
Electrochemical response of nitrite and nitric oxide on graphene oxide nanoparticles doped with Prussian blue (PB) and Fe2O3 nanoparticles
Abstract
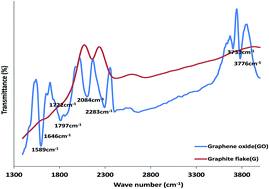
Electrocatalytic behaviour of graphene oxide (GO), iron(III) oxide (Fe2O3) and Prussian blue (PB) nanoparticles and their nanocomposite towards nitrite (NO2−) and nitric oxide (NO) oxidation in neutral and acidic media respectively was investigated on a platinum (Pt) modified electrode. Successful synthesis of these nano materials was confirmed using microscopic and spectroscopic techniques. Successful modification of the electrode was confirmed using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The results showed that the Pt–GO–Fe2O3 and Pt–GO–PB nanocomposite modified electrodes gave a faster electron transfer process in both a 5 mM Ferri/Ferro ([Fe(CN)6]3−/4−) redox probe and 0.1 M phosphate buffer solution (PBS). The Pt–GO–Fe2O3 and Pt–GO–PB electrodes also gave an enhanced NO2− and NO oxidation current compared with bare Pt and the other electrodes studied. Electrocatalytic oxidation of the analyte occurred through a simple diffusion process but were characterized with some level of adsorption. Tafel slopes b of 468.4, 305.2 mV dec−1 (NO2−, NO); and 311.5, 277.2 mV dec−1 (NO2−, NO) were obtained for the analyte at the Pt–GO–Fe2O3 and Pt–GO–PB electrode respectively. The Pt–GO–Fe2O3 limit of detection and sensitivity in NO2− and NO are 6.60 μM (0.0084 μA μM−1) and 13.04 μM (0.0160 μA μM−1) respectively, while those of the Pt–GO–PB electrode are 16.58 μM (0.0093 μA μM−1) and 16.50 μM (0.0091 μA μM−1). The LoD compared favourably with literature reported values. Pt–GO–Fe2O3 gave a better performance to NO2− and NO electrooxidation, good resistance to electrode fouling, a higher catalytic rate constant and lower limit of detection. The adsorption equilibrium constant β and the standard free energy change ΔG0 due to adsorption are 10.29 × 103 M−1 (−22.89 kJ mol−1) and 3.26 × 103 M−1 (−20.04 kJ mol−1) for nitrite and nitric oxide respectively at the Pt–GO–Fe2O3 electrode. An interference study has also been reported. The fabricated sensors are easy to prepare, cost effective and can be applied for real sample analysis of nitrite and nitric oxide in food, water, biological and environmental samples.
 Please wait while we load your content...
Please wait while we load your content...

