
Guanidine-functionalized Fe3O4 magnetic nanoparticles as basic recyclable catalysts for biodiesel production†
Abstract
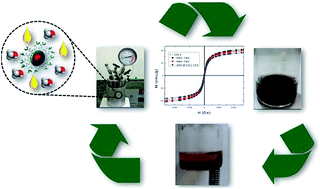
Two organic superbases, 1,5,7-triazabicyclo[4,4,0]dec-5-ene (TBD) and 1,1,3,3-tetramethylguanidine (TMG), were anchored onto silica-coated and uncoated iron oxide nanoparticles, resulting in three (MNP–TBD, MNP–TMG and MNP@SiO2–TBD) recoverable basic nanocatalysts. The nanocatalysts were fully characterized by elemental analysis, infrared and Raman spectroscopies, X-ray diffraction, transmission electron microscopy, N2 adsorption/desorption isotherms, thermogravimetric analysis and magnetic measurements. X-ray diffraction indicated the presence of a spinel-structured iron oxide, and Raman spectroscopy revealed both magnetite and maghemite phases in the prepared nanocatalysts. The average particle sizes of the nanocatalysts were in the range of 11 to 12 nm, and they exhibited superparamagnetic behaviour at 300 K. Infrared spectroscopy indicated the presence of the superbases (TBD and TMG) on the surface of the silica-coated and uncoated iron oxide nanoparticles. The performance of the nanocatalysts was tested in the methanolysis reaction of soybean oil under different conditions. At the end of each reaction, the nanocatalysts were magnetically recovered from the medium, and the product was analysed and quantified by high-performance liquid chromatography (HPLC). MNP–TBD exhibited the best catalytic performance in the first cycle (96% biodiesel conversion); however, MNP@SiO2–TBD exhibited the best reusability.
 Please wait while we load your content...
Please wait while we load your content...

