of the lowest triplet states of the investigated compounds has been based on their zero-field splittings ΔE(ZFS), their radiative emission decay times, and, if available, to the (normalized) emission intensities of the vibrational satellites of metal–ligand character.28,37,72,74,75 Thus, T1 states with ΔE(ZFS) values < 2 cm−1 are assigned to a 3LC(ππ*) character. To have a course guide line, we classify states that show splitting values up to about 10 cm−1 or 20 cm−1 as 3LC(ππ*)/3MLCT(dπ*) states and for the range of about 20 cm−1 < ΔE(ZFS) < 50 cm−1, we use the notation 3MLCT(dπ*)/3LC(ππ*), while states with splittings above about 50 cm−1 are assigned as 3MLCT(dπ*) states.37,72,75 These notations are independently supported by the trends that are found for the radiative rates and the vibrational satellite structures (for details see especially ref. 28, 37, 72 and 75). Obviously, these assignments should only be used for approximate characterizations.
3 Computational
All calculations reported herein were performed with the NWChem code76 version 6.1, 6.3 and 6.5. The compounds depicted in Fig. 1 were first optimized in the ground state using the hybrid B3LYP77 functional and a 6-31G* basis set for the light atoms. For the central metal atom relativistic electronic core potentials of the LANL2DZ type with the corresponding valence orbital basis set were employed.78 Using the same computational methodology, Hay reported metal–ligand bond distances with an error of less than 0.06 Å compared to experimental X-ray data for a series of Ir(III) complexes.56At the optimized geometry, excitation energies were compared for several different functionals. We consider representative functionals for each of the major classes highlighted in the introduction: the semi-local PBE,79 the often used hybrid B3LYP, and the range-separated LC-PBE functional. The latter is based on a partitioning of the Coulomb operator into a short-range and long-range component, dictated by the parameter γ, for example by utilizing the standard error-function erf(x):48,49
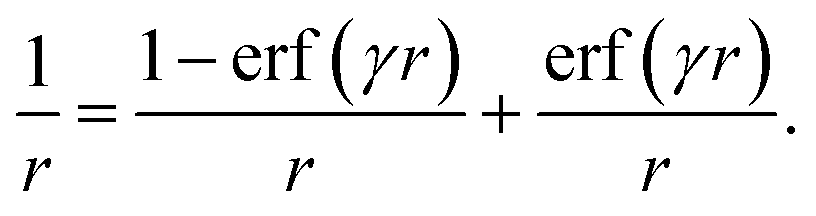 | (4) |
The first term is a Coulomb operator decaying to zero on a length scale of ≈1/γ and is therefore short-ranged (SR), while the second term dominates at large r accounting for the long-range (LR) behavior. This gives rise to a decomposition of the xc energy functional according to
| Exc = ESRx,DFT(γ) + ELRx,HF(γ) + Ec,DFT. | (5) |
In the case of LC-PBE, ESRx,DFT is the short-range form of the gradient-corrected PBE exchange functional,80ELRx,HF denotes the Hartree–Fock exchange energy evaluated with the long-range part of the interaction in eqn (4), while the correlation part Ec,DFT is left unchanged with respect to the usual form of PBE. There are different strategies to determine the value of the parameter γ. One possibility is choosing it to minimize the deviation of various molecular properties with respect to experimental or accurate first principles data on large test sets (see, e.g., ref. 81 and 82), leaving the parameter fixed in further applications. Another route is the system specific tuning proposed by Baer and co-workers,53 which we will follow here. Given that DFT with the exact exchange–correlation functional should obey Koopmans' theorem, one can find the optimal γ for the compound in question by minimizing the following error function:
| ΔIP(γ) = |εHOMOγ − [Etotγ(N) − Etotγ(N − 1)]|, | (6) |
For all mentioned functionals, linear response TDDFT calculations (also known as Casida approach) were performed in the adiabatic approximation.42 Depending on the actual value of γ, the range-separated functionals may incorporate a large fraction of Hartree–Fock exchange. As shown by Tozer83,84 and Brédas,85 triplet instabilities in the ground state may lead to a significant underestimation of triplet excited state energies in such a case, especially in large π-conjugated systems. Since several of the ligands investigated in this study fall into this class, we also perform calculations in the Tamm–Dancoff approximation86 (TDA), which is known to alleviate these problems. Metal atoms were treated at the same level as in the ground state, while for the remaining atoms a split-valence basis set of triple-ζ quality plus polarization functions (6-311G*) was employed. For a subset of the studied compounds also calculations including diffuse functions for the ligands (6-311++G**) and using the triple-ζ basis LANL2TZ(f)87 for the metal have been performed. The results in the ESI (Table S4†) indicate a modest gain in accuracy (<0.1 eV).
Earlier TDDFT studies on transition metal complexes also indicate that solvent effects play a crucial role. For simulations in the gas phase, it was frequently observed that the ligand-to-ligand charge-transfer character is exaggerated, with a concomitant underestimation of excitation energies with respect to experiment.88 Here we employ the COSMO solvent model89 as implemented in NWChem with a dielectric constant of 1.95 corresponding to n-octane. This is the solvent that was used in the majority of measurements that are reported here. In order to estimate the influence of the solvent model we also tested the so-called solvation model based on density (SDM, solvent octane) put forward by Truhlar and co-workers90 and implemented in version 6.5 of NWChem. The corresponding results in the ESI (Table S5†) show only very minor changes (<0.01 eV) with respect to the COSMO model.
Based on Kasha's rule, we limit the investigation of luminescence energies to the lowest singlet and triplet state, respectively. Also, structural relaxation in the excited state is not taken into account. This reflects the experimental situation, where the matrices provide a rigid environment for the emitter molecule. Admittedly, this rigidity does not rule out the possibility of inward relaxations that reduce the occupied volume. Given the difficulties in modelling the precise morphology of the molecular environment, we discard this possibility. In order to estimate 0–0 transitions ΔE0–0, we therefore compute vertical absorption energies ΔEvert at the optimized S0 geometry which are corrected according to:
| ΔE0–0 = ΔEvert + ZPET1 − ZPES0. | (7) |
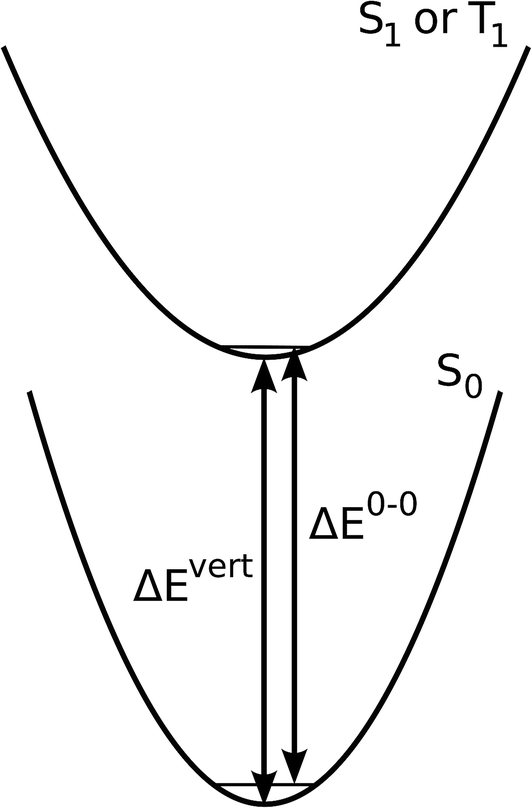 | ||
| Fig. 2 Potential energy surfaces for ground and excited states indicating the key quantities calculated. Depicted is a scenario with vanishing excited state relaxation. | ||
Here ZPE denotes the unscaled B3LYP/6-31G* zero-point energy at the minimum of the T1 and S0 potential energy surfaces, respectively. The T1 geometry optimization was carried out at the unrestricted Kohn–Sham level with the S0 minimum as initial geometry. Singlet excited state geometry optimizations based on TDDFT are computationally still very demanding. In addition, the problem of state crossing is frequently encountered during the optimization, especially for the organo-transition metal complexes discussed here with a relatively dense set of low lying states. Due to the these difficulties in obtaining ZPES1, we applied the correction protocol of eqn (7) also for singlet excited states, coarsely assuming parallelity of the T1 and S1 potential energy surfaces. Results for the correction terms and more computational details are provided in the ESI.†
4 Results and discussion
4.1 Triplet state emission
We start with an examination of the computed triplet emission energies. Table 1 lists the results, which are graphically displayed in Fig. 3. In addition, Table 2 provides a statistical summary for the full set of complexes. All studied exchange–correlation functionals are able to provide the general energetical ordering of the luminescence energies, with large discrepancies in absolute values. Turning first to the results for the local PBE functional, we find that T1 energies are systematically lower than experimental values by 0.35 eV. This underestimation of triplet energies is typical for local functionals, although the actual amount is slightly smaller than reported in earlier benchmark studies on organic chromophores.91–93 The PBE results may be further analyzed by investigating the CI expansion of the excited state |Φ〉 in terms of singly excited determinants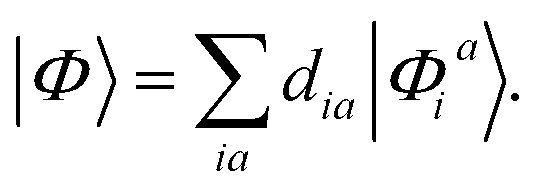 | (8) |
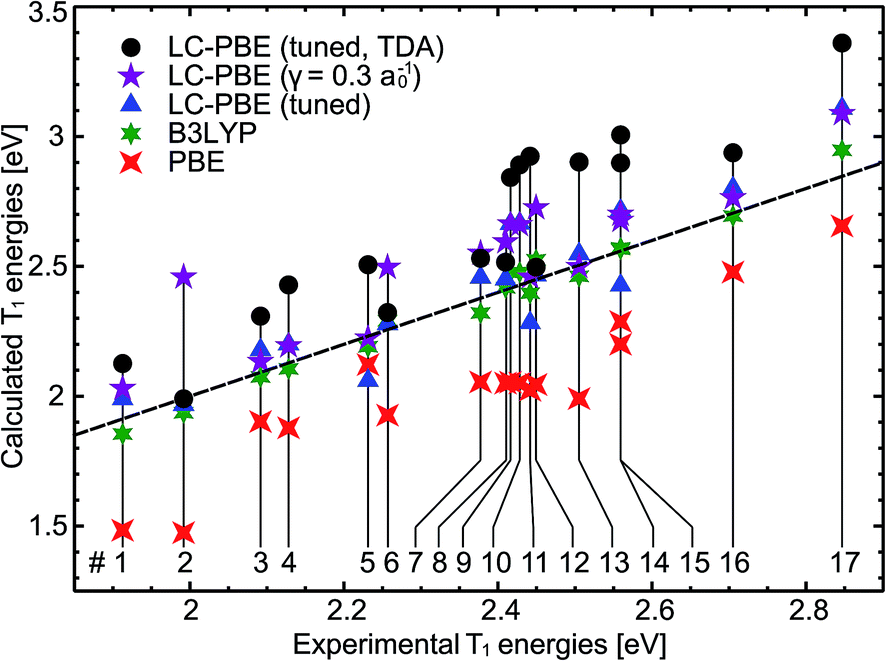 | ||
Fig. 3 Calculated versus experimentally determined triplet emission energies (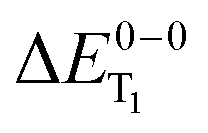 ) for various exchange–correlation functionals. The dashed line indicates perfect agreement. The stated numbers correspond to the complexes depicted in Fig. 1. ) for various exchange–correlation functionals. The dashed line indicates perfect agreement. The stated numbers correspond to the complexes depicted in Fig. 1. | ||
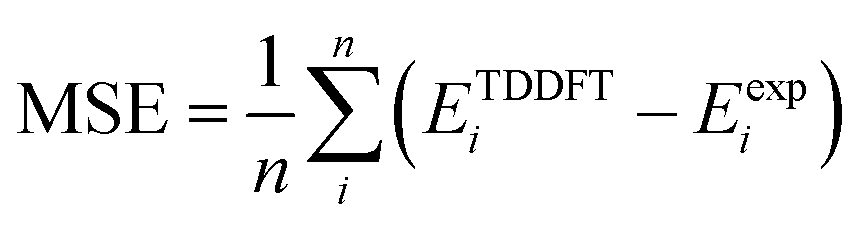 ) and root mean square deviation (
) and root mean square deviation (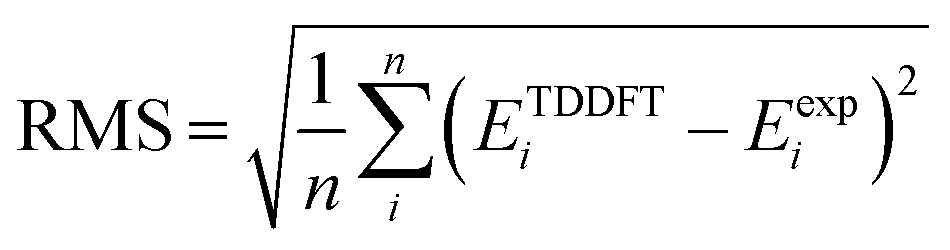 ) of TDDFT energies (ETDDFT) with respect to experiment (Eexp) for the sets presented in Tables 1 and 3. All energies in eV
) of TDDFT energies (ETDDFT) with respect to experiment (Eexp) for the sets presented in Tables 1 and 3. All energies in eV
| PBE | B3LYP | LC-PBE | ||||
|---|---|---|---|---|---|---|
| Tuned | TDA | γ = 0.3a0−1 | ||||
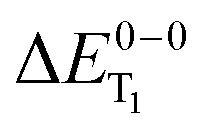
|
MSE | −0.33 | 0.00 | 0.06 | 0.28 | 0.15 |
| RMS | 0.35 | 0.05 | 0.14 | 0.32 | 0.20 | |
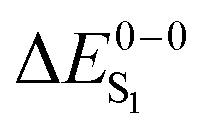
|
MSE | −0.44 | 0.09 | 0.41 | 0.47 | 0.65 |
| RMS | 0.46 | 0.10 | 0.46 | 0.53 | 0.65 | |
| ΔEST | MSE | −0.11 | 0.13 | 0.37 | 0.26 | 0.49 |
| RMS | 0.14 | 0.13 | 0.46 | 0.31 | 0.50 | |
Strictly speaking, TDDFT does not provide direct access to the excited state wave function. As shown by Casida, eqn (8) might still be used for diagnostic purposes as an approximation to the true excited state.42 The coefficients dia are readily available from the solution of the eigenvalue equations in TDDFT. For all complexes studied here, the HOMO–LUMO transition is the dominating excitation for PBE in the expansion of eqn (8) with participation ratios of more than 93%. This allows us to estimate the two electron integral (ia|f↑↑xc − f↑↓xc|ia) according to eqn (1) from the difference of the excitation energy and Kohn–Sham energy gap. We obtain consistent values of roughly 0.1 eV which are smaller than corresponding integrals for π → π* and n → π* transition in typical organic molecules.94 This indicates a small but non-vanishing overlap between the HOMO and LUMO orbitals and hence, a partial charge-transfer character of the transition. These findings are in line with the localization of the frontier orbitals that are depicted in the ESI (Fig. S1 to S17†). The excitations may be broadly classified as a metal–ligand to ligand charge-transfer. A more quantitative measure is obtained from a Löwdin analysis95 of the atomic population (PiA) for a given molecular orbital i:
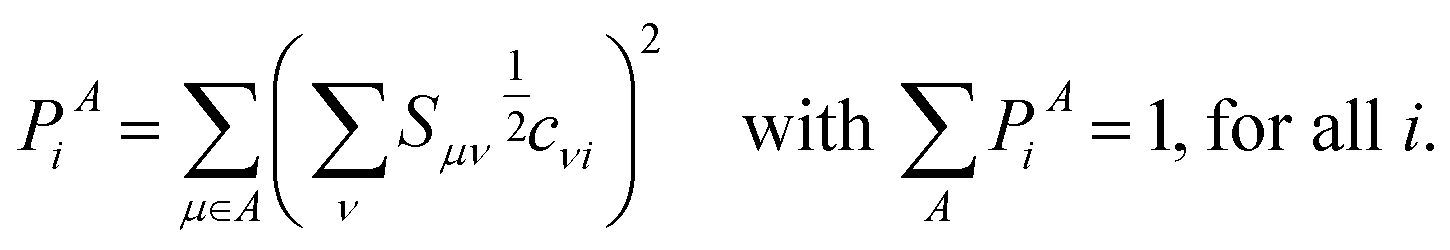 | (9) |
Eqn (9) contains the square root of the overlap matrix S between atom-centered basis functions and the molecular orbital coefficients cvi. As it is well known, the division of molecular density into atomic contributions is not unique. Löwdin populations provide a better estimate for charge distributions than the more common Mulliken scheme for extended basis sets like the ones used in this study.96 They are used here as a simple and robust measure of wave function localization providing consistent results for a given basis set and functional.
In Table 1 the populations of the central metal atom are listed for the PBE and B3LYP functional. As can be seen, the HOMO is delocalized over the central metal atom and one or more ligands, while the LUMO is a π* orbital on the same ligand(s) in the majority of cases (compounds 7 and 17 show some Pt participation in the LUMO). Unexpectedly, the computed d-orbital contribution to the HOMO is larger in PBE than in B3LYP by about 10%. This is surprising as the self interaction error present in PBE (and reduced in B3LYP) gives usually rise to an overly delocalized electron density and exaggerated covalency.97 Given this fact, the charge-transfer character of the triplet states should also be larger for PBE and lead to larger deviations from experiment.
Coming to the results for the B3LYP functional, one observes in Fig. 3 an excellent agreement with the experimental data with a RMS error of only 0.05 eV, which is even smaller than the deviation found for supposedly simpler organic systems (namely hydrocarbons, heterocycles and carbonyl compounds) with this functional.91–93 Compared to the local PBE, a higher number of singly excited determinants mixes into the excited state wave function. For complex 5, as an example, the contribution of the most important determinant drops to 56%.§ This is due to the fraction of exact exchange in B3LYP which leads to larger matrix elements KT and hence also increased coupling of individual single-particle transitions. The Kohn–Sham gap ΔEia likewise increases substantially by 1.3–1.6 eV due to the exact exchange. In the computation of the final emission energies, both effects cancel to a large degree (see eqn (3), KT < 0) such that the B3LYP energies are only modestly larger than the PBE ones.
Based on the Löwdin population analysis mentioned above, we propose a simple guideline to classify the excited states as being ligand-centered, metal-to-ligand charge-transfer like or extensive mixtures of these basics types. To this end we consider the difference density Δn(r) = nS0(r) − nT1(r), which simplifies to Δn(r) ≈ |ψi(r)|2 − |ψa(r)|2 if the triplet state is well described by a single-particle transition from orbital ψi to ψa. A Löwdin projection on the central metal atom M then leads to a measure nCT = PiM − PaM of the fractional number of electrons that take part in the metal to ligand transition (cf.eqn (9)). This quantity may be used to estimate the charge-transfer character of a given excitation according to:
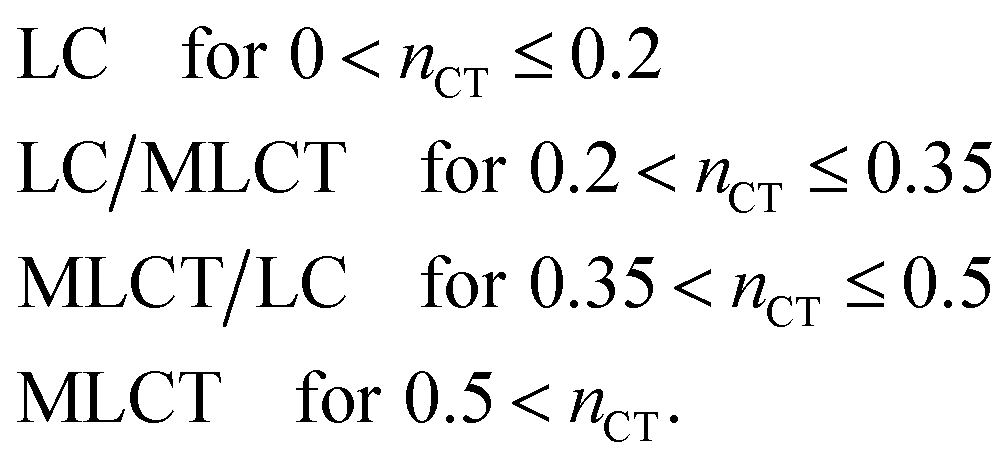 | (10) |
As seen in Table 1, this assignment correlates rather well with the experimentally derived classification and lends further support to the quality of the B3LYP results. With adapted limits for the different excitation types, a similar procedure might also be applied at the PBE level. The agreement with experiment, however, turns out to be inferior in this case. A more refined measure that takes not only the dominant determinant into account could be based on an analysis of the difference density102 or the transition density,103,104 both of which are readily available in DFT/TDDFT. Here we use the parameter nCT as an easily accessible quantity which allows for a quick and pragmatic classification of the excited state.
Despite of the general agreement with measurements in terms of emission energies and excitation character, it should be noted that there are still remaining qualitative discrepancies. The d-orbital participation seems to be exaggerated by DFT in general. According to eqn (10), excitations with nCT up to 0.2 are still classified as 3LC, although the d-orbital participation is experimentally found to be almost neglible.27,28,37,65,75 In line with this, zero field splittings computed by TDDFT including SOC are slightly, but systematically, overestimated with respect to experiment.34 In our study, the neglect of structural relaxation in the excited state might also contribute to the magnified metal character.
Before discussing the performance of the range-separated LC-PBE functional, a short digression on the optimal values for the parameter γ seems to be appropriate. Although γ is practically found by application of the IP criterion (eqn (6)), this parameter should reflect the electronic structure of the system and is expected to depend directly on quantities like the electron density or microscopic dielectric function.105 In fact, a clear dependence of the optimal range-separation parameter with the conjugation length of polymers was found.106 Since the conjugation length strongly influences the optical gap in polymers, it is interesting to verify whether there is a direct link also in the present organo-transition metal compounds. Inspection of Table 1 reveals that there is actually no correlation between the range-separation parameter and emission energies. The values of γ vary between 0.16a0−1 and 0.41a0−1 in a rather arbitrary fashion. The LC-PBE results for the emission energies are on average slightly less accurate than the B3LYP results with a RMS deviation of 0.14 eV (cf., Table 2). Interestingly, the LC-PBE functional with a fixed value of γ = 0.3a0−1 performs only slightly worse than the tuned variant. Only for compound 2 a clear benefit of the tuning procedure is discernible.
4.2 Singlet excitation energies and singlet–triplet gap
For most organo-transition metal complexes, fluorescence is difficult to detect due to the strong spin–orbit coupling and concomitant fast inter-system crossing. Only under suitable conditions, it is possible to identify the 0–0 transition energies of S0 to S1 transitions even at low temperature. Examples are summarized in Table 3. The data result from highly resolved and site-selectively detected excitation spectra measured at T = 1.3 K. However, very frequently, the corresponding transitions are hidden under overlapping transitions that belong to other sites, to other electronic states lying in this energy range, and/or to vibrational and phonon satellites that refer to the lower lying triplet substates. Moreover, the corresponding transitions are lifetime-broadened65 and thus, smeared out. Consequently, the possibility to measure a fairly well resolved 0–0 transition corresponding to S0 to S1 is very restricted.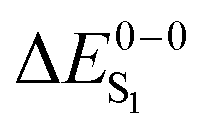 (6-311G*) for different exchange–correlation functionals compared to experiment. All excitation energies are given in eV. The radiative lifetime (τr[s]) is derived from the computed oscillator strengths according to the Strickler–Berg relationship including the effect of the dielectric environment.108,109 References for the experimental data are given in square brackets
(6-311G*) for different exchange–correlation functionals compared to experiment. All excitation energies are given in eV. The radiative lifetime (τr[s]) is derived from the computed oscillator strengths according to the Strickler–Berg relationship including the effect of the dielectric environment.108,109 References for the experimental data are given in square brackets
| # | Compound | PBE | B3LYP | LC-PBE (tuned) | Exp. | |||
|---|---|---|---|---|---|---|---|---|
|
|
τ r |
|
τ r |
|
τ r |
|
||
| 1 | Pt(qol)2 | 1.74 | 2.6 × 10−8 | 2.39 | 8.4 × 10−9 | 2.66 | 5.5 × 10−9 | 2.33 (ref. 65) |
| 2 | Pt(ppy2-tBu2a) | 1.67 | 6.9 × 10−8 | 2.20 | 3.8 × 10−8 | 2.22 | 7.8 × 10−8 | 2.16 (ref. 98) |
| 4 | Pt(2-thpy)2 | 2.08 | 8.0 × 10−7 | 2.64 | 5.0 × 10−8 | 3.12 | 1.2 × 10−8 | 2.54 (ref. 27) |
| 5 | Pt(thpy)(acac) | 2.60 | 4.5 × 10−8 | 3.00 | 1.3 × 10−8 | 3.53 | 3.9 × 10−9 | 2.82 (ref. 99) |
| 7 | Pt(dphpy)(CO) | 2.22 | 2.7 × 10−6 | 2.63 | 8.8 × 10−7 | 2.82 | 8.9 × 10−7 | 2.58 (ref. 107) |
| 14 | Pt(4,6-dFppy)2 | 2.34 | 1.0 × 10−5 | 2.97 | 1.9 × 10−7 | 3.36 | 4.9 × 10−8 | 2.86 (ref. 67) |
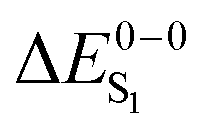
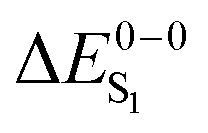
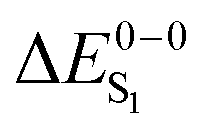
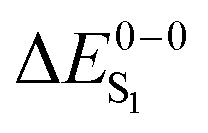
For the theoretical analysis it should also be kept in mind that the 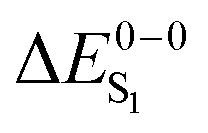 values were approximated using the zero-point energy values of the T1 potential energy surface. Although a statistical analysis should be performed with caution for such a small number of test cases, we also report the average errors for
values were approximated using the zero-point energy values of the T1 potential energy surface. Although a statistical analysis should be performed with caution for such a small number of test cases, we also report the average errors for 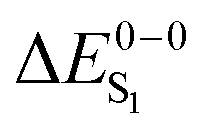 and the singlet–triplet gap
and the singlet–triplet gap 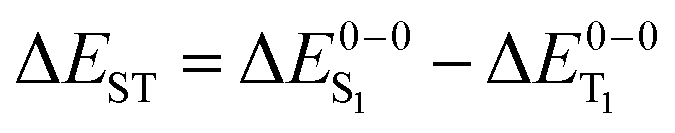 in Table 2. Note that the first excited singlet and triplet state are not necessarily strongly coupled by the spin–orbit interaction.37,72,110 Strictly speaking, the parameter ΔEST is in these cases not crucial for radiative lifetimes and OLED device performance. We include it here to obtain a broader picture of the TDDFT accuracy for organo-transition metal complexes.
in Table 2. Note that the first excited singlet and triplet state are not necessarily strongly coupled by the spin–orbit interaction.37,72,110 Strictly speaking, the parameter ΔEST is in these cases not crucial for radiative lifetimes and OLED device performance. We include it here to obtain a broader picture of the TDDFT accuracy for organo-transition metal complexes.
Turning to the results, the gradient-corrected PBE functional strongly underestimates transition energies, but it does so consistently for both triplets and singlets. As a result, the computed singlet–triplet gap ΔEST is in surprisingly good agreement with the experiment. Considering eqn (1)–(3), this can be traced back to the cancellation of the Kohn–Sham gap, which is too small to provide accurate absolute singlet and triplet energies. The exchange integral (ia|ia) that remains after subtraction is qualitatively correct also at the PBE level. In contrast, the hybrid B3LYP yields reasonable singlet energies with an RMS deviation of 0.10 eV in good agreement with earlier more extensive benchmarks on vertical singlet excitations.111 The singlet–triplet gap is systematically overestimated by only 0.13 eV, which allows for a reliable determination of this important parameter in large scale computational screenings.
Coming to the tuned LC-PBE functional, we find a strong overestimation of singlet energies and a pronounced error for the singlet–triplet gap. These trends are likewise already documented in the literature for vertical excitations of organic compounds.92,93,111 For singlet excitations, the tuning of the LC-PBE functional turns out to be beneficial and reduces the error compared to a fixed range-separation parameter significantly. Since ΔEST is strongly dependent on the amount of HF exchange in the functional, it is not surprising that LC-PBE with 100% long-range unscreened exchange considerably overestimates this quantity with respect to the experiment. Comparison of the radiative lifetimes between the different methods reveals that PBE exhibits shorter values compared to B3LYP and LC-PBE, for 14 the difference even amounts to 2–3 orders of magnitude. In their study of TDDFT oscillator strengths for small organic molecules, Caricato et al.112 find similar trends and show that range-separated functionals (like LC-PBE) provide results close to higher level theory. In summary it can be stated that B3LYP outperforms the other studied functionals also in the description of singlet excited states.
5. Conclusion
The objective of this article was to investigate whether TDDFT in its current implementations is accurate enough to provide a reliable and general description of optical properties of organo-transition metal complexes. Although a statistical evaluation of the results is inevitable in such a context, we also tried to rationalize the general trends by analyzing the structure of the TDDFT eigenvalue equations for the different functionals. The main finding of our study is that the hybrid B3LYP functional is an excellent choice and provides a balanced treatment of LC, LC/MLCT, MLCT/LC, and MLCT excited states. Somewhat surprising, range-separated functionals do not provide a clear advantage even for states with strong intra-molecular charge-transfer character. In addition, the tuning of the range-separation parameter leads to improvements only for singlet excited states. If the interest is only in singlet–triplet gaps, the semi-local PBE functional might serve as a cost effective alternative to B3LYP.With respect to the computational protocol, we find that vibrational contributions in the form of zero-point energies are necessary to match the experimental ΔE0–0 energies, while excited state relaxation is not crucial for emitters embedded in a rigid matrix. The B3LYP average error of only 0.05 eV for triplet emission energies is highly encouraging. This paves the way for further in silico pre-screening of possible OLED materials. We hope that the extended experimental and theoretical data set provided in this article might also be useful also in the development of novel exchange–correlation functionals which aim at a coherent description of valence, Rydberg and charge-transfer excited states in real world materials.
Our results show that charge transfer excitations in the investigated compounds do not pose a significant challenge for TDDFT. This is mainly because transitions that are often characterized as MLCT states actually involve transitions out of a strongly hybridized metal d-orbital. As mentioned in Section 4.1, there are even indications that the calculated d-orbital contribution is overestimated. This calls for further studies which connect calculated metal participations, SOC matrix elements and singlet–triplet gaps with measurements of zero-field splittings and radiative lifetimes. Such a combination of theory and experiment should be extremely helpful in the detailed understanding and further optimization of metal-organic emitters.
Acknowledgements
The authors (H. Y. and T. H.) gratefully acknowledge the “Bundesminsterium für Bildung und Forschung” (BMBF) for funding of our research. T. A. N. gratefully acknowledges support from the Deutsche Forschungsgemeinschaft (GRK 1570).References
- H. Yersin, Highly efficient OLEDs with phosphorescent materials, Wiley-VCH, Weinheim, 2008 Search PubMed.
- C.-M. Che, C.-C. Kwok, S.-W. Lai, A. F. Rausch, W. J. Finkenzeller, N. Zhu and H. Yersin, Photophysical properties and OLED applications of phosphorescent platinum(II) Schiff base complexes, Chem.–Eur. J., 2010, 16(1), 233–247 CrossRef CAS.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Recent progresses on materials for electrophosphorescent organic light-emitting devices, Adv. Mater., 2011, 23(8), 926–952 CrossRef CAS PubMed.
- Physics of Organic Semiconductors, ed. W. Brütting and C. Adachi, Wiley-VCH, Weinheim, 2012 Search PubMed.
- H. Sasabe and J. Kido, Development of high performance OLEDs for general lighting, J. Mater. Chem. C, 2013, 1(9), 1699–1707 RSC.
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, White organic light-emitting diodes: status and perspective, Rev. Mod. Phys., 2013, 85(3), 1245 CrossRef.
- C. Murawski, K. Leo and M. C. Gather, Efficiency roll-off in organic light-emitting diodes, Adv. Mat., 2013, 25(47), 6801–6827 CrossRef CAS PubMed.
- K. Li, G. Cheng, C. Ma, X. Guan, W.-M. Kwok, Y. Chen, W. Lu and C.-M. Che, Light-emitting platinum(II) complexes supported by tetradentate dianionic bis(N-heterocyclic carbene) ligands: towards robust blue electrophosphors, Chem. Sci., 2013, 4(6), 2630–2644 RSC.
- G. Cheng, P.-K. Chow, S. C. Kui, C.-C. Kwok and C.-M. Che, High-Efficiency Polymer Light-Emitting Devices with Robust Phosphorescent Platinum(II) Emitters Containing Tetradentate Dianionic ONCN Ligands, Adv. Mater., 2013, 25(46), 6765–6770 CrossRef CAS.
- W. Brütting, J. Frischeisen, T. D. Schmidt, B. J. Scholz and C. Mayr, Device efficiency of organic light-emitting diodes: Progress by improved light outcoupling, Phys. Status Solidi A, 2013, 210(1), 44–65 CrossRef.
- K. Udagawa, H. Sasabe, C. Cai and J. Kido, Low-driving-voltage blue phosphorescent organic light-emitting devices with external quantum efficiency of 30%, Adv. Mater., 2014, 26(29), 5062–5066 CrossRef CAS PubMed.
- A. Kapturkiewicz, Electrochemiluminescent Systems as Devices and Sensors, John Wiley & Sons, Inc., Hoboken, NJ, USA., 2009, ch. 16, pp. 477–522 Search PubMed.
- C. S. K. Mak, D. T. V. Pentlehner, M. Stich, O. S. Wolfbeis, W. K. Chan and H. Yersin, Exceptional Oxygen Sensing Capabilities and Triplet State Properties of Ir(ppy-NPh2)(3), Chem. Mater., 2009, 21(11), 2173–2175 CrossRef CAS.
- S. J. Payne, G. L. Fiore, C. L. Fraser and J. N. Demas, Luminescence Oxygen Sensor Based on a Ruthenium(II) Star Polymer Complex, Anal. Chem., 2010, 82(3), 917–921 CrossRef CAS.
- X.-D. Wang and O. S. Wolfbeis, Optical methods for sensing and imaging oxygen: materials, spectroscopies and applications, Chem. Soc. Rev., 2014, 43(10), 3666–3761 RSC.
- K. Zeitler, Photoredox catalysis with visible light, Angew. Chem., Int. Ed., 2009, 48(52), 9785–9789 CrossRef CAS PubMed.
- N. Hoffmann, Photochemical reactions as key steps in organic synthesis, Chem. Rev., 2008, 108(3), 1052–1103 CrossRef CAS.
- V. Fernández-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Application of d6 transition metal complexes in fluorescence cell imaging, Chem. Commun., 2010, 46(2), 186–202 RSC.
- B. O'Regan and M. Grätzel, A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films, Nature, 1991, 353, 737 CrossRef.
- M. Nazeeruddin, Michael Graetzel Festschrift, a tribute for his 60th Birthday, Coord. Chem. Rev., 2004, 248, 1161–1164 CrossRef CAS.
- E. I. Mayo, K. Kilså, T. Tirrell, P. I. Djurovich, A. Tamayo, M. E. Thompson, N. S. Lewis and H. B. Gray, Cyclometalated iridium(III)-sensitized titanium dioxide solar cells, Photochem. Photobiol. Sci., 2006, 5(10), 871–873 CAS.
- N. Robertson, Cu(I) versus Ru(II): Dye-Sensitized Solar Cells and Beyond, ChemSusChem, 2008, 1(12), 977–979 CrossRef CAS PubMed.
- N. Robertson, Optimizing dyes for dye-sensitized solar cells, Angew. Chem., Int. Ed., 2006, 45(15), 2338–2345 CrossRef CAS PubMed.
- S. Haneder, E. Da Como, J. Feldmann, J. M. Lupton, C. Lennartz, P. Erk, E. Fuchs, O. Molt, I. Münster, C. Schildknecht and G. Wagenblast, Controlling the radiative rate of deep-blue electrophosphorescent organometallic complexes by singlet–triplet gap engineering, Adv. Mater., 2008, 20(17), 3325–3330 CrossRef CAS.
- M. Maestri, V. Balzani, C. Deuschel-Cornioley and A. Von Zelewsky, Photochemistry and luminescence of cyclometallated complexes, Advances in photochemistry, 1992, 17(S 1) CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Synthesis and characterization of phosphorescent cyclometalated iridium complexes, Inorg. Chem., 2001, 40(7), 1704–1711 CrossRef CAS PubMed.
- H. Yersin and D. Donges, Low-Lying Electronic States and Photophysical Properties of Organometallic Pd(II) and Pt(II) Compounds. Modern Research Trends Presented in Detailed Case Studies, Top. Curr. Chem., 2001, 214, 81–186 CrossRef CAS.
- H. Yersin and W. J. Finkenzeller, Triplet Emitters for Organic Light-Emitting Diodes: Basic Properties, Wiley-VCH, Weinheim, 2008, ch. 1, pp. 1–97 Search PubMed.
- M. K. Nazeeruddin, C. Klein, M. Grätzel, L. Zuppiroli, and D. Berner, Molecular Engineering of Iridium Complexes and their Application in Organic Light Emitting Devices, Wiley-VCH, Weinheim, 2008, ch. 11, pp. 363–369 Search PubMed.
- E. Runge and E. K. U. Gross, Density-functional theory for time-dependent systems, Phys. Rev. Lett., 1984, 52(12), 997–1000 CrossRef CAS.
- C. Ullrich, Time-Dependent Density-Functional Theory: Concepts and Applications, Oxford University Press, USA, 2012 Search PubMed.
- C. A. Ullrich and Z. Yang, A brief compendium of time-dependent density-functional theory, 2013, arXiv:1305.1388.
- F. Wang, T. Ziegler, E. van Lenthe, S. van Gisbergen and E. J. Baerends, The calculation of excitation energies based on the relativistic two-component zeroth-order regular approximation and time-dependent density-functional with full use of symmetry, J. Chem. Phys., 2005, 122(20), 204103 CrossRef PubMed.
- K. Mori, T. Goumans, E. van Lenthe and F. Wang, Predicting phosphorescent lifetimes and zero-field splitting of organometallic complexes with time-dependent density functional theory including spin–orbit coupling, Phys. Chem. Chem. Phys., 2014, 16, 14523 RSC.
- D. Escudero and D. Jacquemin, Computational insights into the photodeactivation dynamics of phosphors for oleds: a perspective, Dalton Trans., 2015,(18), 8346–8355 RSC.
- M. E. Casida, Time-dependent density-functional theory for molecules and molecular solids, J. Mol. Struct.: THEOCHEM, 2009, 914(1), 3–18 CrossRef CAS.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, The Triplet State of Organo-Transition Metal Compounds. Triplet Harvesting and Singlet Harvesting for Efficient OLEDs, Coord. Chem. Rev., 2011, 255(21), 2622–2652 CrossRef CAS.
- D. Escudero and L. González, RASPT2/RASSCF vs. Range-Separated/Hybrid DFT Methods: Assessing the Excited States of a Ru(II)bipyridyl Complex, J. Chem. Theory Comput., 2011, 8(1), 203–213 CrossRef PubMed.
- A. Dreuw, J. L. Weisman and M. Head-Gordon, Long-range charge-transfer excited states in time-dependent density functional theory require non-local exchange, J. Chem. Phys., 2003, 119, 2943 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Failure of time-dependent density functional theory for long-range charge-transfer excited states: The zincbacteriochlorin–bacteriochlorin and bacteriochlorophyll–spheroidene complexes, J. Am. Chem. Soc., 2004, 126(12), 4007–4016 CrossRef CAS PubMed.
- O. Gritsenko and E. J. Baerends, Asymptotic correction of the exchange–correlation kernel of time-dependent density functional theory for long-range charge-transfer excitations, J. Chem. Phys., 2004, 121, 655–660 CrossRef CAS PubMed.
- M. E. Casida, Recent Advances in Density Functional Methods, Part I, ch. Time-dependent Density Functional Response Theory for Molecules, World Scientific, Singapore, 1995, pp. 155–192 Search PubMed.
- I. Vasiliev, S. Öğüt and J. R. Chelikowsky, Ab initio excitation spectra and collective electronic response in atoms and clusters, Phys. Rev. Lett., 1999, 82, 1919–1922 CrossRef CAS.
- F. Jensen, Introduction to computational chemistry, John Wiley & Sons, 2013 Search PubMed.
- J. P. Perdew, R. G. Parr, M. Levy and J. L. Balduz Jr, Density-functional theory for fractional particle number: derivative discontinuities of the energy, Phys. Rev. Lett., 1982, 49(23), 1691 CrossRef CAS.
- A. Görling, Density-functional theory for excited states, Phys. Rev. A: At., Mol., Opt. Phys., 1996, 54(5), 3912 CrossRef.
- M. Hellgren and E. Gross, Discontinuities of the exchange–correlation kernel and charge-transfer excitations in time-dependent density-functional theory, Phys. Rev. A: At., Mol., Opt. Phys., 2012, 85(2), 022514 CrossRef.
- P. M. W. Gill, R. D. Adamson and J. A. Pople, Coulomb-attenuated exchange energy density functionals, Mol. Phys., 1996, 88, 1005–1009 CrossRef CAS.
- A. Savin, Recent Developments and Applications of Modern Density Functional Theory, Elsevier, Amsterdam, 1996, pp. 327–357 Search PubMed.
- T. Leininger, H. Stoll, H.-J. Werner and A. Savin, Combining long-range configuration interaction with short-range density functionals, Chem. Phys. Lett., 1997, 275, 151 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the coulomb-attenuating method (cam-b3lyp), Chem. Phys. Lett., 2004, 393(1), 51–57 CrossRef CAS.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, A long-range correction scheme for generalized-gradient-approximation exchange functionals, J. Chem. Phys., 2001, 115, 3540 CrossRef CAS.
- R. Baer, E. Livshits and U. Salzner, Tuned range-separated hybrids in density functional theory, Annu. Rev. Phys. Chem., 2010, 61, 85–109 CrossRef CAS PubMed.
- S. Refaely-Abramson, S. Sharifzadeh, N. Govind, J. Autschbach, J. B. Neaton, R. Baer and L. Kronik, Quasiparticle spectra from a nonempirical optimally tuned range-separated hybrid density functional, Phys. Rev. Lett., Nov 2012, 109, 226405 Search PubMed.
- T. Stein, L. Kronik and R. Baer, Reliable prediction of charge transfer excitations in molecular complexes using time-dependent density functional theory, J. Am. Chem. Soc., 2009, 131(8), 2818–2820 CrossRef CAS PubMed.
- P. J. Hay, Theoretical Studies of the Ground and Excited Electronic States in Cyclometalated Phenylpyridine Ir(III) Complexes Using Density Functional Theory, J. Phys. Chem. A, 2002, 106(8), 1634–1641 CrossRef CAS.
- M. S. Lowry, W. R. Hudson, R. A. Pascal and S. Bernhard, Accelerated luminophore discovery through combinatorial synthesis, J. Am. Chem. Soc., 2004, 126(43), 14129–14135 CrossRef CAS PubMed.
- L. Petit, P. Maldivi and C. Adamo, Predictions of optical excitations in transition-metal complexes with time dependent-density functional theory: influence of basis sets, J. Chem. Theory Comput., 2005, 1(5), 953–962 CrossRef CAS PubMed.
- A. Abbotto, C. Barolo, L. Bellotto, F. De Angelis, M. Grätzel, N. Manfredi, C. Marinzi, S. Fantacci, J.-H. Yum and M. K. Nazeeruddin, Electron-rich heteroaromatic conjugated bipyridine based ruthenium sensitizer for efficient dye-sensitized solar cells, Chem. Commun., 2008,(42), 5318–5320 RSC.
- J. Bossert and C. Daniel, Electronic absorption spectroscopy of [Ru(phen)2(bpy)]2+, [Ru(phen)2(dmbp)]2+, [Ru(tpy)(phen)(CH3CN)]2+ and [Ru(tpy)(dmp)(CH3CN)]2+ A theoretical study, Coord. Chem. Rev., 2008, 252(23–24), 2493–2503 CrossRef CAS.
- B. Beyer, C. Ulbricht, D. Escudero, C. Friebe, A. Winter, L. González and U. S. Schubert, Phenyl-1H-[1,2,3]triazoles as New Cyclometalating Ligands for Iridium(III) Complexes, Organometallics, 2009, 28(18), 5478–5488 CrossRef CAS.
- D. Escudero, M. Assmann, A. Pospiech, W. Weigand and L. González, Substituent effects on the light-induced C–C and C–Br bond activation in (bisphosphine)(η 2-tolane) Pt(0) complexes. A TD-DFT study, Phys. Chem. Chem. Phys., 2009, 11(22), 4593–4600 RSC.
- N. Tian, D. Lenkeit, S. Pelz, L. H. Fischer, D. Escudero, R. Schiewek, D. Klink, O. J. Schmitz, L. González and M. Schäferling, et al., Structure–Property Relationship of Red-and Green-Emitting Iridium(III) Complexes with Respect to Their Temperature and Oxygen Sensitivity, Eur. J. Inorg. Chem., 2010, 2010(30), 4875–4885 CrossRef.
- H. Wiedenhofer, S. Schützenmeier, A. von Zelewsky and H. Yersin, Characterization of triplet sublevels by highly resolved vibrational satellite structures. Application to Pt(2-thpy)2, J. Phys. Chem., 1995, 99(36), 13385–13391 CrossRef CAS.
- D. Donges, J. K. Nagle and H. Yersin, Intraligand Charge Transfer in Pt(qol)2. Characterization of Electronic States by High-Resolution Shpol'skii Spectroscopy, Inorg. Chem., July 1997, 36, 3040–3048 Search PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Synthesis and characterization of phosphorescent cyclometalated iridium complexes, Inorg. Chem., 2001, 40(7), 1704–1711 CrossRef CAS PubMed.
- A. F. Rausch, Diplomarbeit, Universität Regensburg, 2006.
- W. J. Finkenzeller, T. Hofbeck, M. E. Thompson and H. Yersin, Triplet State Properties of the OLED Emitter Ir(btp)2(acac): Characterization by Site-Selective Spectroscopy and Application of High Magnetic Fields, Inorg. Chem., 2007, 46(12), 5076–5083 CrossRef CAS PubMed.
- R. Bauer, W. J. Finkenzeller, U. Bogner, M. E. Thompson and H. Yersin, Matrix influence on the OLED emitter Ir(btp)2(acac) in polymeric host materials – Studies by persistent spectral hole burning, Org. Electron., 2008, 9(5), 641–648 CrossRef CAS.
- T. Hofbeck and H. Yersin, The triplet state of fac-Ir(ppy)3, Inorg. Chem., 2010, 49(20), 9290–9299 CrossRef CAS PubMed.
- T. Fischer, R. Czerwieniec, T. Hofbeck, M. M. Osminina and H. Yersin, Triplet state properties of a red light emitting [Pt(s-thpy)(acac)] compound, Chem. Phys. Lett., 2010, 486(1–3), 53–59 CrossRef CAS.
- H. Yersin, A. F. Rausch, and R. Czerwieniec, Organometallic emitters for OLEDs: Triplet harvesting, singlet harvesting, case structures, and trends, in Physics of Organic Semiconductors, ed. W. Brütting and C. Adachi, Wiley-VCH, Weinheim, 2012, pp. 371–424 Search PubMed.
- A. Bossi, A. F. Rausch, M. J. Leitl, R. Czerwieniec, M. T. Whited, P. I. Djurovich, H. Yersin and M. E. Thompson, Photophysical Properties of Cyclometalated Pt(II) Complexes: Counterintuitive Blue Shift in Emission with an Expanded Ligand π System, Inorg. Chem., 2013, 52(21), 12403–12415 CrossRef CAS PubMed.
- H. Yersin, Triplet emitters for OLED applications. Mechanisms of exciton trapping and control of emission properties, Top. Curr. Chem., 2004, 241, 1–26 CrossRef CAS.
- H. Yersin, P. Huber and H. Wiedenhofer, Vibrational satellite structures and properties of electronic states of transition metal complexes, Coord. Chem. Rev., 1994, 132, 35–42 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, NWChem: A Comprehensive and Scalable Open-Source Solution for Large Scale Molecular Simulations, Comput. Phys. Commun., 2010, 181(9), 1477–1489 CrossRef CAS.
- A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82(1), 299–310 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393(1–3), 51–57 CrossRef CAS.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, A long-range-corrected time-dependent density functional theory, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128(8), 084106 CrossRef PubMed.
- M. J. Peach, M. J. Williamson and D. J. Tozer, Influence of triplet instabilities in TDDFT, J. Chem. Theory Comput., 2011, 7(11), 3578–3585 CrossRef CAS PubMed.
- M. J. Peach and D. J. Tozer, Overcoming Low Orbital Overlap and Triplet Instability Problems in TDDFT, J. Phys. Chem. A, 2012, 116(39), 9783–9789 CrossRef CAS PubMed.
- J. S. Sears, T. Koerzdoerfer, C.-R. Zhang and J.-L. Brédas, Orbital instabilities and triplet states from time-dependent density functional theory and long-range corrected functionals, J. Chem. Phys., 2011, 135, 151103 CrossRef PubMed.
- S. Hirata and M. Head-Gordon, Time-Dependent Density Functional Theory within the Tamm–Dancoff Approximation, Chem. Phys. Lett., 1999, 314(3), 291–299 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, Revised basis sets for the lanl effective core potentials, J. Chem. Theory Comput., 2008, 4(7), 1029–1031 CrossRef CAS PubMed.
- A. Vlček Jr and S. Zališ, Modeling of Charge-transfer Transitions and Excited States in d6 Transition Metal Complexes by DFT Techniques, Coord. Chem. Rev., 2007, 251, 258–287 CrossRef.
- A. Klamt and G. Schüürmann, Cosmo: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient, J. Chem. Soc., Perkin Trans. 1, 1993, 2, 799–805 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378 CrossRef CAS PubMed.
- M. R. Silva Jr, M. Schreiber, S. P. Sauer and W. Thiel, Benchmarks for electronically excited states: Time-dependent density functional theory and density functional theory based multireference configuration interaction, J. Chem. Phys., 2008, 129, 104103 CrossRef PubMed.
- D. Jacquemin, E. A. Perpete, I. Ciofini and C. Adamo, Assessment of Functionals for TD-DFT Calculations of Singlet–Triplet Transitions, J. Chem. Theory Comput., 2010, 6(5), 1532–1537 CrossRef CAS PubMed.
- A. Dominguez, B. Aradi, T. Frauenheim, V. Lutsker and T. A. Niehaus, Extensions of the time-dependent density functional based tight-binding approach, J. Chem. Theory Comput., 2013, 9(11), 4901–4914 CrossRef CAS PubMed.
- T. A. Niehaus, Entwicklung approximativer Methoden in der zeitabhängigen Dichtefunktional Theorie, PhD thesis, Universität Paderborn, 12 2001, Dissertation.
- P.-O. Löwdin, Adv. Chem. Phys., 1970, 5, 185 Search PubMed.
- J. J. Philips, M. A. Hudspeth, P. M. Browne and J. E. Peralta, Basis set dependence of atomic spin populations, Chem. Phys. Lett., 2010, 495(1), 146–150 CrossRef CAS.
- R. K. Szilagyi, M. Metz and E. I. Solomon, Spectroscopic calibration of modern density functional methods using [CuCl4]2, J. Phys. Chem. A, 2002, 106(12), 2994–3007 CrossRef CAS.
- T. Hofbeck, Radiative und nicht-radiative Desaktivierung von OLED-relevanten Ir(III)- und Pt(II)-Komplexen, PhD thesis, University Regensburg, 2011.
- T. Fischer, PhD thesis, University of Regensburg, 2010.
- H. Wiedenhofer, PhD thesis, University of Regensburg, 1994.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118(11), 4775–4777 CrossRef CAS.
- M. Head-Gordon, A. M. Grana, D. Maurice and C. A. White, Analysis of electronic transitions as the difference of electron attachment and detachment densities, J. Phys. Chem., 1995, 99(39), 14261–14270 CrossRef CAS.
- M. J. Peach, P. Benfield, T. Helgaker and D. J. Tozer, Excitation energies in density functional theory: An evaluation and a diagnostic test, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- C. A. Guido, P. Cortona, B. Mennucci and C. Adamo, On the Metric of Charge Transfer Molecular Excitations: A Simple Chemical Descriptor, J. Chem. Theory Comput., 2013, 9(7), 3118–3126 CrossRef CAS PubMed.
- M. Modrzejewski, Ł. Rajchel, G. Chalasinski and M. M. Szczesniak, Density-dependent onset of the long-range exchange: A key to donor–acceptor properties, J. Phys. Chem. A, 2013, 117(45), 11580–11586 CrossRef.
- T. Körzdörfer, J. Sears, C. Sutton and J.-L. Brédas, Long-range corrected hybrid functionals for π-conjugated systems: Dependence of the range-separation parameter on conjugation length, J. Chem. Phys., 2011, 135, 204107 CrossRef.
- C. Kratzer, PhD thesis, University of Regensburg, 2004.
- S. Strickler and R. A. Berg, Relationship between absorption intensity and fluorescence lifetime of molecules, J. Chem. Phys., 1962, 37(4), 814–822 CrossRef CAS.
- J. Mohanty and W. M. Nau, Refractive index effects on the oscillator strength and radiative decay rate of 2,3-diazabicyclo[2.2.2]oct-2-ene, Photochem. Photobiol. Sci., 2004, 3(11–12), 1026–1031 CAS.
- A. F. Rausch, H. H. Homeier and H. Yersin, Organometallic Pt(II) and Ir(III) Triplet Emitters for OLED Applications and the Role of Spin–Orbit Coupling: A Study Based on High-Resolution Optical Spectroscopy, in Photophysics of Organometallics, Springer, 2010, pp. 193–235 Search PubMed.
- D. Jacquemin, V. Wathelet, E. A. Perpete and C. Adamo, Extensive TD-DFT Benchmark: Singlet–Excited States of Organic Molecules, J. Chem. Theory Comput., 2009, 5(9), 2420–2435 CrossRef CAS.
- M. Caricato, G. W. Trucks, M. J. Frisch and K. B. Wiberg, Oscillator strength: How does TDDFT compare to EOM-CCSD?, J. Chem. Theory Comput., 2010, 7(2), 456–466 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Listings of vertical excitation energies at different levels of theory, zero point energies and plots of relevant molecular orbitals. See DOI: 10.1039/c5ra12962a |
| ‡ It should be noted that even though the Kohn–Sham gap will differ from the quasiparticle gap also for the (unknown) exact Kohn–Sham exchange–correlation functional, charge-transfer excited states can be correctly described also in the framework of local functionals. To this end one has to go beyond a static xc kernel fxcστ which restores both the difference between Kohn–Sham and quasiparticle gap as well as the 1/R stabilization.47 |
| § In such a situation the analysis is better based on the concept of natural transition orbitals introduced by Martin,101 which is not yet implemented in NWChem. |
| This journal is © The Royal Society of Chemistry 2015 |