
Polyacrylonitrile/lignin sulfonate blend fiber for low-cost carbon fiber
Abstract
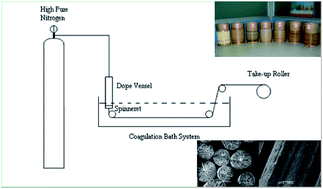
Polyacrylonitrile/lignin sulfonate (PAN/LS) blend fibers were spun via a wet spinning process. The fiber structure, mechanical properties and thermal stability of the precursor fibers were studied by FT-IR, SEM, tensile tester, and TG-DSC. Results indicated that there was no chemical crosslinking between PAN and LS during the process of wet spinning. PAN and LS had good compatibility in the blend fibers. LS could weaken the skin of the blend fibers and reduce the fiber structure defects. The increase of dope concentration could improve the fiber structure and mechanical properties. LS blending with PAN could reduce fiber weight loss in the thermal stabilization process, and most importantly the precursor fibers could be stabilized rapidly without fiber fusion. Through polymer blending and wet spinning, this study provided a promising way to prepare a precursor fiber for carbon fiber.
 Please wait while we load your content...
Please wait while we load your content...

