
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyethyleneimine for copper absorption II: kinetics, selectivity and efficiency from seawater†
Johan B. Lindéna,
Mikael Larsson*a,
Simarpreet Kaura,
William M. Skinnera,
Stanley J. Miklavcicb,
Thomas Nanna,
Ivan M. Kempsona and
Magnus Nydéna
aIan Wark Research Institute, University of South Australia, Mawson Lakes, SA 5095, Australia. E-mail: larsson.mikael@gmail.com; Fax: +61-8-830-23493; Tel: +61-8-830-23493
bPhenomics and Bioinformatics Research Centre, University of South Australia, Mawson Lakes, SA 5095, Australia
First published on 4th June 2015
Abstract
Glutaraldehyde (GA) cross-linked polyethyleneimine (PEI) coatings have previously been reported to effectively and selectively take up copper from seawater relevant concentrations in artificial seawater. We evaluate the copper uptake of such coatings from natural seawater. X-ray photoelectron spectroscopy elemental analysis revealed the coatings to be highly efficient and equally selective for copper uptake in natural seawater, reaching a maximum copper loading of 2 wt% in 48 hours. Similar to observations in artificial seawater we found that zinc was initially accumulated in the coatings, but was exchanged by copper over time. We investigate the spatial distribution of copper in the coatings by time-of-flight secondary ion mass spectrometry (ToF-SIMS), which revealed that copper was evenly distributed in the coating, with the exception of lower concentrations at the coating-water interface. We use synchrotron X-ray absorption studies and Fourier transform infrared (FTIR) spectroscopy to show that the copper–ligand interaction was mediated by Schiff's bases (imines).
Introduction
Materials with the ability to bind copper from seawater could enable remediation of copper-contaminated marine environments as well as the use of the copper for advanced marine applications. Anti-biofouling coatings on vessels are possibly the largest contaminant source of copper to marinas and copper-based anti-biofouling coatings could end up sharing similar regulations to those containing tributyltin (TBT).1–4 The academic and industry communities working on anti-biofouling coatings have so far been unable to respond to the challenge of creating new environmentally friendly and biocide-free alternatives with performance comparable to currently used coatings that release copper as the main biocidal agent.We propose here an anti-biofouling mechanism based on cycles of accumulation and release of copper naturally abundant in seawater to create a biocidal flux of copper across an interface, i.e., a coating that uses the well-documented biocidal effect of copper without any net release of copper into the sea. The mechanism relies on copper-selective polymers for uptake and electrically conducting and active polymers for triggered release. The working principle of the antibiofouling concept comprises four stages: (1) uptake of copper into the coating, (2) electrochemical stimulus for switching the function of the coating into a copper-release state, (3) antibiofouling action of the released copper and (4) electrochemical stimulus for switching back to copper-uptake mode. This would enable a never-ending cycle because of the unlimited access to copper from seawater.
In this work we focused on copper-uptake from seawater, stage (1). For the concept to work, a polymeric material with extreme affinity and selectivity for copper is needed to overcome the effects of extremely low copper concentrations and competing ions in natural seawater.
Materials with extreme copper-uptake efficiency and selectivity also have potential for mining the oceans and pollution remediation. Chouyyok et al.5 showed that chelating diamines could be successfully used for the purpose of extracting copper from polluted natural waters. With respect to mining the oceans we note that the focus has been on polymer-based materials, critically reviewed by Schwochau.6 The main problem with this application is that the process is too energy demanding.7
Polyethyleneimine (PEI)-based materials have been intensively investigated for metal binding.8–10 Removal of many metal ions from wastewaters including Cu2+, Ni2+, Fe2+, Co2+, Zn2+, Cd2+, Pb2+, Cr3+ and Hg2+ has been demonstrated8–13 and they seem to have strong preference for copper,10–12,14 although there is a deviating report.8 Due to its copper selectivity and efficiency, PEI has been used as a spectroscopic detection agent of low parts per billion (ppb) concentrations.15–17 PEI is often attached to carrier particles such as silica,11,12,14,18 porous magnetic powder,10 biomass,19,20 commercial acrylic fibre,21 porous cellulose material,9 agarose beads22,23 poly(methyl methacrylate) microspheres8 and polystyrene-based macroporous cation exchange resin.24
Branched PEI binds metal ions stronger than linear versions25 and typically consists of primary, secondary and tertiary amines in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.7 ratios26 with branching sites every 3–3.5 nitrogen atoms.27 PEI has the advantages of good hydrophilicity, fast rate of adsorption and high adsorption capacity.28,29 Moreover PEI does not chelate alkali or alkaline earth metals at any pH, which is advantageous in water with high salt content as in the case of seawater.18,30
:1:0.7 ratios26 with branching sites every 3–3.5 nitrogen atoms.27 PEI has the advantages of good hydrophilicity, fast rate of adsorption and high adsorption capacity.28,29 Moreover PEI does not chelate alkali or alkaline earth metals at any pH, which is advantageous in water with high salt content as in the case of seawater.18,30
In a recent communication31 we reported on the potential of glutaraldehyde (GA) cross-linked PEI for selective extraction of copper. Nano-thin coatings were shown to accumulate up to 5–13 wt% copper from artificial seawater containing 2–200 ppb copper, e.g., concentrations relevant to polluted harbours and marinas. The work further revealed a time-dependant competitive binding and replacement mechanism between copper and zinc.
In a recent publication we investigated possible scenarios for the replacement mechanism by mathematical modelling.32 We showed that difference in uptake kinetics and equilibrium content of copper and zinc can be explained by a combination of (a) differing ion diffusive rates and (b) an ion exchange process that is superimposed on the independent binding processes. The difference in uptake kinetics could be due to differences in hydrated size of the metal complexes and the ion exchange process likely involves conformational changes in the polymer structure in response to the binding of one of the species.32
In this paper we evaluate the performance of the GA cross-linked PEI coatings in natural seawater and further investigate the competitive binding between copper and zinc. Furthermore, we characterise the copper distribution in the coatings and the copper–ligand chemistry. We determine the uptake of copper and zinc by X-ray photoelectron spectroscopy (XPS) and evaluate the distribution of copper in the coatings with time-of-flight secondary ion mass spectrometry (ToF-SIMS). Finally, we use FTIR and synchrotron X-ray absorption spectroscopy (XAS) to study the chemistry of the copper–PEI interaction.
Experimental
Materials
All chemicals were purchased as analytical or reagent grade and used as received without further purification. Branched polyethyleneimine (50 wt% in H2O, Mn ∼ 60000 and MW ∼ 750000) and glutaraldehyde (25 wt% in H2O) were purchased from Sigma-Aldrich and stored under N2. ZnCl2 was purchased from Scharlau, Scharlab S.L. Copper(II) sulphate pentahydrate (CuSO4·5H2O) and sodium hydroxide (NaOH) pellets were purchased from Chem-Supply Pty Ltd. Sea salts (40 g L−1, Sigma-Aldrich) was used to prepare artificial seawater. Ultrapure water with a resistivity of 18.2 MΩ cm was obtained using a Milli-Q® Advantage A10® water purification system. Seawater was collected at Outer Harbour, Adelaide, South Australia, Australia (coordinates 34°46′49.0′′S 138°28′49.4′′E).
For XAS, copper(II)oxide (CuO), copper(I)oxide (Cu2O), EDTA tetrapotassium salt, ammonia solution, copper(II)nitrate (Cu(NO3)2·3H2O), copper(I)chloride (CuCl), copper(II)chloride (CuCl2) and ethylenediamine were all purchased from Sigma-Aldrich. Ligands were mixed with copper(II)sulphate pentahydrate (CuSO4·5H2O) in Milli-Q water, while the copper salts were mixed with Milli-Q water for liquid standards or microcrystalline cellulose (powder, 20 μm, Sigma-Aldrich) for solid standards.
Ellipsometry
Dry layer thicknesses were determined using a Variable Angle Spectroscopic Ellipsometer (VASE®) and WVASE32® software (J.A. Woollam Co., Inc.) with the thickness of the coatings being modelled as a Cauchy layer on a sublayer with the optical constants of the used silicon wafers.X-ray photoelectron spectroscopy (XPS)
XPS measurements were undertaken using monochromatized Al Kα X-rays (1486.7 eV) at a power of 225 W on a Kratos Axis-Ultra spectrometer (160 eV analyzer pass energy for survey scans, 20 eV for high-resolution scans). The analysis spot size was ∼300 × 700 μm. Core electron binding energies are given relative to an adventitious hydrocarbon C 1s binding energy of 284.8 eV. Two spots per sample were analyzed and averaged to determine elemental composition within a sample. All XPS spectra were processed with CasaXPS (ver. 2.3.16 PR 1.6) data processing software using a Shirley background correction. Atomic ratios of metal to nitrogen provided an evaluation of the coordination of the metals to nitrogen in the coating.Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR)
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra were obtained using a Bruker Hyperion 1000 IR microscope operating with a Bruker Vertex 80 IR spectrometer. The IR microscope was equipped with a liquid nitrogen cooled MCT detector. Samples were prepared on ∼4 cm2 microscope slides (coating type: Ti/Au, thickness: 40 nm/100 nm, DRLI). ATR spectra were collected over 64 scans, with a resolution of 4 cm−1, using a Ge ATR crystal. Spectra of the PEI coatings were recorded using OPUS version 7.0 software in the range of 650–4000 cm−1 and analysed using OMNIC FTIR spectroscopy data analysis software in the range of 1000–4000 cm−1. All IR spectra were processed by auto-correction of baseline and smoothing before subtraction of a control spectrum of a bare gold coated microscope slide to remove any signals from the substrate.Time-of-flight secondary ion mass spectrometry (ToF-SIMS)
A Physical Electronics Inc. PHI TRIFT V nanoTOF (Physical Electronics, Inc., Chanhassen, MN) ToF-SIMS was used for conducting depth profiles through coatings. Sputtering was performed over a 500 × 500 micron area with a 20 kV C60 ion source for 5 second intervals and a source current of 7 nA. Between sputtering, spectra were acquired for 2 minutes with a 30 kV Au+ liquid metal ion gun with a total primary ion dose less than 1010 ions cm−2 to remain within static limits.X-ray absorption near edge spectroscopy (XANES)
XANES was conducted at the XAS beamline at the Australian Synchrotron using a 1.9T Wiggler insertion device and a Si(111) monochromator. Nominal specifications gave an energy resolution of 1.5 × 10−4, beam size of 250 × 250 microns and a flux of 1010 to 1012 ph s−1. Absorption of the incident X-ray beam was monitored by fluorescence with a 100 element germanium detector perpendicular to the incident beam. Except for powder standards, samples were analysed hydrated. Standards in solution were encapsulated in a Perspex mount with kapton foil windows and plunge frozen in liquid nitrogen. PEI coatings, after copper absorption, were quickly rinsed with Milli-Q water and immediately plunge frozen in liquid nitrogen. Cryogenic temperatures were maintained between 5 and 10 K for the duration of analysis. XANES was performed over the Cu-K edge (8979 eV) with the pre-edge region acquired in 10 eV steps before using 0.25 eV steps from 8975 to 9012.5 eV followed by 0.06 steps in K out to 6 K. Data across detector elements were averaged using the ‘Average’ software package and further post-processed with Athena.33,34Preparation of cross-linked PEI coatings
Cross-linked PEI coatings were prepared according to a previously reported method.31 Briefly, the coatings were prepared in three steps on ∼1 cm2 silicon wafer substrates. First a thin layer of PEI was spin coated from a solution in ethanol, thereafter cross-linking was conducted by immersion in GA. The samples were then immersed in a solution of PEI in Milli-Q water before washing and gentle drying using N2 (Scheme 1). The GA cross-linking technique was adapted from Tong et al.35 The average dry layer thicknesses of the coatings were determined in previous work31 to be 8 nm (S.D. 1.1 nm, n = 32) by ellipsometry.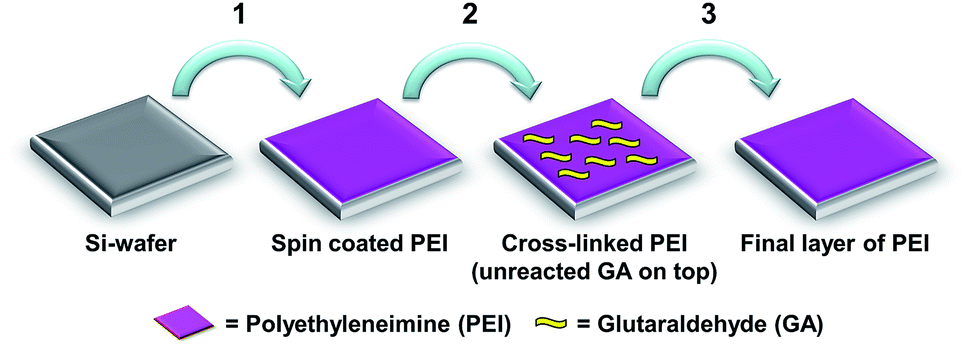 | ||
| Scheme 1 Preparation of cross-linked PEI coatings. Reproduced from ref. 31. | ||
Preparation of solutions
Spiked seawater was prepared by adding 50 L of collected seawater without further purification into a 100 L general plastic storage container. 1.52 mL of 1 mM copper sulphate (CuSO4) solution was added to spike the seawater with ∼2 ppb copper. Non-spiked seawater was prepared by adding 7 L of collected seawater without further purification into a general plastic bucket.Artificial seawater was prepared by mixing 40 g of sea salts per litre of Milli-Q water. 1 mM CuSO4 and ZnCl2 solutions in Milli-Q water were prepared and added to the artificial seawater solution to achieve equimolar concentration of 3.15 μM, which corresponds to 200 ppb for Cu2+ and 206 ppb for Zn2+.
The pH of solutions was determined to ∼8.1 using an ION 700 meter equipped with a pH electrode (Eutech instruments, Singapore).
Copper absorption from artificial seawater and seawater
The volumes used for absorption studies contained more than ten times excess of copper ions relative to the estimated maximum copper uptake by the coatings, estimated as follows: thickness = 15 nm, area = 1 cm2, maximum uptake ratio of copper-to-nitrogen16,36 = 0.25, coating composition = 100% PEI.For absorption from seawater all samples were immersed in the same volume of 50 L, spiked with 2 ppb CuSO4. Three additional samples were immersed separately in 7 L of seawater without added copper for comparison at the longest time point.
For absorption from artificial seawater with added zinc and copper, samples were immersed individually in glass bottles. Samples were removed at pre-determined time points, rinsed with Milli-Q water and dried with N2 before further characterisation. Samples were prepared in triplicate for each time point.
Results and discussion
Copper absorption from seawater
Nano-thin (8 nm, S.D. 1 nm, n = 32) coatings of GA cross-linked PEI have previously been shown to effectively and selectively absorb copper from artificial seawater containing copper in concentrations relevant to contaminated seawater (2–200 ppb).31 Here it was demonstrated that such coatings efficiently accumulated copper from seawater, see Fig. 1. Given the high variability of copper concentration in the sea, 2 ppb copper was added (spiked sample) to ensure a minimum concentration. The uptake was equally effective also for the samples immersed in the non-spiked seawater. Equilibrium absorption was reached after approximately 48 hours, at which time the copper-to-nitrogen ratio was ∼0.02, corresponding to 2 wt% copper in the coating (estimated using calculations described previously31). Compared to the uptake from artificial seawater containing 2 ppb copper, the rate of copper-uptake in natural seawater was slower and the copper-to-nitrogen ratio was 50% lower at equilibrium. This could be due to the presence of competing ligands in the natural seawater.37 Seawater also contains significant amounts of dissolved organic carbon (DOC)38 and it is well-known that polysaccharides and proteins rapidly accumulate on surfaces immersed in seawater, forming a conditioning film.39–41 Thus, it is reasonable to assume that deposition will occur on the PEI coatings over time and possibly influence the copper uptake.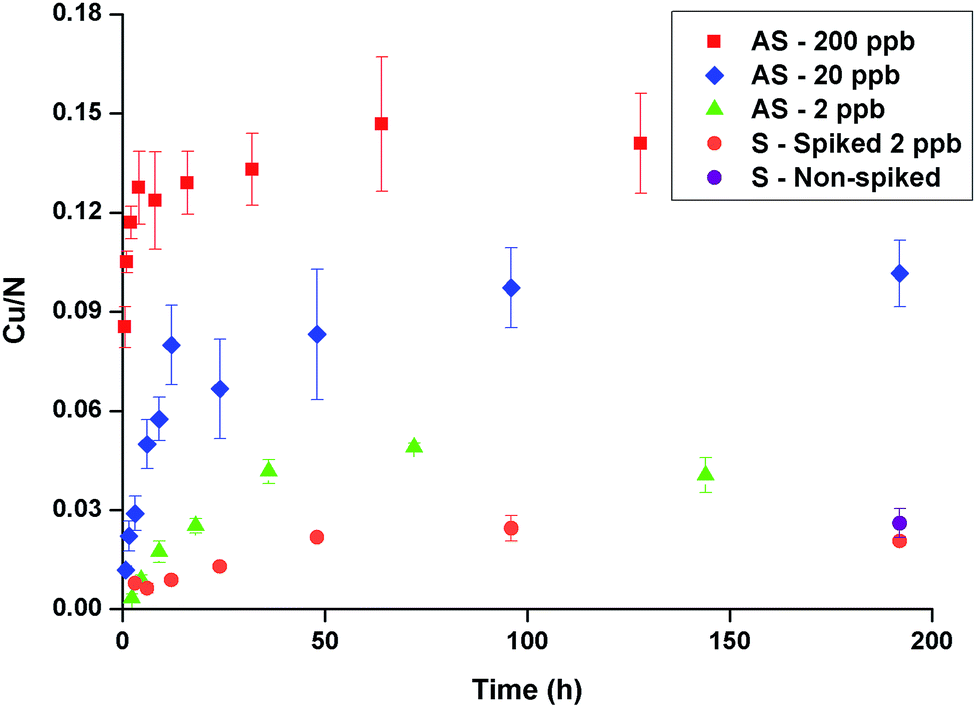 | ||
| Fig. 1 Copper-to-nitrogen ratio vs. time in artificial seawater (AS) spiked with 200–2 ppb CuSO4 or seawater (S) spiked with 2 ppb CuSO4. In addition samples in non-spiked seawater were analysed at 192 h (“Non-spiked”). The data for artificial seawater was adapted from Lindén et al.31 Error bars indicate standard deviation of measurements (n = 3). | ||
In an attempt to investigate the formation of a conditioning film, angle-resolved XPS analysis was conducted on coatings immersed in seawater for 3 and 192 hours. While the carbon atomic percentage did not increase significantly between 3 and 192 hours of immersion (Fig. 2), the carbon-to-nitrogen ratio did, especially amplified in the grazing exit angle measurement. Moreover, the carbon-to-silicon ratio also increased while the nitrogen-to-silicon was constant, which is significant in that we know from previous work31 that the PEI coatings contain sparse distributions of imperfections/holes which result in the Si wafer substrates being “visible” to XPS. This implies that conditioning films were formed over time on the PEI coatings while immersed in seawater.
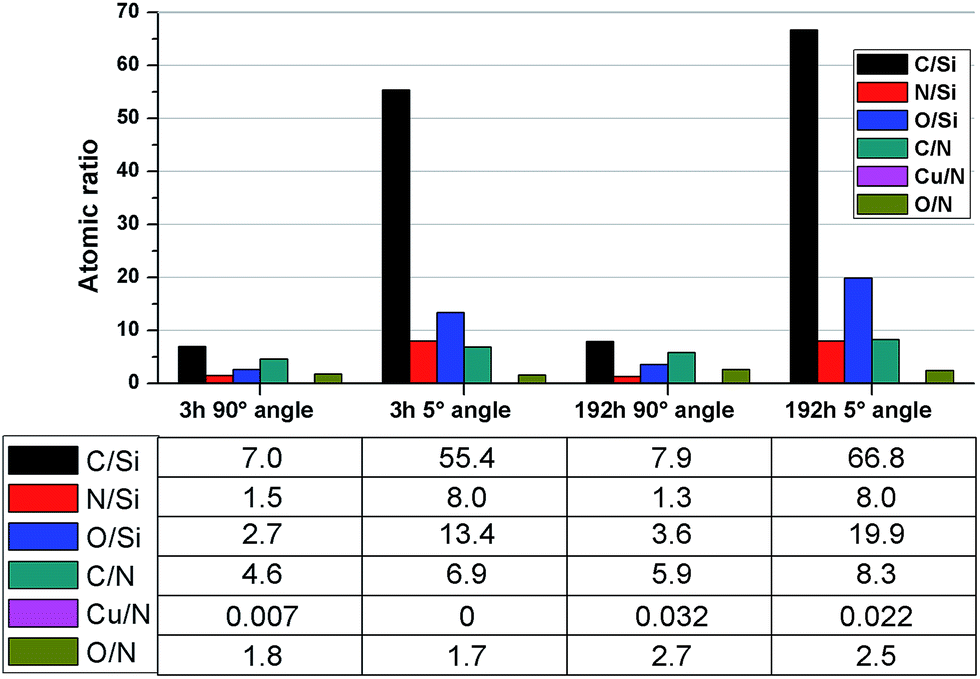 | ||
| Fig. 2 XPS results of atomic ratios after 3 and 192 hours seawater exposure, acquired at 90 and 5 degrees angle. | ||
Copper selectivity
We have previously shown that GA cross-linked PEI selectively accumulated copper from artificial seawater with 12 seawater-relevant metals added. Although zinc absorbed faster it was replaced by copper at longer times.31 As shown in Fig. 3A, the behaviour in seawater was similar to artificial seawater; zinc initially competed for binding sites but was replaced by copper over time.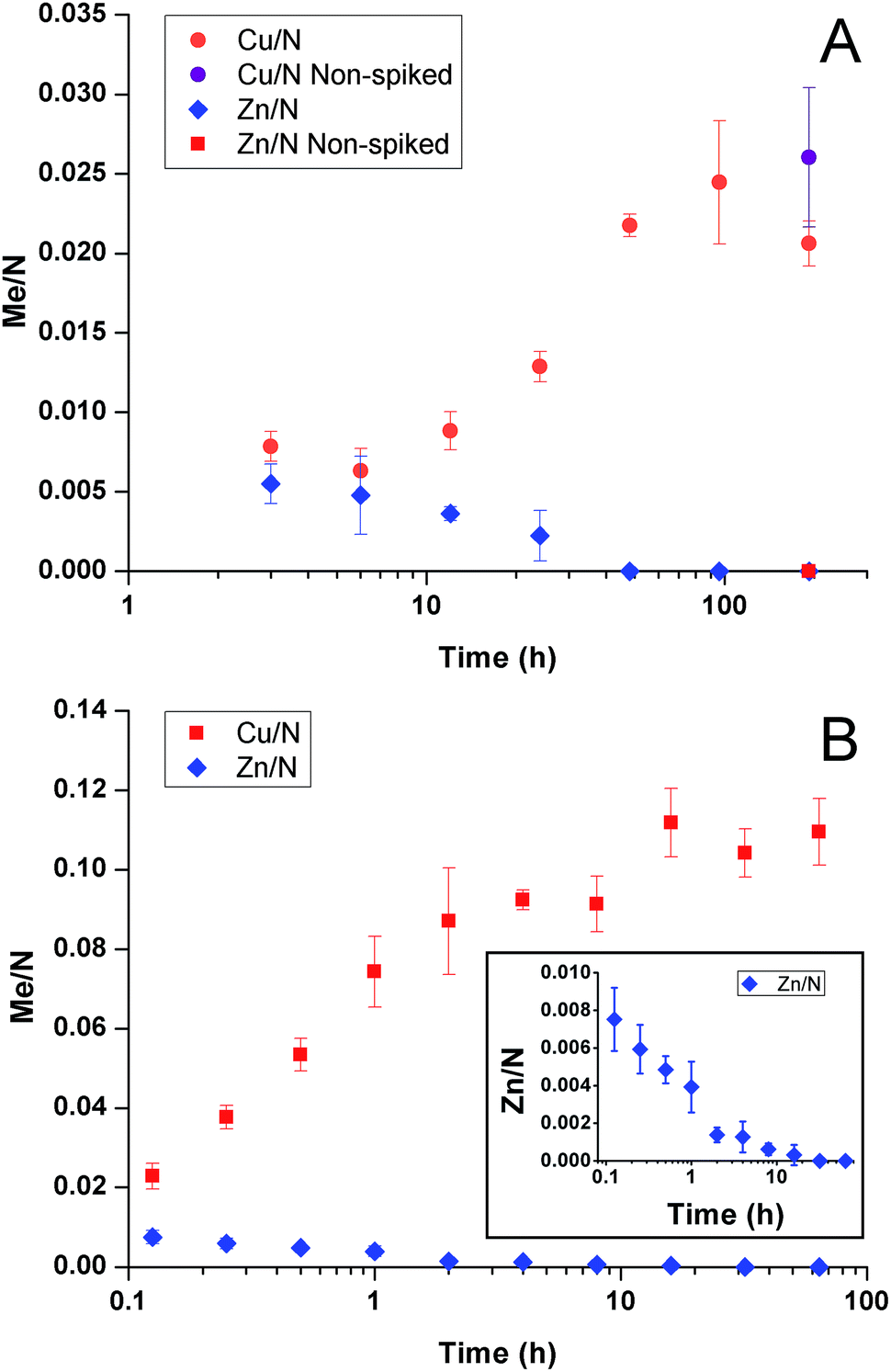 | ||
| Fig. 3 Metal-to-nitrogen ratio as a function of time. (A) After immersion in seawater spiked with 2 ppb CuSO4 and after 192 hours in non-spiked seawater (“Non-spiked”). (B) After immersion in artificial seawater ([Cu2+] = 3.15 μM = 200 ppb, [Zn2+] = 3.15 μM = 206 ppb). Inset showing the zinc-to-nitrogen ratio over time in more detail. Error bars indicate standard deviation of measurements (n = 3). | ||
To study the competitive binding processes in more detail, samples were immersed in artificial seawater with only copper and zinc added to the same molar concentration (3.15 μM = 200 ppb Cu2+, 206 ppb Zn2+) and the metal uptake was evaluated over time, Fig. 3B. Again zinc was exchanged by copper. Thus, the phenomenon was inherent to copper and zinc and did not depend on other metals.
To further study the mechanism behind the competitive binding processes mathematical modelling was used. Fig. 4 shows results of the competitive ion exchange process model described in detail as Scenario I in Miklavcic et al.32 For these results, the diffusion constants of zinc and copper ions were approximately the same, with zinc having a slight kinetic advantage; both ions were assumed to bind with free PEI sites with identical strength (identical binding rate constants). However, as copper ions entrained into the PEI matrix (Fig. 4A) via a forced diffusive process they progressively bind with both free PEI absorption sites as well as with sites already occupied by zinc ions, preferentially replacing the latter. Thus, although zinc diffused into the matrix slightly ahead of copper, forming ZnPEI complexes first, these complexes were destabilised by copper which formed new CuPEI complexes instead. Thus, the conditions assumed led to a transient front of ZnPEI complexes, which moved further and further into the matrix (indicated by the arrow in Fig. 4B). In this figure, the bound ion densities are given as functions of independent variables: y = κx and s = tDmκ2, being dimensionless distance and time, respectively. The spatial variable x represents position in the polymer matrix (zero at the substrate wall and one at the outer extreme of the PEI layer), κ is the Debye constant for the electrolyte and Dm is the maximum diffusion constant of mobile ions. The densities themselves were normalised with respect to the maximum bulk ion density (here being the Cl ion concentration). Full details of the binding and exchange processes are given in Miklavcic et al.32
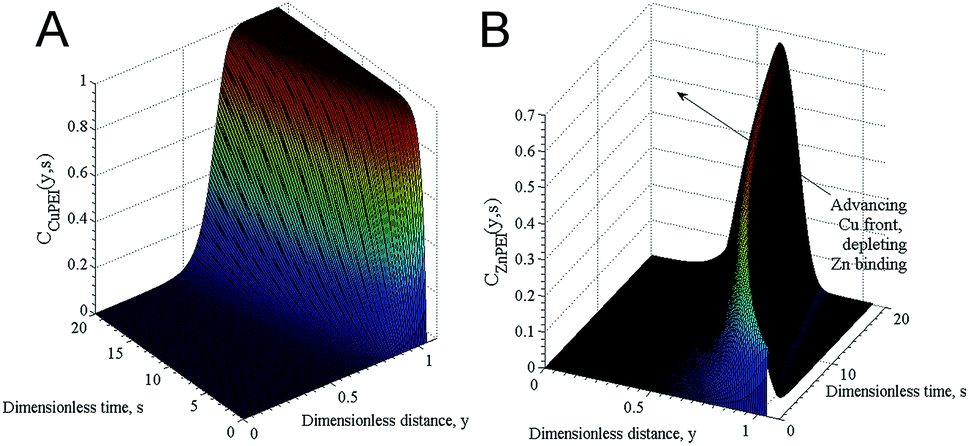 | ||
| Fig. 4 Normalised bound ion densities as a function of dimensionless time and position. (A) Shows the time development of the density of the CuPEI complex, while (B) shows the density corresponding to the ZnPEI complex. Note that (B) has been rotated clockwise 90 degrees to highlight the depletion zone behind an initial front of ZnPEI complexes that results from the replacement of zinc with copper ions. For these results, we assigned diffusion constants of DCu = 2.263 × 10−6 cm2 s−1 and DZn = 2.274 × 10−6 cm2 s−1, ion concentrations cCuCl2 = cZnCl2 = 3 μM with an inert background univalent electrolyte concentration of 1 mM. Using the nomenclature of Miklavcic et al.,32 the equilibrium binding strengths assigned were k1+/k1− = k2+/k2− = 1.25 × 105, with the exchange reaction strength of k3+/k3− = 1.0 × 1012. | ||
Spatial distribution of copper
ToF-SIMS is an extremely surface sensitive analytical tool and will probe the surface 2–3 nm during analysis in each cycle.42 To characterise the spatial distribution of copper in the nano-thin coatings after immersion in seawater, ToF-SIMS was used to generate depth profiles through the coating by interleaving analysis and sputtering cycles. The experiment allowed a component-resolved spatial profile going into the coating and to the silicon substrate. The resulting mass spectra are inherently intricate and the fragmentation patterns can reveal compositional and subtle structural information in complex systems.43,44 It has further been demonstrated to provide information on ligand formation between organic and inorganic materials.45Here, the results from ToF-SIMS depth profiles through PEI coatings exposed to seawater spiked with 2 ppb CuSO4 are revealed and discussed. We have observed a CuNCH+ fragment to be indicative of the copper distribution and how it was primarily associated in the coating. The absolute numbers of ions detected, normalised to the total ion yield, are shown in Fig. 5A. Si+ represents the substrate. The 3 hour time point revealed very little penetration of copper into the coating. After 24 and 192 hours immersion, the copper distribution appeared comparable, with the copper residing in a band below the surface, suggesting a non-copper binding region at the very top, in line with XPS-results and the hypothesis that a conditioning film formed at the coating surface. The composition of the copper-based fragment is suggestive of the copper binding with the polymer via the amine groups. Furthermore, the peak intensity for CuNCH+, can be interpreted in a semi-quantitative way. ToF-SIMS is highly matrix sensitive, however, here the matrix should be identical between each sample and the change in peak intensity should be due to a genuine variation in copper concentration. We estimate that roughly 1 nm of material has been removed in each sputter cycle. At the atomic level this is quite variable however and features in structure as a function of depth lose distinction, as seen in the broad distributions in the depth profile plot (Fig. 5A). A 3D reconstruction of the analysis conducted for the 24 hour time point is given in Fig. 5B. This figure aids in visualisation of the copper distribution in the coating.
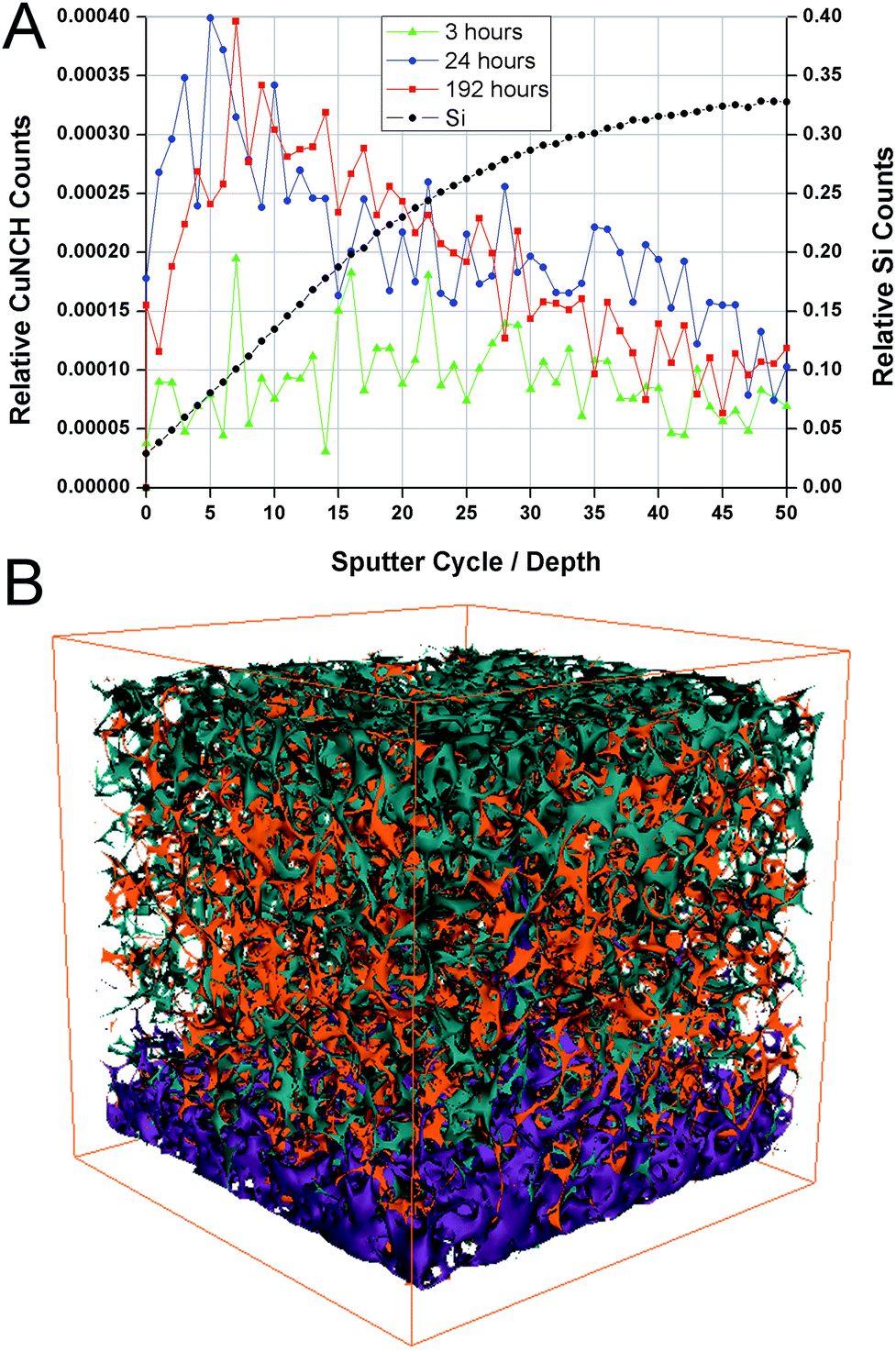 | ||
| Fig. 5 ToF-SIMS depth profiles through PEI coatings on silicon wafer substrate loaded with copper through exposure to seawater. (A) Copper is shown to be incorporated into the bulk of the PEI coating rather than being adsorbed at the surface and was associated with an N-based fragment. The amount of copper absorbed increased with time but had reached equilibrium by 24 hours. (B) A 3D reconstruction of the ToF-SIMS depth profile (conducted for the 24 hour time point) showing the coating via a CH3+ fragment (green) with embedded copper (orange) on the substrate indicated by Si+ (purple). The X and Y axes are 100 microns while the Z axis represents roughly 10 nm, as estimated from thickness determination by ellipsometry.31 | ||
ATR-FTIR analysis of PEI coatings
To provide information on the chemistry of the GA cross-linked PEI and copper binding, ATR-FTIR analysis was conducted before and after cross-linking and after copper uptake from artificial seawater containing 200 ppb copper added in the form of CuSO4, with spectra presented in Fig. 6. C–H stretches were observed for spin coated PEI at 2836 cm−1 and 2947 cm−1.46 After cross-linking, the (CH2)n band at 2947 cm−1 appeared more distinct, indicating the presence of glutaraldehyde.18 An imine band (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching) appeared at 1660 cm−1 after cross-linking, likely from a Schiff's base formed from reaction between GA and PEI primary amines.18,47,48 A band from primary amines (N–H bending) was observed at 1585 cm−1 but disappeared after cross-linking.49 The band that appeared at 1101 cm−1 after exposure to CuSO4 is likely from an overlap of stretching vibrations of S–O (from SO4− as the counter ion to copper) and C–N bonds.50 The band at 1605 cm−1 indicates a copper–amine interaction. It should be mentioned that the complex signals and overlapping bands from different amine groups in PEI make it difficult to quantify the extent of cross-linking and to draw more detailed conclusions about the copper–ligand interactions.
N stretching) appeared at 1660 cm−1 after cross-linking, likely from a Schiff's base formed from reaction between GA and PEI primary amines.18,47,48 A band from primary amines (N–H bending) was observed at 1585 cm−1 but disappeared after cross-linking.49 The band that appeared at 1101 cm−1 after exposure to CuSO4 is likely from an overlap of stretching vibrations of S–O (from SO4− as the counter ion to copper) and C–N bonds.50 The band at 1605 cm−1 indicates a copper–amine interaction. It should be mentioned that the complex signals and overlapping bands from different amine groups in PEI make it difficult to quantify the extent of cross-linking and to draw more detailed conclusions about the copper–ligand interactions.
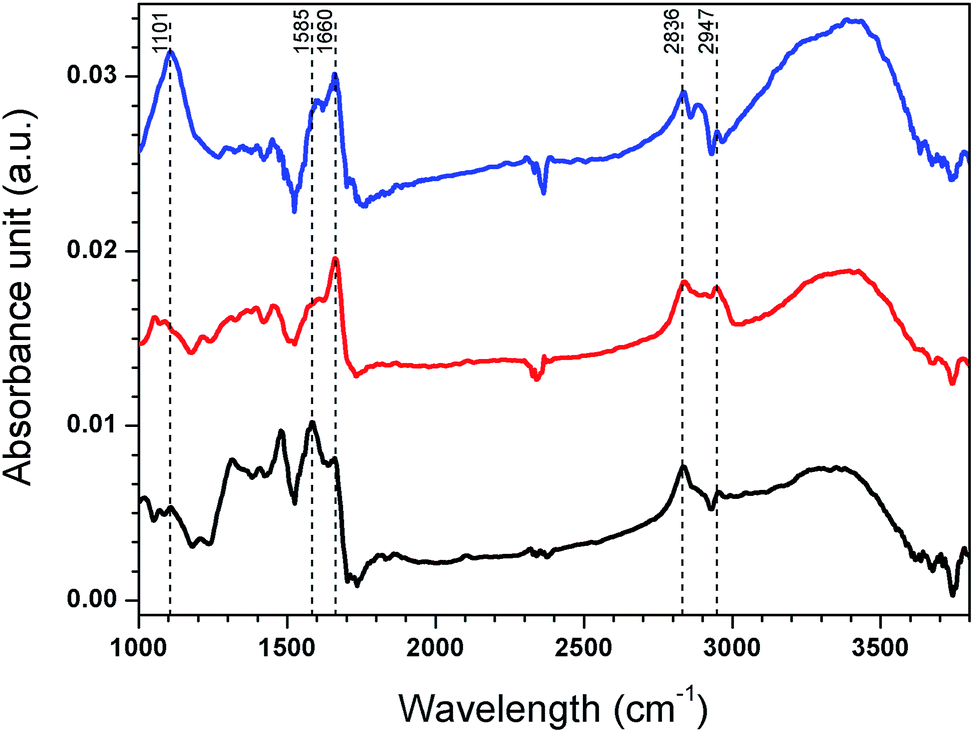 | ||
| Fig. 6 ATR-FTIR spectra of spin coated PEI (black), spin coated PEI after cross-linking (red) and spin coated PEI after cross-linking and copper uptake (blue). | ||
X-ray absorption spectroscopy
In an attempt to further understand the copper binding mechanism and ligand formation, XANES analysis was conducted on copper loaded in the coatings (Fig. 7) and the results were compared to a variety of copper standards (Fig. S1, ESI†).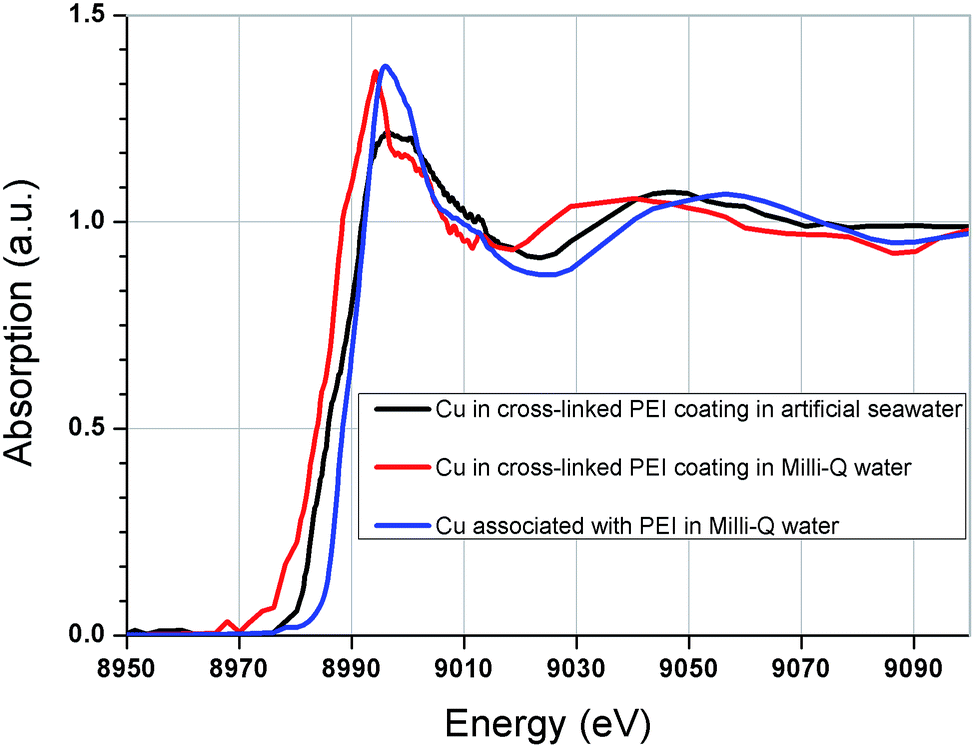 | ||
| Fig. 7 Copper K-edge XANES spectra for copper; in cross-linked PEI coating in artificial seawater, in cross-linked PEI coating in Milli-Q water and associated with PEI in Milli-Q water. | ||
It is apparent that the copper binding environment in the GA cross-linked coatings was significantly different from that of the free PEI in solution, as evidenced by the dissimilarity between the corresponding spectra in Fig. 7. This observation may explain the extreme copper selectivity and affinity of the coatings. The altered copper–ligand environment could be due to the introduction of double bonded amines, Schiff's bases through the glutaraldehyde cross-linking. Furthermore, it was found that the aqueous environment also influenced the state of the bound copper in the coatings. As seen from comparison of the copper spectra between coatings that had been loaded with copper in Milli-Q water solution and coatings that had been loaded with copper in artificial seawater. This could possibly imply differences in counter ions and/or electrolyte charge-driven forces.
The position of the absorption edge implies that copper was bound with an oxidation state of II, which correlates with previous findings by XPS.31 Despite comparison with a suite of 16 standard samples, linear least squares fitting was unable to provide any reasonable fit. In retrospective comparison with literature, a Schiff-base complex appears to produce a comparable spectrum to that found here.51,52 This correlates with the FTIR analysis which showed formation of Schiff's bases (imines) in the coating after cross-linking. It is also consistent with the ToF-SIMS fragmentation suggesting an N-based ligand associated with copper.
Conclusions
We expanded on our previous work on thin glutaraldehyde cross-linked PEI coatings for absorption of copper in artificial seawater by demonstrating the applicability to natural seawater. The coatings were shown to be nearly as effective and equally selective in seawater as in artificial seawater, despite strong indications that a conditioning organic film was formed at the coating surface. FTIR and XANES analyses indicated that GA cross-linking changed the structure from native PEI to a network with a high content of imine nitrogens (Schiff's bases). It seems likely that this structure is responsible for the extraordinary affinity and selectivity of the material towards copper.The findings open the way for extraction and utilisation of naturally abundant copper from seawater. With regard to the proposed new concept in marine anti-biofouling, the presented material enables the first step towards this technology, i.e., the copper uptake. In future work we will move towards development of a mechanism (triggered) for releasing the copper, creating a biocidal flux across the interface, which in combination with uptake would create a continuous cycle. Furthermore, we will continue to build in-depth knowledge regarding the material properties and performance under different conditions. For example, the effect of conditioning polysaccharide films and cross-linking will be investigated.
Acknowledgements
We acknowledge that this research has been financially supported by US DARPA and ARO (Grant number W911NF1310015) and ITEK Ventures Pty Ltd, the commercialisation arm of the University of South Australia. A part of this research was undertaken on the X-ray absorption spectroscopy beamline at the Australian Synchrotron, Victoria, Australia. The authors thank Dr Peter Kappen for assistance with acquisition of the XANES data. We also acknowledge that this work was performed in part at the South Australian node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano and micro-fabrication facilities for Australia's researchers.Notes and references
- K. Schiff, J. Brown, D. Diehl and D. Greenstein, Mar. Pollut. Bull., 2007, 54, 322 CrossRef CAS PubMed.
- K. Schiff, D. Diehl and A. Valkirs, Mar. Pollut. Bull., 2004, 48, 371 CrossRef CAS PubMed.
- T. W. Biggs and H. D'Anna, Mar. Pollut. Bull., 2012, 64, 627 CrossRef CAS PubMed.
- M. Srinivasan and G. Swain, Environ. Manage., 2007, 39, 423 CrossRef PubMed.
- W. Chouyyok, Y. Shin, J. Davidson, W. D. Samuels, N. H. LaFemina, R. D. Rutledge, G. E. Fryxell, T. Sangvanich and W. Yantasee, Environ. Sci. Technol., 2010, 44, 6390 CrossRef CAS PubMed.
- K. Schwochau, in Inorg. Chem., Springer Berlin Heidelberg, 1984, vol. 124, p. 91 Search PubMed.
- U. Bardi, Sustainability, 2010, 2, 980 CrossRef CAS PubMed.
- P. E. Duru, S. Bektas, Ö. Genç, S. Pat
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) r and A. Denizli, J. Appl. Polym. Sci., 2001, 81, 197 CrossRef CAS PubMed.
r and A. Denizli, J. Appl. Polym. Sci., 2001, 81, 197 CrossRef CAS PubMed. - R. R. Navarro, K. Sumi, N. Fujii and M. Matsumura, Water Res., 1996, 30, 2488 CrossRef CAS.
- Y. Pang, G. Zeng, L. Tang, Y. Zhang, Y. Liu, X. Lei, Z. Li, J. Zhang and G. Xie, Desalination, 2011, 281, 278 CrossRef CAS PubMed.
- S. T. Beatty, R. J. Fischer, D. L. Hagers and E. Rosenberg, Ind. Eng. Chem. Res., 1999, 38, 4402 CrossRef CAS.
- S. T. Beatty, R. J. Fischer, E. Rosenberg and D. Pang, Sep. Sci. Technol., 1999, 34, 2723 CrossRef CAS.
- M. Chanda and G. L. Rempel, React. Funct. Polym., 1997, 35, 197 CrossRef CAS.
- R. J. Fischer, D. Pang, S. T. Beatty and E. Rosenberg, Sep. Sci. Technol., 1999, 34, 3125 CrossRef CAS.
- F. Ungaro, G. De Rosa, A. Miro and F. Quaglia, J. Pharm. Biomed. Anal., 2003, 31, 143 CrossRef CAS.
- T. Wen, F. Qu, N. B. Li and H. Q. Luo, Arabian J. Chem., 2013 DOI:10.1016/j.arabjc.2013.06.013.
- Z. Yuan, N. Cai, Y. Du, Y. He and E. S. Yeung, Anal. Chem., 2013, 86, 419 CrossRef PubMed.
- M. Ghoul, M. Bacquet and M. Morcellet, Water Res., 2003, 37, 729 CrossRef CAS.
- S. Deng and Y.-P. Ting, Water Res., 2005, 39, 2167 CrossRef CAS PubMed.
- S. Deng and Y. P. Ting, Environ. Sci. Technol., 2005, 39, 8490 CrossRef CAS.
- M. Chanda and S. A. Pillay, Indian J. Chem. Technol., 2005, 12, 156 CAS.
- W. K. Maketon and K. L. Ogden, IEEE Trans. Semicond. Manuf., 2008, 21, 481 CrossRef.
- L. Steinmann, J. Porath, P. Hashemi and Å. Olin, Talanta, 1994, 41, 1707 CrossRef CAS.
- Y. Chen, B. Pan, H. Li, W. Zhang, L. Lv and J. Wu, Environ. Sci. Technol., 2010, 44, 3508 CrossRef CAS PubMed.
- S. Kobayashi, K. Hiroishi, M. Tokunoh and T. Saegusa, Macromolecules, 1987, 20, 1496 CrossRef CAS.
- BASF, Lupasol® types, Technical Information Sheet 08_0806130e-02, September 2010.
- C. R. Dick and G. E. Ham, J. Macromol. Sci., Chem., 1970, 4, 1301 CrossRef CAS PubMed.
- J. Jia, A. Wu and S. Luan, Phys. Chem. Chem. Phys., 2014, 16, 16158 RSC.
- A. Wu, J. Jia and S. Luan, Colloids Surf., A, 2011, 384, 180 CrossRef CAS PubMed.
- B. L. Rivas and K. E. Geckeler, in Polymer Synthesis Oxidation Processes, Springer, 1992, p. 171 Search PubMed.
- J. B. Lindén, M. Larsson, B. R. Coad, W. M. Skinner and M. Nydén, RSC Adv., 2014, 4, 25063 RSC.
- S. J. Miklavcic, M. Nydén, J. B. Lindén and J. Schulz, RSC Adv., 2014, 4, 60349 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- B. Ravel and M. Newville, Phys. Scr., 2005, 2005, 1007 CrossRef.
- W. Tong, C. Gao and H. Möhwald, Polym. Adv. Technol., 2008, 19, 817 CrossRef CAS PubMed.
- T. D. Perrine and W. R. Landis, J. Polym. Sci., Part A-1: Polym. Chem., 1967, 5, 1993 CrossRef CAS PubMed.
- H. Elderfield, H. D. Holland and K. K. Turekian, The Oceans and Marine Geochemistry, Elsevier Pergamon, 2006 Search PubMed.
- H. Ogawa and E. Tanoue, J. Oceanogr., 2003, 59, 129 CrossRef CAS.
- S. Abarzua and S. Jakubowski, Mar. Ecol.: Prog. Ser., 1995, 123, 301 CrossRef CAS.
- M. E. Callow and R. L. Fletcher, Int. Biodeterior. Biodegrad., 1994, 34, 333 CrossRef CAS.
- D. M. Yebra, S. Kiil and K. Dam-Johansen, Prog. Org. Coat., 2004, 50, 75 CrossRef CAS PubMed.
- D. Briggs, J. C. Vickerman and S. Limited, ToF-SIMS: surface analysis by mass spectrometry, IM; SurfacesSpectra, Chichester, Manchester, 2001 Search PubMed.
- I. M. Kempson, P. Chang, K. Bremmell and C. A. Prestidge, Langmuir, 2013, 29, 15573 CrossRef CAS PubMed.
- I. M. Kempson, A. L. Martin, J. A. Denman, P. W. French, C. A. Prestidge and T. J. Barnes, Langmuir, 2010, 26, 12075 CrossRef CAS PubMed.
- I. M. Kempson, T. J. Barnes and C. A. Prestidge, J. Am. Soc. Mass Spectrom., 2010, 21, 254 CrossRef CAS PubMed.
- N. Khanam, C. Mikoryak, R. K. Draper and K. J. Balkus Jr, Acta Biomater., 2007, 3, 1050 CrossRef CAS PubMed.
- T. Wang, M. Turhan and S. Gunasekaran, Polym. Int., 2004, 53, 911 CrossRef CAS PubMed.
- W.-J. Yang, C.-S. Yang, C.-J. Huang, K.-S. Chen and S.-F. Lin, Enzyme Microb. Technol., 2012, 50, 287 CrossRef CAS PubMed.
- S. Abolmaali, A. Tamaddon and R. Dinarvand, J. Nanopart. Res., 2013, 15, 1 CrossRef.
- Z. Wang and M. W. Urban, Polym. Chem., 2013, 4, 4897 RSC.
- P. M. Dias, L. Kinouti, V. R. Constantino, A. M. Ferreira, M. B. Gonçalves, R. R. d. Nascimento, H. M. Petrilli, M. Caldas and R. C. Frem, Quim. Nova, 2010, 33, 2135 CrossRef CAS PubMed.
- A. Gaur, B. Shrivastava, K. Srivastava, J. Prasad and S. K. Singh, X-Ray Spectrom., 2012, 41, 384 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XANES study of copper speciation in interaction with PEI, Fig. S1. See DOI: 10.1039/c5ra08029k |
| This journal is © The Royal Society of Chemistry 2015 |