
Heterogeneous catalysts for advanced bio-fuel production through catalytic biomass pyrolysis vapor upgrading: a review
Masoud Asadieraghi
,
Wan Mohd Ashri Wan Daud
* and
Hazzim F. Abbas
Department of Chemical Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: masoud.asadieraghi@gmail.com; ashri@um.edu.my; hazzim_f@yahoo.com; Fax: +60 10379675371; Tel: +60 172907256
First published on 18th February 2015
Abstract
Nowadays concerns regarding fossil fuel resources depletion as well as environmental issues attributed to CO2 accumulation in the atmosphere force communities toward utilizing biomass as a substitute fuel source which is environmentally secure and renewable. Pyrolysis bio-oil from biomass comprises varieties of undesirable oxygenate and heavy compounds and has to be treated before feeding to bio-refineries. Catalytic biomass pyrolysis vapor upgrading presently seems to be a techno-economical process toward production of fuel-like components. However, selection of stable and productive catalyst(s) to yield desirable chemicals with low coke formation is a great challenge. The three most important classes of catalysts comprising microporous zeolites, mesoporous catalysts and metal based catalysts are utilized for vapor phase bio-oil upgrading. This study offers a comprehensive review on catalytic biomass pyrolysis vapor upgrading by emphasizing particularly on catalyst types and properties, coke formation over catalysts and catalytic process conditions.
 Masoud Asadieraghi | Masoud Asadieraghi got his bachelor degree in Polymer Engineering at University of Petroleum Industry, Iran in 1998 and his master's degree in Chemical Engineering from Shiraz University, Iran in 2001. Then, he joined to Petrochemical Research and Technology Company, Iran in 2002. Since that time till now he is working as senior researcher there. He received his second master's degree in Master of Science Technology and Management (MaSTeM) from Ferrara University, Italy, in collaboration with LyondellBasell, in 2004. Currently, he is doing PhD under Professor Wan Mohd Ashri Wan Daud supervision working on biomass pyrolysis and in situ bio-oil catalytic upgrading to produce bio-fuel and value added chemicals. |
 Wan Mohd Ashri Wan Daud | Professor Wan Mohd Ashri Wan Daud received his bachelor degree in Chemical Engineering at Leeds University, Leeds, UK in 1991 and his master's degree in Chemical Engineering at the University of Sheffield, Sheffield, UK in 1993. He received his PhD degree in Chemical Engineering at the University of Sheffield in 1996. After nine years as an academic and scientist at the Faculty of Engineering, in 2005, he became Professor of Chemical Engineering. Since 2005 till now he has worked as a Professor of Chemical Engineering at the University of Malaya, Malaysia. His research fields include energy, biomass conversion to bio-fuel, catalysts synthesis, polymerization and separation processes, and hydrogen storage. He has more than 90 publications in Web Science journals. |
 Hazzim F. Abbas | Dr Hazzim Abbas received his BSc in Chemical Engineering from the University of Technology, Iraq in 1989 and his master's degree in chemical Engineering from the University of Baghdad, Iraq in 1996. He achieved his PhD degree in Chemical Engineering at the University of Malaya in 2010 under professor Wan Mohd Ashri Wan Daud's supervision. Dr Hazzim Abbas has seven years of industrial experience in the petrochemical industry. In addition, he has more than twelve years of teaching experience in different levels. Also, he has strong publication records in hydrogen production. Currently, he is working as Assistant Professor in Nizwa University, Oman. |
1. Introduction
Sustainable developments of societies in recent decades lead them to the high consumption of natural fossil fuel resources. Biomass is considered as the only available sustainable energy source of organic carbon which can appropriately substitute petroleum to yield carbon based materials, chemicals and fuels.1Pyrolysis process can be utilized to convert lignocellulosic biomass to liquid fuel.2,3 Fast pyrolysis process, which is distinguished by a high heating rate of particles at a short time,4 has recently attracted the broad attentions and can be considered as one of the most capable technologies which are exploited for the conversion of renewable biomass resources to bio-oil.5–8 The bio-oil derived from depolymerization of cellulose, hemicelluloses and lignin, three main building block of lignocellulosic biomass, is a complex mixture of different oxygenated compounds. A typical bio-oil with broad molecular weight range from 18 to 5000 g mol−1 or even more can contain more than 400 different compounds which most of them are oxygenated. Most of bio-oil deficiencies comprising its low heating value, corrosiveness and instability under long storage time and transportation conditions caused by these oxygenated compounds.9–13
Different approaches were employed targeting bio-oil's quality enhancement consisting: reduced pressure distillation,14 pyrolysis under reactive atmosphere,15–17 high pressure thermal treatment,18 hydro-treatment at high pressure,19 pyrolytic lignin removal,20 pyrolysis vapor upgrading at low pressure,21 and conversion of bio-oil's acidic compounds to esters and ketons over acidic22 and basic23 catalysts, respectively.
Bio-oil upgrading through conventional hydro-treating (HDT) at high pressure could accomplish oxygen removal by high hydrogen consumption, but it will fail to minimize carbon loss. Non-condensable undesirable C2–C3 gases instead liquid C6–C14 hydrocarbons (appropriate for fuel applications) will be resulted from the small molecules during HDT process.24
Bio-oils and the model compounds upgrading investigations showed a considerable decrease in product yield as a result of catalyst deactivation and severe tar and char formation during catalytic upgrading.25,26 Park et al.27 carried out investigation on the catalytic upgrading of biomass pyrolysis vapor over HY and HZSM-5 zeolite catalysts in a fixed bed reactor. Their investigation outcomes, which were compared with the data from Vitolo et al.,28 showed that employing biomass as feedstock instead of bio-oil increased upgraded bio-oil yield by 10 wt%.
The catalytic upgrading of the biomass fast pyrolysis vapor is considered as one of the most promising process to produce upgraded bio-oil. Deoxygenation of the produced bio-oil can be achieved in the presence of selected catalysts to enhance bio-oil properties. Investigations are being conducted towards the design of selective catalysts to achieve production of high added value chemicals (e.g. phenol) or minimizing of the formation of undesirable bio-oil components such as acids and carbonyls.29
Three main important oxygenated compounds families available in bio-oil can be characterized as: (1) aldehyde, ketones and acids (like acetone, acetic acid, acetol, etc.); (2) sugar derived compounds (like levoglucson and furfural); and (3) lignin-derived phenolics.24 Different available components in the bio-oil are illustrated in Fig. 1.30
 | ||
| Fig. 1 Bio-oils (derived from lignocellulosic biomass) chemical composition.30 | ||
Deoxygenation of these components suggests a great challenge. Accordingly, it is important to investigate the role of different catalysts play in the conversion of oxygenated compounds to fuel-like hydrocarbons. In this regard, development of highly durable and selective catalysts will be crucial and can be considered as key to the success for bio-oil upgrading processes at atmospheric condition and in the absence of hydrogen feeding.9
Two important targets in the biomass to bio-fuel conversion researches can be; increase the bio-fuel potential to replace petroleum and its cost competitiveness improvement. These two goals could be attained by minimizing hydrogen consumption and carbon loss. Furthermore, improvement of product quality can assist incorporation of upgraded bio-fuel in petroleum refineries or blending operations.
This review summarizes the recent researches and trends in the bio-oil catalytic vapor cracking/upgrading followed by deoxygenation focusing on catalysts properties and reaction conditions to selectively direct reactions toward production of fuel-like components and valuable chemicals.
2. Catalytic biomass pyrolysis vapor upgrading
The produced bio-oil from fast pyrolysis contains various oxygenated compounds that provide shortcomings to be used as transportation fuel. Although, it can be utilized directly for the purpose of heat and electricity generation. The high oxygen content of bio-oils has the negative effect on the energy density (16–19 MJ kg−1 versus 46 MJ kg−1 for conventional gasoline), and it is caused poor stability as well as low volatility of the liquid bio-oil.1,31 Further, the bio-oils high viscosity and corrosiveness discourage their consumption in internal combustion engines.One of the known solutions to stabilize the bio-oil and decrease its oxygen contents is to blend it with the hydro-treating process feed, even though bio-oil transportation and storage before its blending makes significant problems.31,32 Hydro-treatment, which is the bio-oil treatment at high hydrogen pressure (30–140 bar) and moderate temperature, is likely the most common route to the bio-oil compounds deoxygenation (HDO).33–35 In this method, bio-oil is completely deoxygenated and oxygen is removed in the form of water.
HDO is typically carried out in the presence of NiMo and CoMo catalysts.33 It is worthwhile to mention that Pt and Ru metals exhibit higher hydrogenation activity, although they show lower tolerance of sulfur impurities.36,37
The high hydrogen consumption in the bio-oil HDO process is the main drawback of this technology. Further, high pressure process which leads to high operational cost could be considered as the other disadvantage. One of the main challenges of HDO process is to hydrogenate the aliphatic compounds, whilst avoiding reduction of aromatics. This type of hydrogenation process control is difficult to achieve at high hydrogen pressure required for HDO.
Pyrolysis vapor upgrading can alternatively be carried out before vapor being condensate at atmospheric pressure and 350–500 °C, when vapors are passed through catalyst(s). The pyrolysis vapors need to pass certain stabilizing and oxygen removal processes without external hydrogen supply. At these conditions, vapors components undergo a series of reactions comprising, cracking, aromatization and dehydration. Through these reactions, oxygen is removed in the form of water, CO2 and CO. Consequently, bio-oil is converted into a mixture of aromatic and aliphatic hydrocarbons, although a large fraction of organic carbon reacts to create solid carbonaceous deposits over catalyst named coke.6,11,38–40
Fig. 2 shows schematically this type of process. Following to upgrading process, liquid bio-oil intermediate can be fed to refinery, bio-refinery or petrochemical plants for further treatment toward fuels and/or chemicals production.
 | ||
| Fig. 2 Schematic of pyrolyis process and upgrading – highlighting pyrolysis vapor upgrading. | ||
Zeolites are the most known catalysts used for the bio-oil pyrolysis vapor upgrading. Generally, coke formation over zeolite catalysts is one of the main problems for upgrading process. Investigation showed about 30% (maximum) of carbon in the feed can deposit as coke on the zeolite. It is due to low effective hydrogen available in the bio-oil. There are various oxygenated compounds in the bio-oil and therefore highly oxidized feed need to be converted to hydrocarbons. During this conversion, the excess carbon subsequently deposits as coke on the catalyst(s).41
Chen's effective ratio (H/Ceff.), which is defined as (H–2O)/C (H, O and C are hydrogen, oxygen and carbon moles, respectively), indicates the feedstock effective hydrogen. Generally, a low H/Ceff. ratio of feed is conducted to more coke formation than those having higher ratios.41,42
Considering pyrolysis vapor upgrading approach, due to the similar operating pressure at which pyrolysis and upgrading are carried out, these two processes can be integrated. It is contrary to high pressure HDO process which cannot be easily integrated simultaneously with low pressure pyrolysis. Despite all the advantages associated with this type of upgrading process, there are some drawbacks. The yields of hydrocarbons are somehow modest. Another disadvantage of this technology is irreversible deactivation of catalysts, attributed to the partial de-alumination of zeolite structures in the presence of water (usually found in bio-oils). Researches still undergo toward development of acidic catalysts with better resistance against water.43
By employing high reactive oxygenated compounds (carbonyl, hydroxyl, carboxyl, and ketonic groups) in bio-oils, reactions of C–C bonds formations such as aromatization, aldol condensation and ketonization can be carried out. It means oxygen functionalities potentials can be used to yield high carbon content deoxygenated fuel components instead of their elimination too early.24 Through ketonization, two carboxylic acids are condensed into a larger ketone with the release of stoichiometric amounts of water and CO2. Usually inorganic oxide like Al2O3, TiO2, ZrO2 and CeO2, operating at atmospheric pressure and moderate temperature (300–425 °C), are used as catalyst for these types of reactions.44–48
It is worthwhile to note that ketonization through carboxylic acids consumption can lead to oxygen removal in the form of carbon dioxide and water. Acids almost make about 30 wt% (maximum) of bio-oils and their conversion can improve bio-oils properties and mitigate their corrosiveness and chemical instability.1 As a result, ketonization uses the oxygen functionality of acid groups to produce molecules with high heating value and consequently hydrogen consumption is reduced. Furthermore, through ketonization typical bio-oil components like esters can be condensed. Unlike zeolite catalysts, which their activity is sensitive to water presence, this type of upgrading process can be performed under moderate amounts of water.46,49,50
Upon selective catalytic upgrading and deoxygenation of pyrolysis vapor, depending on the catalyst type, biomass composition and process conditions, different products with improved chemical and physical properties can be yielded. Nowadays, various researches are being conducted toward the design and selection of appropriate solid catalysts for production of high added value chemicals (e.g. phenolic compounds) or molecules with enhanced properties to be used as bio-fuel component. Recent catalytic pyrolysis of the lignocellulosic biomass for the phenolic compounds production has employed different catalysts, comprising alkaline catalysts, K3PO4 and activated carbon.51
Table 1 illustrates what can be anticipated for the characteristics and the compositions between raw pyrolysis oil, hydro-deoxygenated oil (HDO), zeolite cracking oil, and benchmarked crude oil.52
| Pyrolysis oil | HDO | Zeolite cracking | Crude oil | |
|---|---|---|---|---|
| Upgraded bio-oil | ||||
| YOil [wt%] | 100 | 21–65 | 12–28 | — |
| YWater phase [wt%] | — | 13–49 | 24–28 | — |
| YGas [wt%] | — | 3–15 | 6–13 | — |
| YCarbon [wt%] | — | 4–26 | 26–39 | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Oil characteristics | ||||
| Water [wt%] | 15–30 | 1.5 | — | 0.1 |
| pH | 2.8–3.8 | 5.8 | — | — |
| ρ [kg L−1] | 1.05–1.25 | 1.2 | — | 0.86 |
| μ50 °C [cP] | 40–100 | 1–5 | — | 180 |
| HHV [MJ kg−1] | 16–19 | 42–45 | 21–36 | 44 |
| C [wt%] | 55–65 | 85–89 | 61–79 | 83–86 |
| O [wt%] | 28–40 | <5 | 13–24 | <1 |
| H [wt%] | 5–7 | 10–14 | 2–8 | 11–14 |
| S [wt%] | <0.05 | <0.005 | — | <4 |
| N [wt%] | <0.4 | — | — | <1 |
| Ash [wt%] | <0.2 | — | — | 0.1 |
| H/C | 0.9–1.5 | 1.3–2.0 | 0.3–1.8 | 1.5–2.0 |
| O/C | 0.3–0.5 | <0.1 | 0.1–0.3 | ∼0 |
Three categories of catalysts including microporous zeolites, mesoporous catalysts and metal based catalysts have recently attracted the considerations of researchers for the biomass pyrolysis vapor upgrading.
2.1. Microporous zeolite catalysts
Many zeolites have multi-dimensional microporous structure. This micro-porous system permits small molecules of reactants to diffuse in to the zeolite structure, therefore provide molecules access to internal acid sites. The microporous nature provides another essential feature to the zeolites, called shape-selectivity. The micro-pore channels size-restraints can somehow control the formation of unwanted products.41The pores are frequently required to produce sufficiently high surface areas necessary for catalyst high activity. According to the IUPAC definition, porous materials are classified in three main groups; microporous (pore size < 2 nm), mesoporous (2–50 nm), and macroporous (>50 nm) materials.43 Wide varieties of reactions could be catalyzed by zeolites attributed to their shape selectivity. The different zeolites pore size varying from 5 Å to 12 Å affects molecules mass transfer.53
Various types of shape selectivity can be identified depending upon if pore size restricts the reacting molecules entrance, or the product molecules departure, or the creation of certain transition conditions. Selectivity of the reactants achieved while among all the reactant molecules only whom which are small enough can diffuse through the catalyst pores. When parts of the products inside the pores are too large to diffuse out, product selectivity occurs. They are either transformed to smaller molecules or in the worst case block the pores and deactivate the catalyst. Restricted transition state selectivity takes place when particular reactions are avoided because the relevant transition state would need bigger space than available in the cavities. Different pore systems may employ to control the molecular traffic. Reactant molecules may favorably diffuse in the catalyst through one pore, while products leave through the other. So, counter diffusion is minimized.54
One of the most important applications of zeolite catalysts is in fluid catalytic cracking (FCC) process. It provides about 45% of the global gasoline pool through the large hydrocarbon cracking into the gasoline range molecules.55 Zeolites are appropriate catalysts for the biomass pyrolysis vapor/bio-oil upgrading due to containing Lewis and Brønsted acid sites. Reaction selectivity toward desired product can be controlled using acid site's strength and density distribution. Among all zeolites applications, catalytic conversion of oxygenates to hydrocarbons particularly has drawn the attentions. Most known is the methanol conversion to gasoline (MTG) over HZSM-5 catalyst.56
Biomass in the past decade has been considered as an important renewable resource of transportation fuels and its catalytic conversion over zeolites has been widely employed. Through several researches, a broad range of zeolites including HZSM-5, Y zeolite and beta zeolite have been investigated using bio-oils or biomass as feedstock. These studies indicated that the zeolite addition into the pyrolysis reactor could significantly increase the formation of aromatics. CO2, CO, water, tar and coke were also formed during this process.57 The majority of these investigations resulted that HZSM-5 catalyst gave the highest aromatics yield.58 In Huber et al.59 patented investigations, glucose pyrolysis in the presence of HZSM-5 catalyst, maximum aromatics was yielded at catalyst Si/Al ratio of 60 and 600 °C. Agblevor60,61 patented fractional pyrolytic process, wherein the biomass materials were selectively converted into desired products in the presence of HZSM-5 catalyst, eliminating potential secondary and post-pyrolysis processing steps. He showed that the biomass lignin fraction could be converted to phenolic components with low char production when pryolysis and catalytic processes were carried out simultaneously. Due to the considerable demethoxylation and demethylation, the molecular mass distribution of the fractional catalytic process product was about half of the conventional pyrolysis without catalyst.
Zeolite catalysts could be modified by incorporation of metals. Incorporation of Co or Ni transition metals (1–10 wt%) into HZSM-5 catalyst indicated significant effect on the performance of the parent HZSM-5 catalyst. Compared to the Co3O4, NiO modified catalysts showed more reactivity towards increasing the gaseous products and decreasing the organic phase. All the metal-modified catalysts showed remarkable reactivity towards production of phenols and aromatics, although exhibited limited reactivity toward water production. These are attributed to different hydrocarbon conversion reactions, comprising dehydrogenation, cracking and aromatization/cyclization reactions, which are catalyzed by Brønsted acid sites of the zeolite. In addition, water production enhancement was due to increased decarboxylation/dehydration of the oxygenated compounds on the zeolite acid sites.62,63 Catalytic conversion of particle board biomass over microporous zeolite catalyst exhibited that impregnation of 1 wt% Ga on HZSM-5 through incipient-wetness technique enhanced catalyst selectivity toward aromatic production. It is attributed to the dehydrocyclization of bio-oil intermediate products. Ga incorporation to the zeolite caused reduction of acid sites numbers. Although the selectivity towards aromatics was improved, but it caused lower degree of bio-oil deoxygenation (lower water yield).64
| Biomass/feed | Catalyst | Reactor(s) type | Operating conditions | Carrier gas | Analysis method(s) | Comments – highlighted points | Ref. |
|---|---|---|---|---|---|---|---|
| Wood | HZSM-5 | Fluidized bed | T = 450–475 °C, P = atm | Nitrogen | GC FTIR | (1) Low coke formation and prolong catalyst activity. (2) The bio-syncrude oils were low in oxygen, less viscous, less acidic, stable, and high in energy density | 58 |
| White oak wood | Ca–Y zeolite (β-zeolite) | Fluidized bed | T = 500 °C, P = atm | Nitrogen | GC-MS | (1) Catalysts successfully reduced oxygenates, producing aromatics and increasing bio-oil C/O ratio (5.9/1). (2) Bio-oil yield decreased due to carbon deposition on catalysts. (3) The Ca–Y zeolite deactivated less quickly possibly due to the presence of Ca2+ ions in place of Brønsted acid sites | 137 |
| Wood pine | H–β-zeolite | Fluidized bed | T = 450 °C, P = atm | Nitrogen | GC-MS | (1) In comparison to non catalytic pyrolysis, bio oil water contents increased (from 5.4 wt% to about 13 wt%) due to formation of polyaromatic hydrocarbons, gas yield fairly remained constant, char formation increased, bio-oil yield decreased | 138 |
| Aspen wood | X,Y-Zeolite, ZSM-5 and its modified with (co, Fe, Ni, Ce, Ga, Cu, Na) | Fixed bed | T = 400–600 °C, P = atm | He–Ar | MBMS | (1) ZSM-5 showed best performance while larger pore zeolites showed less deoxygenation activity. (2) Highest bio-oil (16 wt%) yielded from nickel-substituted ZSM-5 zeolite. (3) ZSM-5 catalyst in a fixed bed pyrolyzer and at 500 °C showed good deoxygenation | 65 |
| Beech wood | HZSM-5 | Fixed bed | T = 500 °C, P = atm | Nitrogen | GC-MS | (1) HZSM-5 zeolite, led to increase of water in the bio-oil via dehydration reactions and decrease of organics, increase of gases and coke due to decarbonylation, decarboxylation, dealkylation, cracking and aromatization reactions. (2) H-ZSM-5 reduced the organics oxygen content (from 41.68% to 30.45%) by decreasing the concentration of acids, ketones and phenols in the bio-oil | 21 and 66 |
| Pine | Zelites (beta, Y, ferrierite), NH4-beta-25, NH4–Y-12, NH4-Fer-20 | Dual-fluidized bed reactor | Reactor 1, T = 490 °C, P = atm, reactor 2, T = 450 °C, P = atm | Nitrogen | GC-MS, TEN3 gas analyzer | (1) First FBR used as pyrolyzer while second one as upgrading reactor. (2) Water and CO formation were increased over all zeolites while CO2 formation increased in some extent. (3) Beta zeolite was the most active in the de-oxygenation reactions followed by Y and ferrierite zeolites | 95 |
| Woody biomass, energy crops, agricultural residues | Zeolites, H-mordenite H-ZSM-5, H-Y, H-Beta, H-ferrierite | Pyroprob | T = 550 °C, P = atm | He | GC-MS | (1) The H-ZSM-5 catalyst was the most effective catalyst at producing aromatic hydrocarbons from the oxygen-rich vapors. (2) The structure and Si/Al ratio of the catalysts played a major role in their abilities to effectively deoxygenate the pyrolysis vapors and produce aromatic hydrocarbons | 67 |
| Maple wood glucose, furan | ZSM-5 | Fixed bed | T = 600 °C, P = atm | He | GC-MS | (1) Different ZSM-5 catalysts properties (silica-to-alumina ratio, mesoporosity and removal of external surface acid site) on the yield and aromatic hydrocarbons distribution. (2) Glucose catalytic fast pyrolysis yielded max. aromatics (∼42%) and min. coke formation (∼32%) at Si/Al = 30 | 139 |
| Oak | β-Zeolite, Y-zeolite (CaY) | Fast pyrolysis @ fluidized bed, upgrading @ fixed bed | T = 500 °C @ fluidized bed, T = 425 °C @ fixed bed, P = atm | Nitrogen | GC-MS | (1) Both catalysts efficiently deoxygenated a significant fraction of the pyrolytic vapor stream to produce aromatic hydrocarbons, but CaY offered a superior ability to produce aromatics, compared to β-zeolite. (2) The higher-moisture vapors cracking on the upstream of the pyrolysis system resulted in slower coke formation and more naphthalenes | 69 |
• HZSM-5 zeolite catalysts showed very good performance. It yielded bio-oil with low oxygen contents, less acidic, less viscous and stable with high energy content.21,58,65–67
• HZSM-5 zeolite catalysts led to water increase in the bio-oil via dehydration reactions, and enhancement of organics, aromatic hydrocarbons and gaseous products caused by decarbonylation, dealkylation, decarboxylation, cracking, and aromatization reactions. Coke formation over catalysts was also increased during catalytic upgrading.21,58,66,67
• Pore size and Si/Al ratio played important role on HZSM-5 catalyst performance, product distribution and selectivity. Metal substituted HZMS-5 enhanced bio-oil yield and properties.21,65–68
• β-Zeolite, Y-zeolite and ferrierite zeolite showed very good performance in the bio-oil deoxygenation and aromatic compounds production. β-Zeolite showed high activity in the de-oxygenation reactions followed by Y and ferrierite zeolites. Ca–Y-zeolite deactivated less quickly and offered a superior ability to produce aromatics, compared to β-zeolite.58,60,69
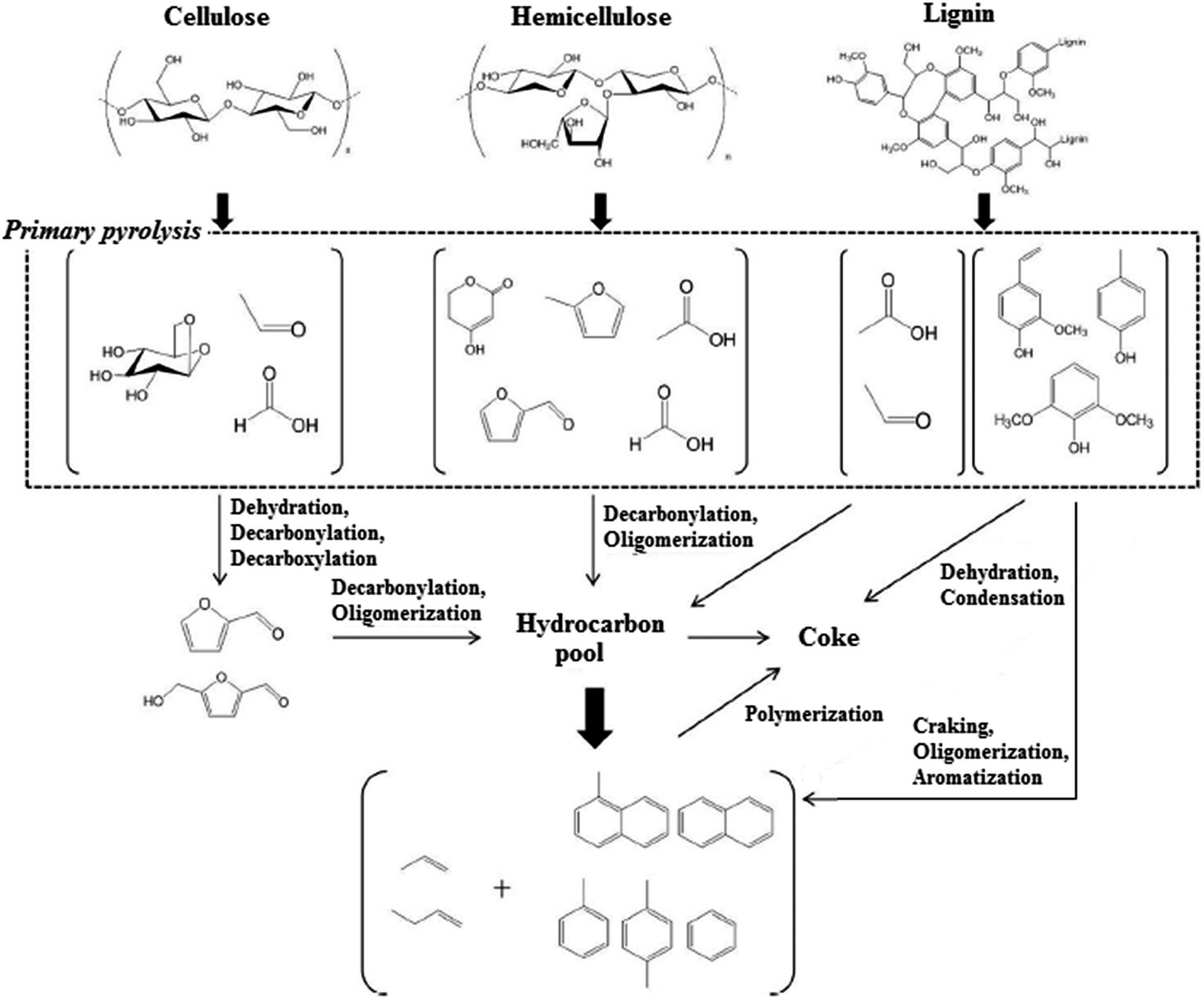 | ||
| Fig. 3 Reaction pathways for pyrolysis and catalytic pyrolysis vapor upgrading of lignocellulosic biomass over HZSM-5 catalyst. Adapted from Wang et al.70 | ||
The biomass oxygenated organic compounds over zeolite catalysts at 350 °C to 500 °C passed through decarboxylation, cracking, alkylation, polymerization, condensation and aromatization reactions. When acidic zeolite catalysts like HZSM-5 was employed, dehydration was the dominant mechanism. Under this condition, the yielded products was a mixture of low molecular weight olefins and aromatic hydrocarbons.71
During biomass pyrolysis and catalytic upgrading, the major product from cellulose pyrolysis was levoglucsoan, which could produce smaller furanic compounds through decarbonylation, decarboxylation or dehydration reactions.72 These furans then could diffuse into the acidic zeolite pores to produce olefins and aromatics through oligomerization and decarbonylation reactions. On the other hand, double hydrated xylose, the predominant product from hemicellulose pyrolysis could diffuse together with other low molecular weight molecules like acetic acid, furaldehyde, acetol and formic acid into zeolite pores without any further reaction. Lignin pyrolysis initially yielded monomeric phenolic components, which showed very low reactivity over HZSM-5 catalyst. Phenols acid-dehydration conducted to the formation of large amounts of cokes, whereas phenols cracking generated aromatics. Alkyl-phenols cracking to produce olefins might be another intermediate to yield aromatic compounds. Their investigation also showed that the aromatics yield of three main building block of lignocellulosic biomasses increased in the following order: lignin ≪ hemicellulose < cellulose. Moderately higher temperature indicated lower coke generation and higher aromatics yield for three components of biomasses. It was attributed to the higher desorption of the coke precursors and generation of lower molecular weight oxygenated components during pyrolysis and upgrading. Lignin, among the three biomass components, had the most complicated structure and phenolic molecules produced from its thermal degradation were prone to the coke and char formations, which could decrease the carbon efficiency for the biomass pyrolysis and catalytic upgrading. Therefore, product distribution of the biomass pyrolysis and catalytic upgrading was highly depending on the biomass composition.
2.2. Mesoporous catalysts
To eliminate the possibility of secondary reactions, which enhance the coke formation and consequently catalyst deactivation caused by a slow mass transport to and away from the catalytic center, suitable catalyst should have all advantages of microporous zeolite while provide additional diffusion pathways for larger molecules as shown in Fig. 4.73 | ||
| Fig. 4 Schematic illustration of a secondary pore system to enable diffusion of large molecules within microporous zeolites. These mesopores can be created as intercrystalline pores in nanozeolite aggregates (right) or may be formed as intracrystalline voids within zeolite single crystals (left). Adapted from Moller et al.73 | ||
Since the pyrolysis vapor comprised various components with different sizes and molecular weight, porosity can play an important role toward production of desired products. Macroporous and mesoporous materials can be selected as the first choice for catalytic process in the presence of large molecules.74 However, while size selectivity is desired, pores need to have a defined structure and be narrow enough to provide reagents; products and/or transition state selectivity. Macroporous materials, due to the high exposure of active site to substrates, restrict the reaction pathway toward selective reaction. To overcome this type of drawback, recently mesoporous materials with highly ordered structures have attracted the attentions.75
Biomass catalytic pyrolysis by the use of different acid catalysts has been employed to improve the bio-oil quality through deoxygenation reactions. Catalysts deactivation through coke formation is one of the main problems during deoxygenation. Coking is mostly caused by the phenolic compounds condensation. Further, the bio-oil components having large molecular volume cannot diffuse to the active sites placed inside the zeolite pores. Therefore, deoxygenation process of the bio-oil components is obstructed. To enhance the molecular transport and to prevent the pore blockage by coke generation, mesoporosity formation into the zeolite catalysts seems to be promising approach. Mesopores presence in the zeolite crystalline framework would be equivalent to external surface enhancement. It makes a large number of pore openings accessible to the large molecules. Shortened diffusion path length and enlarged external surface area would ease the coke precursors mass transfer from the micropores to the external surface of zeolite catalyst and consequently prevent its quick deactivation.76 Therefore, catalytic performance of catalyst is enhanced.77,78
Isomerization and aromatization of 1-hexene over micro-/mesoporous HZSM-5 catalyst (alkali-treated) indicated similar phenomena. The mechanism for catalytic stability improvement of alkali-treated HZSM-5 catalyst is illustrated in Fig. 5. As can be seen, due to the mesopores and micropores interconnection, the diffusion path in the micropores is considerably shortened. So, the isomerization and aromatization products or even precursors of coke (here naphthalene as the representative), which are created in the micropores, can diffuse out of the pores before deposition. It leads to the coke deposition in the mesopores of HZSM-5 catalyst and prevents micropores blockage. Consequently, the improvement of the stability of the catalyst in isomerization and aromatization can be attained attributed to the reduced diffusion path and coke formation in the mesopore structure.76,79
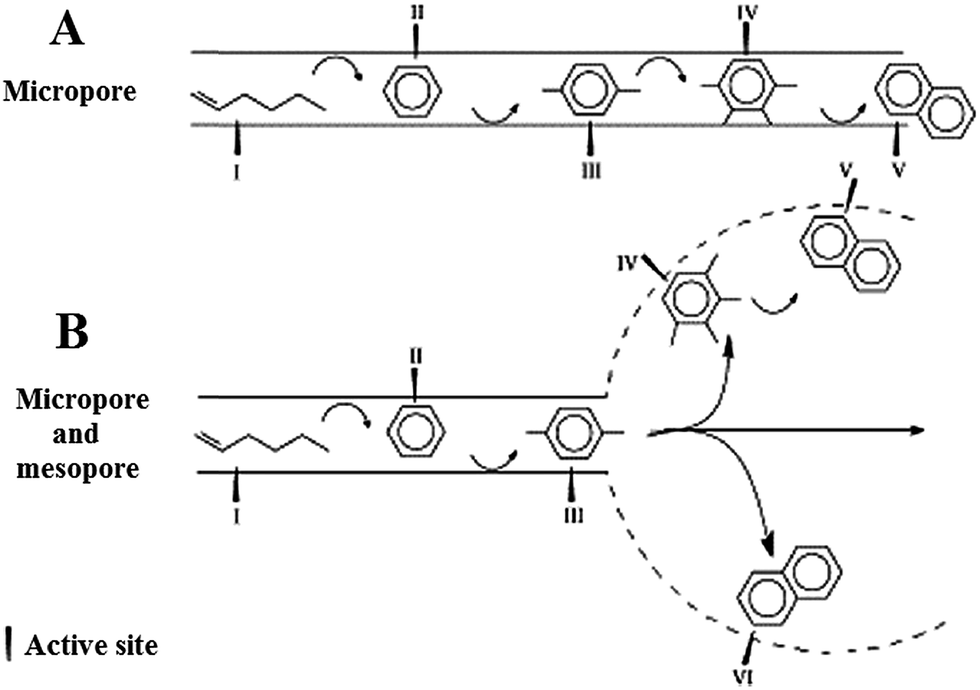 | ||
| Fig. 5 Mechanism for catalytic stability enhancement of the alkali-treated HZSM-5 zeolite with micro-mesopore porosity. Adapted from Li et al.79 | ||
The dual templating method (a) uses the same basis that was already proven to be a very successful route in the synthesis of the microporous zeolites. In this method, a sacrificial scaffold is used to create mesoporosity during crystallization and can be eliminated from the zeolite framework without loss of its final structural characteristics. Based on the physiochemical nature of the secondary templates, they can be divided to hard and soft templates. Soft templates also can be categorized to amphiphilic surfactants derivatives, macromolecular polymers and silylating agents. Applying multifunctional templates (b) produce micro- and mesopore structure in the zeolites at the same time through a single templating molecule. The third method (c) simplify the zeolite synthetic requirements and save additional cost of the production by directing the process toward synthesis of nanozeolite aggregates and mesoporous network.
2.2.2.1. Mesoporosity generation through desilication. Post-synthetic desilication of pre-synthesis zeolite catalyst can be used to induce intracrystalline mesopores. Local dissolution of zeolite frameworks in a basic solution (like NaOH) is a known strategy for mesoporosity creation.83 Fig. 6 depicts schematically the effect of Al content on the desilication treatment of MFI zeolites in alkali solution.84
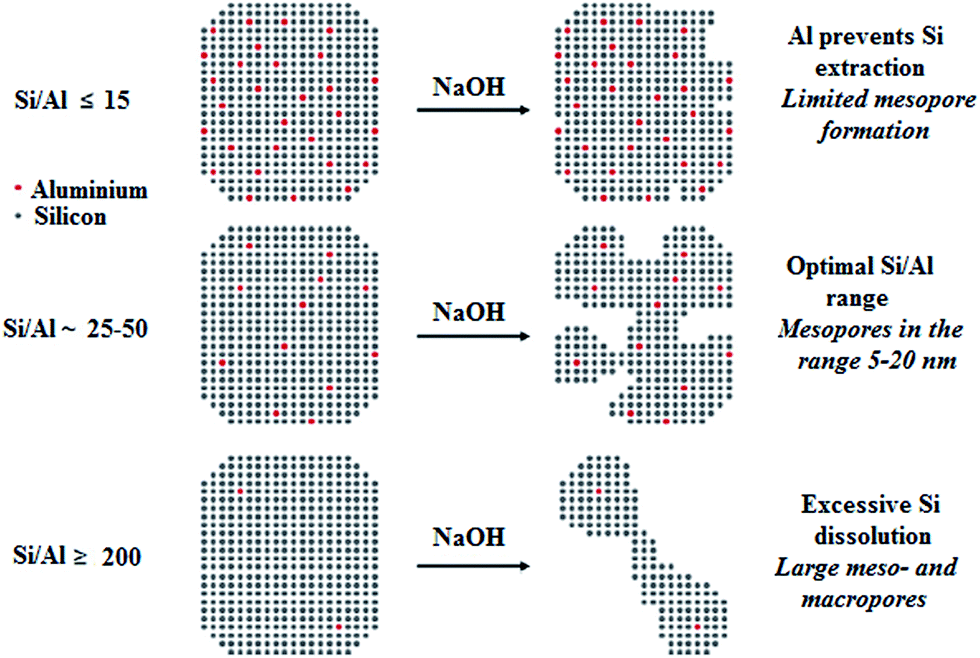 | ||
| Fig. 6 Schematic illustration of the effect of Al content on the desilication treatment of MFI zeolites in alkali solution. Adapted from Groen et al.84 | ||
The desilication process is more effective for zeolites with high silica (Si/Al > 20) than high Al because SiO-removal, which is bonded directly to Al, is very difficult.78 The zeolites desilication can be easily carried out at low concentration of alkali metal hydroxide. Mesoporosity generation highly depends on the Al distribution and concentration within zeolite crystals. Al-rich textures almost remain unchanged, while silica-rich textures are easily leached out to produce large mesopores. Investigations showed that the Si/Al molar ratios in the range of 25–50 were most favorable for the uniform mesoporosity development and keeping the HZSM-5 crystal morphology.84 HZSM-5 with Si/Al molar ratio less than 20 was very difficult to desilicate. Under mild basic conditions its framework was insoluble, while strong basic condition totally destroyed its zeolite framework. Alternatively, zeolites with high Si/Al molar ratio (>50) exhibited unselective and excessive dissolution generating too large pores.78 Highly uniform mesopores could be generated within the zeolite frameworks by the addition of cetyltrimethylammonium bromide (CTAB) surfactant to the desilication medium.85 This surfactant could contribute to the local desilication process, making micelles joint with base in the partly desilicated zeolite. The modified zeolite catalyst through this process showed that the acidic properties of the resultant zeolite changed slightly during desilication process.
2.2.2.2. Mesoporosity generation through dealumination. During the decades selective dealumination has been utilized because it was understood that production of zeolite with higher ratio of Si/Al could create stable zeolites with higher strength acid sites. Generally, during calcination some parts of alumina species are removed from the zeolite structure, when it is less stable. Hydrolysis of the Si–O–Al bonds creates defect sites, therefore extra-framework alumina species can be eliminated. Using extra steam, which is commonly used for zeolite Y dealumination, increases the hydrolysis severity. Then, ultra-stable Y zeolite with higher Si/Al ratio (USY), which is used as cracking catalyst in FCC (fluidized catalytic cracking) process, can be produced.80 Amorphous alumina residues extraction is then performed by diluted nitric acid or oxalic acid. Hence, cavities and pores with broad sizes between 2 and 50 nm are generated.73,76
Apart from Y zeolite, dealumination can be applied to ferrierite, mordenite and beta zeolites mostly by direct leaching with more concentrated acids. Depends on the nature of zeolites, different acids such as oxalic, acetic, tartaric, nitric, hydrochloric and sulfuric were utilized with various concentration (even 6.0 M HCl). For example, in a comparative investigation of three different structures, dealumination of beta zeolite was easier than mordenite, whereas HZSM-5 was almost unaffected under similar situations. Furthermore, beta zeolite dealumination conducted to higher loss of crystallinity, whereas mordenite indicated considerable mesopore volume.86
Principally, aluminum extraction from the zeolite structure inevitably conducted to a change in Si/Al ratio, and consequently the acidity, while mesopores are created at the same time. In this conditions, understanding the effects of mesoporosity on changes of zeolite catalytic activity seems to be difficult. This type of complication may be one the main reason which mesopore generation through leaching process has been recently carried out by desilication process instead.73,80
The recent catalytic biomass to bio-fuel conversion investigations, conducted under different conditions over mesoporous catalysts, are summarized in Table 3. In this regard, the following key features could be concluded:
| Biomass/feed | Catalyst | Reactor(s) type | Operating conditions | Carrier gas | Analysis method(s) | Comments – highlighted points | Ref. |
|---|---|---|---|---|---|---|---|
| Laminaria japonica | Nano Al-MCM-48, meso-MFI zeolites (Si/Al = 20) | Pyroprob | T = 500 °C, P = atm | He | GC-MS | (1) Meso-MFI showed higher catalytic decomposition ability than nano Al-MCM-48. (2) Meso-MFI produced high yields of aromatics, phenolics, and gases due to its strong acidic sites which accelerated cracking of pyrolyzed bio-oil molecules | 90 |
| Poplar wood | Pd/SBA-15 (Pd: 0.79 wt% – 3.01 wt%) | Pyroprob | T = 600 °C, P = atm | He | GC-MS | (1) Lignin cracked to phenols without side chains of carbonyl and unsaturated C–C. (2) The anhydrosugars were almost completely eliminated, and the furans were decarbonylated to form light compounds. (3) Linear aldehydes were significantly decreased, while the acids were slightly decreased. Linear ketones without the hydroxyl group, methanol, and hydrocarbons were all increased | 91 |
| Radiata pine sawdust | Mesoporous MFI, 5 wt% Ga/meso-MFI | Fixed bed | T = 500 °C, P = atm | Nitrogen | GC-TCD, GC-FID | (1) Meso MFI exhibited the highest activity due to synergic effect of a high porosity and strong acidic property, mainly in the deoxygenation. (2) High acidity induced decreasing of organic fraction. This drawback solved by incorporation of gallium in to the catalyst. It then led to less cracking, increase of organic fraction (aromatics) and low coke formation | 92 |
| Beech wood | MSU-S (aluminosilicate mesostructures) | Fixed bed | T = 500 °C, P = atm | Nitrogen | Not available | (1) Compared to non-catalytic pyrolysis, MSU-S led to low organic phase and high coke and char. MSU-S was selective toward PAHs and heavy fractions, while they produced almost no acids, alcohols and carbonyls, and very few phenols | 94 |
| Miscanthus | Al-MCM-41, Al-MCM-48, meso-MFI, Pt/meso-MFI | Fixed bed | T = 450 °C, P = atm | Nitrogen | GC, GC-MS | (1) Mesoporous Al-MCM-41 and Al-MCM-48 catalysts indicated high selectivity toward the production of phenolics while meso-MFI, which possesses strong acid sites, showed high selectivity to aromatics. (2) Loading of Pt on meso-MFI zeolite, which has both mesopores and high acidity, promoted cracking and dehydrogenation and resulted enhanced deoxygenation and aromatization | 93 |
| Spruce wood | Al-MCM-41 mesoporous catalysts | Pyroprob | T = 450 °C, P = atm | He | GC-MS | (1) The lack of levoglucosan was the most important catalytic effect on the products. (2) MCM-41 pore size enlargement and transition metal incorporation reduced the yield of acetic acid and water among pyrolysis products | 87 |
| Beech wood | Al-MCM-41 mesoporous catalysts | Fixed bed | T = 500 °C, P = atm | Nitrogen | GC-MS | (1) Higher amount of coke formation observed in comparison to zeolites. It likely was due to decarbonylation, decarboxylation, dealkylation, cracking and aromatization reactions | 95 |
| Beech wood | Al-MCM-41, metal–Al-MCM-41 | Fixed bed | T = 500 °C, P = atm | Nitrogen | GC-MS | (1) Higher Al content (low Si/Al ratio) and the consequent higher surface acidity of the catalyst resulted in an increase in the yields of the high value aromatic compounds. (2) By metals incorporation into the mesoporous catalysts, the levels of phenols remained high, while the levels of both hydrocarbons and PAHs were low. (3) Fe–Al-MCM-41 and Cu–Al-MCM-41 together with the parent material with the lower Si/Al ratio were the optimum for the production of phenols | 88 |
• Compare to nano Al-MCM-48, meso-MFI showed higher catalytic activity and higher yield of aromatic, phenolic and gaseous components thanks to its strong acidic sites and high porosity, which accelerated cracking reactions. High acidity caused decreasing of organic fraction. Incorporation of Ga to meso-MFI led to less cracking, increasing of aromatic components and coke formation diminishing.90,92
• Mesoporous Al-MCM-48 and Al-MCM-41 catalysts showed high selectivity toward phenolic compounds, while meso-MFI (which possesses strong acid sites) indicated high selectivity toward aromatic components production. Pt incorporation to meso-MFI catalyst promoted dehydrogenation and cracking then conducted to enhanced aromatization and deoxygenation. Enlargement of MCM-41 pore size and loading of transition metals to it reduced acetic acid and water yield among the pyrolysis products.87,93
• Al-MCM-41 catalyst led to decarbonylation, decarboxylation, dealkylation, cracking and aromatization reactions. Higher coke formation, compared to zeolite catalysts, could be strong evidence of mentioned reactions. Higher Al content or in the other world lower Si/Al ratio caused an increase in the yield of the aromatic components. Incorporation of metals (like Fe and Cu) to Al-MCM-41 enhanced the phenols yield and decreased the level of both hydrocarbons and poly-aromatic hydrocarbons (PAHs).88,95
2.3. Metal based catalysts
As explained within the context, there are several methods for the bio-oil upgrading comprising catalytic upgrading, steam reforming and hydrogenation. Among all these, the latter is the most widely used commercial process for the bio-oil upgrading and conducting hydro-deoxygenation reactions. Through these reactions, some components like aldehydes can be converted into stable chemicals like alcohols and hydrocarbons with high heating value and low oxygen contents.89,96,97Mostly noble metal catalysts (e.g., Pt, Ru and Pd) at high temperature and pressure are employed to carry out hydro-deoxygenation reactions. These types of reactions often suffer from catalyst deactivation and clogging of the reactor at high temperatures. To overcome these problem, an approach for the bio-oil deoxygenation into high yield commodity products was employed. In this approach, hydro-processing of the bio-oil performed over supported metal catalysts (Ru/C and Pt/C) followed by conversion over zeolite catalyst (HZSM-5). Using this strategy, drawbacks associated with the conventional hydrogenation processes were overcome by operating the process at moderate temperature (≤250 °C), at which no reactor plugging or catalyst coking was observed. Alternatively, the bio-oil upgrading at atmospheric pressure is the other promising strategy to overcome the aforementioned problems.98–100
Some metal based catalysts like Fe, Zn, Al, and Mg can participate in organic reactions as strong reductants. For instance, Zn and Fe are commonly used for reducing nitro compounds to amines. Further, Zn is a key metal catalyst in the conversion of carbonyl groups (e.g. aldehyde and ketones) into methylene groups. These reactions are usually carried out with a high selectivity and yield at ambient temperature and pressure in acidic conditions.101
Bio-oil is a mixture of many oxygenated compounds like aldehydes, ketones and acids, which conduct bio-oil toward instability and corrosiveness. Therefore, the use of mentioned metal based catalysts can effectively enhance the bio-oil quality. Contrary to conventional hydrogenation process, low pressure pyrolysis vapor upgrading process over mentioned catalysts can be conducted without the presence of other catalysts and additional hydrogen gas.101
Catalytic cascade approach for the biomass pyrolysis vapor upgrading has recently attracted the attentions of researchers. The idea is to maximize carbon efficiency during bio-oil quality enhancement. In this regard, instead of oxygen functionalities (carbonyl, carboxylic, ketonic, and hydroxyl groups) elimination too early, their high reactivity is utilized to conduct C–C bond formation reactions, including aldol condensation and ketonization. Metal oxide catalysts are mostly efficient in catalyzing carboxylic acids ketonization, but reducible oxides like ceria can even catalyze the small aldehyde ketonization.24 Fig. 7 depicts the proposed reaction mechanism of small aldehyde (propanal) conversion over Ce0.5Zr0.5O2 catalyst. The contribution of two major reactions comprising ketonization and aldol condensation could be observed in the network, involving several condensation steps. Furthermore, there were also different side reactions that could take place in parallel. It is well understood that aldol condensation can happen on both basic and acid sites. In the mixed oxide catalysts, the oxygen anions can behave as either Brønsted or Lewis base sites, while the exposed cations are Lewis acid sites.56,102
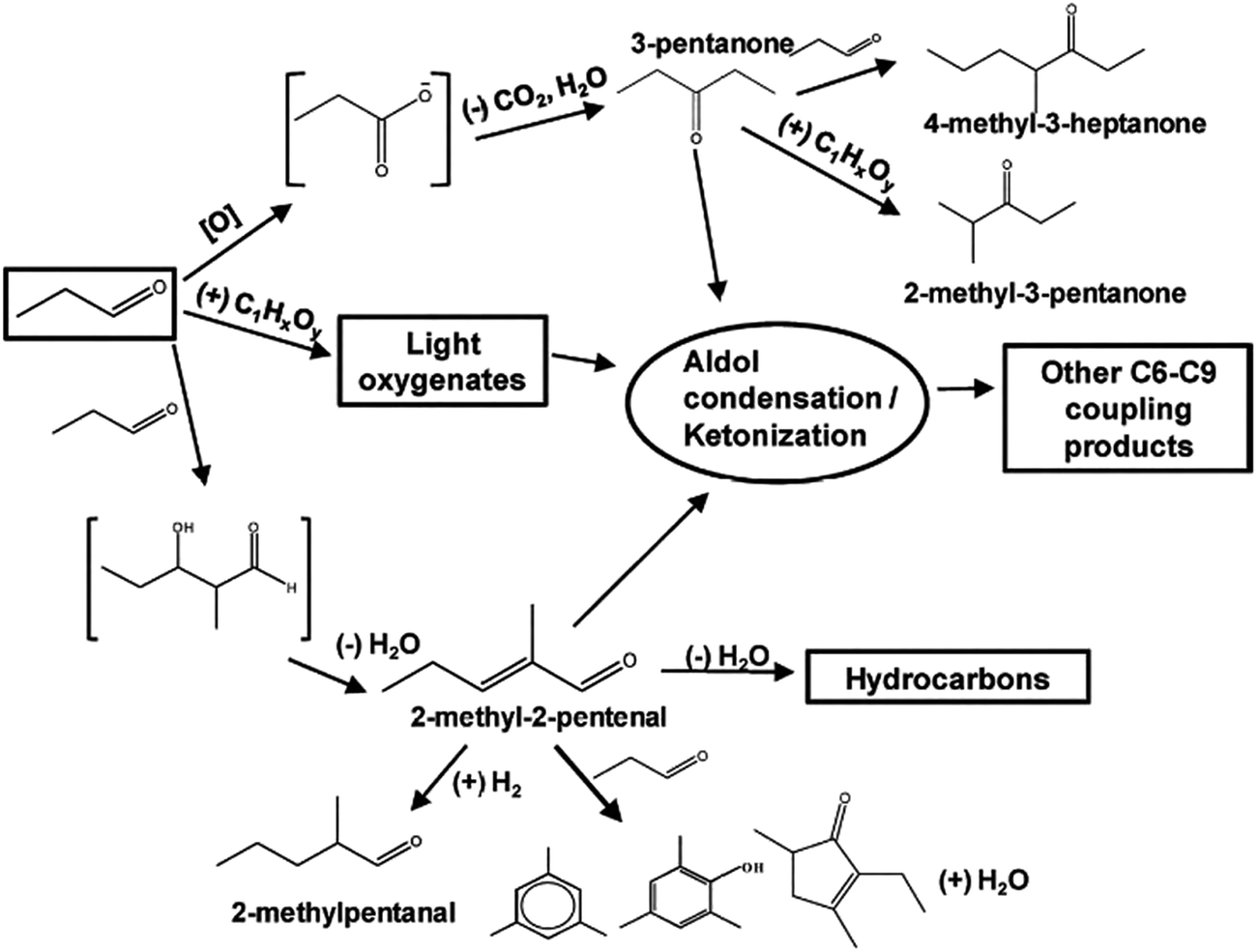 | ||
| Fig. 7 Proposed reaction mechanism for propanal conversion over Ce0.5Z0.5O2 – adapted from Gangadharan et al.102 | ||
Zeolites are also effective catalysts for C–C bond formation, but the selectivity is toward aromatics formation. For instance, propanal can be selectively converted to C7–C9 aromatics through aldol condensation over HZSM-5 catalyst.24
Pacific Northwest National Laboratory (PNNL) in USA has recently focused on the pyrolysis vapor upgrading with the objectives of maximizing carbon efficiency and minimizing hydrogen consumption. They employed a new concept based on chemical looping and utilizing metal oxide catalysts to selectively eliminate oxygen from the pyrolysis vapors without hydrogen feeding.103 The concept is shown in Fig. 8.
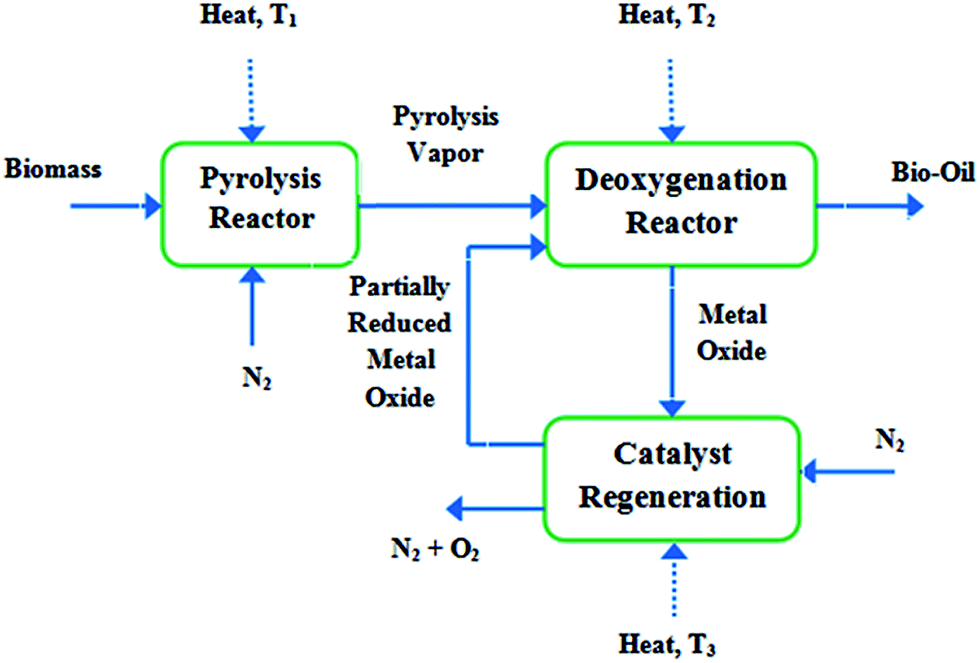 | ||
| Fig. 8 Schematic of the chemical looping deoxygenation (over metal oxide catalysts) concept (T3> T1> T2).103 | ||
The pyrolysis vapors react with the partially reduced metal oxide (MeOx-1) while they pass over the deoxygenation catalyst. The metal oxide is oxidized (MeOx) while the pyrolysis vapors are reduced (deoxygenated). To reduce the catalyst (MeOx-1) having the ability to be recycled back to the reactor, the metal oxide is heated under N2 stream at a higher temperature. Model compound experiments and theoretical calculations identified some promising metal oxide catalysts for such type of vapor phase deoxygenation.103 A similar investigation was patented by Lissianski et al.104 where pyrolyzing the biomass was performed in the presence of a transition metal, using microwave energy.
In an earlier study, Sanna and Andrersen105 suggested new catalysts for the biomass (wheat spent grains) conversion into deoxygenated bio-oil in a fluidized bed reactor. They used two Mg-rich activated olivine (ACOL) and activated serpentine (ACSE), and alumina (ALU) as catalyst. A considerable reduction of oxygen content in the bio-oil was observed in following order: ACOL > ACSE > ALU. Particularly, compared to ACOL which was able to remove about 40 wt% of the original oxygen from the bio-oil, ACSE and ALU decreased it to less than 20–30 wt%. The oxygenated compounds of the bio-oil interacted in the catalyst's active sites with the metallic species and produced C5–C6 components through decarboxylation.
The catalytic vapor upgrading, which is an attractive process with lots of advantages, has been widely investigated employing acidic zeolites. Nevertheless, zeolite catalysts suffer from fast coke deposition and PAHs formation during upgrading. Furthermore, MCM-41 based mesoporous catalysts exhibited crucial disadvantages; high production cost and poor hydrothermal stability.106 Due to the advantages associated with metal based catalysts and in order to likely resolve zeolites and mesoporous catalysts problems, several researches recently have been performed on varieties of metal based catalysts, which some of them are summarized in Table 4.
| Biomass/feed | Catalyst | Reactor(s) type | Operating conditions | Carrier gas | Analysis method(s) | Comments – highlighted points | Ref. |
|---|---|---|---|---|---|---|---|
| Cotton seed | Magnesium oxide | Fixed bed | T = 400–700 °C, P = atm | Nitrogen | GC-MS | (1) Following to the catalytic pyrolysis, almost all of the long chain alkanes and alkenes have been converted to lower molecular weight material of the short chain and alkyl substituted forms. Oxygen content also decreased. (2) Catalyst increasing, decreased liquid yield while increased gas and char yields | 107 |
| Euphorbia rigida | Alumina | Fixed bed | T = 550 °C, P = atm | Nitrogen-steam | GC-MS | (1) Using steam atmosphere instead of nitrogen during pyrolysis produced less paraffins, enriched ketones, carboxylic acids and triterpenoid compounds, while decreased phenol formation. (2) Yield and composition of the oil were depended on the catalyst ratio and pyrolysis atmosphere | 15 |
| Poplar wood | Titanium oxide, zirconium oxide | Pyroprob | T = 600 °C, P = atm | He | GC-MS | (1) Good thermal stability compared to mesoporous silicates and MCM. (2) Incorporation of Pd to catalysts, exhibited very promising effects to convert the lignin-derived oligomers to monomeric phenols. Favored reducing the aldehydes and sugars, while increasing the ketones, acids and cyclopentanones. (3) TiO2 + ZrO2 catalysts reduced the acids deeply, and moreover increased the hydrocarbons. So, yielded bio-oil had improved fuel properties | 111 |
| Corncob | Calcium oxide | Fixed bed-TGA | T = 25–1000 °C, P = atm, Ramp = 90 K min−1 | Nitrogen | FTIR | (1) The pyrolysis vapor composition changed markedly in the presence of CaO; the molality of acids, phenols and carbonyl compounds decreased, while the molality of hydrocarbons increased; CaO was very effective in deacidification and the conversion of acids promoted the formation of hydrocarbons | 112 |
| White pine | Calcium oxide | Fluidized bed | T = 520 °C, P = atm | Nitrogen | GC-MS | (1) CaO/pine ratio increasing, decreased LG, formic acid, acetic acid, and D-allose, guaiacol while increased phenol, ortho-cresol, para-cresol, and meta-cresol. (2) Furfural, furfuryl alcohol, hydroxymethyl furfural as dehydration product increased with CaO/Pine ratio increasing. It was due to dehydration reactions | 113 |
| Beech wood | MgO, NiO, alumina, zirconia/titania, tetragonal zirconia, titania, silica alumina | Fixed bed | T = 500 °C, P = atm | Nitrogen | GC-MS | (1) The most interesting catalyst was zirconia/titania formulation with the highest surface area (85 m2 g−1), which yielded organic liquid products with reduced oxygen and higher aromatics content compared to the non-catalytic runs | 66 |
| Pine sawdust | CuO, Fe2O3, ZnO, CoO | Pyroprob | T = 525 °C, P = atm | He | GC-MIP-AED | (1) The most interesting catalysts were CuO which exhibited the highest yields (49%) in semi-volatile compounds. (2) Mixed metal oxide catalysts (Fe2O3, and mixed metal oxides containing copper and cobalt) and ZnO reduced the proportion of heavy fraction in the bio-oil (from 22% to 15%) with a limited decrease in its yield | 114 |
| Palm oil fruit bunch (EFB) oil palm fronds (OPF) | Boric oxide (B2O3) | Fixed bed | T = 400 °C, P = atm | Nitrogen | GC-MS, NMR | (1) Boric oxide had a deoxygenation effect leading to elimination of hydroxyl and methoxy group from the bio-oil compound. This was associated with a significant suppression in the gas yield and a substantial increase in char yield. (2) The catalytic mechanism of boric oxide suggests that the addition of boric oxide promotes the removal of hydroxyl groups with the generation of alkyl substituted compounds with reduced oxygen content | 115 |
| Wood chips of Canadian white pine | Na2CO3/γ-Al2O3 (20 wt%) | Fixed bed | T = 500 °C, P = atm | Nitrogen | GC-MS | (1) Catalytic vapor upgrading in the presence of Na2CO3/γ-Al2O3 resulted in high level of selective deoxygenation (12.3 wt%) compared to non-catalytic bio-oil (42 wt%). (2) The bio-oil produced from catalytic trial had comparable properties to those of fuel oil, for instance neutral in terms of acidity/basicity (pH = 6.5) and having high energy density (37 MJ kg−1 of bio-oil compared to 40 MJ kg−1 of fuel oil) | 108 |
| Wheat spent grains (WSG), brewers spent grain (BSG) | Alumina sand (Al2O3 content of 91%) | Fluidized bed | T = 460 °C, P = atm | Nitrogen | GC-MS | (1) Although the higher energy content retained in the bio-oils produced at 520 °C, but the bio-oils yielded at lower temperature (460 °C) indicated better quality in terms of low oxygen, lower aromatics and nitrogen content. This condition should be favored for the pyrolysis process. (2) The high O/C ratio of the bio-chars at 460 °C evidenced that the produced chars at this temperature might have retained oxygen from the bio-oils | 140 |
Some of the key features of metal oxide catalysts used for bio-oil vapor phase upgrading can be noted as follows:
• Several metal oxides including MgO, NiO, alumina, zirconia/titania, zirconia and titania were used as catalyst. Deoxygenation occurred up to some extents in compare to non-catalytic pyrolysis, but zirconia/titania showed the most interesting deoxygenation and highest yield of aromatic compounds. Alumina showed that bio-oil yield and composition depended on pyrolysis atmosphere and the catalysis/feed ratio. The bio-oil produced over Na2CO3/γ-Al2O3 had comparable properties to those of fuel oil.15,66,107–110
• TiO2 and ZrO2 investigations indicated that they both had very good heat stability compare to mesoporous catalysts. TiO2 + ZrO2 mixed oxides resulted high hydrocarbon yield and significantly decreasing of acid contents.111
• Reaction over CaO yielded bio-oil with low acid, carbonyl and phenols contents while the molality of hydrocarbons increased. CaO/feed ratio increasing enhanced dehydration reactions.112,113
• CuO exhibited very interesting results to yield semi-volatile compounds and high bio-oil yield. Boric oxide promoted hydroxyl group removal with generation of alkyl groups which consequently reduced oxygen contents.114,115
2.4. Catalyst deactivation
The catalyst deactivation is one of the challenging issues in catalytic biomass pyrolysis vapor upgrading. It is not only caused by coke formation, but also the strong adsorption of the oxygenate components on the surface of catalyst support. Generally two types of cokes are formed over catalyst during biomass catalytic vapor upgrading. One, which has thermal origin, is called thermal coke and the other, which has catalytic origin, is called catalytic coke. Thermal coke, which is often formed over outside of catalyst's particle, is due to phenolic compounds polymerization. Catalytic coke, which is mostly deposited inside the catalyst's channels, is caused by aromatization, oligomerization condensation and cyclization of oxygenate components.116–120 Catalyst characteristics and biomass feedstock properties can influence catalyst deactivation and coke formation. | ||
| Fig. 9 Ammonia temperature programmed desorption (TPD) for the fresh (solid line) and spent (dotted line) catalyst.121 | ||
In zeolites, coke and tar deposits, which block the catalyst pores and cover its acid sites, significantly are influential on the catalyst activity and selectivity reduction.118 Dealumination of the zeolite catalysts (like HZSM-5) in the presence of steam was reported by several researchers. It can be conducted to catalyst acidity lost and irreversible deactivation.121
Physical characteristics of the zeolite catalyst comprising pore shape, pore size and crystallite size can highly affect the coke formation.125 Catalytic upgrading of pine wood in a fluidized bed reactor using HBeta-25, HY-12, HZSM-5 and HMOR-20 (Mordenite) catalysts showed that coke formation was fairly low for both Mordenite and HZSM-5. Spent Y zeolite (HY-12) exhibited the highest coke content. This was possibly due to the highest initial surface area and large cavities in the structure of Y zeolite, which allowing bigger molecules to diffuse to the inner part of zeolite.126
Catalytic pyrolysis, using different zeolite catalysts having small (ZK-5, SAPO-34), medium (Ferrierite, ZSM-23, MCM-22, SSZ-20, ZSM-11, ZSM-5, IM-5, TNU-9) and large (SSZ-55, Beta zeolite, Y zeolite) pore size, were studied by Jae et al.53 Compared to zeolites with large pore size, small pore size produced less coke. The least coke formation was resulted from medium pore size with moderate internal pore space catalysts.
In addition, coke formation over catalyst can be influenced by zeolite crystallite size. Small crystallites showed much slower deactivation and less coke formation compared to large crystallites. This was due to the shorter diffusion path and quicker removal of products from the catalyst's channels. So, products did not have sufficient time to be converted to coke precursors and coke.127
Increasing H/Ceff. mole ratio in oxygenated bio-oil favored the formation of olefins and aromatics and attenuates coke formation.131 Investigations showed that feedstocks with hydrogen to carbon effective ratio (H/Ceff.) less than 1.0 were difficult to upgrade over a HZSM-5 catalyst due to quick deactivation (coke formation) of the catalyst.132 The H/Ceff. ratio of the petroleum based feedstocks varies from 1 to 2, whereas that of the biomass feeds are only from 0 to 0.3. Therefore, the biomass contained hydrogen deficient molecules, and approaches for the biomass and its derived feedstocks transformation must consider their H/Ceff. ratio. Co-feeding of alcohol (like methanol) and biomass is one of the possible ways to enhance H/Ceff. ratio.29 Zhang et al.133 showed that methanol co-feeding with biomass (pine wood) at H/Ceff. ratio of 1.25 increased the aromatics yield and decreased coke formation over HZSM-5 catalyst. Fig. 10 depicts a kinetic scheme suggested for the bio-oil/biomass pyrolysis vapor and methanol mixture conversion into coke and hydrocarbons over HZSM-5 catalyst.
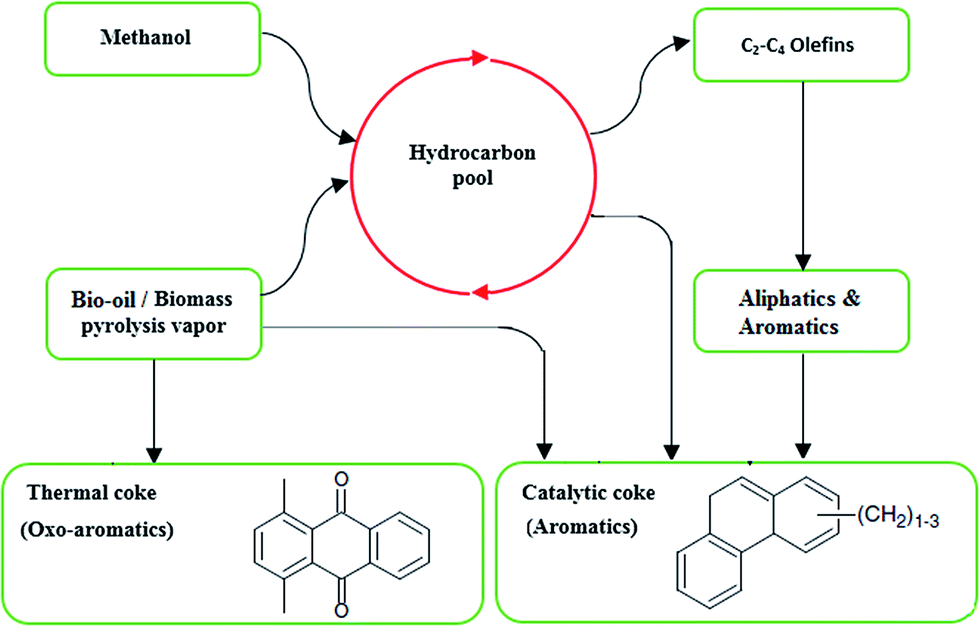 | ||
| Fig. 10 Kinetic scheme suggested for the bio-oil/biomass and methanol mixture conversion into hydrocarbons and coke on HZSM-5 catalysts. Adapted from Valle et al.119 | ||
The quantity and composition of the deposited coke on the HZSM-5 catalyst showed the significance of catalyst acidity for the formation of catalytic and thermal coke fractions. The major fraction of the produced coke was possibly due to the polymerization of the products derived from the biomass components pyrolysis (mostly lignin). Mostly, two fractions of coke were formed on the catalyst. The fraction of coke which was burned at low temperature was formed by condensation–degradation of lignin based oxygenated compounds. This type of coke was deposited on macro- and mesoporous structure of the zeolite catalyst matrix. The other one, which was burned at higher temperature and being deposited on the catalyst's micropores, was formed by condensation reactions activated by the acid sites. Formation of this type of coke was considerable in pure methanol catalytic conversion. Methanol addition to the pyrolysis vapor decreased the coke formation attributed to the attenuation of the phenolic compounds (lignin originated) polymerization and their deposition on the catalyst. According to the literature, pure methanol catalytic conversion on HZSM-5 catalyst formed non-oxygenated aromatics and aliphatic hydrocarbons as major components.119,120
Lignin derived phenolic compounds like anisole and guaiacol and mostly those with multiple oxygen functionalities (–OH, –OCH3, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) are the major deactivating components. The presence of alkalis, as well as N- and likely S-containing compounds in the biomass, improve catalyst deactivation.134,135
O) are the major deactivating components. The presence of alkalis, as well as N- and likely S-containing compounds in the biomass, improve catalyst deactivation.134,135
| Biomass/feed | Catalyst | Reactors type | Operating conditions | Carrier gas | Analysis method | Comments – highlighted points | Ref. |
|---|---|---|---|---|---|---|---|
| Lignin | HZSM-5 | Py-GC/MS pyrolyzer | T = 650 °C, P = atm | He | GC-MS | (1) The partial deoxygentaion of the lignin aromatic units produced simple phenols, which could be potential sources of catalyst deactivation and coke formation. (2) HZSM-5 used for catalytic pyrolysis of biomass feedstocks might be more quickly deactivated by feedstocks containing a higher proportion of H-lignin which could produce a higher concentration of simple phenols | 135 |
| Swedish pine-wood | HZSM-5 | Fixed bed, L = 700 mm, D = 20 mm | T = 600 °C, P = atm | Nitrogen | GC/MS | (1) Beginning from the first regeneration, a gradual decrease of the regenerated catalyst activity was observed up to an irreversible poisoning after the fifth upgrading–regenerating cycle. (2) The active acid sites in the upgrading reactions were presumed to be preferentially Brønsted acid sites that were gradually deactivated by the repeated regeneration treatments. (3) The regeneration of the catalyst was performed by heating the spent catalyst, once it was washed with acetone and dried, in a furnace at 500 °C in the presence of air for 12 h | 28 |
| Oak | β-Zeolite, Y-zeolite (CaY) | Fast pryolysis @ fluidized bed | Pyrolysis, T = 500 °C, P = atm | Nitrogen | GC-MS | (1) Catalysts could be fully reactivated by ex situ regeneration by heating in an oven at 500 °C in air atmosphere. (2) Cracking of the higher-moisture pyrolysis vapors found on the upstream end of the pyrolysis system resulted in slower coke formation and more naphthalenes. (3) The cracking of the less-moisture-laden, downstream pyrolysis vapors with greater concentrations of medium to large molecules, led to greater C-rich hydrocarbon product. This was richer in monocyclic benzene compounds, but it resulted in faster catalyst deactivation by coking | 69 |
| Upgrading @ fixed bed | Upgrading, T = 425°, P = atm | ||||||
| Pine (pinus insignis) sawdust | Ni-HZSM-5 (1 wt% of nickel) | Pyrolysis @ spouted-bed reactor | Pyrolysis, T = 450 °C, P = atm | Nitrogen | GC-MS | (1) The co-feeding of methanol with crude bio-oil had a significant effect of reducing coke deposition and catalyst deactivation, which was the main cause of catalyst deactivation it was attributable to two factors: (a) coke formation from methanol was lower than that from oxygenate components in the bio-oil; and (b) the water generation in the dehydration of methanol on the catalyst acid sites contributed to inhibiting coke formation. Nevertheless, a very high content of methanol in the feed also had an unfavorable effect on the conversion of the bio-oil contained in the bio-oil/methanol mixture, a feed of 40 wt% bio-oil/60 wt% methanol was suitable for balancing these two effects | 136 |
| Upgrading @ fluid bed reactor | Upgrading, T = 450 °C, P = atm | ||||||
| White oak wood | Ca_Y zeolite (β zeolite) | Pyroprob-GC/MS | T = 500 °C, P = atm | Nitrogen | GC-MS | (1) Decreasing of bio-oil yield due to carbon deposition of catalyst surface observed. (2) The Ca–Y zeolite appeared to be deactivated less quickly, possibly as a result of the presence of Ca2+ ions in place of Brønsted acid sites, which allowed for more oxygen removal following decarboxylation pathways. This allowed for slower coke formation because slightly more hydrogen was conserved in the pyrolysis products | 137 |
| Beech wood | H-ZSM-5 (Si/Al = 25), Al-MCM-41 (Si/Al = 30) | Fixed bed | T = 500 °C, P = atm | Nitrogen | GC-MS | (1) Strongly acidic HZSM-5 zeolite, led to increase of water in the bio-oil via dehydration reactions and decrease of organics, increase of gases and coke due to decarbonylation, decarboxylation, dealkylation, cracking and aromatization reactions. (2) Higher amount of coke deposited on the Al-MCM-41 mesoporous catalyst compared to HZSM-5 zeolite | 21 |
| Beech wood | MSU-S, Al-MCM-41 | Fixed bed | T = 500 °C, P = atm | Nitrogen | — | (1) The high selectivity towards aromatics, PAHs and coke, as well as the higher production of propylene in the pyrolysis gases with the MSU-S catalysts, compared to Al-MCM-41, suggested that the former possessed stronger acid sites | 94 |
Some key aspects in this regard are as following:
• Feedstock (biomass and lignin) pyrolysis product deoxygenation over HZSM-5 catalyst indicated that higher content of H-lignin, which could produce higher concentration of phenols, could cause quick catalyst deactivation. An irreversible poisoning was observed after some regeneration cycles due to the Brønsted acid sites deactivation. Strong acidic HZSM-5 zeolite catalyst led to coke formation due to dealkylation, decarboxylation, decarbonylation, aromatization and cracking reactions. The methanol co-feeding indicated a significant effect on coke deposition reduction on Ni-HZSM-5 catalyst during bio-oil upgrading.21,28,135,136
• Cracking of the high moisture pyrolysis vapors resulted slower catalyst deactivation and coke formation on β- and Y-zeolites. Incorporation of Ca to Y-zeolite caused more oxygen removal and slower catalyst deactivation. It is due to conservation of slightly more hydrogen in pyrolysis products.69,137
• MSU-S catalyst high selectivity toward aromatics, PAHs and coke formation (catalyst deactivation) were attributed to its stronger acid sites in comparison to Al-MCM-41 catalyst. Compared to HZSM-5 zeolite, Al-MCM-41 showed higher tendency toward coke formation and catalyst deactivation.21,94
3. Conclusion and outlook
Catalytic biomass pyrolysis vapor upgrading process to enhance the bio-oil quality indicates immense potential to convert renewable biomass to bio-fuel. Fast pyrolysis, which is known as a promising process to convert pretreated biomass to bio-oil, is affected by the biomass types and reaction conditions. Catalytic vapor phase upgrading is aiming to treat the fast pyrolysis vapor before condensation. It recently has attracted the attentions of bio-fuel researchers due to the prominent techno-economical characteristics of this type of upgrading in comparison with conventional hydro-deoxygenation (HDO) process. Despite HDO process which consumes high hydrogen quantity and requires complicated equipment working at high pressure, this upgrading approach is carried out at atmospheric condition without hydrogen feeding. The produced bio-oils yields and qualities are strongly dependant on catalysts types and properties (e.g. structure, acidity and pore size), reaction conditions and feed type.Three most important classes of catalysts including microporous zeolites, mesoporous catalysts, and metal based catalysts are used for vapor phase bio-oil upgrading. Among zeolite catalysts, HZSM-5 (which possesses a three dimensional pore structure, high acidity and shape selectivity) indicates the superior performance in deoxygenation, aromatic compounds production and resistance to coke formation. However, to keep the catalyst activity and selectivity for long time, deactivation through dealumination as well as coke deposition need to be minimized. The product distribution and coke formation amount over catalyst are strong function of catalyst shape selectivity and acidity. Shape selectivity of the zeolite catalyst are mostly influenced by pore size and shape as well as crystallite size. Mesoporous catalysts having pore diameter larger than 2 nm can resolve large molecules mass transfer limitation, associated with microporous zeolites. Contrary to mesoporous MCM-41 based catalysts, which have lower thermal stability and acidity compared to zeolite based catalysts, mesoporous MFI catalysts simultaneously possess high acidity as well as large pore diameter to overcome mass transfer limitation (which is micro-zeolites drawback). On the other hand, metal based catalysts, exhibit high acidity and outstanding resistance to coke deposition; therefore they can be reputable catalysts for bio-oil upgrading.
Efforts to transform lignocellulosic biomass to intermediate and base chemicals for the biofuels production have been fruitful to a considerable extent in recent years. In the next decades, it will be expected to find more techno-economical processes which can employ advanced catalytic processes to convert biomasses from various resources into fine chemicals, base chemicals and fuels. We will be approaching to more sustainable and renewable economy, although further efforts will be required. Biofuel upgrading technologies still need development to create cost-competitive products with acceptable productivity and selectivity. Promising improvement on heterogeneously catalyzed transformation of lignocellulosic biomasses to fuel like and value added chemicals with low coke formation over catalysts has attracted intensive attention in the past few years and breakthroughs have been attained up to some extent. It might be proper to mention that conversion of biomasses to desired chemicals with low coke formation, high selectivity and yield remains in its infancy until there are considerable developments in heterogeneous catalysts. Biomasses catalytic pyrolysis vapor upgrading through a cascade system of different catalysts (micropore zeolites, mesopore and metal based catalysts), that includes several consecutive steps for various bio-oil fractions upgrading, seems to be a promising thermochemical conversion/upgrading technology. Each individual mentioned catalysts or their employment in a cascade system indicated high potential for industrialization, although bio-oil upgrading through a cascade systems of catalysts most probably in the near future would attract the researchers' attentions.
Acknowledgements
The authors acknowledge the financial support from the High Impact Research (HIR) Grant (no. UM.C/HIR/MOHE/ENG/11 from the Ministry of Higher Education, Malaysia), Chemical Engineering Department, Engineering Faculty, University of Malaya, Malaysia.References
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- Y.-B. Huang, Z. Yang, J.-J. Dai, Q.-X. Guo and Y. Fu, RSC Adv., 2012, 2, 11211 RSC.
- M. Asadieraghi and W. M. A. Wan Daud, Energy Convers. Manage., 2014, 82, 71–82 CrossRef CAS PubMed.
- R. H. Venderbosch and W. Prins, Biofuels, Bioprod. Biorefin., 2010, 4, 178–208 CrossRef CAS.
- L. Ingram, D. Mohan, M. Bricka, P. Steele, D. Strobel, D. Crocker, B. Mitchell, J. Mohammad, K. Cantrell, J. Charles and U. Pittman, Energy Fuels, 2008, 22, 614–625 CrossRef CAS.
- D. Mohan, J. Charles, U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- J. Zhang, H. Toghiani, D. Mohan, J. Charles, U. Pittman and R. K. Toghiani, Energy Fuels, 2007, 21, 2373–2385 CrossRef CAS.
- A. V. Bridgwater, J. Anal. Appl. Pyrolysis, 1999, 51, 3–22 CrossRef CAS.
- J. C. Hicks, J. Phys. Chem. Lett., 2011, 2, 2280–2287 CrossRef CAS.
- D. Chiaramonti, A. Oasmaa and Y. Solantausta, Renewable Sustainable Energy Rev., 2007, 11, 1056–1086 CrossRef CAS PubMed.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- D. C. Elliott, D. Beckman, A. V. Bridgwater, J. P. Diebold, S. B. Gevert and Y. Solantausta, Energy Fuels, 1991, 5, 399–410 CrossRef CAS.
- Q. Lu, W.-Z. Li and X.-F. Zhu, Energy Convers. Manage., 2009, 50, 1376–1383 CrossRef CAS PubMed.
- J.-L. Zheng and Q. Wei, Biomass and Bioenergy, 2011, 35, 1804–1810 CrossRef CAS PubMed.
- E. Pütün, F. Ateş and A. E. Pütün, Fuel, 2008, 87, 815–824 CrossRef PubMed.
- S. Thangalazhy-Gopakumar, S. Adhikari, R. B. Gupta, M. Tu and S. Taylor, Bioresour. Technol., 2011, 102, 6742–6749 CrossRef CAS PubMed.
- Z. Zhang, Q. Wang, P. Tripathi and C. U. Pittman Jr, Green Chem., 2011, 13, 940–949 RSC.
- F. d. M. Mercader, M. J. Groeneveld, S. R. A. Kersten, R. H. Venderbosch and J. A. Hogendoorn, Fuel, 2010, 89, 2829–2837 CrossRef CAS PubMed.
- Y. Wang, T. He, K. Liu, J. Wu and Y. Fang, Bioresour. Technol., 2012, 108, 280–284 CrossRef CAS PubMed.
- A. G. Gayubo, B. Valle, A. T. Aguayo, M. Olazar and J. Bilbao, J. Chem. Technol. Biotechnol., 2010, 85, 132–144 CrossRef CAS.
- S. Stephanidis, C. Nitsos, K. Kalogiannis, E. F. Iliopoulou, A. A. Lappas and K. S. Triantafyllidis, Catal. Today, 2011, 167, 37–45 CrossRef CAS PubMed.
- X. Junming, J. Jianchun, S. Yunjuan and L. Yanju, Biomass Bioenergy, 2008, 32, 1056–1061 CrossRef PubMed.
- L. Deng, Y. Fu and Q.-X. Guo, Energy Fuels, 2009, 23, 564–568 CrossRef CAS.
- D. E. Resasco, J. Phys. Chem. Lett., 2011, 2, 2294–2295 CrossRef CAS.
- J. D. Adjaye and N. N. Bakhshi, Fuel Process. Technol., 1995, 45, 161–183 CrossRef CAS.
- J. D. Adjaye and N. N. Bakhshi, Fuel Process. Technol., 1995, 45, 185–202 CrossRef CAS.
- H. J. Park, J.-I. Dong, J.-K. Jeon, K.-S. Yoo, J.-H. Yim, J. M. Sohn and Y.-K. Park, J. Ind. Eng. Chem., 2007, 13, 182–189 CAS.
- S. Vitolo, B. Bresci, M. Seggiani and M. G. Gallo, Fuel, 2001, 80, 17–26 CrossRef CAS.
- M. Asadieraghi and W. M. A. Wan Daud, Energy Convers. Manage., 2015, 92, 448–458 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106 Search PubMed.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- A. Bridgwater, S. Czernik, J. Diebold, D. Meier, A. Oasmaa, C. Peacocke, J. Piskorz and D. Radlein, The Fast Pyrolysis of Biomass: A Handbook, CPL Press, Newbury, 2008 Search PubMed.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef CAS.
- Y. Yamada, M. Segawa, F. Sato, T. Kojima and S. Sato, J. Mol. Catal. A: Chem., 2011, 346, 79–86 CrossRef CAS PubMed.
- T.-S. Kim, S. Oh, J.-Y. Kim, I.-G. Choi and J. W. Choi, Energy, 2014, 68, 437–443 CrossRef CAS PubMed.
- J. Wildschut, F. H. Mahfud, R. H. Venderbosch and H. J. Heeres, Ind. Eng. Chem. Res., 2009, 48, 10324–10334 CrossRef CAS.
- J. Wildschut, M. Iqbal, F. H. Mahfud, I. M. Cabrera, R. H. Venderbosch and H. J. Heeres, Energy Environ. Sci., 2010, 3, 962–970 CAS.
- J. D. Adjaye, S. P. R. Katikaneni and N. N. Bakhshi, Fuel Process. Technol., 1996, 48, 115–143 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2610–2618 CrossRef CAS.
- R. K. Sharma and N. N. Bakhshi, Energy Fuels, 1993, 7, 306–314 CrossRef CAS.
- E. Taarning, C. M. Osmundsen, X. Yang, B. Voss, S. I. Andersen and C. H. Christensen, Energy Environ. Sci., 2011, 4, 793–804 CAS.
- N. Y. Chen, T. F. Degnan Jr and L. R. Koenig, Chemtech, 1986, 16, 506–511 CAS.
- R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
- K. M. Dooley, A. K. Bhat, C. P. Plaisance and A. D. Roy, Appl. Catal., A, 2007, 320, 122–133 CrossRef CAS PubMed.
- C. A. Gaertner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, J. Catal., 2009, 266, 71–78 CrossRef CAS PubMed.
- C. A. Gärtner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, ChemSusChem, 2009, 2, 1121–1124 CrossRef PubMed.
- C. A. Gaertner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, Ind. Eng. Chem. Res., 2010, 49, 6027–6033 CrossRef CAS.
- T. S. Hendren and K. M. Dooley, Catal. Today, 2003, 85, 333–351 CrossRef CAS.
- M. Gliński, W. Szymański and D. Łomot, Appl. Catal., A, 2005, 281, 107–113 CrossRef PubMed.
- R. Klimkiewicz, E. Fabisz, I. Morawski, H. Grabowska and L. Syper, J. Chem. Technol. Biotechnol., 2001, 76, 35–38 CrossRef CAS.
- J. Kim, Bioresour. Technol., 2015, 178, 90–98 CrossRef CAS PubMed.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS PubMed.
- S. M. Csicsery, Pure Appl. Chem., 1986, 58, 841–856 CrossRef CAS.
- H. K. G. Ertl, F. Schüth and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley-VCH, 2nd edn, 2008 Search PubMed.
- M. Asadieraghi, W. M. A. Wan Daud and H. F. Abbas, Renewable Sustainable Energy Rev., 2014, 36, 286–303 CrossRef CAS PubMed.
- A. T. To and D. E. Resasco, Appl. Catal., A, 2014, 487, 62–71 CrossRef CAS PubMed.
- O. Mante and F. Agblevor, Biomass Convers. Biorefin., 2011, 1, 203–215 CrossRef CAS PubMed.
- G. W. Huber, Y.-T. Cheng, T. Carlson, T. Vispute, J. Jae, G. Tompsett, US Pat., 2009/0227823 A1, 2009.
- F. A. Agblevor, US Pat., 2009/0165378 A1, 2009.
- F. A. Agblevor, US Pat., 2010/0212215 A1, 2010.
- E. F. Iliopoulou, S. D. Stefanidis, K. G. Kalogiannis, A. Delimitis, A. A. Lappas and K. S. Triantafyllidis, Appl. Catal., B, 2012, 127, 281–290 CrossRef CAS PubMed.
- A. A. Lappas, S. Bezergianni and I. A. Vasalos, Catal. Today, 2009, 145, 55–62 CrossRef CAS PubMed.
- S. J. Choi, S. H. Park, J.-K. Jeon, I. G. Lee, C. Ryu, D. J. Suh and Y.-K. Park, Renewable Energy, 2013, 54, 105–110 CrossRef CAS PubMed.
- R. French and S. Czernik, Fuel Process. Technol., 2010, 91, 25–32 CrossRef CAS PubMed.
- S. D. Stefanidis, K. G. Kalogiannis, E. F. Iliopoulou, A. A. Lappas and P. A. Pilavachi, Bioresour. Technol., 2011, 102, 8261–8267 CrossRef CAS PubMed.
- D. J. Mihalcik, C. A. Mullen and A. A. Boateng, J. Anal. Appl. Pyrolysis, 2011, 92, 224–232 CrossRef CAS PubMed.
- S. Vichaphund, D. Aht-ong, V. Sricharoenchaikul and D. Atong, Renewable Energy, 2014, 65, 70–77 CrossRef CAS PubMed.
- D. J. Mihalcik, A. A. Boateng, C. A. Mullen and N. M. Goldberg, Ind. Eng. Chem. Res., 2011, 50, 13304–13312 CrossRef CAS.
- K. Wang, K. H. Kim and R. C. Brown, Green Chem., 2014, 16, 727 RSC.
- M. S. Talmadge, R. M. Baldwin, M. J. Biddy, R. L. McCormick, G. T. Beckham, G. A. Ferguson, S. Czernik, K. A. Magrini-Bair, T. D. Foust, P. D. Metelski, C. Hetrick and M. R. Nimlos, Green Chem., 2014, 16, 407–453 RSC.
- D. Shen, R. Xiao, S. Gu and K. Luo, RSC Adv., 2011, 1, 1641 RSC.
- K. Moller and T. Bein, Chem. Soc. Rev., 2013, 42, 3689–3707 RSC.
- E. P. Önal, B. B. Uzun and A. E. Pütün, Fuel Process. Technol., 2011, 92, 879–885 CrossRef PubMed.
- S. Dutta, RSC Adv., 2012, 2, 12575 RSC.
- Z. L. Hua, J. Zhou and J. L. Shi, Chem. Commun., 2011, 47, 10536–10547 RSC.
- S. Stefanidis, K. Kalogiannis, E. F. Iliopoulou, A. A. Lappas, J. M. Triguero, M. T. Navarro, A. Chica and F. Rey, Green Chem., 2013, 15, 1647 RSC.
- K. Na, M. Choi and R. Ryoo, Microporous Mesoporous Mater., 2013, 166, 3–19 CrossRef CAS PubMed.
- Y. Li, S. Liu, Z. Zhang, S. Xie, X. Zhu and L. Xu, Appl. Catal., A, 2008, 338, 100–113 CrossRef CAS PubMed.
- G. Agostini, C. Lamberti, L. Palin, M. Milanesio, N. Danilina, B. Xu, M. Janousch and J. A. van Bokhoven, J. Am. Chem. Soc., 2009, 132, 667–678 CrossRef PubMed.
- M.-C. Silaghi, C. Chizallet and P. Raybaud, Microporous Mesoporous Mater., 2014, 191, 82–96 CrossRef CAS PubMed.
- F. Schmidt, M. R. Lohe, B. Büchner, F. Giordanino, F. Bonino and S. Kaskel, Microporous Mesoporous Mater., 2013, 165, 148–157 CrossRef CAS PubMed.
- M. Ogura, S.-y. Shinomiya, J. Tateno, Y. Nara, M. Nomura, E. Kikuchi and M. Matsukata, Appl. Catal., A, 2001, 219, 33–43 CrossRef CAS.
- J. C. Groen, J. C. Jansen, J. A. Moulijn and J. Pérez-Ramírez, J. Phys. Chem. B, 2004, 108, 13062–13065 CrossRef CAS.
- R. Chal, C. Gerardin, M. Bulut and S. van Donk, ChemCatChem, 2011, 3, 67–81 CrossRef CAS.
- M. D. González, Y. Cesteros and P. Salagre, Microporous Mesoporous Mater., 2011, 144, 162–170 CrossRef PubMed.
- J. Adam, M. Blazsó, E. Mészáros, M. Stöcker, M. H. Nilsen, A. Bouzga, J. E. Hustad, M. Grønli and G. Øye, Fuel, 2005, 84, 1494–1502 CAS.
- E. Antonakou, A. Lappas, M. H. Nilsen, A. Bouzga and M. Stöcker, Fuel, 2006, 85, 2202–2212 CrossRef CAS PubMed.
- G. Fogassy, N. Thegarid, Y. Schuurman and C. Mirodatos, Energy Environ. Sci., 2011, 4, 5068–5076 CAS.
- J.-K. Lee Hw Fau-Jeon, S. H. Jeon Jk Fau-Park, K.-E. Park Sh Fau-Jeong, H.-J. Jeong Ke Fau-Chae and Y.-K. Chae Hj Fau-Park, Nanoscale Res. Lett., 2011, 6, 6–500 Search PubMed.
- Q. Lu, Z. Tang, Y. Zhang and X.-f. Zhu, Ind. Eng. Chem. Res., 2010, 49, 2573–2580 CrossRef CAS.
- H. J. Park, H. S. Heo, J.-K. Jeon, J. Kim, R. Ryoo, K.-E. Jeong and Y.-K. Park, Appl. Catal., B, 2010, 95, 365–373 CrossRef CAS PubMed.
- H. J. Park, K.-H. Park, J.-K. Jeon, J. Kim, R. Ryoo, K.-E. Jeong, S. H. Park and Y.-K. Park, Fuel, 2012, 97, 379–384 CrossRef CAS PubMed.
- K. S. Triantafyllidis, E. F. Iliopoulou, E. V. Antonakou, A. A. Lappas, H. Wang and T. J. Pinnavaia, Microporous Mesoporous Mater., 2007, 99, 132–139 CrossRef CAS PubMed.
- A. Aho, N. Kumar, A. V. Lashkul, K. Eränen, M. Ziolek, P. Decyk, T. Salmi, B. Holmbom, M. Hupa and D. Y. Murzin, Fuel, 2010, 89, 1992–2000 CrossRef CAS PubMed.
- C. Rioche, S. Kulkarni, F. C. Meunier, J. P. Breen and R. Burch, Appl. Catal., B, 2005, 61, 130–139 CrossRef CAS PubMed.
- H. Zhang, R. Xiao, D. Wang, G. He, S. Shao, J. Zhang and Z. Zhong, Bioresour. Technol., 2011, 102, 4258–4264 CrossRef CAS PubMed.
- Y. Tang, W. Yu, L. Mo, H. Lou and X. Zheng, Energy Fuels, 2008, 22, 3484–3488 CrossRef CAS.
- W. Yu, Y. Tang, L. Mo, P. Chen, H. Lou and X. Zheng, Bioresour. Technol., 2011, 102, 8241–8246 CrossRef CAS PubMed.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- W.-J. Liu, X.-S. Zhang, Y.-C. Qv, H. Jiang and H.-Q. Yu, Green Chem., 2012, 14, 2226–2233 RSC.
- A. Gangadharan, M. Shen, T. Sooknoi, D. E. Resasco and R. G. Mallinson, Appl. Catal., A, 2010, 385, 80–91 CrossRef CAS PubMed.
- NABC, http://www.nabcprojects.org/pdfs/pyrolysis_vapors_upgrading.pdf, accessed October 2013.
- V. V. Lissianski, R. G. Rizeq and S. P. Singh, US Pat., 2012/0029252 A1, 2012.
- A. Sanna and J. M. Andresen, ChemSusChem, 2012, 5, 1944–1957 CrossRef CAS PubMed.
- H. Park, J.-K. Jeon, D. Suh, Y.-W. Suh, H. Heo and Y.-K. Park, Catal. Surv. Asia, 2011, 15, 161–180 CrossRef CAS.
- E. Pütün, Energy, 2010, 35, 2761–2766 CrossRef PubMed.
- T. S. Nguyen, M. Zabeti, L. Lefferts, G. Brem and K. Seshan, Bioresour. Technol., 2013, 142, 353–360 CrossRef CAS PubMed.
- A. Imran, E. A. Bramer, K. Seshan and G. Brem, Fuel Process. Technol., 2014, 127, 72–79 CrossRef CAS PubMed.
- J. Payormhorm, K. Kangvansaichol, P. Reubroycharoen, P. Kuchonthara and N. Hinchiranan, Bioresour. Technol., 2013, 139, 128–135 CrossRef CAS PubMed.
- Q. Lu, Y. Zhang, Z. Tang, W.-z. Li and X.-f. Zhu, Fuel, 2010, 89, 2096–2103 CrossRef CAS PubMed.
- D. Wang, R. Xiao, H. Zhang and G. He, J. Anal. Appl. Pyrolysis, 2010, 89, 171–177 CrossRef CAS PubMed.
- Y. Lin, C. Zhang, M. Zhang and J. Zhang, Energy Fuels, 2010, 24, 5686–5695 CrossRef CAS.
- C. Torri, M. Reinikainen, C. Lindfors, D. Fabbri, A. Oasmaa and E. Kuoppala, J. Anal. Appl. Pyrolysis, 2010, 88, 7–13 CrossRef CAS PubMed.
- X. Y. Lim and J. M. Andrésen, Fuel Process. Technol., 2011, 92, 1796–1804 CrossRef CAS PubMed.
- I. s. Graça, J.-D. Comparot, S. b. Laforge, P. Magnoux, J. M. Lopes, M. F. Ribeiro and F. Ramôa Ribeiro, Energy Fuels, 2009, 23, 4224–4230 CrossRef.
- I. Graça, J. D. Comparot, S. Laforge, P. Magnoux, J. M. Lopes, M. F. Ribeiro and F. R. Ribeiro, Appl. Catal., A, 2009, 353, 123–129 CrossRef PubMed.
- P. A. Horne and P. T. Williams, J. Anal. Appl. Pyrolysis, 1995, 34, 65–85 CrossRef CAS.
- B. Valle, P. Castaño, M. Olazar, J. Bilbao and A. G. Gayubo, J. Catal., 2012, 285, 304–314 CrossRef CAS PubMed.
- A. G. Gayubo, B. Valle, A. T. Aguayo, M. Olazar and J. Bilbao, Energy Fuels, 2009, 23, 4129–4136 CrossRef CAS.
- T. R. Carlson, Y.-T. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145–161 CAS.
- H. J. Park, H. S. Heo, J.-K. Jeon, J. Kim, R. Ryoo, K.-E. Jeong and Y.-K. Park, Appl. Catal., B, 2010, 95, 365–373 CrossRef CAS PubMed.
- Y. Ni, A. Sun, X. Wu, G. Hai, J. Hu, T. Li and G. Li, Microporous Mesoporous Mater., 2011, 143, 435–442 CrossRef CAS PubMed.
- J. Kim, M. Choi and R. Ryoo, J. Catal., 2010, 269, 219–228 CrossRef CAS PubMed.
- H. Ben and A. J. Ragauskas, RSC Adv., 2012, 2, 12892 RSC.
- A. Aho, N. Kumar, K. Eränen, T. Salmi, M. Hupa and D. Y. Murzin, Fuel, 2008, 87, 2493–2501 CrossRef CAS PubMed.
- T. Q. Hoang, X. Zhu, L. L. Lobban, D. E. Resasco and R. G. Mallinson, Catal. Commun., 2010, 11, 977–981 CrossRef CAS PubMed.
- M. Ibáñez, B. Valle, J. Bilbao, A. G. Gayubo and P. Castaño, Catal. Today, 2012, 195, 106–113 CrossRef PubMed.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, B. Valle and J. Bilbao, J. Chem. Technol. Biotechnol., 2005, 80, 1244–1251 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Prieto and J. Bilbao, Energy Fuels, 2004, 18, 1640–1647 CrossRef CAS.
- H. Zhang, Y.-T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297–2307 CAS.
- N. Chen, T. Degnan and L. Koenig, Chemtech, 1986, 16, 506–511 CAS.
- H. Zhang, T. R. Carlson, R. Xiao and G. W. Huber, Green Chem., 2012, 14, 98 RSC.
- I. Graça, J. M. Lopes, M. F. Ribeiro, M. Badawi, S. Laforge, P. Magnoux and F. Ramôa Ribeiro, Fuel, 2012, 94, 571–577 CrossRef PubMed.
- C. A. Mullen and A. A. Boateng, Fuel Process. Technol., 2010, 91, 1446–1458 CrossRef CAS PubMed.
- B. Valle, A. G. Gayubo, A. s. T. Aguayo, M. Olazar and J. Bilbao, Energy Fuels, 2010, 24, 2060–2070 CrossRef CAS.
- C. A. Mullen, A. A. Boateng, D. J. Mihalcik and N. M. Goldberg, Energy Fuels, 2011, 25, 5444–5451 CrossRef CAS.
- A. Aho, N. Kumar, K. Eränen, T. Salmi, M. Hupa and D. Y. Murzin, Process Saf. Environ. Prot., 2007, 85, 473–480 CrossRef CAS.
- A. J. Foster, J. Jae, Y.-T. Cheng, G. W. Huber and R. F. Lobo, Appl. Catal., A, 2012, 423–424, 154–161 CrossRef CAS PubMed.
- A. Sanna, S. Li, R. Linforth, K. A. Smart and J. M. Andresen, Bioresour. Technol., 2011, 102, 10695–10703 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |