
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dendron conjugation to graphene oxide using click chemistry for efficient gene delivery†
Kishor
Sarkar
a,
Giridhar
Madras
 a and
Kaushik
Chatterjee
*b
a and
Kaushik
Chatterjee
*b
aDepartment of Chemical Engineering, Indian Institute of Science, Bangalore 560012, India
bDepartment of Materials Engineering, Indian Institute of Science, Bangalore 560012, India. E-mail: kchatterjee@materials.iisc.ernet.in; Tel: +91-80-22933408
First published on 1st June 2015
Abstract
Owing to its large surface area and rapid cellular uptake, graphene oxide (GO) is emerging as an attractive candidate material for delivery of drugs and genes. The inherent sp2 π–π interaction of GO helps to carry drugs and single stranded RNA (ssRNA) but there is no such interaction with double stranded DNA (dsDNA). In this work, a polyamidoamine (PAMAM) dendron was conjugated with nano GO (nGO) through “click” chemistry to improve the DNA complexation capability of GO as well as its transfection efficiency. The DNA complexation capability of GO was significantly enhanced after dendronization of GO yielding spherical nanosized (250–350 nm) particles of the dendronized GO (DGO)/pDNA complex with a positive zeta potential. The transfection efficiency of GO dramatically increased after conjugation of the PAMAM dendron. Transfection efficiency of 51% in HeLa cells with cell viability of 80% was observed. The transfection efficiency was significantly higher than that of polyethyleneimine 25 kDa (27% efficiency) and also surpassed that of lipofectamine 2000 (47% efficiency). The uptake of the DGO/pDNA complex by the caveolae mediated endocytosis pathway may significantly contribute to the high transfection efficiency. Thus, dendronized GO is shown to be an efficient gene carrier with minimal toxicity and is a promising candidate for use as a nonviral carrier for gene therapy.
1. Introduction
Gene therapy is an emerging therapeutic tool of modern medicine promising to treat a variety of genetic disorders and other diseases.1,2 The lack of an efficient yet safe carrier is a significant impediment in the field. High toxicity, immunogenicity, tumorigenicity, a lack of targeting efficiency and high cost of production of viral vectors have underscored the need to develop safe and efficient alternative nonviral vectors.3,4 Cationic macromolecules such as polyethyleneimine (PEI),5 poly-L-lysine,6 polyamidoamine (PAMAM) dendrimers7etc. are some of the widely studied nonviral carriers. Despite the emergence of several carriers, there is a need for novel efficient and safe nonviral carriers for successful gene therapy.Recently, graphene oxide (GO) and functionalized GO have gained tremendous attention in biomedical applications such as biosensors, bio-imaging, tissue engineering, drug and gene delivery due to their superior water dispersibility, high surface to volume ratio, low production cost and scope for facile functionalization.8–11 GO has been widely used for drug delivery due to favourable sp2 π–π interaction between GO and the hydrophobic drugs.12 GO has also been used for imaging.13,14 Thus, the use of GO for gene delivery offers an additional advantage of potentially monitoring cellular uptake and synergistically utilize its other properties for theranostics.
The π–π interaction of GO can carry single stranded RNA (ssRNA). But, there is no such interaction with double stranded DNA (dsDNA) and hence GO is unable to transport dsDNA.15 Few attempts on utilizing modified GO for delivery of plasmid DNA (pDNA) have been reported recently but the transfection efficiency was poor. Kim et al.16 prepared low molecular weight branched PEI-conjugated-GO using 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide hydrochloride (EDC) and observed transfection efficiency comparable to that of PEI (25 kDa). Chen et al.17 conjugated high molecular weight (25 kDa) branched PEI with GO through EDC and the transfection efficiency of GO–PEI was also equivalent to that of PEI (25 kDa). Tripathi et al.18 reported higher transfection efficiency (∼33%) after conjugation of linear PEI (25 kDa) with GO. The toxicity of graphene is a general concern for its use in biomedical applications. However, the reports are contradictory and cytotoxicity depends on the concentration, size and functionalization.19,20 GO is reported to be minimally toxic at low concentration.21 Thus, it appears that GO may be used in vivo at low concentration with minimal toxicity although further investigations of long term effects are warranted.
We hypothesize that conjugation of PAMAM to GO can significantly improve the DNA binding capacity and transfection efficiency of GO. Amine terminated PAMAM dendrimer has been widely used for drug/gene delivery because of its well defined architecture, high surface charge density, low polydispersity and biodegradability.22 However, severe toxicity of PAMAM dendrimer limits its widespread application for drug/gene delivery. The toxicity and transfection efficiency are dependent on the generation of PAMAM dendrimer.23,24 Higher generation dendrimers (>3.0G) show better transfection efficiency but induce higher toxicity whereas, low generation dendrimers (<3.0G) show lower transfection efficiency with higher cell viability.25,26 Thus, GO modified with low generation PAMAM can yield a carrier with good transfection efficiency with low toxicity.
In this work, we report on the synthesis of PAMAM dendrimer conjugated GO (dendronized GO) by reacting focal point PAMAM dendrimer with GO through “click” chemistry for DNA delivery. We used low generation PAMAM dendrimers (1.0, 2.0 and 3.0G) to conjugate with GO by “click” chemistry. The use of “click” chemistry offers many advantages for bioconjugation including high specificity with minimal by-products.27 Importantly, a major limitation of EDC conjugation used previously for PEI modification of GO is the lack of specificity,12,15,28 which may result in a crosslinked mass of many GO flakes conjugated to a single PEI molecule. Such crosslinking can be minimized by “click” chemistry utilized herein. Moreover, the strategy presented here can minimize non-covalent electrostatic interactions between GO and the polycationic chains. The dendronized GO (DGO)/pDNA complexes were characterized. Gene transfection efficiency and cytotoxicity of DGO were assessed in HeLa cells. The rapid intracellular kinetics and cellular uptake pathways of the DGO/pDNA complex were characterized to elucidate the mechanisms underlying high transfection efficiency.
2. Experimental sections
2.1. Materials
Pristine graphite flake (100 mesh), propargylamine (98%), methyl acrylate (99%), ethylenediamine (ReagentPlus®, ≥99%), 3-chloropropylamine hydrochloride (98%), sodium azide (ReagentPlus®, ≥99.5%), sodium ascorbate (≥98%), N,N-dimethylformamide (anhydrous, 99.8%), ethidium bromide (EtBr), branched PEI (25 kDa), 2,4,6-trinitrobenzene sulfonic acid solution (5% w/v) and 3-(4,5-dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide) (MTT) were purchased from Sigma-Aldrich. Chloroacetic acid, methyl alcohol, copper(II) sulphate and sodium hydroxide were obtained from S. D. Fine Chemicals. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), agarose and sodium salt of 2-(N-morpholino) ethanesulfonic acid (MES) were purchased from SRL Pvt. Ltd. Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), 0.25% trypsin and penicillin–streptomycin were purchased from Life Technologies. Enhanced green fluorescence protein (EGFP) encoding plasmid DNA (pEGFP-N1, 4.7 kbp) was propagated in Escherichia coli (E. coli) and isolated by pDNA isolation Kit (Midiprep, Qiagen, USA). All other reagents were used without any purification.2.2. Synthesis of PAMAM dendrimer
PAMAM dendrimers were synthesized according to the previous report with slight modification.29 Briefly, 5.87 g (68 mmol) methyl acrylate (MA) and 20 ml methanol were taken in 250 ml round bottom three neck flask and put on an ice bath. 1.5 g (27.2 mmol) propargylamine dissolved in 80 ml methyl alcohol was added drop wise to the above mixture over 1 h under nitrogen atmosphere at 0° C with constant stirring. The reaction mixture was stirred for another 30 min in ice and then continued at room temperature for 48 h. The excess solvent was removed by rotary evaporator under reduced pressure at 40° C to get light yellow colored ester terminated 0.5G (half generation) PAMAM dendrimer (5.85 g, 94.5%).To a stirred solution of ethylenediamine (40 g, 0.67 mol) in 80 ml methyl alcohol, 5 g (22 mmol) 0.5G PAMAM dissolved in 20 ml methyl alcohol was added drop wise over 30 min under nitrogen atmosphere at 0° C. The reaction was allowed to continue at 0° C for another 30 min and further for 96 h at room temperature under nitrogen atmosphere. The excess solvent was removed by rotary evaporation at 40° C using toluene–methanol mixture (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) to obtain a light yellow colored amine terminated 1.0G (full generation) PAMAM dendrimer (6.0 g, 96.3%). The above two consecutive steps were repeated to synthesize 2.0G and 3.0G PAMAM dendrimers as shown in Fig. 1a. The selected NMR data of 0.5, 1.0, 1.5, 2.0 and 3.0G PAMAM dendrimers are given in ESI.†
:1 volume ratio) to obtain a light yellow colored amine terminated 1.0G (full generation) PAMAM dendrimer (6.0 g, 96.3%). The above two consecutive steps were repeated to synthesize 2.0G and 3.0G PAMAM dendrimers as shown in Fig. 1a. The selected NMR data of 0.5, 1.0, 1.5, 2.0 and 3.0G PAMAM dendrimers are given in ESI.†
 | ||
| Fig. 1 Schematic diagram for (a) synthesis of focal point PAMAM dendrimer and (b) synthesis of DGO by “click” chemistry and cellular uptake of dendronized GO/pDNA complex. | ||
2.3. Synthesis of nano graphene oxide (nGO)
GO was synthesized from pristine graphite flakes by modified Hummers method.30 Briefly, 1.0 g graphite flakes were taken in a 1000 ml beaker. 57.0 ml concentrated H2SO4 and 6.5 ml H3PO4 were added to the graphite flakes with constant stirring for 8 h. 5.6 g KMnO4 was gradually added to the above mixture under ice and the reaction was continued for another 48 h. 30% H2O2 (10 ml) and 140 ml distilled water were added to the reaction mixture to terminate the reaction. The reaction mixture was washed with 1.0 N HCl solution and distilled water several times by repeating centrifugation and dispersion methods. Finally, the synthesized GO was dried at 40° C under vacuum for 48 h. The dried GO was dispersed in distilled water by bath sonication at concentration of 3 mg ml−1 for 1 h. The GO solution was then ultrasonicated at 400 W for 1 h under ice to obtain nGO and stored at 4° C for future use.2.4. Synthesis of aminopropyl azide
2.0 g (15.4 mmol) chloropropylamine hydrochloride was added in a 100 ml round bottom flask containing 4.0 g (61.6 mmol) sodium azide dissolved in 30 ml distilled water and the reaction mixture was stirred at 75° C for 18 h. The reaction mixture was then concentrated by rotary evaporator under reduced pressure and KOH pellet was added to the concentrated mixture followed by extraction using diethyl ether (3 × 10 ml). The organic layer was finally dried over anhydrous sodium sulphate followed by rotary evaporation to get colorless volatile aminopropyl azide (1.43 g, 93%). 1H NMR (CDCl3) δH: 3.38 ppm (2H, t, N3−C![[H with combining macron]](https://www.rsc.org/images/entities/char_0048_0304.gif) 2−), 2.81 ppm (2H, t, −C2−NH2), 1.74 ppm (2H, m, −CH2−C2−CH2−), 1.25 ppm (2H, bs, −CH2−N2). 13C NMR (CDCl3) δC: 49.23 ppm (N3−
2−), 2.81 ppm (2H, t, −C2−NH2), 1.74 ppm (2H, m, −CH2−C2−CH2−), 1.25 ppm (2H, bs, −CH2−N2). 13C NMR (CDCl3) δC: 49.23 ppm (N3−![[C with combining macron]](https://www.rsc.org/images/entities/char_0043_0304.gif) H2−), 39.37 ppm (−H2−NH2), 32.45 ppm (−CH2−H2−CH2−).
H2−), 39.37 ppm (−H2−NH2), 32.45 ppm (−CH2−H2−CH2−).
2.5. Synthesis of azide functionalized nGO
16.6 ml nGO solution (3 mg ml−1) was taken in a 100 ml round bottom flask and the solution was diluted to 50 ml to make 1 mg ml−1 final concentration using 5 mM MES buffer (pH = 5.5). Then, 0.27 g EDC and 0.16 g NHS were added to the nGO solution followed by addition of 0.5 g aminopropyl azide and the reaction was carried out at room temperature for 24 h. The reaction product was purified by dialysis for 3 days using SnakeSkin dialysis tubing (3.5 kDa MWCO, Thermo Scientific) and finally freeze dried for 3 days to get azide functionalized GO.2.6. Synthesis of PAMAM conjugated GO
10 mg nGO azide was dispersed in 12 ml DMF–H2O (5:1 v/v) solvent mixture by bath sonication for 1 h. After that, 98.6 mg (0.4 mmol) CuSO4 and 158.4 mg (0.8 mmol) sodium ascorbate was added to the above solution followed by addition of PAMAM dendrimers of different generations (1.0, 2.0 and 3.0G) and the reaction was carried out at 50° C for 24 h under nitrogen atmosphere. Finally, the reaction mixture was dialyzed against distilled water for 3 days followed by lyophilization for another 3 days to get the DGO. The percentage of primary amine groups of DGO was determined by TNBS assay according to the previous report using glycine for the standard calibration curve.31
2.7. Characterization
The synthesis of dendrimers and DGO was characterized by Fourier transform infrared (FTIR) spectrophotometer (100 FTIR, Perkin Elmer) using attenuated total reflection (ATR) attachment at a frequency range of 4000–600 cm−1. Proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were carried out at 400 MHz (Bruker NMR) NMR spectrometer. All half and full generation PAMAM dendrimers were dissolved in CDCl3 and D2O, respectively, with tetramethylsilane (TMS) as internal reference. Atomic force micrographs were acquired using NanoWizard®3a NanoScience AFM (JPK Instruments AG, Germany) in tapping mode by depositing the GO and DGO solution (10 μg ml−1) on a freshly cleaved mica surface and dried in ambient temperature for overnight. Transmission electron microscopy (TEM) images of GO and DGO deposited on copper grid were captured using FEI Tecnai T20 U-TWIN TEM at 200 kV. X-Ray Photoelectron Spectroscopy (XPS) and Raman spectroscopy were carried out in Axis Ultra DLD XPS system (Kratos Analytical Ltd, UK) and LabRAM HR Evolution (HORIBA Scientific), respectively.2.8. Preparation and characterization of GO/pDNA complex
nGO/pDNA and DGO/pDNA complexes were prepared according to a previous study.32 At first, both GO and DGO solution were prepared in 5 mM MES buffer (pH 6.5) by bath sonication at a concentration of 1 mg ml−1. pDNA stock solution was diluted in 25 mM sodium sulphate solution at a concentration of 100 μg ml−1. Subsequently, equal volume of nGO or DGO solution and pDNA solution were mixed at different weight ratios (1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) and vortexed for 20 s followed by incubation at room temperature for 30 min to prepare GO/pDNA or DGO/pDNA complex (GOplex). The formation of GOplex was confirmed by agarose gel electrophoresis. The GOplexes at different weight ratios having 0.5 μg DNA in each weight ratio were loaded in 0.8% agarose gel containing ethidium bromide (10 μg ml−1) as DNA visualizer. The gel was run in 1× TAE running buffer at 100 V for 40 min and subsequently the gel picture was taken by a gel documentation system (MyECL Image, Thermo Scientific). The GOplex formation was further confirmed by ethidium bromide (EtBr) assay according to a previous report.33 pDNA/EtBr complex was first prepared at a mole ratio of 10:1 (pDNA:EtBr mole ratio). nGO or DGO solution were mixed with pDNA/EtBr complex at different weight ratios with respect to pDNA (1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) and incubated for 30 min at room temperature. The fluorescence intensity of the resulting mixture solution at excitation and emission wavelengths of 510 and 605 nm, respectively was recorded by a multimode microplate reader (Synergy HT, BioTek).
The hydrodynamic size of nGO/pDNA and DGO/pDNA complexes was measured by Zetasizer Nano ZS particle size analyzer (Malvern Instruments, UK). Prior to particle size analysis, GOplexes were prepared at different weight ratios (GO or DGO/pDNA weight ratios of 1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) as discussed above and diluted to 3 ml in filtered (0.22 μm Axiva syringe filter paper, Axiva Sichem Biotech, India) double distilled water. The zeta potential of GO and DGO before and after complexation with pDNA and nGO-N3 was measured using the same instrument. The morphology and particle size of GOplex was further observed by atomic force microscopy (AFM).
2.9. In vitro toxicity assay
Cell viability of nGO and DGO (1.0, 2.0 and 3.0G) at different concentrations (5, 10, 20, 30, 40, 50, 100 and 200 μg ml−1) on HeLa cell was carried out by MTT assay. 5 × 103 cells in DMEM medium containing 10% fetal bovine serum (FBS) were seeded in each well of a 96 well plate and cultured in a humidified chamber supplying 5% CO2 at 37° C (Thermo Scientific) for 24 h. GO or DGO solutions at different concentrations were added to the cells in serum free medium and incubated at 37° C in CO2 incubator. After 4 h, GO or DGO containing medium was replaced by fresh medium containing serum and incubated further for 44 h in the CO2 incubator. The medium was removed and MTT solution (5 mg ml−1) diluted in 100 μl serum free medium (1 mg ml−1 working concentration) was added to each well. After incubation for 4 h, the medium containing MTT solution was replaced by 100 μl DMSO to dissolve the formazan crystals. Finally, the absorbance of the solution was measured at 570 nm using a microplate reader and the cell viability (%) was calculated by the following equation:where, OD570(Control) and OD570(Sample) are the measurements of the untreated and treated cells, respectively. All measurements were the mean of three measurements.

The in vitro toxicity assay of nGO/pDNA and DGO (1.0, 2.0 and 3.0G)/pDNA complexes at different weight ratios (1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) was also carried out in HeLa cell as mentioned above.
2.10. In vitro transfection
The transfection efficiency of nGO/pDNA and DGO/pDNA complexes at different weight ratios was carried out in HeLa cells. Before transfection, 1 × 105 cells in complete DMEM medium containing 10% FBS were seeded in each well of a 24 well plate. After 24 h, freshly prepared GOplexes at different weight ratios (5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) containing 1 μg pDNA in each weight ratio in serum free medium were added to the cells and incubated for 4 h in a CO2 incubator at 37° C. The transfection medium was replaced by fresh serum-containing medium and further incubated for 44 h. Lipofectamine 2000 (LF 2K, Life Technologies) and PEI (25 kDa) were used as the positive controls. pDNA and PAMAM (3.0G) were used as negative controls. The GFP (green fluorescence protein) expression of the transfected cells were observed by fluorescence microscope (Olympus IX53, Japan). The transfection efficiency of nGO and DGO was further quantified by FACS analysis (Becton–Dickinson) using 1 × 104 cells per analysis.
2.11. Endocytosis pathway
Low temperature or different endocytic inhibitors were used to determine the cellular uptake pathways of GOplexes. The transfection of DGO (3.0G)/pDNA complex at weight ratio of 30:1 was carried out at 4° C during the 4 h transfection period to inhibit energy dependent endocytosis. Also, cells were pre-incubated with genistein (100 mg ml−1), amiloride (10 mg ml−1), or chlorpromazine (10 mg ml−1) for 1 h prior to transfection to inhibit caveolae, macro-pinocytosis and clathrin mediated endocytosis pathways, respectively. Thereafter, DGO (3.0G)/DNA complex at weight ratio of 30:1 containing 1 μg DNA was added to the treated cells and incubated for 4 h. The transfection efficiency was determined by flow cytometry after 44 h post transfection.
2.12. Intracellular distribution
The intracellular distribution of DGO (3.0G)/pDNA complex at weight ratio of 30:1 was observed by labelling pDNA with Cy3 according to manufacturer's protocol (Label IT® Tracker™ intracellular nucleic acid localization kit, Mirus Bio, USA). Before transfection, 5 × 104 cells (HeLa cell) were seeded on four chambered glass slides (Biofil, South Korea). After 24 h, DGO (3.0G)/pDNA complex at weight ratio of 30:1 containing 1 μg Cy3-labeled pDNA for 4 h. LF 2K/pDNA complex was used as positive control. After 4 h, the transfection medium was removed and the cells were washed with PBS followed by fixing with 3.7% (w/v) formaldehyde for 30 min. The nucleus was then stained with diamidino phenylindole (DAPI) for 10 min and the cells were covered with cover slip with mounting medium containing antifading agent and sealed with colorless nail polish. The intracellular distribution of GOplex was imaged by confocal laser scanning microscopy (CLSM; Leica TCS Sp5).
2.13. Statistical analysis
All the data are presented as the average ± the standard deviation. One-way ANOVA was used to calculate statistical differences and differences were considered significant for p values less than 0.05.3. Results and discussion
3.1. Synthesis of dendronized GO
DGO was synthesized by reaction of alkyne terminated focal point PAMAM dendrimers (1.0, 2.0 and 3.0G) with azide functionalized nGO through “click” chemistry as shown in Fig. 1. Full generation (amine terminated) focal point PAMAM dendrimers (1.0, 2.0 and 3.0G) were synthesized by repeating the Michael addition reaction followed by amidation reaction using propargyl amine as starting material, as shown in Fig. 1a. Fig. S1† shows the FTIR spectra of synthesized PAMAM dendrimers (0.5, 1.0, 1.5 and 2.0G). A strong peak at 1735 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of ester) indicates the presence of ester group (–COOCH3) in half generation (ester terminated) PAMAM dendrimer (0.5G). A weak stretching peak at 2102 cm−1 (inset) and a medium stretching peak at 3279 cm−1 correspond to –C
O stretching of ester) indicates the presence of ester group (–COOCH3) in half generation (ester terminated) PAMAM dendrimer (0.5G). A weak stretching peak at 2102 cm−1 (inset) and a medium stretching peak at 3279 cm−1 correspond to –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– stretching and C–H stretching of –CCH group, respectively indicating the presence of alkyne group in 0.5G PAMAM dendrimer. After amidation reaction of 0.5G PAMAM with ehthylene diamine, the absorption peak for ester group disappeared and two new peaks appeared at 1644 cm−1 and 1554 cm−1 equivalent to the CO stretching of amide (–CO–NH–) group and N–H bending of primary amine (–NH2) group, respectively. The appearance of other new peaks at 3351 and 3289 cm−1 related to hydrogen bonded N–H stretching and free N–H stretching, respectively of primary amine group indicates the synthesis of 1.0G PAMAM dendrimer (full generation). The reappearance and disappearance of the absorption peak at 1731 cm−1 for ester group in the successive steps indicating the synthesis of 1.5, 2.0 (Fig. 2), 2.5 and 3.0G PAMAM dendrimers. The synthesis of PAMAM dendrimers was further confirmed by 1H and 13C NMR spectra as described in Section 1 of ESI.†
C– stretching and C–H stretching of –CCH group, respectively indicating the presence of alkyne group in 0.5G PAMAM dendrimer. After amidation reaction of 0.5G PAMAM with ehthylene diamine, the absorption peak for ester group disappeared and two new peaks appeared at 1644 cm−1 and 1554 cm−1 equivalent to the CO stretching of amide (–CO–NH–) group and N–H bending of primary amine (–NH2) group, respectively. The appearance of other new peaks at 3351 and 3289 cm−1 related to hydrogen bonded N–H stretching and free N–H stretching, respectively of primary amine group indicates the synthesis of 1.0G PAMAM dendrimer (full generation). The reappearance and disappearance of the absorption peak at 1731 cm−1 for ester group in the successive steps indicating the synthesis of 1.5, 2.0 (Fig. 2), 2.5 and 3.0G PAMAM dendrimers. The synthesis of PAMAM dendrimers was further confirmed by 1H and 13C NMR spectra as described in Section 1 of ESI.†
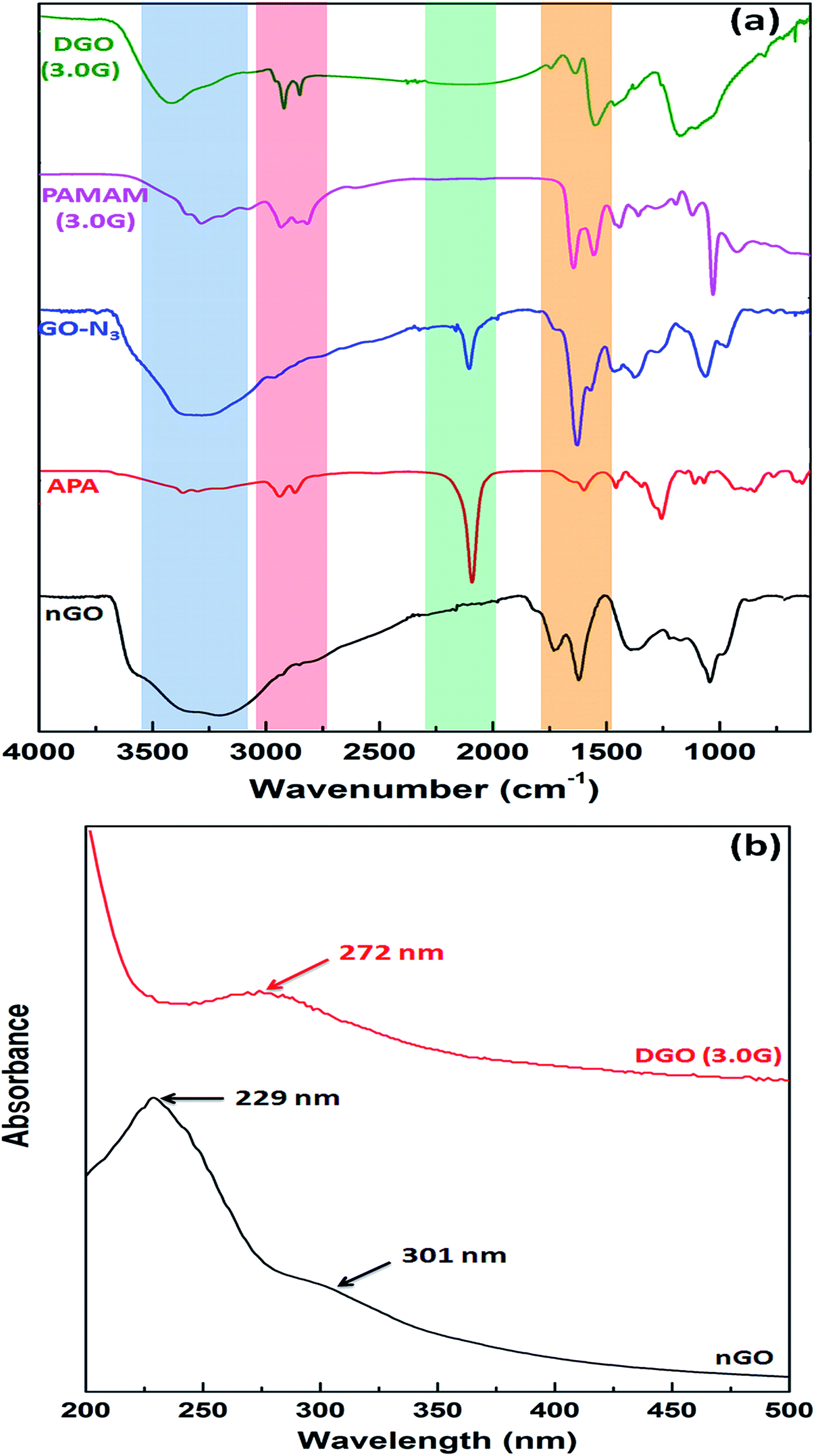 | ||
| Fig. 2 (a) FTIR spectra of synthesized nGO, aminopropyl azide (APA), azide functionalized GO (GO-N3), PAMAM dendrimer (3.0G) and dendronized GO (3.0G), and (b) UV-vis spectroscopy of nGO and DGO (3.0G). | ||
nGO was prepared by oxidation of pristine graphite followed by ultra sonication. Fig. S2a† shows the X-ray diffraction pattern of pristine graphite and nGO. Pristine graphite shows characteristic strong diffraction peak at 2θ = 26.5° corresponding to 002 plane having d-spacing of 0.335 nm. This peak disappeared after oxidation of pristine graphite and a new diffraction peak appeared at 2θ = 10.8° with higher d-spacing of 0.818 nm confirming the formation of GO.34 The particle size distribution of nGO is shown in Fig. S2b† and the average particle of nGO was 180 ± 55 nm. The size and morphology of nGO was observed by AFM as shown in Fig. S2c and d,† respectively. The AFM image shows that the flake diameter of nGO was about 160–230 nm and the flake thickness was about 0.6–0.8 nm (Fig. S2e†) consistent with previous reports.35
GO showed characteristic absorption peaks at 3150–3500 cm−1 (broad peak of hydrogen bonded O–H stretching), 1726 cm−1 (CO stretching of –COOH), 1621 cm−1 (CC stretching of aromatic ring), 1043 cm−1 (C–O stretching of –C–O–C–) and 1378 cm−1 (in plane O–H bending) as shown in Fig. 2a.36 A strong absorption peak at 2092 cm−1 and a absorption peak at 3300 cm−1 are corresponding to azide (–N3) and primary amine (–NH2) groups indicating the synthesis of aminopropyl azide by reaction of chloropropyl amine and sodium azide. After reaction of GO with aminopropyl azide, two new absorption peaks appeared at 2104 cm−1 and 1630 cm−1 analogous to the azide group (–N3) and CO stretching of amide group (–CONH–), respectively and the disappearance of CO stretching peak for carboxylic acid of GO suggesting the synthesis of azide functionalized GO (GO-N3) as shown in Fig. 1b. PAMAM dendrimer of 3.0G showed strong absorption peaks at 1647 cm−1 and 1552 cm−1 for CO stretching of amide (–CONH–) and N–H bending of primary amine (–NH2) groups, respectively, along with N–H stretching of primary amine group at 3287 cm−1. The strong absorption peak at 2104 cm−1 for azide group of azide functionalized graphene oxide (GO-N3) disappeared after reaction with focal point PAMAM dendrimer through “click” chemistry. The intensity of C–H stretching (–CH2–) peak at 2920 cm−1 and 2850 cm−1 also increased significantly compared to that of GO-N3. Along with this, three new peaks appeared at 1742 cm−1, 1635 cm−1 and 1547 cm−1 related to triazole ring, CO stretching of amide group and N–H bending of primary amine group, respectively confirming the synthesis of DGO. In contrast to other conjugation techniques the chemical specificity imparted by “click” chemistry utilized herein can overcome potential challenges such as crosslinking of many nGO flakes by a single polycationic chain and electrostatic interactions between nGO and the polycation.
Fig. 2b shows the UV-vis spectra of nGO and DGO (3.0G). GO shows characteristic strong absorption peak at 229 nm and a broad shoulder at 301 nm due to the π–π* transition of aromatic CC and carboxylic acid group's CO, respectively.37 But the absorption peak of GO shifted to 272 nm after reaction with PAMAM dendrimer corresponding to the triazole ring formed during the reaction of GO-N3 and focal point PAMAM dendrimer through “click” chemistry.
1H NMR spectra further supports the synthesis of dendronized GO as shown in Fig. S3.† PAMAM dendrimer (3.0G) showed the characteristic peaks at 2.25–2.51 ppm (−C2−CONH−), 2.52–2.58 ppm (−N−C2−CH2−), 3.20 ppm (−C2−NH2), 3.29 ppm (CHC−C2−) and 3.36 ppm (−C2−CH2−NH2). After reaction of PAMAM dendrimer with GO-N3 through “click” chemistry, new peaks appeared at 2.70 ppm, 2.86 ppm, 3.2 ppm and 3.45–3.53 ppm (inset) related to the respective protons of PAMAM dendrimers.
The synthesis of GO followed by DGO was further confirmed by Raman and X-ray photoelectron spectroscopies, as shown in Fig. 3a and b, respectively. Fig. 3a shows the Raman spectra of pristine graphite, GO and DGO (3.0G). Pristine graphite shows the typical Raman band at 1583 cm−1 due to the E2g phonon of sp2 hybridized carbon atom of graphite, called G band and a small deflection at 1355 cm−1 corresponding to D band, which is associated with structural defects. The intensity of G band increased significantly and shifted to higher frequency (1606 cm−1) due to the presence of isolated double in the oxidized GO.38 The D band became more prominent and shifted to lower value (1548 cm−1) suggesting generation of small sized in-plane sp2 grains during oxidation of graphite to form GO. The intensity ratio (ID/IG) of the D band (ID) and the G band (IG) determines the amount of defect and disorder in graphite structure.39 The ID/IG value of pristine graphite was only 0.48 that indicates highly ordered and defect free structure.40 The ID/IG value of GO increased significantly from 0.48 to 1.08 during the oxidation of graphite suggesting the formation of large defects in GO structure. The intensity ratio further increased from 1.08 to 1.11 in DGO and the G band shifted to lower frequency (1598 cm−1) compared to GO. This indicates further structural defects after reaction of PAMAM dendrimer with GO.
 | ||
| Fig. 3 (a) Raman spectra of pristine graphite, nGO and DGO (3.0G) and X-ray photoelectron spectra of nGO and DGO (3.0G). | ||
Fig. 3b shows the XPS wide scan spectra of GO and DGO (3.0G). GO shows two intense peaks at binding energies of 285 and 532 eV for C 1s and O 1s, respectively indicating the presence of carbon and oxygen as major elements in GO. A new peak appeared at binding energy of 401 eV for N 1s (core-level spectra in inset) after reaction of PAMAM dendrimer suggesting that PAMAM dendrimer was successfully conjugated with GO through “click” chemistry.
The surface morphology of GO and DGO was characterized by TEM as shown in Fig. S4.† GO shows typical wrinkled morphology and transparent thin flake (Fig. S4a†). However, the morphology of GO flake changed after surface modification by dendrimer. After conjugation of PAMAM dendrimers with GO through “click” chemistry, small black spots (1.0G PAMAM, Fig. S4b†) and comparatively large black spots (3.0G PAMAM, Fig. S4c†) formed on the GO flake due large size of 3.0G PAMAM dendrimer compared to that of 1.0G PAMAM dendrimer suggesting the successful conjugation of PAMAM dendrimer on GO surface.
Table 1 also summarizes the change in particle size and thickness of GO flakes after dendronization. The diameter and thickness of unmodified GO flakes were 206 ± 46 nm and 0.6 ± 0.2 nm, respectively. Both the diameter and thickness of GO flakes increased progressively with increasing the generation of PAMAM dendrimer due to increasing the size of the PAMAM dendrimer from low generation to higher generation accordingly. Unmodified GO showed negative zeta potential (−41.5 ± 3.4 mV) as expected due to presence of negatively charged carboxylic acid functional groups. The zeta potential was −0.9 ± 3.8 mV after modification of nGO with the azide group. This reduction in anionic character of nGO-N3 is a significant advantage of the chemical synthesis route proposed in this study. It minimizes electrostatic interactions between unmodified nGO and cationic polymers that may be significant in other techniques such as EDC conjugation. The interactions between PAMAM dendrimers and the modified nGO can thus primarily be attributed to covalent interactions between them. The zeta potential became positive (5.4 ± 2.4 mV) after conjugation of 1.0G PAMAM dendrimer and the value of zeta potential increased gradually with increasing the generation of PAMAM dendrimer due to increasing the content of primary amine groups from 2.5 ± 0.5% (DGO 1.0G) to 6.4 ± 1.1% (DGO 2.0G) to 12.8 ± 1.5% (DGO 3.0G). The zeta potential values of DGO (2.0G) and DGO (3.0G) were 18.3 ± 5.4 and 38.6 ± 3.6 mV, respectively.
| Sample | % NH2a | Diameterb (nm) | Thicknessc (nm) | Zeta potential (mV) |
|---|---|---|---|---|
| a Determined by TNBS assay. b Determined by dynamic light scattering. c Determined by atomic force microscopy. | ||||
| nGO | — | 206 ± 46 | 0.6 ± 0.2 | −41.5 ± 3.4 |
| DGO (1.0G) | 2.5 ± 0.5 | 226 ± 15 | 1.6 ± 0.5 | 5.4 ± 2.4 |
| DGO (2.0G) | 6.4 ± 1.1 | 254 ± 18 | 2.5 ± 0.8 | 18.3 ± 5.4 |
| DGO (3.0G) | 12.8 ± 1.5 | 284 ± 24 | 4.0 ± 1.0 | 38.6 ± 3.6 |
3.2. Preparation and characterization of GO/pDNA complex
nGO/pDNA and DGO/pDNA complexes (GOplexes) at different weight ratios (1:1, 5:1, 10:1, 15:1, 20:1, 25:1, and 30:1) were prepared by mixing of equal volume of nGO or DGO solution and pDNA solution through electrostatic interaction between the positively charged carrier and negatively charged DNA.33 The formation of GOplexes was confirmed by agarose gel electrophoresis assay, as shown in Fig. 4a–d. Fig. 4a shows the agarose gel image of nGO/pDNA complexes at different weight ratios. It is found that nGO has no complexation capability with pDNA at all weight ratios as expected due to absence of any cationic functional groups. Previous studies also showed that GO has no complexation capability with pDNA due its negative zeta potential value.16 The DNA complexation capability of GO started after conjugation of PAMAM dendrimer with GO due to the positive zeta potential of DGO as shown in Table 1. DGO (1.0G) started to complex almost all pDNA at high weight ratio of 30:1 due to its low zeta potential (5.4 ± 2.4 mV). The DNA complexation capability of DGO increased with increase in the generation of PAMAM dendrimer due to enhancement of zeta potential. DGO (2.0G) and DGO (3.0G) complexed all pDNA at 15:1 and 5:1 weight ratio, respectively, where the electrophoretic mobility of pDNA was completely retarded after complexation with positively charged DGO. The number of primary amine groups increased with increase in the generation of PAMAM dendrimer increasing the zeta potential and as a result this facilitated the efficient DNA complexation at low weight ratio.
 | ||
| Fig. 4 Agarose gel electrophoresis assay image of (a) nGO/pDNA, (b) DGO (1.0G)/pDNA, (c) DGO (2.0G)/pDNA and (d) DGO (3.0G)/pDNA complexes at different N/P ratios of 1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1, and (e) EtBr assay of nGO and dendronized GO (1.0, 2.0 and 3.0G) at different weight ratios. | ||
The DNA complexation capability of GO and DGO was further characterized by EtBr assay as shown in Fig. 4e. In this assay, EtBr was complexed with pDNA at a molar ratio of 1:10 and gave maximum fluorescence intensity due to intercalation of EtBr molecule into the double helix of DNA.41 After addition of any polycation externally to the EtBr/pDNA complex, the fluorescence intensity will come down gradually with increase in concentration of cationic molecule due to replacement of EtBr molecule by foreign cationic molecule indicating the formation of cationic molecule/pDNA complex. Fig. 4e shows that the fluorescence intensity of EtBr/pDNA complex did not change because of inefficient DNA complexation by unmodified GO. The fluorescence intensity decreased gradually with increase in the amount of DGO (1.0G) and reached minimum fluorescence intensity at 30:1 weight ratio. But, DGO (3.0G) most efficiently replaced the EtBr molecules from EtBr/pDNA complex at very low weight ratio of 5:1 and consequently the fluorescence intensity decreased significantly at this weight ratio due to its highest zeta potential among the DGOs. At higher weight ratio, the change in fluorescence intensity remained almost unchanged because nearly all EtBr molecules were replaced by DGO (3.0) molecules.
3.3. Particle size and zeta potential of GOplex
Hydrodynamic diameter and zeta potential of nGO/pDNA and DGO/pDNA complexes at different nGO or DGO to pDNA weight ratios were determined by DLS as shown in Fig. 5a and b, respectively. From Fig. 5a, it is found that the particle size of nGO/pDNA complexes did not change at all weight ratios and the average particle size of nGO/pDNA complexes was 250 ± 40 nm which was almost equal to the size of nGO only. In agarose gel and EtBr assay, it was found that the unmodified GO has no DNA binding capability at all weight ratios and as a result the nGO/pDNA complexes at all weight ratios showed the diameter of nGO flakes alone. DGO (1.0G)/pDNA complexes showed larger size (∼950 nm) at low weight ratio (5:1) due to inefficient binding of DGO (1.0G) with pDNA. The hydrodynamic diameter gradually decreased with increase in the weight ratio. However, the size was quite large compared to that of DGO (2.0) and DGO (3.0). DGO (3.0G) formed smaller particles among all the dendronized GO due to its highest cationic charge. DGO (3.0G) formed smallest particles (∼285 nm) with pDNA at the weight ratio 20:1 and then the particle size increased marginally with further increase in weight ratio putatively due to repulsion of excessive charge offered by DGO.42 Chen et al.17 synthesized PEI conjugated GO by carbodiimide method and they observed similar trends in particle size of PEI–GO/pDNA complexes at different weight ratios. The particle size of PEI–GO/pDNA complexes was in the range of 250–500 nm at the weight ratio of 1.3:1 to 20:1. It is suggested that the particle size up to 500–700 nm can easily be taken up by cells.43 Therefore, it is envisaged that the size of DGO/pDNA complex will be suitable for cellular uptake and consequently may be lead to high transfection efficiency.
 | ||
| Fig. 5 (a) Average particle size and (b) zeta potential of nGO/pDNA and DGO (1.0, 2.0 and 3.0G)/pDNA complexes at different weight ratios of 1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1 determined by DLS. | ||
Zeta potential of carrier/DNA complex is an important parameter for cellular uptake. The overall positive zeta potential of the complex will help the attachment of complex with negatively charged cellular membrane through electrostatic interactions facilitating for cellular uptake through endocytosis.44 As shown in Fig. 5b, nGO/pDNA complexes at all weight ratios showed negative zeta potential due to incomplete complexation of nGO with pDNA. DGO (1.0G)/pDNA complexes also showed negative zeta potential at lower weight ratio but the value of zeta potential changed from higher negative to lower negative value with increase in weight ratio. The positive zeta potential (5.3 ± 1.4 mV) was obtained at weight ratio of 30:1 indicating complete DNA complexation, which is also showed in agarose gel and EtBr assay. DGO (3.0G) was more efficient among the all dendronized GOs and it showed positive zeta potential (5.6 ± 2.6 mV) at very low weight ratio (5:1). The zeta potential of DGO (3.0G)/pDNA complexes increased with increase in weight ratio up to the weight ratio of 20:1 and then reached plateau after the weight ratio of 20:1. The zeta potential of DGO (3.0G)/pDNA complexes at 20:1 and 30:1 was 25.3 ± 2.3 and 27.3 ± 1.3 mV, respectively whereas the maximum zeta potential of DGO (2.0G)/pDNA complexes at 30:1 was 20.5 ± 3.4 mV. In a previous study42 a similar trend in zeta potential of dendronized chitosan/pDNA complexes was observed. In another study, Kim et al.16 also obtained negative zeta potential of GO/pDNA complexes at all N/P ratios (nitrogen to phosphate ratios) from 2:1 to 20:1 ratios with constant positive zeta potentials with branched PEI–GO/pDNA complexes after the N/P ratio of 5:1.
The morphology and particle size of DGO (3.0G) before and after complexation with pDNA was characterized by AFM, as shown in Fig. 6. Fig. 6a, c and e show the AFM images of DGO (3.0G) before complexation with pDNA. The particle diameter and thickness of DGO (3.0G) before complexation were 150 ± 45 nm and 4.0 ± 1.0 nm, respectively. Both the particle diameter and thickness of DGO (3.0G) increased after complexation with pDNA at weight ratio of 20:1 and the sizes were 170 ± 35 and 6 ± 1 nm, respectively (Fig. 6b, d and f). Along with these, the morphology of DGO (3.0G0/pDNA complex was spherical in shape, which was different from DGO before complexation with pDNA (Fig. 6d).
 | ||
| Fig. 6 Atomic force micrographs of DGO (3.0G) at (a) low magnification and (c) high magnification, and DGO (3.0G)/pDNA complex at weight ratio of 20:1 at (b) low magnification and (d) high magnification. Line graphs of (e) DGO (3.0) and (f) DGO (3.0G)/pDNA complex at weight ratio of 20:1. | ||
3.4. In vitro cytotoxicity assay
The in vitro toxicity of nGO and DGO at different concentrations (5, 10, 20, 30, 40, 50, 100 and 200 μg ml−1) and weight ratios (1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1) before and after complexation with pDNA, respectively was assessed by MTT assay against HeLa cells as shown in Fig. 7. Fig. 7a shows the percent cell viability of nGO and DGOs at different concentrations. nGO was less toxic compared to that of dendronized GOs at all concentrations. nGO showed >80% cell viability at high concentration (200 μg ml−1) which corroborates findings of previous studies.45 The toxicity of nGO may be attributed to damage to cell membrane by sharp edges of graphene sheets.46 The toxicity of nGO increased after dendronization and the cell viability of DGO decreased with increase in the generation of PAMAM dendrimer. DGO (1.0G) showed lowest toxicity among the all dendronized GOs. However, the cell viability was above 60% in the presence of DGO (3.0G) at high concentration (200 μg ml−1). The toxicity of dendronized GOs increased due to the positive charge of DGO. It is reported that the toxicity of polycation is related to its surface positive charge which interacts with the negatively charged plasma membrane and consequently impairs the function of cellular membrane through aggregation on the cell membrane.47 The toxicity of DGO (3.0G) decreased after complexation with pDNA (Fig. 7b) due to reduction in zeta potential of DGO (3.0G) from 38.6 ± 2.6 mV to 27.3 ± 1.3 mV after complexation with pDNA at the weight ratio of 30:1 through charge neutralization by negatively charged pDNA. However, the toxicity increased for all DGO/pDNA complexes with increase in the weight ratio although the cell viability of DGO (3.0G)/pDNA complexes was above 80% even at high weight ratio of 30:1.
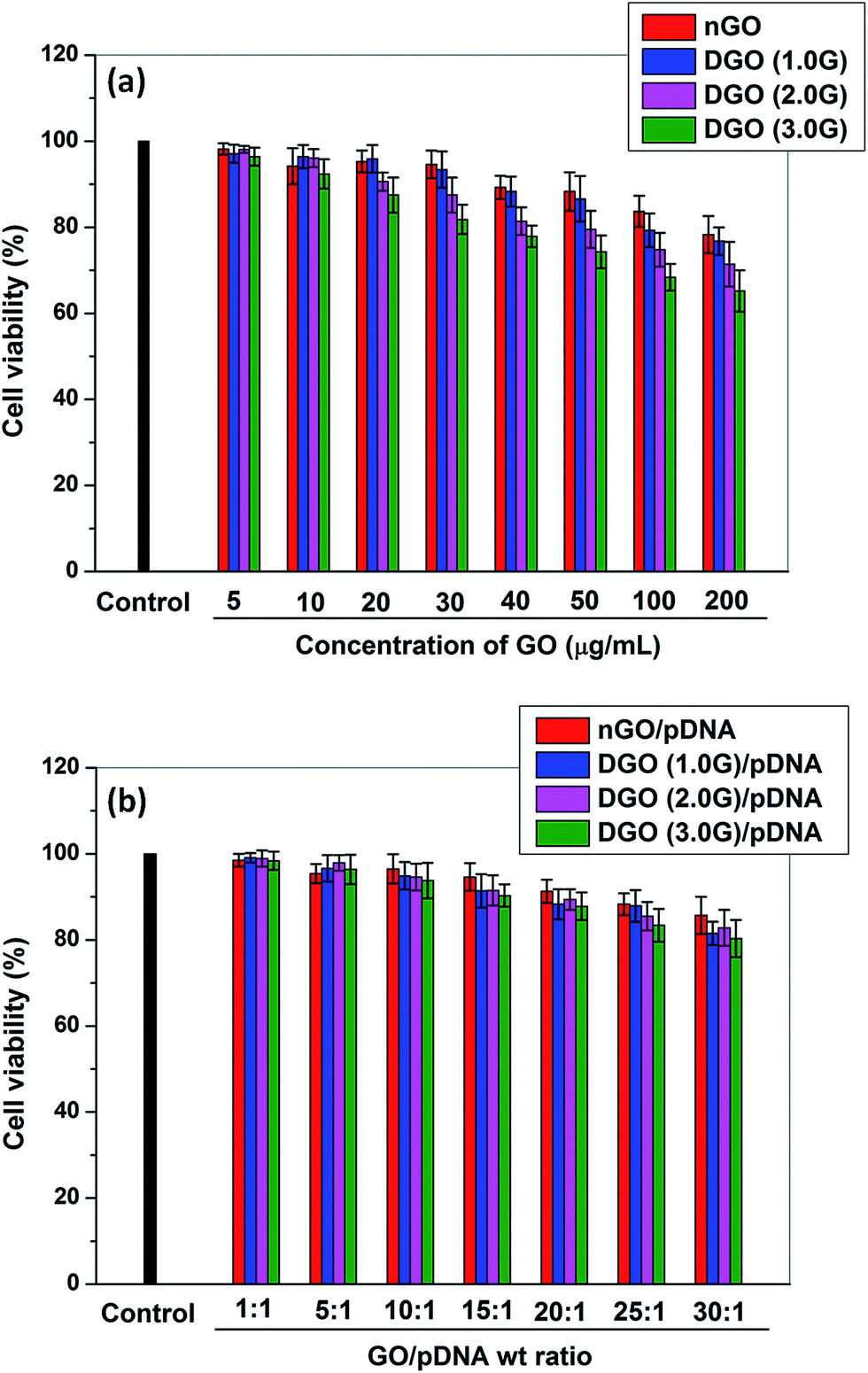 | ||
| Fig. 7
In vitro toxicity assay of (a) nGO and DGO (1.0, 2.0 and 3.0G) at different concentrations of 5, 10, 20, 30, 40, 50, 100 and 200 μg ml−1, and (b) nGO/pDNA and DGO (1.0, 2.0 and 3.0G)/pDNA complexes at different weight ratios of 1:1, 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1 against HeLa cell. | ||
3.5. In vitro transfection efficiency
The transfection efficiency of nGO/pDNA and DGO/pDNA complexes at different weight ratios from 5:1 to 30:1 using EGFP pDNA was observed in HeLa cells. PEI (25 kDa)/pDNA complex at N/P ratio of 10 and LF 2K were used as positive control. As shown in Fig. 8, nGO/pDNA and PAMAM (3.0G)/pDNA complexes at weight ratio of 30:1 did not show any transfection efficiency as expected. It is shown above that nGO has poor complexation ability with pDNA. The transfection efficiency of GO improved after dendronization of GO although the transfection efficiency of DGO increased with increase in the generation of dendrimer. DGO (3.0G0/pDNA complex at weight ratio of 30:1 showed largest number of GFP positive cells and the number of GFP positive cells was even higher that of LF 2K.
 | ||
| Fig. 8 Fluorescence micrographs of transfected HeLa cell by nGO/pDNA, PAMAM (3.0G)/pDNA, DGO (1.0, 2.0 and 3.0G)/pDNA complexes at weight ratios of 30:1 and LF 2K/pDNA complex containing 1 μg of pDNA in each formulation. The scale bar is 10 mm. | ||
The transfection efficiency of GOplexes was further quantified by FACS analysis, as shown in Fig. 9a. It was observed that nGO/pDNA complexes at all weight ratios did not show any transfection as noted in fluorescence micrographs. DGO (1.0G)/pDNA complex at weight ratio of 30:1 showed only 10% transfection efficiency whereas DGO (2.0G)/pDNA and DGO (3.0G)/pDNA complex showed 35% and 51% transfection efficiency, respectively at the same weight ratio. The transfection efficiency of DGO (3.0G)/pDNA complex at the weight ratio 30:1 was significantly higher that of PEI (25 kDa)/pDNA complex (28%). It was higher than that of LF 2K/pDNA complex (47%) although the differences were not statistically significant. Thus, the transfection efficiency of GO significantly increased after conjugation of PAMAM dendrimer to GO and it scaled with increase in both weight ratio as well as generation of the dendrimer. Previously, Tripathi et al.18 prepared linear PEI-conjugated-GO (LP–GO) for gene delivery and observed approximately 33% transfection efficiency in HeLa cells at N/P ratio of 97:1. In another study, Liu et al.48 synthesized PAMAM dendrimer (4.0G) conjugated GO through oleic acid using carbodiimide and they also obtained 18.3% transfection efficiency in HeLa cells. In comparison to these reported studies, the synthesized DGO (3.0G) in this work showed markedly higher transfection efficiency and therefore, it is a significant improvement over currently available GO-based gene carriers.
 | ||
| Fig. 9 (a) Representative flow cytometric analysis of GFP-expressing cells after 48 h post transfection by nGO/pDNA and DGO (1.0, 2.0 and 3.0G)/pDNA complexes at different weight ratios of 5:1, 10:1, 15:1, 20:1, 25:1 and 30:1, PEI/pDNA complex at N/P ratio 10 and LF/pDNA complexes containing 1 μg of pDNA in each formulation against HeLa cell, and (b) relative transfection efficiency determined by FACS analysis of DGO (3.0G)/pDNA complex at the weight ratio of 30:1 against HeLa cell after treating with various endocytotic inhibitors for 1 h before transfection containing 1 μg of pDNA followed by further incubation for 48 h. | ||
3.6. Mechanism of cellular uptake
The transfection efficiency of nonviral vector not only depends on the complexation capability with pDNA but also depends on the cellular uptake mechanism. It is reported that nonviral vectors enter into the cell through one or more of the following endocytosis pathways including clathrin, caveolae and macro-pinocytosis mediated endocytosis pathways.49,50 To determine the pathway for transfection of DGO, we measured transfection of DGO (3.0G)/pDNA complex at weight ratio of 30:1 in the presence of the following endocytotic inhibiting drugs and at low temperature (Fig. 9b). Chlorpromazine (CPZ) is known to inhibit clathrin mediated endocytosis by preventing the recycling of clathrin proteins,51 genistein (GEN) inhibits tyrosine kinase involved in caveolae mediated endocytosis,52 whereas amiloride (AMIL) inhibits macro-pinocytotic uptake.53 In addition, low temperature (4° C) inhibits energy dependent pathway, that is, overall endocytosis pathway.54 It was observed that the transfection efficiency significantly decreased at low temperature (4° C) suggesting that DGO (3.0G)/pDNA complex enters into the cell by energy dependent pathway, that is, endocytosis pathway was involved during cellular uptake. Furthermore, the transfection efficiency was inhibited by the presence of chlorpromazine and genistein. But genistein inhibited 67% transfection efficiency whereas chlorpromazine inhibited only 25%. Therefore, it appears that the cellular uptake of DGO (3.0G)/pDNA complex occurred primarily by caveolae mediated endocytosis pathway supported by clathrin mediated pathway whereas the macro-pinocytosis was not involved in the cellular uptake. It is reported that the particles of size <200 nm are internalized into the cell through the clathrin mediated endocytosis pathway and the caveolae mediated endocytosis is involved in uptake of particles >200 nm.55 It has also been reported that the clathrin mediated endocytosis pathway involves the degradative acidic lysosomes during nuclear transportation and this is the main drawback associated with this pathway leading to lower transfection efficiency. In contrast, caveolae mediated pathway avoids the lysosome compartment in nuclear transportation resulting in higher transfection efficiency.44 Recently, Zhi et al.56 prepared multifunctional GO nanocomplex composed of PEI/poly(sodium 4-styrenesulfonates) (PSS)/GO (PPG) for the delivery of adriamycin along with siRNA. They also showed that the multifunctionalized GO nanocomplexes were internalized primarily by caveolae mediated endocytosis pathway during cellular uptake in MCF-7 cells.
3.7. Intracellular distribution
After cellular uptake, intracellular distribution of carrier/DNA complex is an important parameter for efficient transgene expression because the carrier/DNA complex has to overcome several obstacles in the form of transport and degradative mechanisms in the cell. For intracellular distribution of DGO (3.0G)/pDNA complex at weight ratio of 30:1, the nucleus of HeLa cell was stained with DAPI and the plasmid DNA was labeled with Cy3 and finally observed using CLSM after 4 h of transfection as shown in Fig. 10. PEI (25 kDa) and LF 2K were used as positive controls. Fig. 10a1 shows that the numbers of red particles were much higher, that is, more pDNA was carried out by DGO (3.0G) in to the cell compared to that of either PEI (Fig. 10b1) or LF 2K (Fig. 10c1) after 4 h of transfection. It is also observed that more numbers of pDNA localized in the nucleus by DGO (3.0G) within 4 h of tranfection whereas most of pDNA remained far away from the nucleus resulting in low transfection efficiency for PEI. On the other hand, LF 2K was able to transfect fewer pDNA into the nucleus compared to that of DGO (3.0G). It has been reported that the transfection of both branched and linear PEI/DNA complex may involve either clathrin or caveolae mediated endocytosis but clathrin mediated endocytosis plays a critical role for transfection of PEI/DNA complex.57 Uptake of lipoplex (liposome/pDNA complex) is mainly through clathrin mediated pathway.58 The uptake of DGO (3.0)/pDNA complex reported in this study is largely through the caveolae mediated endocytosis pathway, which helps the DGO/DNA complex to avoid the degradative endosomal compartment facilitating more effective transport to the nucleus and results in high transfection efficiency compared to both PEI (25 kDa) and LF 2K.
 | ||
| Fig. 10 CLSM images of cellular uptake by HeLa cells after 4 h of transfection with DGO (3.0G)/Cy3-labeled pDNA complex at the weight ratio of 30:1 (a1–a3), PEI/Cy3-labeled pDNA complex at N/P ratio of 10 (b1–b3) and LF 2K/Cy3-labeled pDNA complex (c1–c3) containing 1 μg pDNA in each formulation. For each panel, images from left to right show Cy3 labelled pDNA (red), cell nuclei stained by DAPI (blue), and overlay of the two images. Scale bar is 25 μm. | ||
4. Conclusion
DGO was synthesized by reaction of focal point PAMAM dendrimer of different generations (1.0, 2.0 and 3.0G) with azide functionalized nGO through “click” chemistry. The synthesis of DGO was confirmed by FTIR, NMR, XPS, Raman spectroscopy and TEM analysis. The DNA complexation capability of nGO was significantly improved after conjugation of PAMAM dendrimer with nGO. DGO formed spherical nano sized complexes with pDNA through electrostatic interaction having particle diameter of 250–350 nm with positive zeta potential. The transfection efficiency of nGO was significantly increased from 1.2% to 51% in HeLa cells with minimal toxicity and also surpassed the transfection efficiency of both LF 2K (47% efficiency) and high molecular weight branched PEI (27% efficiency). Caveolae mediated endocytosis pathway is shown to play an important role underlying the high transfection efficiency of DGO. Therefore, DGO could be developed as an efficient and safe novel nonviral vector in gene therapy applications.Acknowledgements
This work was funded by the Department of Science and Technology (DST), India. K.S. was supported by the D. S. Kothari fellowship (BSR/EN/13-14/0005) from the University Grants Commission (UGC), India. K.C. acknowledges the Ramanujan fellowship from DST.References
- E. Kennington, Nat. Rev. Drug Discovery, 2009, 8, 275 CrossRef CAS PubMed.
- P. G. Coune, B. L. Schneider and P. Aebischer, Cold Spring Harbor Perspect. Med., 2012, 2, a009431 Search PubMed.
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- J. W. Wiseman, C. A. Goddard, D. McLelland and W. H. Colledge, Gene Ther., 2003, 10, 1654–1662 CrossRef CAS PubMed.
- D. Y. Kwoh, C. C. Coffin, C. P. Lollo, J. Jovenal, M. G. Banaszczyk, P. Mullen, A. Phillips, A. Amini, J. Fabrycki, R. M. Bartholomew, S. W. Brostoff and D. J. Carlo, Biochim. Biophys. Acta, Gene Struct. Expression, 1999, 1444, 171–190 CrossRef CAS.
- C. S. Braun, J. A. Vetro, D. A. Tomalia, G. S. Koe, J. G. Koe and C. R. Middaugh, J. Pharm. Sci., 2005, 94, 423–436 CrossRef CAS PubMed.
- K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- S. Goenka, V. Sant and S. Sant, J. Controlled Release, 2014, 173, 75–88 CrossRef CAS PubMed.
- S. Kumar and K. Chatterjee, Nanoscale, 2015, 7, 2023–2033 RSC.
- S. Kumar, S. Raj, E. Kolanthai, A. K. Sood, S. Sampath and K. Chatterjee, ACS Appl. Mater. Interfaces, 2015, 7, 3237–3252 CAS.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243–9257 CrossRef CAS PubMed.
- Y. Wang, Z. Li, D. Hu, C.-T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274–9276 CrossRef CAS PubMed.
- X. Ma, H. Tao, K. Yang, L. Feng, L. Cheng, X. Shi, Y. Li, L. Guo and Z. Liu, Nano Res., 2012, 5, 199–212 CrossRef CAS.
- Z. Tang, H. Wu, J. R. Cort, G. W. Buchko, Y. Zhang, Y. Shao, I. A. Aksay, J. Liu and Y. Lin, Small, 2010, 6, 1205–1209 CrossRef CAS PubMed.
- H. Kim, R. Namgung, K. Singha, I.-K. Oh and W. J. Kim, Bioconjugate Chem., 2011, 22, 2558–2567 CrossRef CAS PubMed.
- B. Chen, M. Liu, L. Zhang, J. Huang, J. Yao and Z. Zhang, J. Mater. Chem., 2011, 21, 7736–7741 RSC.
- S. K. Tripathi, R. Goyal, K. C. Gupta and P. Kumar, Carbon, 2013, 51, 224–235 CrossRef CAS PubMed.
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Boggild, S. Borini, F. H. L. Koppens, V. Palermo, N. Pugno, J. A. Garrido, R. Sordan, A. Bianco, L. Ballerini, M. Prato, E. Lidorikis, J. Kivioja, C. Marinelli, T. Ryhanen, A. Morpurgo, J. N. Coleman, V. Nicolosi, L. Colombo, A. Fert, M. Garcia-Hernandez, A. Bachtold, G. F. Schneider, F. Guinea, C. Dekker, M. Barbone, Z. Sun, C. Galiotis, A. N. Grigorenko, G. Konstantatos, A. Kis, M. Katsnelson, L. Vandersypen, A. Loiseau, V. Morandi, D. Neumaier, E. Treossi, V. Pellegrini, M. Polini, A. Tredicucci, G. M. Williams, B. H. Hong, J.-H. Ahn, J. M. Kim, H. Zirath, B. J. van Wees, H. van der Zant, L. Occhipinti, A. Di Matteo, I. A. Kinloch, T. Seyller, E. Quesnel, X. Feng, K. Teo, N. Rupesinghe, P. Hakonen, S. R. T. Neil, Q. Tannock, T. Lofwander and J. Kinaret, Nanoscale, 2015, 7, 4598–4810 RSC.
- A. Bianco, Angew. Chem., Int. Ed., 2013, 52, 4986–4997 CrossRef CAS PubMed.
- Y. Chang, S.-T. Yang, J.-H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- K. Sarkar and P. P. Kundu, Int. J. Biol. Macromol., 2012, 51, 859–867 CrossRef CAS PubMed.
- A. Janaszewska, K. Mączyńska, G. Matuszko, D. Appelhans, B. Voit, B. Klajnert and M. Bryszewska, New J. Chem., 2012, 36, 428–437 RSC.
- T. C. K. Heiden, E. Dengler, W. J. Kao, W. Heideman and R. E. Peterson, Toxicol. Appl. Pharmacol., 2007, 225, 70–79 CrossRef PubMed.
- H. Wang, H.-B. Shi and S.-K. Yin, Exp. Ther. Med., 2011, 2, 777–781 CAS.
- N. S. Templeton, Gene and Cell Therapy: Therapeutic Mechanisms and Strategies, Revised and Expanded, CRC Press, 2003 Search PubMed.
- W. Tang and M. L. Becker, Chem. Soc. Rev., 2014, 43, 7013–7039 RSC.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 CrossRef CAS PubMed.
- K. Sarkar, A. Chatterjee, G. Chakraborti and P. P. Kundu, Carbohydr. Polym., 2013, 98, 596–606 CrossRef CAS PubMed.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- P. Cayot and G. Tainturier, Anal. Biochem., 1997, 249, 184–200 CrossRef CAS PubMed.
- K. Sarkar, R. Srivastava, U. Chatterji and P. Kundu, J. Appl. Polym. Sci., 2011, 121, 2239–2249 CrossRef CAS PubMed.
- K. Sarkar, M. Debnath and P. P. Kundu, Carbohydr. Polym., 2013, 92, 2048–2057 CrossRef CAS PubMed.
- K. Zhang, Y. Zhang and S. Wang, Sci. Rep., 2013, 3, 3448 Search PubMed.
- R. Jin, X. Ji, Y. Yang, H. Wang and A. Cao, ACS Appl. Mater. Interfaces, 2013, 5, 7181–7189 CAS.
- M. Naebe, J. Wang, A. Amini, H. Khayyam, N. Hameed, L. H. Li, Y. Chen and B. Fox, Sci. Rep., 2014, 4, 4375 Search PubMed.
- J. Li, C.-y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426–8430 RSC.
- A. Kaniyoor, T. T. Baby and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 8467–8469 RSC.
- L. G. Cançado, A. Jorio, E. M. Ferreira, F. Stavale, C. Achete, R. Capaz, M. Moutinho, A. Lombardo, T. Kulmala and A. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
- L. Wei, F. Wu, D. Shi, C. Hu, X. Li, W. Yuan, J. Wang, J. Zhao, H. Geng and H. Wei, Sci. Rep., 2013, 3, 2636 Search PubMed.
- P. O. Vardevanyan, A. P. Antonyan, M. A. Parsadanyan, M. A. Shahinyan, L. A. Hambardzumyan, M. A. Torosyan and A. T. Karapetian, J. Braz. Chem. Soc., 2012, 23, 2016–2020 CrossRef CAS PubMed.
- K. Sarkar and P. Kundu, Carbohydr. Polym., 2013, 98, 495–504 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- A. F. Adler and K. W. Leong, Nano Today, 2010, 5, 553–569 CrossRef CAS PubMed.
- S. Some, A. R. Gwon, E. Hwang, G.-h. Bahn, Y. Yoon, Y. Kim, S.-H. Kim, S. Bak, J. Yang, D.-G. Jo and H. Lee, Sci. Rep., 2014, 4, 6314 CrossRef CAS PubMed.
- A. M. Jastrzębska, P. Kurtycz and A. R. Olszyna, J. Nanopart. Res., 2012, 14, 1–21 CrossRef PubMed.
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS.
- X. Liu, D. Ma, H. Tang, L. Tan, Q. Xie, Y. Zhang, M. Ma and S. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 8173–8183 CAS.
- A. El-Sayed and H. Harashima, Mol. Ther., 2013, 21, 1118–1130 CrossRef CAS PubMed.
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Pharmacol. Rev., 2006, 58, 32–45 CrossRef CAS PubMed.
- X. Sun, V. K. Yau, B. J. Briggs and G. R. Whittaker, Virology, 2005, 338, 53–60 CrossRef CAS PubMed.
- L. Pelkmans, Biochim. Biophys. Acta, Mol. Cell Res., 2005, 1746, 295–304 CrossRef CAS PubMed.
- M. Koivusalo, C. Welch, H. Hayashi, C. C. Scott, M. Kim, T. Alexander, N. Touret, K. M. Hahn and S. Grinstein, J. Cell Biol., 2010, 188, 547–563 CrossRef CAS PubMed.
- U. Langel, Cell-penetrating peptides: processes and applications, CRC press, 2010 Search PubMed.
- J. Rejman, V. Oberle, I. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- F. Zhi, H. Dong, X. Jia, W. Guo, H. Lu, Y. Yang, H. Ju, X. Zhang and Y. Hu, PLoS One, 2013, 8, e60034 CAS.
- K. von Gersdorff, N. N. Sanders, R. Vandenbroucke, S. C. De Smedt, E. Wagner and M. Ogris, Mol. Ther., 2006, 14, 745–753 CrossRef CAS PubMed.
- J. Rejman, A. Bragonzi and M. Conese, Mol. Ther., 2005, 12, 468–474 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07004j |
| This journal is © The Royal Society of Chemistry 2015 |