
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-high dispersion NMR reveals new levels of detail†
Juan A.
Aguilar
*a,
Peter
Kiraly
b,
Ralph W.
Adams
b,
Mickaëlle
Bonneau
a,
Elizabeth J.
Grayson
a,
Mathias
Nilsson
b,
Alan M.
Kenwright
a and
Gareth A.
Morris
b
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: j.a.aguilar@durham.ac.uk
bSchool of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK
First published on 5th June 2015
Abstract
Pure shift NMR techniques can provide exquisite resolution, enabling chemists to analyse samples that would otherwise require unrealistically high magnetic fields. Although powerful, such techniques remain little used, in part because of the false perception that they are unsuitable for daily use. The present paper shows that this need not be the case and that high quality results can be produced routinely even for dilute and highly complex samples.
Every day very large numbers of 1H NMR spectra are acquired both in academia and industry: 1H NMR is the main workhorse of chemical structure determination. One common problem with 1H NMR spectra is that signals overlap. The low dispersion of proton signals and the extra space needed to accommodate signal multiplicity often conspire to make such spectra hard to interpret. One remedy is to use two-dimensional NMR methods. Experiments such as 1H–13C HSQC1 that exploit the large dispersion of the carbon dimension provide the best resolving power for most molecules. Unfortunately, many samples remain difficult to analyse even with these methods. Typical problems that are frequently encountered are those in which structural similarity between different parts of a molecule is present. Carbohydrates, complexes, lipids, aromatic systems and oligomers are typical examples. Naturally, the presence of multiple species makes the problem even worse. Some cases lead to such difficulties that analysis is often not even attempted.
There is little that can be done to deal with the poor chemical shift range of protons, but, fortunately, signal dispersion can be improved by collapsing proton multiplets into singlets using “pure shift” methods.2,3 This approach can be highly efficient: to achieve similar resolution by the usual means of increasing the magnetic field would require fields that are way beyond current engineering capabilities.4,5 However, in spite of the great performance advantage of pure shift methods, their potential remains largely untapped; they are as yet relatively little used. They are two reasons for this. The first is that there is a mismatch between what a chemist considers a typical sample and the samples that have been used to demonstrate pure shift methods. Most samples run by NMR services catering to chemists are not particularly concentrated, tend to be impure (only end products tend to be pure), often contain unknowns, and are sometimes unstable. In contrast, samples used for demonstrating the performance of new NMR techniques tend to be concentrated, pure, of known composition, and stable. The second reason is the common assumption that pure shift experiments are complex and time-consuming, and not suitable for routine service use.
The purpose of this paper is to show, as for example in Fig. 1–3 below, that this assumption is both incorrect and unfortunate: many problems that could be solved using pure shift methods go unnoticed. It is precisely in routine NMR facility use that such methods can make a major impact on research projects that require frequent NMR analysis.7 Furthermore, it is not true that all pure shift experiments require excessively long times. The sensitivity of current hardware means that most samples can be run within modest time allocations. As will be shown here, impressive results can be produced routinely even with samples of uncertain composition, often dilute, sometimes unstable, and always complex. The target audience is that of researchers dealing with small- to medium-sized molecules.
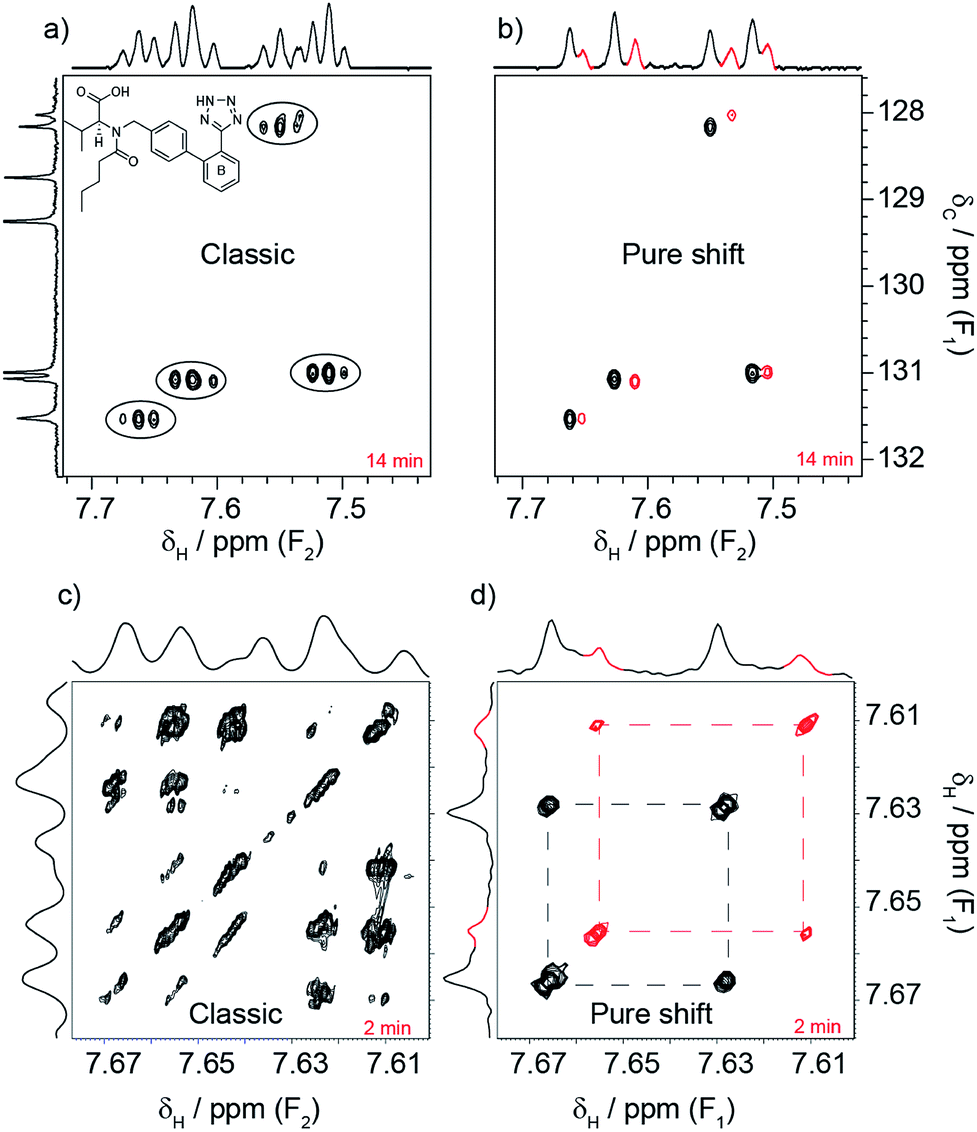 | ||
| Fig. 1 These spectra illustrate how pure shift NMR can reveal a level of detail that conventional experiments lack. The sample is an anti-hypertensive drug, valsartan. There is little indication in the classic HSQC spectrum (a) that the spin system of ring B is duplicated. This becomes clear when the proton splittings are removed in the pure shift version (b). The extra spin system is shown in red. The same improvement applies to the COSY ((c) conventional and (d) pure shift) and the 1H (top and side of the COSY) spectra. Note that the spin system of (c) and (d) is only part of that shown in (a) and (b). Many such cases are likely to go unnoticed in everyday investigations. | ||
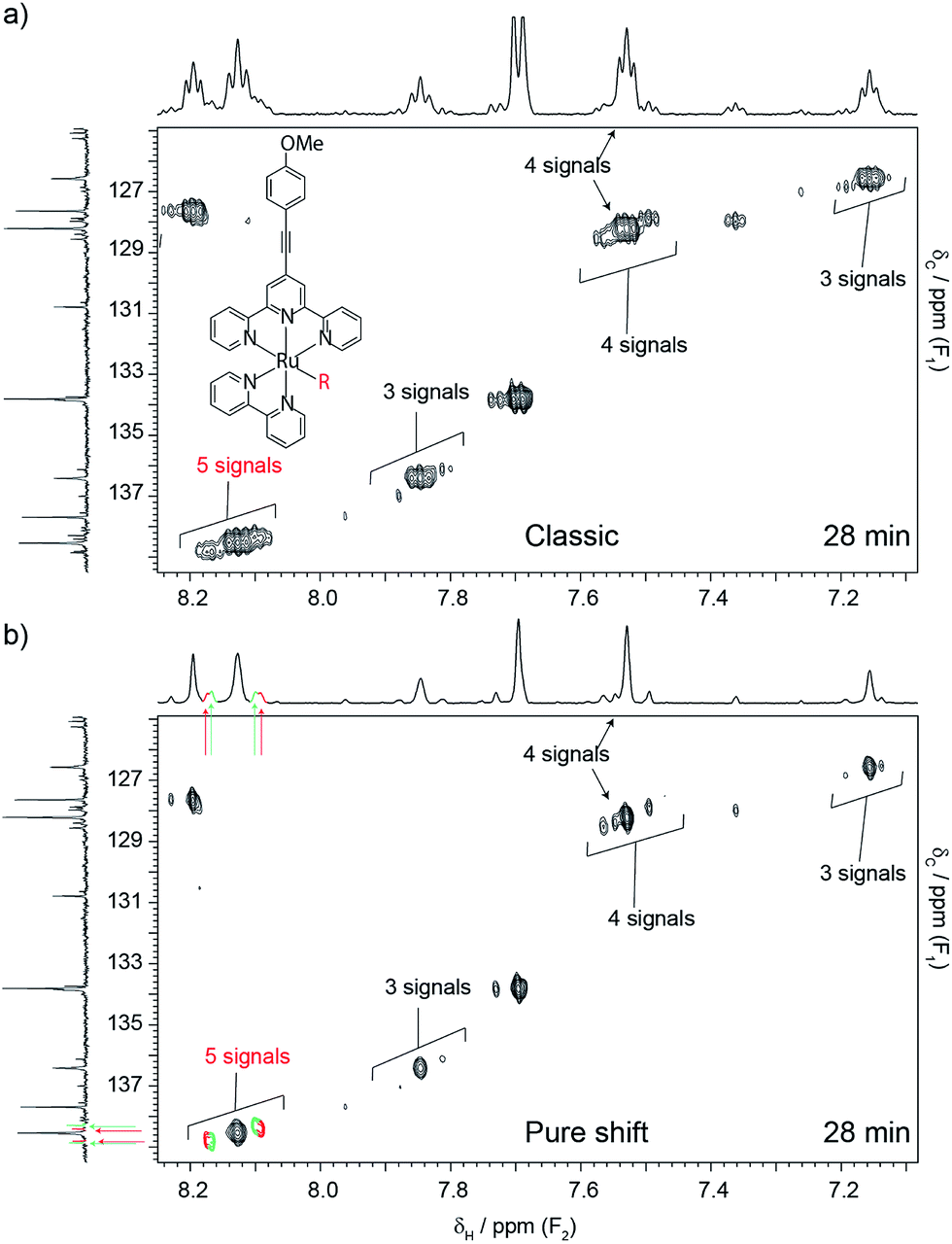 | ||
| Fig. 2 Conventional (a) and pure shift (b) HSQC spectra of a metallo-photo-catalyst whose structure is depicted in (a). The number and nature of species present in solution was of interest because the activity of the catalyst depends on the replacement of the labile group (–R, originally a triflate), with weak chelators. It was also of interest because degradation products compromise the performance of the catalyst. Replacement of the labile group is characterised by patterns of peaks that shadow one another, due to small chemical shift changes. On the other hand, degradation products produce signals that deviate from such patterns. The shadowing pattern can be seen in both experiments, but the pure shift version (b) reveals more species that its conventional counterpart (a). Each spectrum took 28 min to acquire, while the carbon spectrum took 3 h. The total catalyst concentration was 7.5 mM, made up of multiple species. | ||
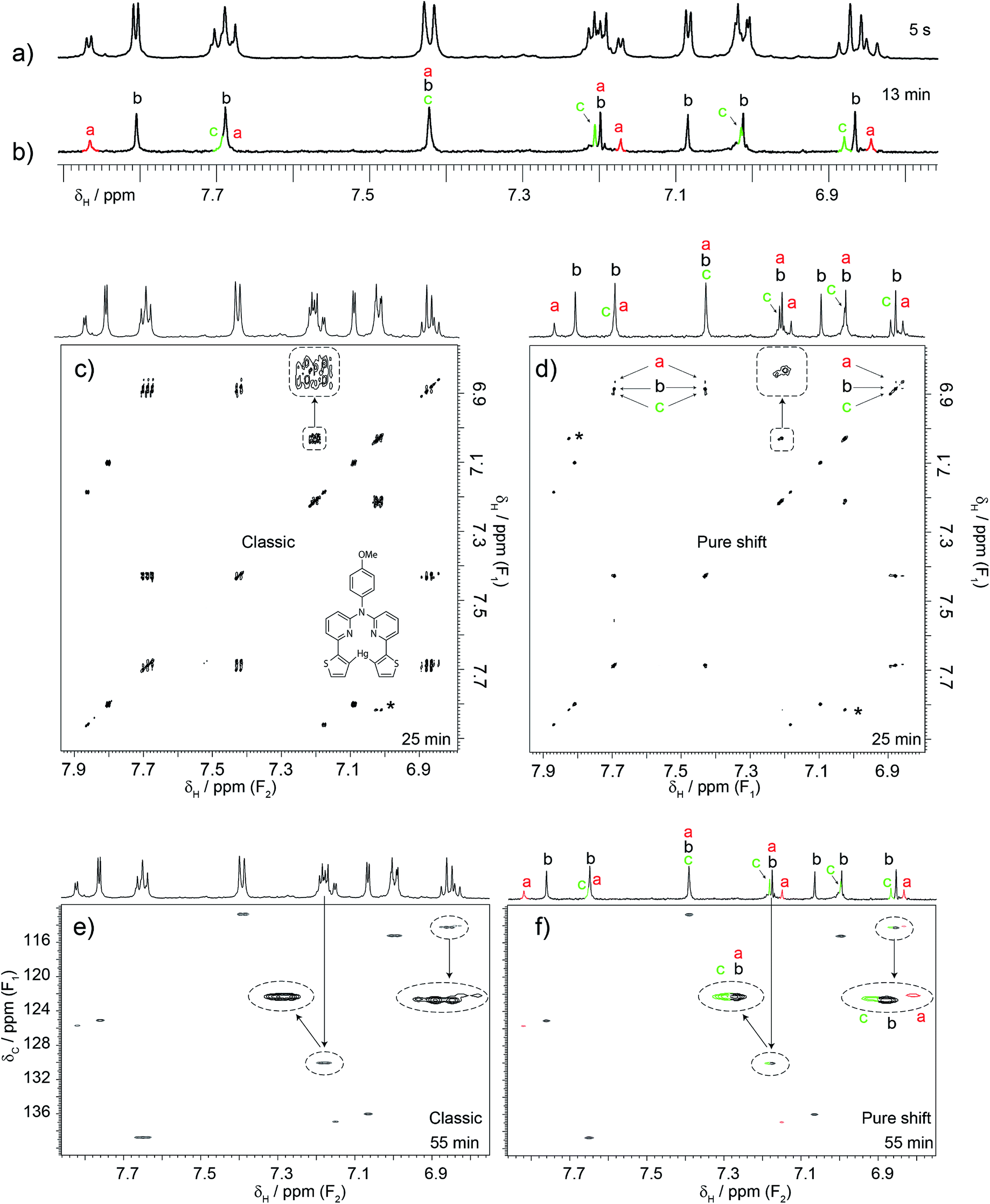 | ||
| Fig. 3 An example of how the proposed combination of experiments was used to solve a problem involving a dilute sample containing unexpected components. A researcher was analysing the 1H, COSY and HSQC spectra of a sample containing the expected mercury complex shown in (c). The researcher was only suspicious of the 7.85 ppm signal but a colleague, suspecting that this was not the only problem, acquired a pure shift 1H spectrum. This revealed the presence of more signals than could be attributed to the expected product. Some signals were doubled or even tripled, indicating that the sample contained at least three species. This led to the acquisition of pure shift COSY and HSQC experiments. These revealed that other signals that appear as singlets in the one-dimensional pure shift spectrum were mixtures of overlapping signals. Notice that their correlation patterns are quite similar even in the HSQC spectra, thus indicating structural relatedness. An extended analysis indicated the presence of the uncoordinated ligand and two complexes. The free ligand was actually present but in exchange with at least one of the complexes. It was this dynamic process that prevented the researcher from detecting the presence the free ligand. Note that the combination of pure shift 1H, COSY and HSQC made the analysis possible. The pure shift experiments took a total of 1.5 h of spectrometer time although only one should have sufficed. Notice how a little more than one hour of spectrometer time made an important impact on this particular investigation. The total concentration was 1.6 mM but three species are present. The cross-peak labelled with an asterisk is a folded peak. Note that the labels of the axes in (c) are correct, F1 and F1, since covariance NMR derives the frequency of both dimensions from t1 data. | ||
The examples presented here are all drawn from an NMR service that caters to typical synthetic chemists. All the samples formed part of real investigations, and had to be analysed within the time-constrained schedule of such a service. In all cases, sample composition turned out to be more complex than initially anticipated, and in some cases, there was no indication that pure shift methods were necessary. Three experiments were used, a one-dimensional pure shift 1H, a pure shift COSY and a modified real-time pure shift 1H–13C HSQC experiment. One-dimensional spectra were produced using a Zangger–Sterk5 (ZS) pulse sequence, taking the precaution of reducing the bandwidth to be decoupled to cover only signals of interest. It has to be kept in mind that sensitivity deceases as decoupling bandwidth increases. The recently reported PSYCHE method6 could have been used to advantage, had it been available before the manuscript was in production, as it has higher sensitivity and does not require a compromise to be made between sensitivity and decoupling bandwidth. The COSY experiment used a single quantum constant-time sequence instead of the previously reported multiple-quantum filtered experiment,8 as it is more sensitive. PSYCHE-TOCSY9 could also been used following similar arguments to the 1D experiment. Both constant-time COSY and PSYCHE-TOCSY are less sensitive than their traditional counterparts, but this is normally not a problem for the target audience provided that large constant times in the first case and long adiabatic pulses in the second are avoided. Combining these experiments with covariance processing10 further simplifies spectra, as in the examples presented, but this is optional. For HSQC, a variant of a previous version11 was used differing only in that, in this version, adiabatic pulses were used to improve the off-resonance performance of the carbon pulses. Unlike other pure shift experiments, this one is more sensitive than its conventional counterpart. All pulse sequences can be found in the ESI.† Other pure shift experiments exist that require far longer acquisition times than their conventional analogues. In some situations those will be necessary. The proposed set of experiments can then be used to assess the severity of the problem and whether the aforementioned ones are worth running.
Valsartan, an angiotensin II receptor antagonist, is a good example of why it is helpful to acquire pure shift spectra routinely. The drug was being investigated to determine whether its conformations in solution could predict the existence of polymorphs in solid formulations. Part of the investigation focused on the spin system shown in Fig. 1a. The spin system is duplicated due to the presence of different conformers, but this duplication went unnoticed even when a combination of conventional experiments was used (1H, 1H–1H COSY, 13C{1H} and 1H–13C HSQC). In contrast, such duplication became clear when pure shift variants were used (same figure). 20 min was all that was needed to break the problem using the pure shift experiments.
Time restrictions are a fact of life for NMR facilities, and users often struggle to solve the problem at hand under typical time constraints. The problem is aggravated when samples are suspected of being unstable, as in the next example. Here, the object of the investigation was the photo-catalyst of Fig. 2a. There was an interest in determining how many different complexes existed in solution, as the efficiency of the catalyst depends on the nature and stability of such complexes. All the complexes were expected to have the same structure, apart from one labile group (–R). This group was initially a triflate, but was expected to be replaced with other species present in solution. This type of problem is very difficult because all the species are very similar, and produce signals that are very close to one another. The utility of the catalyst also depended on its stability, so the need to identify possible degradation products further complicated matters. Cases such as the present one can be analysed using 1H–13C HSQC experiments, because signals from similar complexes produce patterns only slightly displaced from one another while degradation products tend to produce at least some signals with significant deviations. The problem is that conventional 1H–13C HSQC can only be successful if the carbon dimension is very well digitised, which is very time-consuming. In this particular case, adequate digitisation would have required 4 to 5 h of spectrometer time. This was not available because little time was left after three hours of spectrometer time had already been consumed acquiring a 13C{1H} spectrum that did not provide much insight. In contrast, a pure shift HSQC spectrum provided the necessary resolving power within 28 min, showing that at least five very similar species were present. Notice how major peaks are shadowed by minor ones, producing the repetitive pattern noted earlier (see Fig. 2b.) It is remarkable that this was accomplished in spite of using standard equipment, the short time available, and the low concentration of the sample, 7.5 mM shared among the different species. This was possible because while the resolving power of the pure shift HSQC experiment increases only linearly with experiment duration in the carbon domain, the large resolution improvement afforded by pure shift acquisition comes at no cost in experiment time. A variety of similar examples from our NMR service can be seen in the ESI† section.
Although in the previous case a single experiment was enough, it is generally better to use the three proposed experiments together. A case that typifies this situation is that of Fig. 3. Here another researcher was making a biomedical imaging probe12 that required formation of the mercury complex shown. The unexpected form of the signal at 7.85 ppm led to the measurement of a 1D pure shift 1H spectrum. Unexpectedly, this revealed that some signals were doubled or tripled, indicating that the sample contained at least three species (see for example the 6.8–6.9 ppm cluster of peaks). The task was then to determine the identities of these species. It was necessary to correlate and group those extra signals, but this was not possible using conventional COSY. The double pure shift COSY spectrum revealed that some signals that appear as singlets in the pure shift 1H spectrum actually originate from mixtures of signals that show similar correlation patterns. This becomes clear when all three complementary pure shift experiments are used in conjunction. Further analysis indicated the presence of the free ligand and two complexes, meaning that the sample was unsuitable for its intended use. However, the experiments also indicated that the problem could be solved by repeating the reaction with a higher mercury to ligand ratio. The reaction was attempted and indeed a single product was obtained. The take-home message here is not that pure shift experiments revealed some hidden signals, but that they allowed analysis at a level of detail that made it possible to identify an unsuspected problem with the synthesis, and subsequently optimise the synthesis to avoid the problem. Again, this was done with a sample that contained less than 1 mg mL−1 of material (i.e. sub-mM concentrations) using a conventional (not cryogenically-cooled) probe. All the experiments took the same time to run as their classic counterparts, with the exception of the pure shift 1H 1D spectrum, which took 13 min. The pure shift HSQC was left to run for 55 min, but 30 would have been sufficient, and the constant-time COSY took 25 min. To put these experiments in context, a standard 13C{1H} spectrum was acquired. Chemists typically rely on such experiments to provide resolving power, yet after 4 h the resulting spectrum (included in the ESI†) was barely usable.
Conclusions
In all the cases presented, pure shift methods revealed a level of detail that was unobtainable using conventional NMR experiments. In all cases, the pure shift experiments fitted well within the typical schedule of the NMR service, and indeed in some cases required less time than conventional experiments. The potential of pure shift experiments as enablers of better and more efficient investigations is only going to grow in the near future, and there is much to be gained by early adoption. Used in conjunction with emerging time-saving approaches such as non-uniform sampling13 and with high sensitivity probes,14,15 such experiments are poised to deliver a minor revolution in NMR spectroscopy.Acknowledgements
The authors are indebted to the Durham University NMR service, R. Goodwill, Prof P. J. Low, Prof P. G. Steel and Prof J. A. G. Williams. This work was supported by the Engineering and Physical Sciences Research Council (grant numbers EP/I007989/1, and EP/L018500/1). Dr Kiraly thanks the European Commission for a Marie Curie Intra-European Fellowship, RTACQ4PSNMR, no. PIEF-GA-2013-625058. The authors also thank the Universities of Durham and Manchester for support.References
- G. Bodenhausen and D. J. Ruben, Chem. Phys. Lett., 1980, 69, 185 CrossRef CAS.
- N. H. Meyer and K. Zangger, ChemPhysChem, 2014, 15, 49 CrossRef CAS PubMed.
- R. W. Adams, eMagRes, 2014, 3, 295, DOI:10.1002/9780470034590.emrstm1362.
- K. Zangger and H. Sterk, J. Magn. Reson., 1997, 124, 486 CrossRef CAS.
- J. A. Aguilar, S. Faulkner, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2010, 122, 3901 CrossRef PubMed.
- J. A. Aguilar, J. Cassani, M. Delbianco, R. W. Adams, M. Nilsson and G. A. Morris, Chem.–Eur. J., 2015, 21, 6623 CrossRef CAS PubMed.
- M. Foroozandeh, R. W. Adams, N. J. Meharry, D. Jeannerat, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2014, 53, 6990 CrossRef CAS PubMed.
- J. A. Aguilar, A. A. Colbourne, J. Cassani, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2012, 51, 6460 CrossRef CAS PubMed.
- M. Foroozandeh, R. W. Adams, M. Nilsson and G. A. Morris, J. Am. Chem. Soc., 2014, 136, 11867 CrossRef CAS PubMed.
- R. Brüschweiler and F. Zhang, J. Chem. Phys., 2004, 120, 5253 CrossRef PubMed.
- L. Paudel, R. W. Adams, P. Király, J. A. Aguilar, M. Foroozandeh, M. J. Cliff, M. Nilsson, P. Sándor, J. P. Waltho and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 11616 CrossRef CAS PubMed.
- W. A. Tarran, G. R. Freeman, L. Murphy, A. M. Benham, R. Kataky and J. A. G. Williams, Inorg. Chem., 2014, 53, 5738 CrossRef CAS PubMed.
- M. Mobli and J. C. Hoch, Prog. Nucl. Magn. Reson. Spectrosc., 2014, 83, 21 CrossRef CAS PubMed.
- H. Kovacs, D. Moskau and M. Spraul, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 131 CrossRef CAS PubMed.
- Y. Liu, M. D. Green, R. Marques, T. Pereira, R. Helmy, R. T. Williamson, W. Bermel and G. E. Martin, Tetrahedron Lett., 2014, 55, 5450 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (1) The 13C{1H} spectrum acquired to serve as a reference for the example of Fig. 3. (2) Pure shift NMR spectra of selected samples submitted to the Durham NMR service. (3) Pulse sequences and macros used in the present publication. The real-time HSQC pulse sequence for Agilent spectrometers. The constant-time COSY pulse sequence for Agilent spectrometers. The 1D Zangger–Sterk pulse sequence for Agilent spectrometers. The reconstruction macro necessary to assemble the raw data produced by the Zangger–Sterk pulse sequence. The raw data files used to produce the spectra shown in the figures are available from DOI: 10.15128/s1784k78s. See DOI: 10.1039/c5ra10192a |
| This journal is © The Royal Society of Chemistry 2015 |