
Structural feature evolution – from fluids to the solid phase – and crystal morphology study of LASSBio 1601: a cyclohexyl-N-acylhydrazone derivative†
Fanny Nascimento Costaa,
Tiago F. da Silvabc,
Eduardo Miguez B. Silvad,
Regina C. R. Barrosoe,
Delson Brazf,
Eliezer J. Barreirobc,
Lídia Moreira Limabc,
Francesco Punzog and
Fabio Furlan Ferreira *a
*a
aCentro de Ciências Naturais e Humanas (CCNH), Universidade Federal do ABC (UFABC), Av. dos Estados, 5001, Santo André, SP 09210-580, Brazil. E-mail: fabio.furlan@ufabc.edu.br
bLASSBio, Institute of Biomedical Sciences, Federal University of Rio de Janeiro (UFRJ), Av. Carlos Chagas Filho, 373, Rio de Janeiro, RJ 21941-902, Brazil
cGraduate Program of Chemistry, Institute of Chemistry, Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
dMacromolecules Institute (IMA), Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
ePhysics Institute, State University of Rio de Janeiro (UERJ), Rio de Janeiro, RJ, Brazil
fProgram of Nuclear Engineering (PEN/COPPE), Federal University of Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
gDipartimento di Scienze del Farmaco, Sezione Chimica, Università degli Studi di Catania, Catania 95125, Italy
First published on 27th April 2015
Abstract
LASSBio-1601, a cyclohexyl-N-acylhydrazone derivative, was synthesized as part of a research program to develop a series of anti-inflammatory and analgesic compounds. A complete knowledge of the structure, including stereochemistry, is essential to lead optimization in drug discovery. In this work different techniques were used to obtain detailed information on the evolution of the structural characteristics of this compound from fluids to the solid state, in order to shed some light on the conformation of the molecule in different physical states. By quoting the aforementioned structural analysis, a crystal morphology prediction, compared with the experimentally inferred SEM images, has been performed to analyze potentially alternative routes useful for pharmaceutical tableting.
Introduction
Several approaches have been used for the discovery of new drug prototypes and for this purpose Medicinal Chemistry has developed different strategies to design new hits. One of these approaches consists in designing new molecules on the basis of some already validated drugs and with this aim researchers have gathered efforts to develop new bioactive compounds.1–3 An important step in the fulfillment of this task is the identification of the pharmacophore group, a structural framework that comprises stereoelectronic properties and three-dimensional characteristics necessary to the complexation of ligands to a specific target receptor4 and, consequently, to initiate a set of activities. An example of such group may be found in the N-acylhydrazone (NAH) moiety, already shown to be related to a series of biological activities such as analgesic, anti-inflammatory, platelet aggregation inhibition activities,5–7 cardiovascular actions,8–10 among others.In this context, LASSBio-1601 was planned and synthesized as part of a research program to develop a series of compounds with anti-inflammatory activities by structural modification of the LASSBio-294 prototype.9–11
The chemistry of N-acylhydrazones has been the subject of much interest in recent years and many papers and reviews concerning the use of hydrazone derivatives in different areas have been published.12–15 Due to the concomitant presence of amide and imine functions, NAH compounds may exist as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double-bond stereoisomers (E/Z) and as syn/anti-periplanar conformers about the amide CO–NH bond.16,17 A complete knowledge of the structure, including stereochemistry, is essential to lead optimization in drug discovery. For this reason, in this work, different techniques as well as an in silico study were used to better understand the conformational and configurational aspects of the LASSBio-1601 compound. The X-ray powder diffraction analysis, coupled with several other different techniques, such as NMR, HPLC and GC-MS analyses, was particularly important to confirm the geometric configuration and other conformational aspects of LASSBio-1601 in solid phase as well as in fluids.
N double-bond stereoisomers (E/Z) and as syn/anti-periplanar conformers about the amide CO–NH bond.16,17 A complete knowledge of the structure, including stereochemistry, is essential to lead optimization in drug discovery. For this reason, in this work, different techniques as well as an in silico study were used to better understand the conformational and configurational aspects of the LASSBio-1601 compound. The X-ray powder diffraction analysis, coupled with several other different techniques, such as NMR, HPLC and GC-MS analyses, was particularly important to confirm the geometric configuration and other conformational aspects of LASSBio-1601 in solid phase as well as in fluids.
Moreover, we carried out a detailed crystal morphology prediction on the basis of our structural findings. In fact, pharmaceutical industries are interested in the opportunity to predict crystal morphologies, including crystal habits as well as particles size, shape, flow characteristics, density, hygroscopicity, compressibility and compaction, in order to understand and characterize the physical and chemical properties of drugs, excipients, and their powder mixtures, aiming the optimization of their use in the solid dosage forms.18
It is well known that the crystal habit as well as a noncrystalline amorphous form may affect the drug stability, dissolution rate, flow and mechanical properties, bioavailability, and their ability to mix with excipients.19 Mechanical properties such as flowability, miscibility, particle strength, and cohesiveness, mainly due to the surface free energy (Esurf) effect that results in particle aggregation, often vary among different polymorphs, crystal shapes, or habits.20–25 Moreover, the amorphous form of a drug has the lowest melting point and usually the fastest dissolution rate, but it is most likely to react or degrade.19 Hence, identifying the desired form to design a reliable process for its consistent production could be critical for a successful drug development.26,27
Experimental section
Material
The compound (E)-N′-benzylidenecyclohexanecarbohydrazide, herein referred to as LASSBio-1601, was synthesized in the Laboratory of Evaluation and Synthesis of Bioactive Substances – LASSBio®, as part of a strategy to develop a series of cyclohexyl-N-acylhydrazone derivatives with pharmacological properties. The compound was planned on the basis of principles of molecular simplification and classical bioisosterism of LASSBio-294, a cardioactive derivative.11The pharmacophoric character of the N-acylhydrazone (NAH), which seems to contribute to the desired pharmacological activities, was preserved.28,29 Fig. 1 shows the rational design of LASSBio-1601. Initially, the structural planning of this compound involved the molecular simplification30 of LASSBio-294 derivative replacing the 1,3-benzodioxole ring by the cyclohexyl group.31 This replacement was planned in order to simplify the structure of the original compound and to verify the eventual effect to desired bioactivities. We performed the replacement of the methylenedioxyphenyl ring of the original compound (LASSBio-294) by a cyclohexyl one improving the hydrophobicity of this new NAH derivative (LASSBio-1601). The presence of the cyclohexyl group also provides high conformational freedom to this compound. Subsequently, the classical bioisosterism was employed, thus replacing the thiophenyl by a phenyl ring.
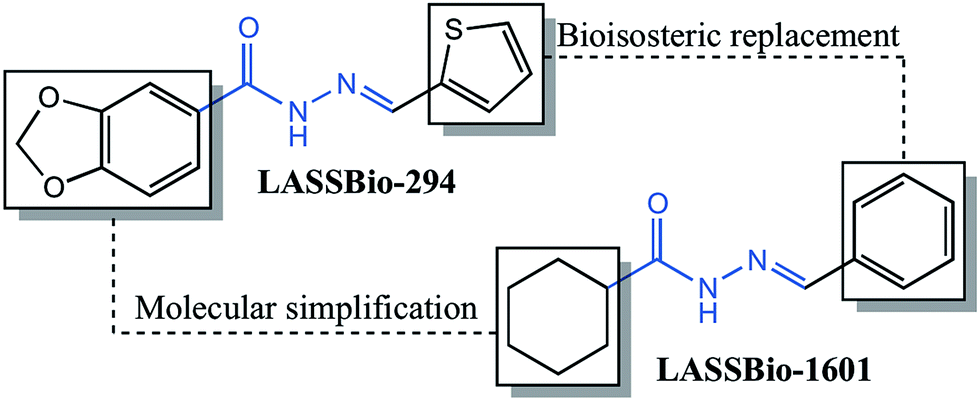 | ||
| Fig. 1 Conceptual design of cyclohexyl-N-acylhydrazone derivative: LASSBio-1601. | ||
The cyclohexyl-N-acylhydrazone compound (LASSBio-1601), was obtained by two synthetic steps in good yields. The first synthesis step consisted in the hydrazinolysis of methyl cyclohexanecarboxalate followed by condensation with thiophene-2-carbaldehyde. LASSBio-1601 was obtained as a white solid in 99% yield and m.p. 163–165 °C in full agreement with previous reports.32 Scheme 1 describes the synthesis steps of LASSBio-1601.
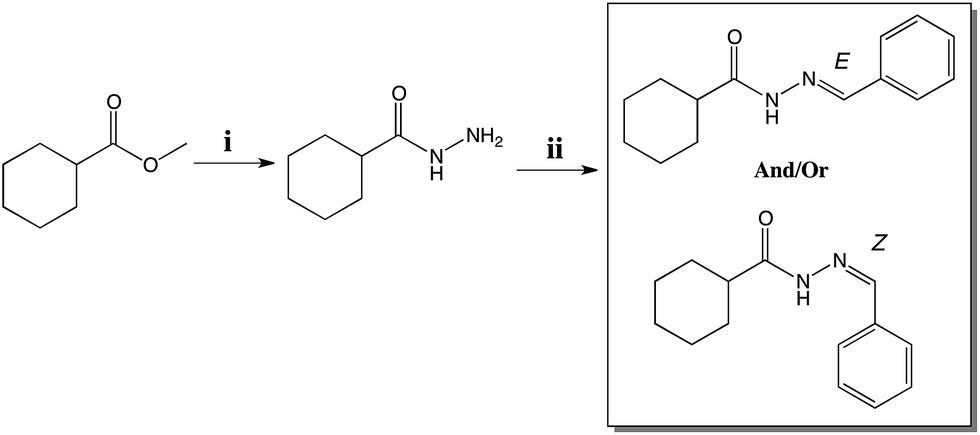 | ||
| Scheme 1 Synthesis of LASSBio-1601. Reagents and conditions: (i) NH2 NH2·H2O 64%, EtOH, reflux, 4 h, 79% (ii) benzaldehyde, EtOH, HCl (cat.), room temperature, 91 min, 99% yield. | ||
However, according to Palla and coworkers,16,17 due to the condensation step of hydrazides and aromatic aldehydes, NAH compounds may exist as CN double-bond stereoisomers (E/Z) and as syn/anti-periplanar conformers about the amide CO–NH bond.
X-ray powder diffraction
The sample was gently hand-grinded in an agate mortar, in order to get a fine powder suitable for X-ray powder diffraction analysis, and packed between two 0.014 mm thick cellulose-acetate foils in a sample holder that was held spinning during data collection.X-ray powder diffraction data were collected at room temperature on a STADI-P powder diffractometer (Stoe®, Darmstadt, Germany) in transmission geometry by using a Kα1 (λ = 1.54056 Å) wavelength emitted by a Cu anode and selected by a curved Ge (111) crystal, with a tube voltage of 40 kV and a current of 40 mA. The diffracted intensities were collected by a silicon microstrip detector, Mythen 1K (Dectris®, Baden, Switzerland), in the range from 2° to 60°, with step sizes of 0.015° and 60 s of integration time at each 1.05°.
Nuclear magnetic resonance (NMR)
NMR spectra were determined in deuterated chloroform or dimethyl sulfoxide containing ca. 1% tetramethylsilane as an internal standard, using a 200/50 MHz Bruker DPX-200, 400/100 MHz Varian 400-Mr, 300/75 MHz Varian Mercury VX and 500/125 MHz Varian VNMRS-500 spectrometers. NMR 1H (200 MHz, DMSO, TMS) δ (ppm): 11.28/11.08 (s, 1H, CONH–), 8.18/7.98 (s, 1H, HCN), 7.66 (m, 2H, H9/H13), 7.43 (m, 3H, H10/H11/H12), 3.10/2.19 (m, 1H), 1.72 (m, 5H, cyclohexyl eq), 1.32 (m, 5H, cyclohexyl ax). NMR 13C [50 MHz, DMSO, TMS] δ [ppm]: 176.92/171.68 (NCO), 145.90/142.28 (HCN), 135.72 (1C), 129.79 (1C), 128.84 (2C), 126.89 (2C), 43.88 (1C), 29.01 (2C), 25.66 (3C). IR [εmax, KBr] ε (cm−1): 3177 (NH); 2927 (CH3); 1663 (CO); 1610 (CN). Anal. calc. for C14H18N2O: C, 63.92; H, 6.56; N, 8.28. Found: C, 63.54; H, 6.34; N, 8.28.
Reversed-phase high-performance liquid chromatography (HPLC)
The HPLC experiment was conducted using Kromasil 100-5C18 (4.6 mm 6250 mm) and a Shimadzu SPD-M20A detector at 254 nm wavelength. The solvent system for HPLC purity analyses was 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 acetonitrile:phosphate buffer solution at pH = 7. The isocratic HPLC mode was used, and the flow rate was 1.0 mL min−1.
:30 acetonitrile:phosphate buffer solution at pH = 7. The isocratic HPLC mode was used, and the flow rate was 1.0 mL min−1.
Gas chromatography-mass spectrometry
GC-MS analyses were recorded on a Shimadzu QP5000 GC/MS instrument (Shimadzu Co., Kyoto, Japan) (5% phenylmethyl silicone column, 30 m × 0.25 mm ID, 0.25 μm film thickness: programmed column temperature from 110 °C to 290 °C, 5 °C min−1). Injector and interface temperatures were set for 300 and 280 °C, respectively.In silico studies
The evaluation of the molecular modeling of LASSBio-1601 was performed in Spartan 8.0 program.33 The geometry evaluations were performed by molecular mechanics force field (Merck Molecular Force Field – MMFF),34 semi-empirical and ab initio simulations. Subsequently, the optimized geometries were used ab initio (Hartree–Fock; 6-31G*) to proceed calculating solvation energy, charges and energies of orbitals.The study of Overhauser effect (NOE) was performed by using the MSpin program.35 For this end the most stable conformers of LASSBio-1601 (E configuration with respect to the CN bond) and its correspondent diastereoisomer Z were designed on the Spartan 8.0 program. With this data, the NOE calculation was performed and correlated with the experimental results of LASSBio-1601.
Crystal morphology prediction
The crystal morphology predictions were performed using a preliminary equilibration protocol, by means of the Discover module included in the Material Studio 7.0 package of Accelrys©, adopting the molecular mechanics approximation and the Compass Force Field (FF).36 The cif file containing all the crystallographic information was used as the input for an energy minimization, carried out with the Smart Minimizer method. The morphology protocol itself is based on the GM method. For this purpose, the calculations were performed allowing a minimum interplanar distance (dhkl) of 0.800 Å without setting any limit neither to the values of the three Miller indices nor to the overall number of growing faces.Hartman37 and Perdock,38,39 in order to account for the energy involved in the process related to the definition of a list of potential grown faces and their ability and tendency to grow, introduced the periodic bond chain (PBC) theory, giving rise to the first methodological attempt to rationalize the influence of the energy related to the interactions between growth units. As a consequence, it was possible to give rise to the classification of crystal faces on the basis of the number of PBC belonging to each given adjacent hkl layer – a slice – while the crystallization energy is considered a constant for a given crystal. According to the literature40–42 there are two main contributions to the crystallization energy, (Ecr): the energy of each slice (Eslice), i.e. the energy resulting from the lateral interaction of each formula unit within a slice and the attachment energy (Eatt) related to the energy released as a consequence of the vertical interaction of the formula unit with an underlying slice.43 This is summarized as:
| Ecr = Eslice + Eatt | (1) |
It was demonstrated39 that
| Gr ∝ Eatt | (2) |
The search for possible solvent accessible voids was performed using the VOID algorithm44 setting a grid of 0.20 Å and a probe radius of 1.20 Å.
Results and discussion
In order to evaluate this hypothesis, 1H and 13C nuclear magnetic resonance (NMR) spectroscopy were used. Concerning the structural characterization, the 1H and 13C-NMR spectra revealed the duplication of some signals (Fig. 2 and Fig. 3, respectively), leading to the supposition of a mixture of stereoisomers or conformers.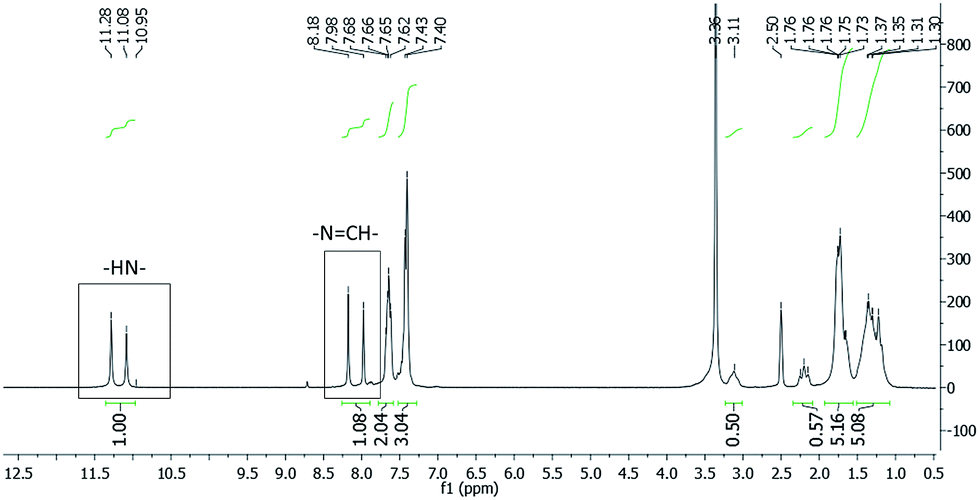 | ||
| Fig. 2 1H-NMR spectra of LASSBio-1601 (200 MHz, DMSO-d6/TMS). The imine hydrogen (–NCH–) and the hydrogen atom attached to nitrogen amide (–NH–) are indicated. | ||
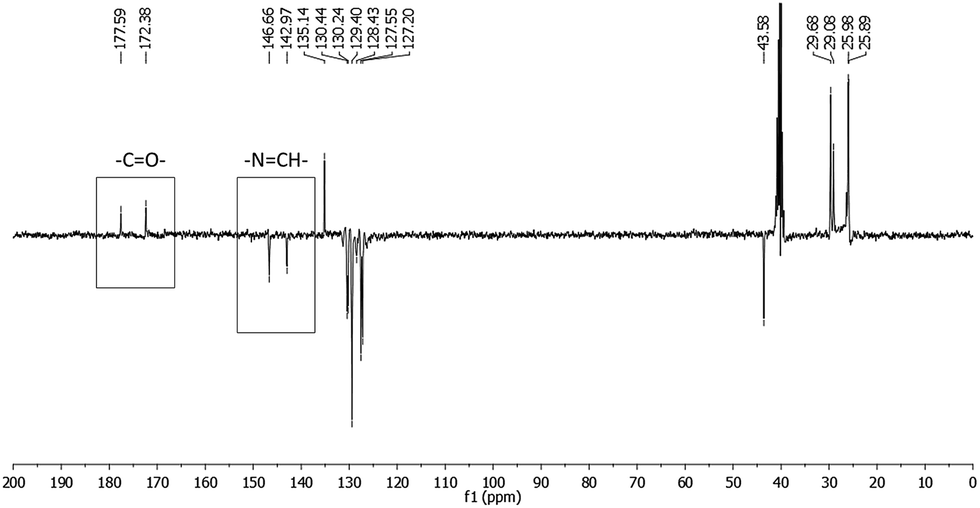 | ||
| Fig. 3 13C-NMR spectra (APT) of LASSBio-1601 (75 MHz, DMSO-d6/TMS). The carbon of carbonyl group (–CO–) and carbon of imide group (–NCH–) are indicated. | ||
The 1H-NMR experiment was performed using two solvents with different polarities – dimethyl sulfoxide-d6 (DMSO-d6) and deuterated chloroform (CDCl3) (ESI†). The relative polarities of both DMSO and chloroform are, respectively, 0.444 and 0.259. These values are dimensionless, meaning that DMSO solvent exhibits 44.4% of the solvent polarity of water and chloroform solvent exhibits 25.9%.45
Due to the polarity of the solvent, a change of the signal integration ratio of the duplicated NCH peaks in the 1H-NMR spectrum was observed. When the CDCl3 solvent was used in place of DMSO-d6 the ratio between the pair of duplicated signals changed from 54/46 (equivalent) to 14/86. This fact suggests an isomeric interconversion at room temperature. Thus an in silico study using the SPARTAN 08 program33 was performed in order to evaluate the imine double bond rotational barrier. A higher energy barrier (61 kcal mol−1) was observed. This value is almost three times higher than the one cited in the literature for a compound that featured this interconversion.46 Furthermore, only one compound was detected in the reversed-phase high-performance liquid chromatography (HPLC) experiment, indicating the presence of only one diastereoisomer with purity of 98.90%. Thus, all these results suggested the presence of conformers.
Considering these conformers we carried out a 1H-NMR experiment in DMSO-d6 at 90 °C (see ESI†). This experiment showed the coalescence of signals, previously duplicated. The reversibility of these changes was verified when the experimental temperature was returned to 25 °C. This result is in agreement with the literature47 and reinforces the assumption of the conformers presence.
Another approach used to evaluate the presence of the two conformers was the synthesis of LASSBio-1782, by reacting cyclohexanecarbohydrazide with acetophenone in order to eliminate the possibility of formation of E/Z stereoisomers.16,17 Since LASSBio-1782 has a symmetrical plane, E/Z geometric isomers about the imide double bond could not exist. Thus, we carried out a 1H-NMR experiment (Fig. 4) to verify this assumption. As a result we observed the duplication of signals concerning the hydrogen attached to nitrogen amide (–NH–: 9.73 and 8.81 ppm). This latter chemical shift can be due to an additional anisotropic effect caused by the aromatic rings. This result once again supported the presence of conformers.
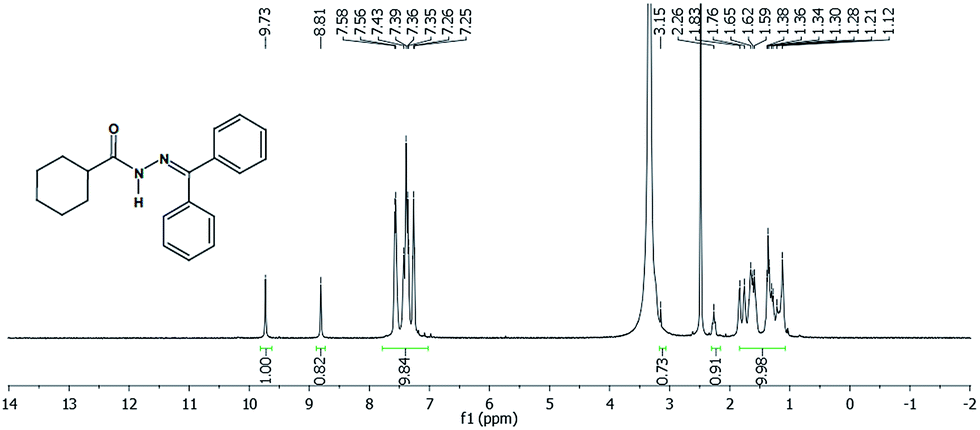 | ||
| Fig. 4 LASSBio-1782 1H-NMR spectra (300 MHz, DMSO-d6/TMS). | ||
A cyclohexyl ring is nonplanar and conformationally flexible and may exist in chair or non-chair conformations (e.g. twist boat).48 Thus, to evaluate the possibility of chair conformations of the cyclohexane ring of LASSBio-1601, a complementary parameter related to hydrogen atoms of the cyclohexyl chain was observed (1H-NMR experiment at 500 MHz) – the three-bond coupling constant (3J). Thus, the duplicated signals related to the hydrogen atom bonded to a tertiary carbon of the cyclohexane ring (Ht) and its coupling (illustrated in ESI†) were observed. According to the literature, the 3J coupling constant between two axial hydrogen atoms (1Haxial–2Haxial or 1Haxial–6Haxial) of cyclohexane derivatives is normally found between 8 to 14 Hz, whereas 3J couplings between axial–equatorial or equatorial–equatorial hydrogen atoms of cyclohexane derivatives are normally found from 1 to 6 Hz.49,50 The value of the 3J coupling constant was found to be about 11 Hz for LASSBio-1601, for both duplicated signals related to Ht. This result allowed us to suggest that the duplication is coming from the amide conformers in solution, once the duplicated signals related to Ht are attributed only to the axial position. Thus, it can be inferred that the duplication of signals at 1H-NMR is not due to chair conformers.
In order to assign the different signals and to analyze the conformation of the cyclohexane ring we collected the 1H gradient correlation spectroscopy (gCOSY) spectrum at 500 MHz. The signal from the equatorial hydrogen atoms would be deshielded, by an anisotropic effect typical of cyclohexane, due to the proximity of the carbonyl of the NAH scaffold correlated with the respective geminal hydrogen by gCOSY. The axial hydrogen atoms would be more blocked, with larger width, W, due to the presence of the coupling J3Jaxial–axial (∼11 Hz), also geminally correlated (Fig. 5). These pieces of information well match with previously reported data.51
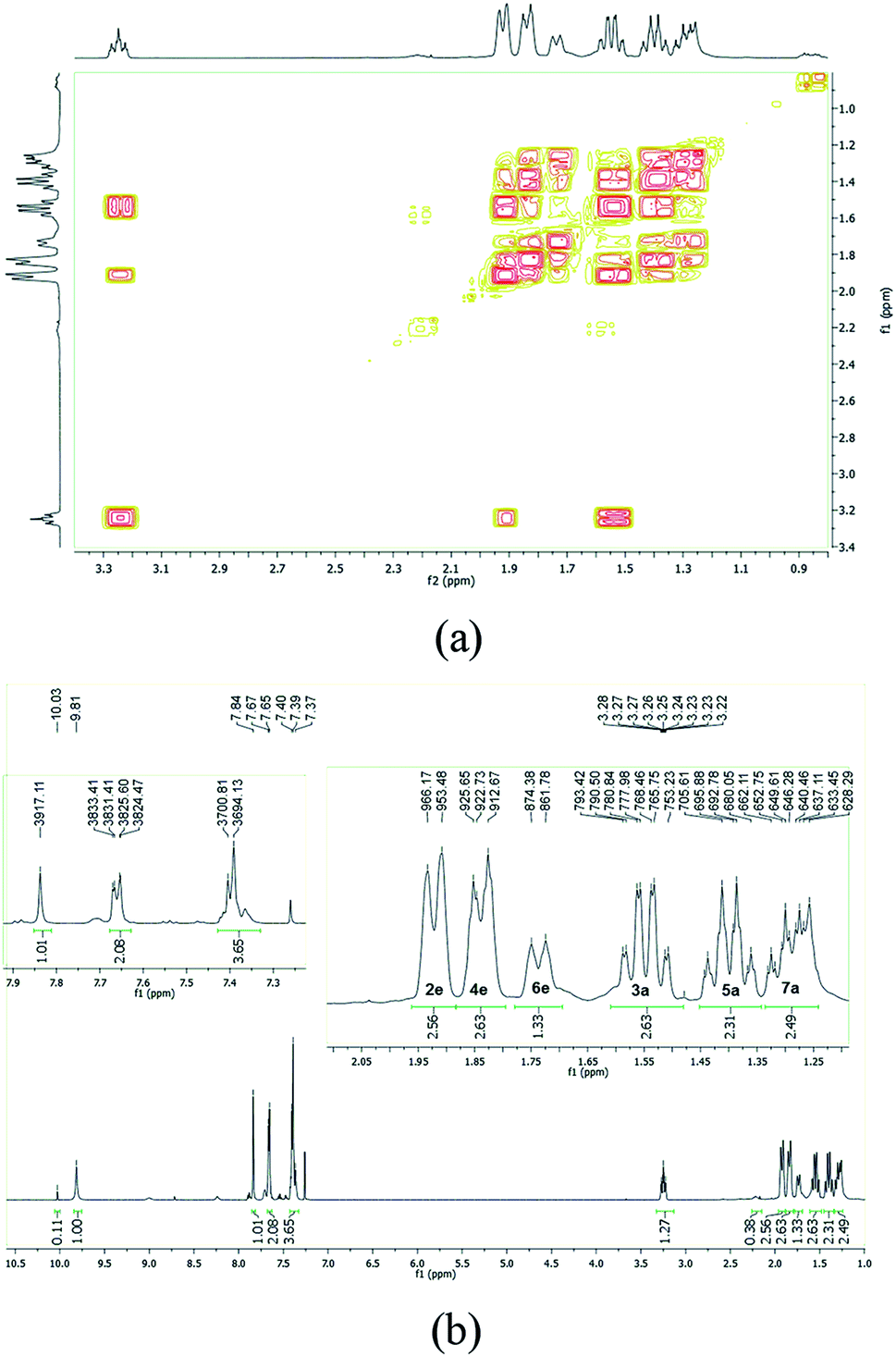 | ||
| Fig. 5 (a) LASSBio-1601 1H gCOSY NMR spectrum (500 MHz, CDCl3) displaying the correlation between cyclohexyl hydrogen atoms that have couplings relations among themselves (500 MHz, CDCl3). (b) The assignment of 1H-NMR signals of the hydrogen atoms of cyclohexyl chain. The labels 2e, 4e, 6e correspond to equatorial hydrogen atoms and the axial hydrogen atoms are indicated by 3a, 5a and 7a. | ||
The last hypothesis to be investigated is the one in which the duplication of the hydrogen signals would be due to the presence of syn/anti-amide conformers. Thus, to accomplish this task, geometry optimization and conformational analyses of LASSBio-1601 were performed by means of molecular modeling using once again the SPARTAN program33 parameterized to solvation through SM8 function. The five more stable conformers (Fig. S5 in ESI†) in different solvents (vacuum, DMSO and water) were selected. Considering a Boltzmann distribution, the syn/anti-amide conformer ratio in the system were calculated and correlated with the 1H-NMR experiment (see Table 1). The results showed that in vacuum predominates the syn amide conformation (>90%). When the evaluation was conducted using DMSO as solvent a trend to conformational equilibrium was observed with 58.6% of syn and 36.6% of anti-conformer. This result is very similar to the values measured in the aforementioned 1H-NMR solvent induced experiment. However, when the polarity of the system increases, using water as solvent, the anti-amide conformation (65%) predominates thereby demonstrating the influence of polarity on the system. This behaviour is interpreted, since the atoms of the NAH scaffold are more available for interactions, on the basis of the formation of hydrogen bonds with water molecules, in the anti-conformation. Thus, the higher polarity of the solvent, the more favored will be the anti-conformer of LASSBio-1601 on conformational equilibrium, thus confirming the landscape experimentally observed in the 1H-NMR spectrum.
| Amide rotamers | Δrel. E (kcal mol−1) | Boltzmann dist (%) |
|---|---|---|
| Vacuum | ||
| syn | 0.00 | 93.2 |
| syn | 1.66 | 5.6 |
| syn | 3.00 | 0.6 |
| anti | 3.08 | 0.5 |
| anti | 5.02 | 0.1 |
|
||
| DMSO | ||
| syn | 0.00 | 58.6 |
| anti | 0.28 | 36.6 |
| syn | 2.01 | 2.0 |
| anti | 2.27 | 1.3 |
| anti | 2.52 | 0.8 |
|
||
| Water | ||
| anti | −2.45 | 65.5 |
| syn | −1.87 | 24.5 |
| anti | −1.10 | 6.7 |
| anti | −0.29 | 1.7 |
| syn | 0.00 | 1.0 |
As these results suggested the presence of amide conformers in solution, we decided to proceed with differential nuclear Overhauser effect (differential NOE) experiments to evaluate the relative configuration of the imine double bond to better understand the pharmacodynamic profile. When the imine hydrogen was irradiated it was possible to assess the spatial proximity between amide and imine hydrogen, which complies with a relative E configuration. Due to the high conformational freedom of NAH function this result was checked and compared with the one arisen from an in silico methodology. Calculations using the Mspin 1.2 software35 were performed to simulate the NOE effect at a radius of 5 Å around the irradiated hydrogen. For this simulation, more stable E and Z stereoisomers, obtained through a process of geometric optimization (Hartree–Fock 6-31G*) by molecular mechanics followed by conformational analysis using the SPARTAN 8.0 software,33 were selected. The theoretical results for imine hydrogen irradiation showed the preferential intensity increase of the signal related to the amide hydrogen (∼31%) followed by an intensity increase of the signal related to hydrogen, which is in the ortho position of the aromatic ring (∼19%) (Fig. 6). These results demonstrate the spatial proximity between the amide and imine hydrogen of LASSBio-1601, which suggests the relative configuration E.
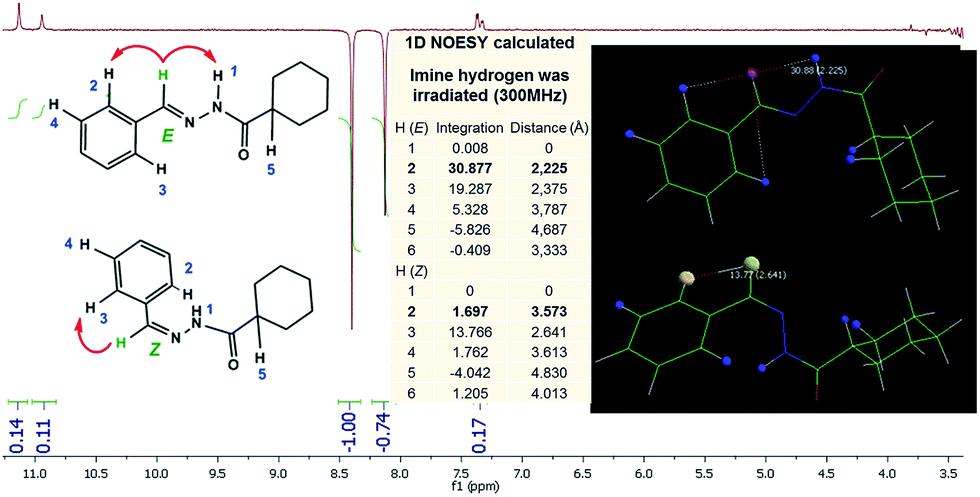 | ||
| Fig. 6 LASSBio-1601 NOEdiff spectra for irradiation of imine hydrogen (300 MHz; DMSO-d6/TMS) and LASSBio-1601 NOEdiff theoretical (300 MHz; 5 Å; Mspin 1.2). | ||
In order to confirm the last outcome a mass spectrometry analysis was performed. Using gas chromatography-mass spectrometry, Pereira and coworkers52 indicated that pirazolic-N-acylhydrazones derivatives present relative configuration E of imine double bond (CN) since a molecular fragment was observed as result of McLafferty rearrangement.53 Likewise, LASSBio-1601 was evaluated by mass spectrometry (ESI†). As a result of this evaluation it was identified a fragment consistent with the McLafferty rearrangement (m/z = 127) indicating the relative configuration E for this compound. There is therefore a perfect agreement between the LASSBio-1601 configuration in liquid and in the gas phase.
Although with the previous experiments we could infer the structural properties of LASSBio-1601, the crystallography can unambiguously determine the relative configuration and evidenced conformational attributes in solid phase. The single crystal X-ray diffraction is the most powerful approach for the determination of structural information at the atomic level. Nevertheless, it is important to recall that the requirement for a single crystal sample of appropriate size and quality imposes a natural limitation on the scope of this technique. An attempt to use the single-crystal X-ray diffraction methodology was performed but it failed since we could not obtain a single crystal of LASSBio-1601 with adequate qualities. Due to the fact that polycrystalline forms are detected by X-ray diffraction, the powder diffraction has played a central role in structural physics, chemistry and materials science.54 Every crystalline solid phase has a unique X-ray powder diffraction (XRPD) pattern, which can form the basis for its identification and structure determination.55,56 In this case, the most direct approach is to use X-ray powder diffraction data.57 The availability of reliable procedures for determining crystal structures directly from X-ray powder diffraction data has led to significant advances in the scope and power of techniques in this field.58–62 In this work we used it for unequivocally elucidate the crystal structure of LASSBio-1601.
X-ray powder diffraction data and a simulated annealing algorithm implemented into the DASH software63 were used to determine the crystal structure of LASSBio-1601, on the basis of previous procedures.64–67 The hydrogen atoms were introduced into calculated positions considering the orbital geometry using the Mercury software program68 and their Biso values were constrained to be 1.2 times the value of the respective atoms to which they are connected. Their factional coordinates were refined taking into account a macro implemented in Topas-Academic that “rides” (restrains the distances between hydrogen and neighbouring atoms) the H atoms to the ones they are associated. Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin 14.10.27.0, 2014, ChemAxon (http://www.chemaxon.com). Thus, a Rietveld refinement69,70 of the final crystal structure was conducted using the Topas-Academic v.5 (ref. 71) software program providing a satisfactory fit as shown in Fig. 7.
 | ||
| Fig. 7 Plot from the final Rietveld refinement. Black crosses represent observed data, the red line indicates the calculated pattern and the blue line represents the difference between the observed and calculated patterns. Magenta vertical bars at the bottom indicate Bragg reflections. | ||
LASSBio-1601 crystallized in the triclinic space group (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), with unit cell dimensions a = 4.7774(3) Å, b = 10.8468(6) Å, c = 15.7986(11) Å, α = 52.351(5)°, β = 99.385(5)°, γ = 99.707(3)°, V = 637.03(8) Å3. The goodness of fit indicator and R-factors were, respectively: 1.115, Rexp = 2.866%, Rwp = 3.197% and RBragg = 0.537%. The crystal structure of LASSBio-1601 is comprised by two formula units per unit cell (Z = 2), accommodating one molecule in the asymmetric unit (Z′ = 1), as represented in Fig. 8.
), with unit cell dimensions a = 4.7774(3) Å, b = 10.8468(6) Å, c = 15.7986(11) Å, α = 52.351(5)°, β = 99.385(5)°, γ = 99.707(3)°, V = 637.03(8) Å3. The goodness of fit indicator and R-factors were, respectively: 1.115, Rexp = 2.866%, Rwp = 3.197% and RBragg = 0.537%. The crystal structure of LASSBio-1601 is comprised by two formula units per unit cell (Z = 2), accommodating one molecule in the asymmetric unit (Z′ = 1), as represented in Fig. 8.
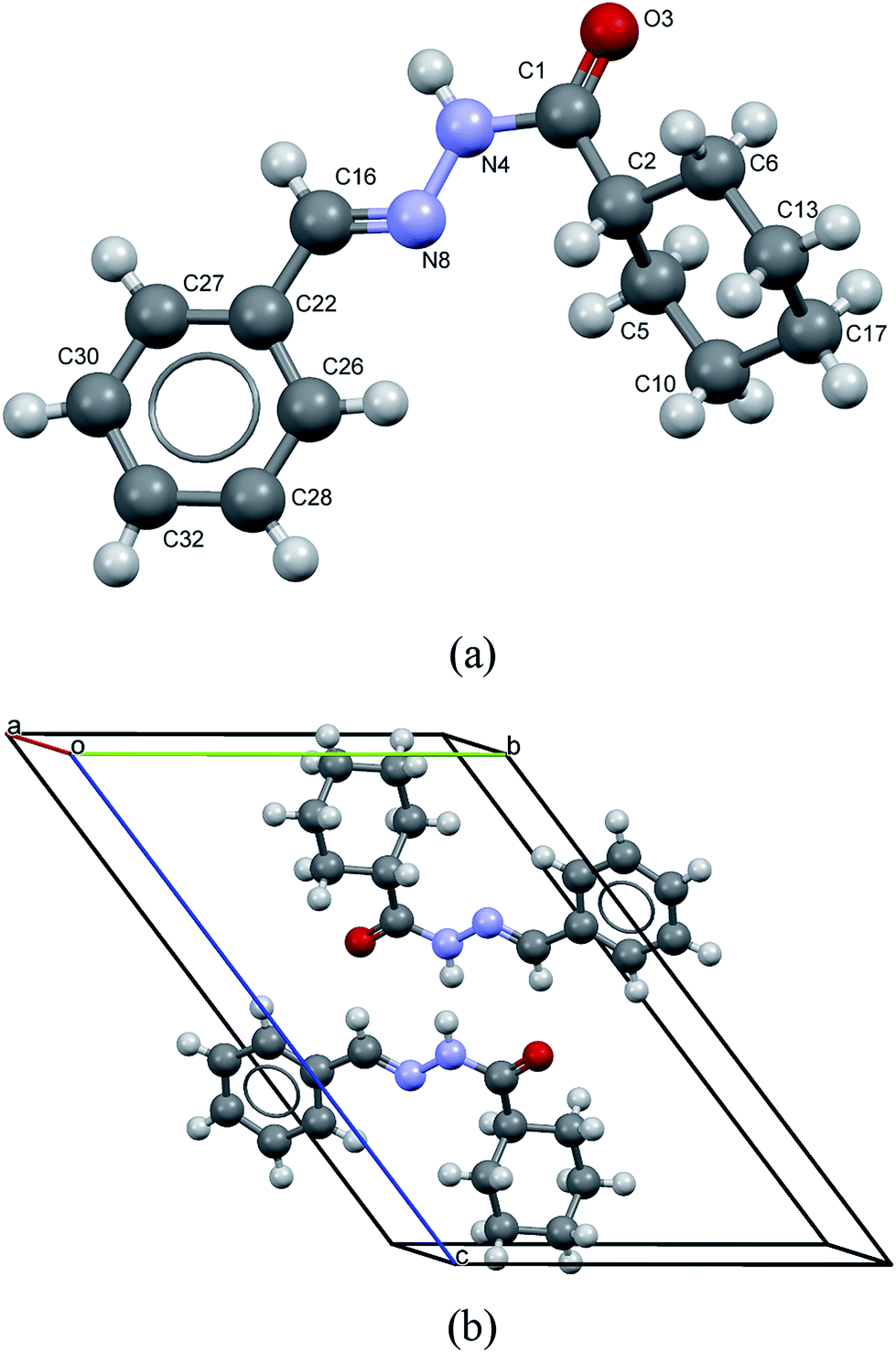 | ||
| Fig. 8 (a) View of molecule of LASSBio-1601 showing the non-hydrogen atoms labeling. (b) Packing of molecules in the crystal. LASSBio-1601 is comprised by two formula units per unit cell. | ||
Short hydrogen bonds between atoms N(4)–H(9)⋯O(3) (D⋯A = 2.94(3) Å and D–H⋯A = 163(2)°; where “D” and “A” are, respectively, hydrogen donor and acceptor) as represented by cyan lines in Fig. 9, seem to contribute to build a network of molecular aggregates. The possible complementary π-stacking interactions between N-acylhydrazone scaffold with phenyl ring (C(26)–N(8) = 3.66(2) Å) also are shown in Fig. 9 (represented by green line). These interactions contribute for the organization of the space arrangement in the unit cell. The geometry of the crystal structure was validated by means of a Mogul check. All bond distances and angles are in accordance to the corresponding values found in the CSD database.72 Also, in order to check the consistency of the obtained results for space group choice, unit cell parameters, bond distances, angles and torsions, the PLATON software program was used.73 The ADDSYM routine did not detect any obvious extra crystallographic symmetry. Crystal data as well as details of the structure determination are displayed in Table 2.
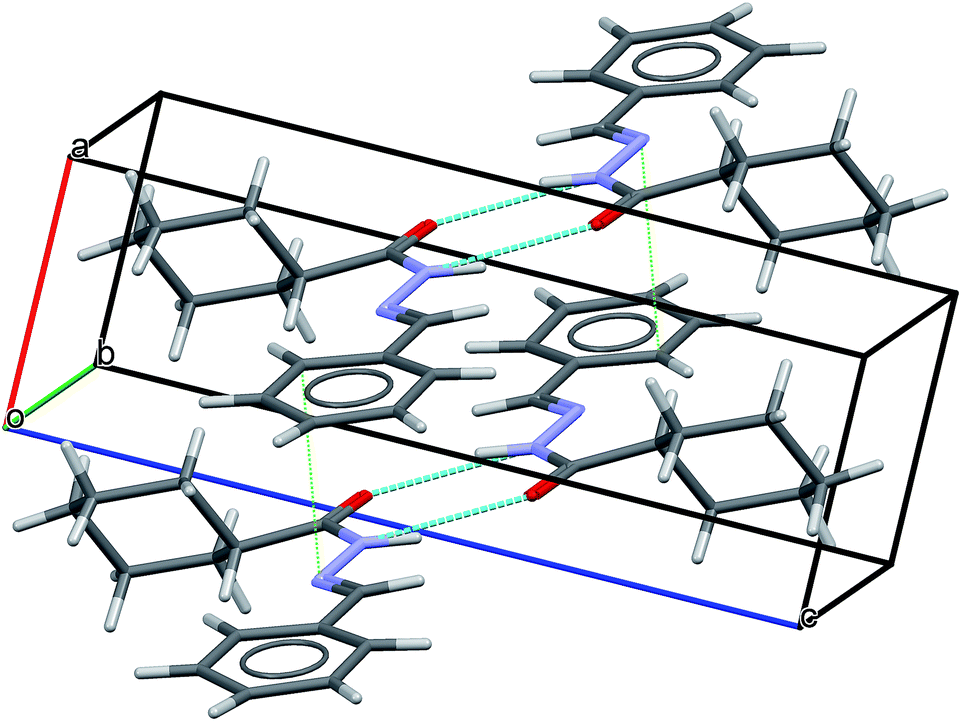 | ||
| Fig. 9 LASSBio-1601 arrangement displaying intermolecular hydrogen bonds (cyan lines) as well as possible complementary π-stacking interactions (green dotted lines). | ||
| Chemical formula | C14H18N2O |
| Formula weight (g mol−1) | 230.31 |
| Crystal system | Triclinic |
| Space group | P (no. 2) |
| a, b, c (Å) | 4.7774(3), 10.8468(6), 15.7986(11) |
| α, β, γ (°) | 52.351(5), 99.385(5), 99.707(3) |
| Volume (Å3) | α, β, γ (°) |
| Z, Z′ | 2, 1 |
| ρcalc (g cm−3) | 1.2007(2) |
| μ (cm−1) | 6.03 |
| T (K) | 298 |
|
|
| Data collection | |
| Diffractometer | STADI P |
| Monochromator | Ge(111) |
| Wavelength (Å) | 1.54056 |
| 2θ range (°) | 2–60 |
| Step size (°) | 1.05 |
| Time per step (s) | 60 |
|
|
| Refinement | |
| Number of data points | 3990 |
| Number of contributing reflections | 410 |
| Number of restraints | 41 |
| Number of refined parameters | 124 |
| Rexp (%) | 2.866 |
| Rwp (%) | 3.197 |
| RBragg (%) | 0.537 |
| χ2 | 1.105 |
The structural determination of LASSBio-1601 compound was conducted validating the previous investigations about relative configuration of imine double bond and conformation properties. It was determined the relative configuration E about the imine double bond. Furthermore, as described previously, the preference for chair conformer attached on the equatorial position was identified. The preference for syn amide conformer in solid-phase was also evidenced. Finally, the unit cell obtained allowed us to observe a hydrogen bond interaction between the carbonyl oxygen and the amide nitrogen and the possible π-stacking interaction of phenyl groups of LASSBio-1601 molecules.
These findings are in very good agreement with the structural features highlighted in solution thus evidencing no relevant changes in the molecular geometry passing from gas to liquid to solid phase.
The scanning electron microscopy (SEM) image was obtained in order to analyse the habit of the formed crystals. This experiment was conducted using a JSM-6010LA microscope, from JEOL® (Tokyo, Japan), located at the Multiuser Experimental Center of Federal University of ABC (CEM/UFABC). Fig. 10 shows the acquired image of some LASSBio-1601 crystals, exhibiting a needle-like shape and relatively smooth surfaces. In general, the crystals have thicknesses up to 5 μm and longer lengths.
 | ||
| Fig. 10 Scanning electron microscopy image of some LASSBio-1601 crystals exhibiting needle-like shapes and relatively smooth surfaces. | ||
A crystal morphology prediction has been carried out as described in the Experimental section, and its results were directly compared with the above reported images inferred by means of SEM. As reported in Fig. 11 and Table 3, the so resulted crystal habit is in a very good agreement with the experimental morphology.
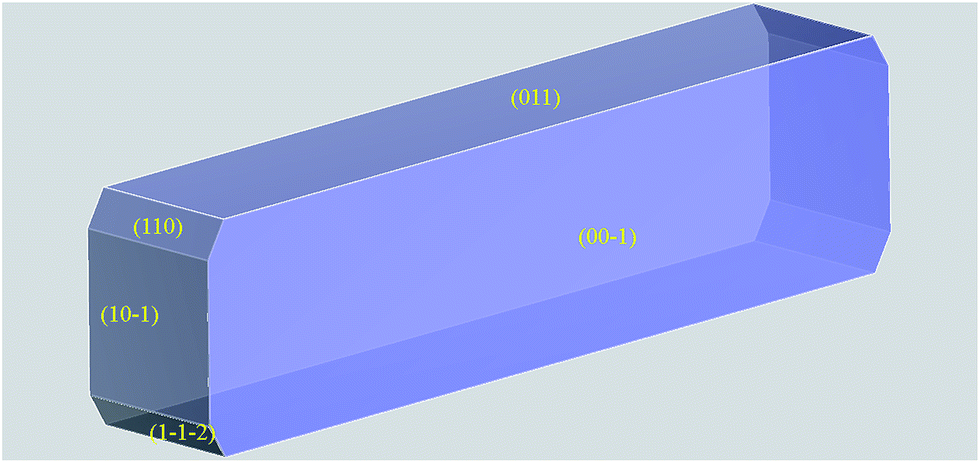 | ||
| Fig. 11 Crystal morphology prediction of LASSBio-1601. Miller indices of the MI faces are reported. (00) MI face is reported for geometrical reasons; please bear in mind that it is symmetry related to (001) one reported in Table 3. | ||
| hkl | Multiplicity | dhkl (Å) | Eatt total (kcal mol−1) | Total facet area (%) |
|---|---|---|---|---|
| (001) | 2 | 12.4718 | −10.6159 | 46.88 |
| (011) | 2 | 10.6302 | −12.5256 | 38.16 |
| (10) |
2 | 4.5099 | −34.3542 | 10.23 |
| (110) | 2 | 3.9706 | −33.4848 | 2.55 |
(1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) ) |
2 | 4.3602 | −38.8629 | 2.19 |
By using the GM approach, we take into account the energetics in the system, i.e. Eatt. It is on this basis that we can interpret the apparently inconsistent and swapped TFA percentages for (110) and (1): the higher (1) Eatt values are in fact counterbalanced by the greater interplanar distance, dhkl. This effect would have been probably underestimated by a pure geometrical approach, such as the BFDH one.74
As reported in the Experimental section the calculations were performed in vacuo while the “real” crystals were grown interacting with solvents. For this reason, we decided to perform an analysis of the potential influence of the crystallization solvents over the final crystal shape. This study will give us key information on one hand about the reasons why some MI faces could be even overgrown or underestimated with respect to the calculation; on the other hand, it will provide useful hints about the solubility of these crystals.75–78
For this purpose, in Fig. 12 we analyzed the results of the computed morphology, drawing the hkl plane corresponding to the MI crystal faces reported in the habit, simply taking into account which kind of groups are outcropping from the considered face. The structure shows a high packing index (67.3%)79 and no residual solvent accessible voids. As a consequence we can assume LASSBio-1601 is unable to host solvents without modifying its crystal structure. As a consequence, the attachments of solvent molecules must take place directly from the outer part of the considered faces. This is true whatever crystallization mechanism is considered, neither the classical nucleation theory nor the two-step mechanism.80,81
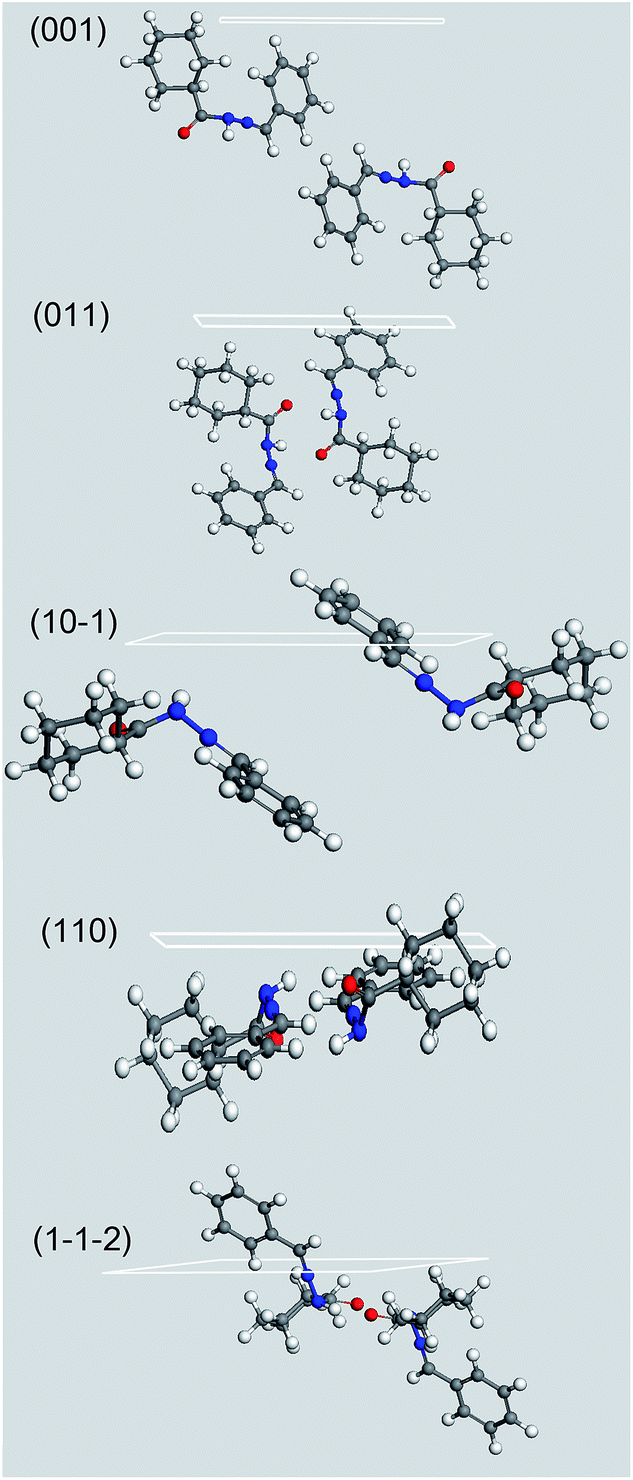 | ||
| Fig. 12 Crystal morphology prediction of LASSBio-1601. Miller indices of the MI faces are reported. (00) MI face is reported for geometrical reasons; please bear in mind that it is symmetry related to (001) one reported in Table 3. | ||
It is evident that, whilst (001) and (011) MI faces, which represent more than the 85% of the crystal surface, are populated almost exclusively by non polar groups, the other MI faces show some minor presence of polar functional groups. The growth of these slightly polar MI faces could have been also further hindered by the use of the polar solvents employed during the compound synthesis – hydrazine and ethyl alcohol – thus lending an additional elongation to the crystals to the detriment of their apical growth. This effect would have lent an additional anisotropicity to the crystal, magnifying the needle like shape of the crystal. On the other hand, if needed, the choice of a slightly non-polar environment could have produced the opposite effect. This could therefore be key information for alternative routes to a successful and stable pharmaceutical tableting.82
Conclusions
In this work, different experimental approaches revealed the nature of duplicated signals observed in NMR spectra of LASSBio-1601 compound is due to the presence of syn/anti-amide conformers in solution.On the basis of the differential NOE experiment and gas chromatography-mass spectrometry the relative configuration E of the imine double bond was suggested. The crystal structure determination of LASSBio-1601 was conducted, by means of XRPD, validating the previous investigations. Thus, we could unambiguously determine the relative configuration E and evidenced conformational attributes, in solid state. The preference for chair conformer attached on the equatorial position was identified, as well as the preference for syn amide conformer.
The crystalline arrangement allowed us to observe hydrogen bond interactions between the carbonyl oxygen and the amide nitrogen as well as the complementary π-stacking interactions of phenyl groups of LASSBio-1601 molecules, which contribute to the organization of the LASSBio-1601 molecules within the unit cell. The SEM images confirmed the crystal morphology prediction, which in turn allowed us to make suggestions about the possible effect of solvents on the final crystal shape.
The present work highlighted the presence of significant conformation retention for LASSBio-1601 passing from gas to liquid to solid phase. This structural information is not only relevant for LASSBio-1601 tableting, as already specified, but also for a more detailed study of its pharmacological activity.
Acknowledgements
We thank the financial support provided by the São Paulo State Foundation (FAPESP, grant nr. 2008/10537-3), the National Council for Scientific and Technological Development (CNPq, grants no. 305186/2012-4, 477296/2011-4), INCT-INOFAR (BR, #573.564/2008-6), CAPES (grant no. 88881.062195/2014-01), FAPERJ and UFABC.Notes and references
- L. M. Lima and E. J. Barreiro, Curr. Med. Chem., 2005, 12, 23 CrossRef CAS
.
- R. Morphy and Z. Rankovic, J. Med. Chem., 2005, 48, 6523 CrossRef CAS PubMed
- R. Morphy, C. Kay and Z. Rankovic, Drug Discovery Today, 2004, 9, 641 CrossRef CAS
- L. Pol-Fachin, C. A. Fraga, E. J. Barreiro and H. Verli, J. Mol. Graphics Modell., 2010, 28, 446 CrossRef CAS PubMed
- A. C. Cunha, J. M. Figueiredo, J. L. M. Tributino, A. L. P. Miranda, H. C. Castro, R. B. Zingali, C. A. M. Fraga, M. C. l. B. V. de Souza, V. F. Ferreira and E. J. Barreiro, Bioorg. Med. Chem., 2003, 11, 2051 CrossRef CAS
- L. M. Lima, F. S. Frattani, J. L. Dos Santos, H. C. Castro, C. A. Fraga, R. B. Zingali and E. J. Barreiro, Eur. J. Med. Chem., 2008, 43, 348 CrossRef CAS PubMed
- G. A. Silva, L. M. Costa, F. C. Brito, A. L. Miranda, E. J. Barreiro and C. A. Fraga, Bioorg. Med. Chem., 2004, 12, 3149 CrossRef CAS PubMed
- A. G. Silva, G. Zapata-Sudo, A. E. Kummerle, C. A. Fraga, E. J. Barreiro and R. T. Sudo, Bioorg. Med. Chem., 2005, 13, 3431 CrossRef CAS PubMed
- R. T. Sudo, G. Zapata-Sudo and E. J. Barreiro, Br. J. Pharmacol., 2001, 134, 603 CrossRef CAS PubMed
- G. Zapata-Sudo, R. T. Sudo, P. A. Maronas, G. L. M. Silva, O. R. Moreira, M. I. S. Aguiar and E. J. Barreiro, Eur. J. Pharmacol., 2003, 470, 79 CrossRef CAS
- H. Gonzalez-Serratos, R. Chang, E. F. Pereira, N. G. Castro, Y. Aracava, P. A. Melo, P. C. Lima, C. A. Fraga, E. J. Barreiro and E. X. Albuquerque, J. Pharmacol. Exp. Ther., 2001, 299, 558 CAS
- A. E. Kümmerle, J. M. Raimundo, C. M. Leal, G. S. da Silva, T. L. Balliano, M. A. Pereira, C. A. de Simone, R. T. Sudo, G. Zapata-Sudo, C. A. Fraga and E. J. Barreiro, Eur. J. Med. Chem., 2009, 44, 4004 CrossRef PubMed
- C. D. Duarte, E. J. Barreiro and C. A. M. Fraga, Mini-Rev. Med. Chem., 2007, 7, 1108 CrossRef CAS
- L. Mazur, K. N. Jarzembska, R. Kamiński, K. Woźniak, E. Pindelska and M. Zielińska-Pisklak, Cryst. Growth Des., 2014, 14, 2263 CAS
- A. B. Lopes, E. Miguez, A. E. Kümmerle, V. M. Rumjanek, C. A. M. Fraga and E. J. Barreiro, Molecules, 2013, 18, 11683 CrossRef PubMed
- G. Palla, C. Pelizzi, G. Predieri and C. Vignali, Gazz. Chim. Ital., 1982, 112, 339 CAS
- G. Palla, G. Predieri, P. Domiano, C. Vignali and W. Turner, Tetrahedron, 1986, 42, 3649 CrossRef CAS
- M. H. Shariare, F. J. Leusen, M. de Matas, P. York and J. Anwar, Pharm. Res., 2012, 29, 319 CrossRef CAS PubMed
- N. J. Babu and A. Nangia, Cryst. Growth Des., 2011, 11, 2662 CAS
- H. G. Brittain, J. Pharm. Sci., 2012, 101, 464 CrossRef CAS PubMed
- S.
K. Niazi, Handbook of Pharmaceutical Manufacturing Formulations: Compressed Solid Products, InformaUK Ltda, London, 2nd edn, 2009 Search PubMed
- S. C. Gad, Pharmaceutical manufacturing handbook: production and processes, John Wiley & Sons, Inc, New jersey and Canada, 2008 Search PubMed
- S. X. Yin and J. A. Grosso, Curr. Opin. Drug Discovery Dev., 2008, 11, 771 CAS
- D. S. S. Coombes, C. R. A. Catlow, J. D. Gale, M. J. Hardy and M. R. Saunders, J. Pharm. Sci., 2002, 91, 1652 CrossRef CAS PubMed
- N. Rasenack and B. W. Müller, Int. J. Pharm., 2002, 244, 45 CrossRef CAS
- N. Variankaval, A. S. Cote and M. F. Doherty, AIChE J., 2008, 54, 1682 CrossRef CAS PubMed
- M. Müller, U. Meier, D. Wieckhusen, R. Beck, S. Pfeffer-Hennig and R. Schneeberger, Cryst. Growth Des., 2006, 6, 946 Search PubMed
- H. J. C. Bezerra-Netto, D. I. Lacerda, A. L. P. Miranda, H. M. Alves, E. J. Barreiro and C. A. M. Fraga, Bioorg. Med. Chem., 2006, 14, 7924 CrossRef CAS PubMed
- P. C. Lima, L. M. Lima, K. C. da Silva, P. H. Leda, A. L. de Miranda, C. A. Fraga and E. J. Barreiro, Eur. J. Med. Chem., 2000, 35, 187 CrossRef CAS
- E. J. Barreiro, Quim. Nova, 2002, 25, 1172 CrossRef PubMed
- E. J. Barreiro, C. A. M. Fraga, A. L. P. Miranda and C. R. Rodrigues, Quim. Nova, 2002, 25, 129 CrossRef CAS PubMed
- S. Olsen and E.-M. Enkemeyer, Chem. Ber., 1948, 81, 359 CrossRef CAS PubMed
- I. Wavefunction, Irvine, CA, USA.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490 CrossRef CAS
- MestreSpin, Mestrelab Research, Santiago de Compostela, A Coruña, Spain.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338 CrossRef CAS
- P. Hartman, J. Cryst. Growth, 1980, 49, 166 CrossRef CAS
- P. Hartman and P. Bennema, J. Cryst. Growth, 1980, 49, 145 CrossRef CAS
- P. Hartman and W. G. Perdok, Acta Crystallogr., 1955, 8, 49 CrossRef
- R. Docherty, G. Clydesdale, K. J. Roberts and P. Bennema, J. Phys. D: Appl. Phys., 1991, 24, 89 CrossRef CAS
- F. Punzo, Cryst. Growth Des., 2011, 11, 3512 CAS
- F. Punzo, J. Mol. Struct., 2013, 1032, 147 CrossRef CAS PubMed
- Z. Berkovitch-Yellin, J. Am. Chem. Soc., 1985, 107, 8239 CrossRef CAS
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, WILEY-VCH Verlag GmbH & Co, Marburg, Germany, 3rd edn, 2003 Search PubMed
- E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of organic compounds, Wiley-Interscience, New York, USA, 1994 Search PubMed
- E. Wyrzykiewicz and D. Prukała, J. Heterocycl. Chem., 1998, 35, 381 CrossRef CAS PubMed
- J. A. Hirsch, in Topics in Stereochemistry, ed. N. L. Allinger and E. L. Eliel, John Wiley & Sons, Inc., New York, USA, 1967, vol. 1, ch. 199, pp. 199–222 Search PubMed
- E. A. Basso, P. R. Oliveira, J. Caetano and I. T. A. Schuquel, J. Braz. Chem. Soc., 2001, 12, 215 CrossRef CAS PubMed
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. A. Vyvyan, Introduction to spectroscopy, Brooks/Cole-Thomson Learning, New York, USA, 2001 Search PubMed
- S. E. Biali and I. Columbus, J. Org. Chem., 1993, 58, 7029 CrossRef
- A. S. Pereira, F. A. Violante, F. R. Aquino Neto, J. N. Cardoso, C. A. M. Fraga and E. J. Barreiro, Anal. Lett., 1998, 31, 719 CrossRef CAS PubMed
- F. W. McLafferty, Anal. Chem., 1956, 28, 303 CrossRef
- A. K. Cheetham, Structure determination from powder diffraction data: an overview, ed. W. I. F. David, K. Shankland, L. B. McCusker and C. Bärlocher, International Union of Crystallography/Oxford Science Publications, New York, 1st edn, 2006 Search PubMed
- A. W. Hull, J. Am. Chem. Soc., 1919, 41, 1168 CrossRef CAS
- J. W. Shell, J. Pharm. Sci., 1963, 52, 24 CrossRef CAS PubMed
- K. D. M. Harris and E. Y. Cheung, Chem. Soc. Rev., 2004, 33, 526 RSC
- A. K. Cheetham, Structure determination from powder diffraction data: an overview, International Union of Crystallography/Oxford Science Publications, New York, 1st edn, 2006 Search PubMed
- A. J. Florence, N. Shankland, K. Shankland, W. I. F. David, E. Pidcock, X. L. Xu, A. Johnston, A. R. Kennedy, P. J. Cox, J. S. O. Evans, G. Steele, S. D. Cosgrove and C. S. Frampton, J. Appl. Crystallogr., 2005, 38, 249 CrossRef CAS
- K. D. M. Harris, M. Tremayne and B. M. Kariuki, Angew. Chem., Int. Ed., 2001, 40, 1626 CrossRef CAS
- J. I. Langford and D. Louër, Rep. Prog. Phys., 1996, 59, 131 CrossRef CAS
- A. Menden, Croat. Chem. Acta, 1998, 71, 615 Search PubMed
- W. I. F. David, K. Shankland, J. van de Streek, E. Pidcock, W. D. S. Motherwell and J. C. Cole, J. Appl. Crystallogr., 2006, 39, 910 CrossRef CAS
- F. N. Costa, D. Braz, F. F. Ferreira, T. F. da Silva, E. J. Barreiro, L. M. Lima, M. V. Colaço, L. Kuplich and R. C. Barroso, Radiat. Phys. Chem., 2014, 95, 292 CrossRef CAS PubMed
- F. N. Costa, F. F. Ferreira, T. F. da Silva, E. J. Barreiro, L. M. Lima, D. Braz and R. C. Barroso, Powder Diffr., 2013, 28, S491 CrossRef CAS
- F. F. Ferreira, S. G. Antonio, P. C. P. Rosa and C. O. Paiva-Santos, J. Pharm. Sci., 2010, 99, 1734 CAS
- F. F. Ferreira, A. C. Trindade, S. G. Antonio and C. O. Paiva-Santos, CrystEngComm, 2011, 13, 5474 RSC
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41 Search PubMed
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151 CrossRef CAS
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS
- A. A. Coelho, J. Evans, I. Evans, A. Kern and S. Parsons, Powder Diffr., 2011, 26, s22 CrossRef CAS
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS
- J. A. P. Sato, F. N. Costa, M. D. da Rocha, E. J. Barreiro, C. A. M. Fraga, F. Punzo and F. F. Ferreira, CrystEngComm, 2015, 17, 165 RSC
- A. R. Lazo Fraga, F. F. Ferreira, G. M. Lombardo and F. Punzo, J. Mol. Struct., 2013, 1047, 1 CrossRef CAS PubMed
- G. M. Lombardo, A. Rescifina, U. Chiacchio, A. Bacchi and F. Punzo, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 172 CAS
- G. Li Destri, A. Marrazzo, A. Rescifina and F. Punzo, J. Pharm. Sci., 2013, 102, 73 CrossRef CAS PubMed
- G. Li Destri, A. Marrazzo, A. Rescifina and F. Punzo, J. Pharm. Sci., 2011, 100, 4896 CrossRef CAS PubMed
- A. I. Kitaigorodsky, Molecular Crystals and Molecules, ed. E. M. Loebl, Academic Press, Inc, New-York, 1973 Search PubMed
- P. G. Vekilov, Cryst. Growth Des., 2007, 7, 2796 CAS
- P. G. Vekilov, Cryst. Growth Des., 2010, 10, 5007 CAS
- V. Waknis, E. Chu, R. Schlam, A. Sidorenko, S. Badawy, S. Yin and A. S. Narang, Pharm. Res., 2014, 31, 160 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Some 1H-NMR spectra, high performance liquid chromatography, in silico study of LASSBio-1601 conformers distribution in different solvents, mass spectrum and tables for atomic structural parameters as well as some selected bond lengths and bond angles. CCDC 1040619. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra02696b |
| This journal is © The Royal Society of Chemistry 2015 |