
Calorimetric evaluation indicates that lignin conversion to advanced biofuels is vital to improving energy yields†
Abstract
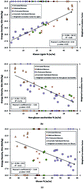
Energy density measurements using bomb calorimetry were applied along with mass yields to calculate energy yields from combinations of individual processes and lignocellulosic feedstocks. Sample preparation and the calorimetric method were fine-tuned for the biofuel process pathway prior to measuring the energy density of liquid fuels and catalysts and solid biomass types (untreated, pelletized, pretreated, and enzymatically hydrolyzed). To statistically establish the method, correlations between biomass composition and energy densities were tested. Strong correlations with lignin, hemicellulose, and ash concentrations were observed and statistically validated (Pearson's coefficient, r = 0.92 and −0.81, respectively). Finally, energy densities were applied along with mass yields on a process pathway including ionic liquid pretreatment (6 L) and saccharification (2 L) of three feedstocks. From switchgrass, eucalyptus, and mixed feedstocks, mass yields of 54.4, 62.0, and 61.7% led to energy yields that were observed to be 59.2, 55.9, and 61.0%, respectively. The disparity in change in mass and energy yields between switchgrass and eucalyptus was identified to have originated from the varied lignin removal during pretreatment. The overall energies recovered from 600 g of switchgrass, eucalyptus, and mixed feedstocks, were 9.8, 10.3, and 10.1 MJ, respectively. Calorimetry can promptly evaluate an integrated multi-process pathway to convert a discrete or mixed feedstock to sugars and other metabolites and eventually to advanced biofuels that can either be hydrocarbons or a mixture thereof. In this particular study, calorimetry and mass yields indicated that lignin removal led to lower energy yield to liquid fuels.
 Please wait while we load your content...
Please wait while we load your content...

