
Gallic acid: a versatile antioxidant with promising therapeutic and industrial applications
Abstract
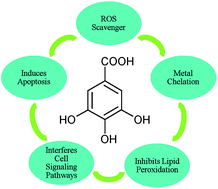
Oxidative stress, a result of an overproduction and accumulation of free radicals, is the leading cause of several degenerative diseases such as cancer, atherosclerosis, cardiovascular diseases, ageing and inflammatory diseases. Polyphenols form an important class of naturally occurring antioxidants, having innumerable biological activities such as anticancer, antifungal, antibacterial, antiviral, antiulcer and anticholesterol, to name a few. Among various polyphenols, gallic acid (3,4,5-trihydroxybenzoic acid), a naturally occurring low molecular weight triphenolic compound, has emerged as a strong antioxidant and an efficient apoptosis inducing agent. Starting from the bioavailability and the biosynthetic pathway of gallic acid, this review includes various in vitro, in vivo and in silico studies providing the mode of action, radical scavenging activity, ability to inhibit lipid peroxidation, maintenance of endogenous defense systems and metal ion chelation by this triphenolic molecule, along with a comprehensive overview of factors responsible for its high antioxidant activity. Gallic acid derivatives have also been found in a number of phytomedicines with diverse biological and pharmacological activities, including radical scavenging, interfering with the cell signaling pathways and apoptosis of cancer cells. The diverse range of applications of this simple polyphenol is due to a fine amalgam between its antioxidant and prooxidant potential. The existing literature on this dual behavior of gallic acid and its derivatives is reviewed here. This is followed by an account of their potential clinical and industrial applications.
- This article is part of the themed collection: Chemistry for Medicine: Special Collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

