
Synthesis and electrochemical performance of Li4Ti5O12/TiO2/C nanocrystallines for high-rate lithium ion batteries†
Abstract
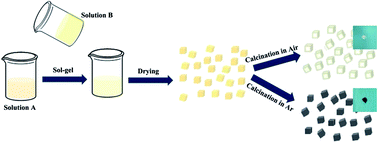
A Li4Ti5O12/TiO2/carbon (Li4Ti5O12/TiO2/C) nanocrystalline composite has been successfully synthesized by a facile sol–gel method and subsequent calcination treatment. Moreover, pure Li4Ti5O12, Li4Ti5O12/C and Li4Ti5O12/TiO2 composites have also been synthesized for comparison. All the samples present a nanocrystalline structure with a uniform size distribution, and abundant phase interfaces can be detected for the Li4Ti5O12/TiO2 and Li4Ti5O12/TiO2/C composites. Electrochemical measurements demonstrate that the resultant Li4Ti5O12/TiO2/C composite exhibits a superior rate capability and cycle stability in comparison with pure Li4Ti5O12, Li4Ti5O12/C and Li4Ti5O12/TiO2 composites, which can be attributed to a high grain boundary density, abundant phase interfaces and in situ formed carbon, for storing extra lithium ions, enhancing rapid lithium insertion/extraction kinetics and improving the electron transport rate.
 Please wait while we load your content...
Please wait while we load your content...

