
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anti-bacterial surfaces: natural agents, mechanisms of action, and plasma surface modification
K.
Bazaka
*ab,
M. V.
Jacob
b,
W.
Chrzanowski
c and
K.
Ostrikov
acd
aSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology, Brisbane, Australia. E-mail: kateryna.bazaka@qut.edu.au; kostya.ostrikov@qut.edu.au
bCollege of Science, Technology and Engineering, James Cook University, Townsville, Australia. E-mail: katia.bazaka@jcu.edu.au; mohan.jacob@jcu.edu.au; Fax: +61 7 4781 5177; Tel: +61 7 4781 4494
cFaculty of Pharmacy, University of Sydney, Sydney, Australia. E-mail: wojciech.chrzanowski@sydney.edu.au
dCSIRO Materials Science and Engineering, Sydney, Australia. E-mail: kostya.ostrikov@csiro.au
First published on 21st May 2015
Abstract
Strategies that confine antibacterial and/or antifouling property to the surface of the implant, by modifying the surface chemistry and morphology or by encapsulating the material in an antibiotic-loaded coating, are most promising as they do not alter bulk integrity of the material. Among them, plasma-assisted modification and catechol chemistry stand out for their ability to modify a wide range of substrates. By controlling processing parameters, plasma environment can be used for surface nano structuring, chemical activation, and deposition of biologically active and passive coatings. Catechol chemistry can be used for material-independent, highly-controlled surface immobilisation of active molecules and fabrication of biodegradable drug-loaded hydrogel coatings. In this article, we comprehensively review the role plasma-assisted processing and catechol chemistry can play in combating bacterial colonisation on medically relevant coatings, and how these strategies can be coupled with the use of natural antimicrobial agents to produce synthetic antibiotic-free antibacterial surfaces.
 K. Bazaka | Dr Kateryna Bazaka is an ARC DECRA Fellow with Health and Biomedical Technologies, Queensland University of Technology, Australia. Kateryna is a recipient of the Australian Institute of Nuclear Science and Engineering Postgraduate Award, the Queensland Government Smart Women Smart State Award, the Science and Innovation Awards for Young People in Agriculture Fisheries and Forestry, two Endeavour Research Fellowships, the Inaugural Advanced Manufacturing Cooperative Research Centre Student Prize, an AINSE Gold Medal, and an author of 1 monograph and 40 refereed journal papers. Her research focuses on nanoscale processing of materials and living matter for biomedical and electronic applications. |
 M. V. Jacob | Associate Professor Mohan Jacob is currently the Associate Dean Research Education for the College of Science, Technology and Engineering, James Cook University. University of Delhi awarded him PhD in 1999 in Electronic Science. He published over 150 peer reviewed articles. He developed methods to precisely characterize the dielectric properties of materials at cryogenic temperatures and microwave frequencies. His main research interests also includes the development of polymer thin films and graphene from sustainable sources using plasma enhanced chemical vapor deposition, environmentally friendly biomaterials and electronic and biomedical devices. |
 W. Chrzanowski | Dr Wojciech Chrzanowski joined the University of Sydney in 2010 and he has established the Bio-interface group and Bio-nano-characterisation laboratory within the Faculty of Pharmacy. His research is balanced between basic and translation sciences. He develops multifunctional surfaces and new examination approaches to interrogate biological responses to biomaterials at nanoscale. Outcomes of his research inform the design of new biologically active materials for implantable devices and drug delivery. For a series of publications describing significant advances in nanobiomedical sciences and biointerfaces he was invited to present over 50 seminars, and lectures at Universities in the USA, Japan, Australia, UK, and Korea. He received three fellowships (UCL, Tokyo University and Chubu University). Dr Chrzanowski published over 100 peer-reviewed publications (last 10 years). His publications attracted 1300 citations and his h-index is 20. He is also an inventor on 4 patents. |
 K. Ostrikov | Professor Kostya (Ken) Ostrikov is a Science Leader, ARC Future Fellow, Chief Research Scientist with CSIRO's Manufacturing Flagship and a Professor with the Institutes of Future Environments and Health and Biomedical Technologies of the Queensland University of Technology, Australia. His achievements include the Pawsey (2008) medal of the Australian Academy of Sciences, the Walter Boas (2010) medal of the Australian Institute of Physics, Building Future Award (2012), the recent NSW Science and Engineering Award (2014), 8 prestigious fellowships in 6 countries, 3 monographs, and more than 430 refereed journal papers. His research on nanoscale control of energy and matter contributes to the solution of the grand challenge of directing energy and matter at the nanoscale, a challenge that is critical for the development of renewable energy and energy-efficient technologies for a sustainable future. |
1. Introduction
In the last twenty years, significant progress has been made in the development of biomaterials and implantable devices, which are characterised by superior biocompatibility, desired integration with peri-implant tissues, controlled fouling with host cell and biomolecules, and which cause minimal acute or chronic inflammation. Numerous modification techniques have been developed to ensure satisfactory clinical performance of these devices by improving their biocompatibility with cells/tissues, by tailoring chemical composition and mechanical properties for specific application. To illustrate, metals such as titanium (Ti) are frequently modified by grain refinement to improve mechanical properties and enhance osteoblast attachment; abrasive-blasted to modify topography and thus improve osseointegration; polished mechanically and chemically to achieve smooth surface morphology to reduce integration with tissues and ease the removal of the devices (short-term implants); passivated/oxidised to improve corrosion resistance and enhance bioinertness; and coated with biomolecules, e.g. proteins and DNA fragments, and other biologically active species for guided cell attachment and integration with host tissues, to name but a few.At the same time, the susceptibility of the implant surface to bacterial colonisation and biofilm formation remains a major problem that is most commonly dealt with by means of prophylaxis with systemic antibiotics. Although administration of broad-spectrum systemic antibiotics is effective in preventing biomaterial-associated infection arising from pathogens introduced into the peri-implant space in the course of surgery or post-operative care, the practice is far less effective in dealing with late haematogenous infections. In the case of the latter, bacteria from an inflammation site elsewhere in the body can enter the blood stream and thus be transferred to the implant surface. In the absence of antibacterial agent, there is little to stop the pathogen from attaching to the surface and initiating a biofilm formation. The colonisation occurs quickly and is rarely detected clinically in time to prevent biofilm formation. Once formed, the biofilm affords the pathogenic cells necessary protection against flow detachment, opsonisation, and the harmful effects of host antimicrobial molecules and systemic antibiotics.1 In biofilm state, the expression of genes and metabolic activity in bacterial cells may also differ from that of their planktonic counterparts, which may lessen sensitivity of sessile bacteria to certain antimicrobials designed to target the pathogen's metabolism.2–4 Oftentimes, even significantly higher doses of systemic antimicrobials are insufficient to clear the biofilm, and implant replacement is required.
Although sound hospital practices ensure the rate of implant-associated infections remains relatively low, the ever increasing volume and variety of biomaterials and medical devices implanted globally results in a substantially large number of infections. Furthermore, increasing human life expectancy and emphasis on active lifestyle is associated with a growing number of revision surgeries, and these are known to have a significantly higher infection rate. With the growing issue of hospital acquired and multi-drug resistant microorganisms,5 there is a strong need to engineer biomaterials that retard microorganism colonisation in the first place.
2. Trends in surface modification
Microbial attachment can be effectively mitigated by introducing an antimicrobial agent throughout the bulk of the material, e.g. silver can be blended into bulk polymeric materials, alloyed into metallic biomaterials, or introduced into glass/ceramic materials.6–8 While the nature of the resultant material ensures the long-lasting antimicrobial effect, the addition of the antibacterial agent may negatively impact on the fundamental properties, stability or processability of the material. In comparison, surface modification can be applied to existing biomaterials, with little impact on such bulk properties as mechanical strength and stability under in vivo conditions, and is often more cost and time effective.9,10When selecting appropriate surface modification approach, there are many material- and application-based considerations that need to be addressed. From processing point of view, the choice of the appropriate method is based on its compatibility with the type of biomaterial, i.e. polymeric, metallic, ceramic or composite; its stability, i.e. temperature sensitivity, solubility, mechanical robustness, etc.; its physical structure, e.g. porosity, and dimension, e.g. bulk or thin film; to name a few.
Financial cost, ease of integration, and scalability of potential modification techniques also need to be considered. For instance, chemical vapour deposition of vertically aligned carbon nanotube forests consumes more time, energy and resources than template-based fabrication of polymer structures. The former is also more difficult to scale up or translate into continuing processing. At the same time, nanoscale materials, such as nanotubes or graphene sheets offer unique and highly valuable properties, such as extreme mechanical strength and durability, electrical and thermal conductivities, and highly adjustable chemical reactivity. Indeed, although relatively easy to fabricate, polymer structures are more fragile and fail easily under load or wear conditions.
From application perspective, general considerations include the intended use, e.g. whether the surface will be subjected to load, wear, flow or harsh chemical environment, as well as the length for which antimicrobial activity is required. The proposed application also places restrictions on the type of antimicrobial activity, for example antibiofouling surfaces may be desirable for urinary tract catheters, but they will not be appropriate for materials where tissue regeneration is required. In general, a biomaterial with excellent bactericidal activity but poor compatibility with host biomolecules, cells and tissues is unlikely to find broad clinical use.
Even a non-cytotoxic coating aimed at preventing bacterial adhesion may change the density or porosity of the underlying material, with significant consequences for attachment, differentiation and metabolic activity of target mammalian cells. Changing surface topography of the biomaterial may also change the availability of specific chemical functionalities at its surface, or reconfigure their 3D confirmation. It is therefore important to understand the interdependence of surface chemistry and physics in order to adequately predict the resultant biological performance with respect to bacteria and mammalian cells.11
The type of antimicrobial agent, its ability to withstand processing conditions, and maintain its antimicrobial potency in the final conformation under physiological conditions will also affect the choice of modification methodology.
2.1 Physico-chemical modifications
For a number of years, control over the attachment and biofilm formation of microorganisms was achieved using specific surface chemistries. This is hardly surprising, as molecular recognition is acknowledged as one of the key factors in determining not only cell–surface interactions, but also many biological functions within the cell itself. These chemistries can be imparted onto the surface by a variety of means, including plasma-assisted techniques, such as plasma (thermal) spraying, plasma immersion ion implantation, and plasma deposition, gas dynamic cold spraying, chemical and physical vapour deposition, and sol–gel. Hydrotropic nanostructues, such as carbon and halloysite nanotubes can also be used to control fouling.12 The key challenge in using these methods is in ensuring that the treatment process and/or the resultant surface chemistry do not undermine the biocompatibility, performance and degradation behaviour of the biomaterial in vitro and in vivo.With the development of novel data acquisition, analysis and visualisation tools, our understanding of cell–surface dynamics have evolved to include the physical as well as the chemical properties of biomaterials as key factors that can regulate biological responses of cells and tissues.13 As a result, several modification strategies have been developed that rely on the synergistic effect of chemistry, e.g. hydrophobic moieties, and surface morphology, e.g. hierarchical arrangement of nano- and micro-features, to prevent microbial attachment and biofilm formation (Fig. 1).14 Laser ablation, abrasive blasting, physical vapour deposition, self-assembly, evaporation and ion assisted deposition are among the frequently used physical modification techniques.
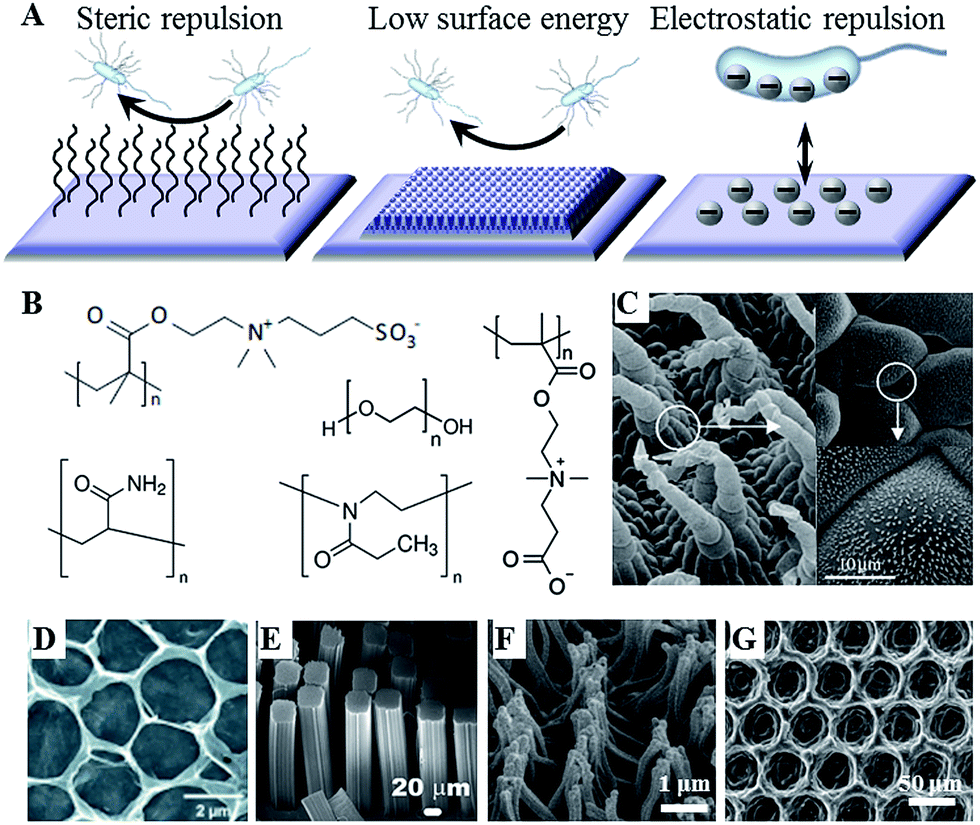 | ||
| Fig. 1 (A) Antifouling strategies for biofilm management. (B) Commonly used hydrophilic chemistries, e.g. poly(ethylene glycol), poly(methyl oxazoline), polyacrylamide, and zwitterionic poly(carboxybetaine methacrylate) and poly(sulfobetaine methacrylate). (C–G) Natural and artificial superhydrophobic surfaces. (C) The hierarchical structure of Salvinia spp. hairs, composed of the multicellular hair with small rodlet-like wax crystals on top.15 (D) Macroporous graphene oxide film (CA = 152°).16 (E) Gecko-inspired setae made of micropatterned carbon nanotube bundles (CA = 155°).17 (F) Perfluoropolyether polymer hairs (CA = 171°).18 (G) Epoxy/γ-Al2O3 nanoparticle composite (CA = 160°).19 Reproduced with permission from ref. 15–20. | ||
Nanostructured surfaces with surface chemistry-independent antimicrobial effect have also been reported.21Fig. 2 shows the nanopattern on the surface of Clanger cicada (Psaltoda claripennis) wings which allows the surfaces to kill bacteria on contact based solely on its physical surface structure. Although unable to prevent microbial attachment,22 the direct contact between the attached cells and highly ordered arrays of surface nanopillars resulted in cell membrane stretching and eventual rupturing, where the adsorption behaviour of bacterial cells and their sensitivity to the material surfaces depended both on the geometry of the pillars and the mechanical properties of the cells, especially cell rigidity.21 In spite of substantial advances in our understanding of how the physical properties of materials determine cell–surface dynamics at nano-, molecular- and atomic scales, this field requires considerable further development.
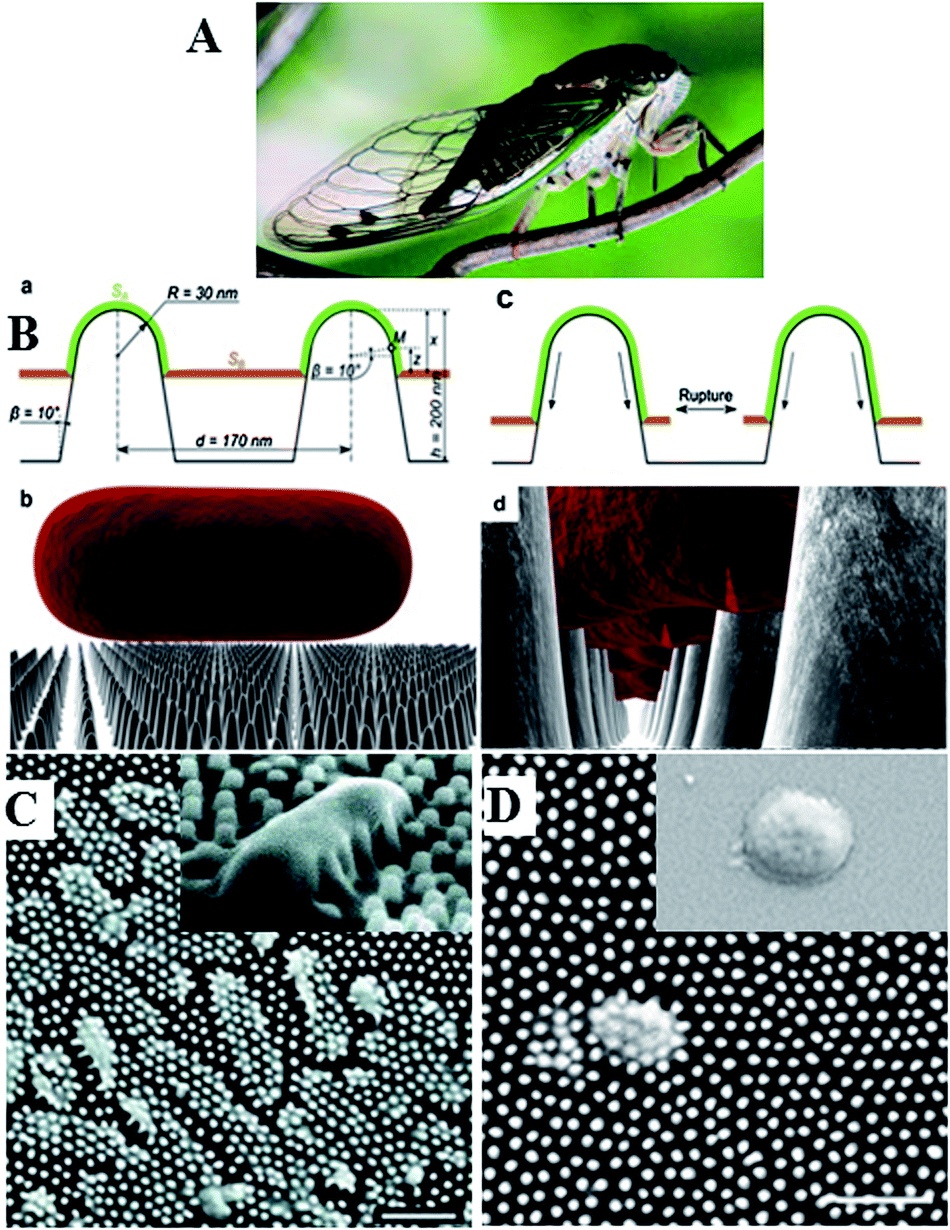 | ||
| Fig. 2 The unique surface morphology of wing surface enables Clanger cicada (A) to resist bacterial colonisation. (B) Proposed mechanism of chemistry-independent contact killing of bacteria on cicada wing surface. (C and D) SEM images of clinically relevant pathogens on the surface of a cicada wing. Pseudomonas aeruginosa (C) and Branhamella catarrhalis (D) cells are clearly penetrated by the nanopillar structures on the wing surface, with cells sinking between the nanopillars (C, inset). On the wing surfaces, bactericidal effect observed for all tested Gram-negative microorganisms, regardless of cell morphology. On glass (D, inset) under equivalent incubation conditions, no killing effect was observed. Reproduced with permission from ref. 21, 22 and 25. | ||
Surface physical properties can be used to enhance the antimicrobial effect. For instance, physical disruption of cell membranes have been demonstrated as an essential contributor to antimicrobial efficacy of copper surfaces, where the damage to cell envelope facilitated further damage by copper ions,23 and of copper containing nanoparticles, where particles also acted as physical carriers of copper into the cells.24
2.2 Biocide-based strategies
Broadly, the antimicrobial agent can either be entrapped in the coating to be released in some predefined fashion upon interaction with its operational environment and/or stimuli, e.g. drug eluting hydrogels and coatings, or immobilised on the surface of the implant to prevent bacterial attachment and/or kill the attached cells on contact, e.g. covalently attached polymer brushes, conventional antibiotics and antimicrobial peptides.There is merit to both strategies. The main advantage of non-leaching systems is in the confinement of the cytotoxic effect to the surface of the implant, thus minimising the potentially harmful interactions between the agent and host tissues, e.g. damage to host cells in peri-implant milieu or accumulation of the antimicrobial liver, spleen, and brain.26,27 Given that the antimicrobial agent is not depleted over time, the effect is sustained for longer. Furthermore, the issue of bacterial cells being exposed to sub-inhibitory concentrations of the antibiotic is avoided. This minimises the chance these bacteria will develop resistance to the drug in use.28
With the efficacy of many antimicrobials relying on a combination of chemical functionality and spatial conformation, covalent immobilisation is more conducive to attaining specific molecular orientation of the agent on the surface. As such, the availability of specific chemistries and structural motifs characteristic of the antimicrobial in suspension can be maintained.29 Nevertheless, prolonged exposure to the physiological environment may result in the concealment of the antimicrobial chemical and physical features of the surface, e.g. through adsorption of host biomolecules or accumulation of killed bacterial cells and their fragments.30 Furthermore, a broader variety of antimicrobial agents can be entrapped in the release- or leach-systems, and their concentration and release rate controlled to ensure bacterial inhibition further away from the implant surface into the peri-implant space. The challenge that is common to drug-release and non-leaching antimicrobial systems is the control over the quality of adhesion between the active agent and the underlying biomaterial surface.
The example in Fig. 3 shows the use of traditional antibiotics as biocidal agents. For antibiotic-sensitive strains of bacteria, these coatings provide an effective means of combating infection. However, a growing emergence of bacteria with antibiotic resistance, particular in hospital settings, resulted in a growing interest in alternative therapeutic concepts and agents. Ideally, these alternative agents should lead to the elimination of bacteria, and have a mode of action that would be sufficiently dissimilar to systemic antibiotics to avoid promoting cross-resistance.
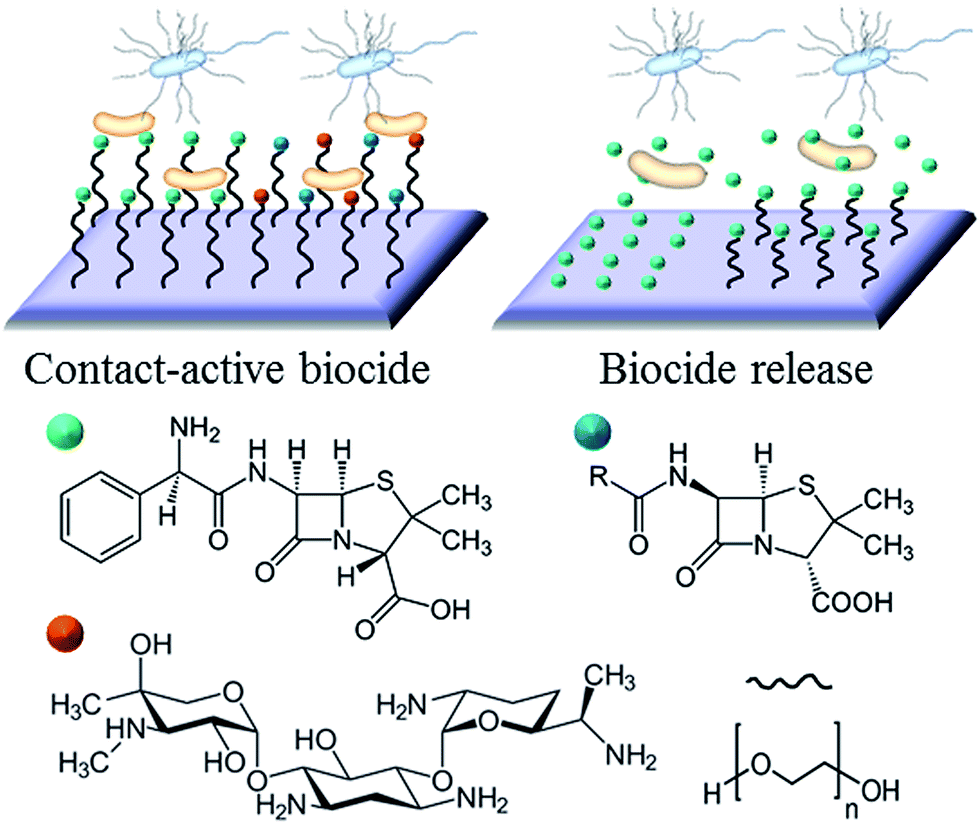 | ||
| Fig. 3 Principles of bactericide contact and release coatings based on conventional antibiotics (penicillin, ampicillin, and gentamicin). Antibiotics can be used individually or in combination. Active agents can be physically adsorbed onto the surface or covalently conjugated to the polymer chain (in this example, PEG). | ||
2.3 Aim and article organisation
The aim of this article is to review two types of highly-versatile modification chemistries, namely (i) highly reactive plasma chemistry and (ii) catechol chemistry that can be applied to a wide range of substrate materials to produce a variety of antifouling, biocide-releasing and contact kill surfaces. A particular focus of this review is the potential use of plasma and catechol chemistries as enabling technologies for surface modification based on natural antimicrobial compounds.Section 2 will provide a broad perspective on surface modification of biomaterials for controlling microbial attachment and biofilm formation, giving examples of desirable surface properties and the methods that are used to attain these properties. Given the vast variety of materials, applications and surface modification methodologies, the fully exhaustive coverage of the relevant existing knowledge is outside the scope of this review.
Section 3 will discuss two broad classes of natural antimicrobial compounds, specifically secondary plant metabolites and antimicrobial peptides, as a viable alternative to conventional systemic antibiotics.
Section 4 will review catechol chemistries inspired by the distinctive water-resistant, material-independent adhesive abilities of many sessile aquatic organisms.31 This section will discuss the relevance of this chemistry to both the assembly of well-adhering releasing hydrogels and for the design of substrate-independent adhesive coatings that can serve as a base layer for further functionalization, such as covalent binding of natural antimicrobial agents.
Section 5 will review general principles of plasma assisted surface modification, as a technique that can be used for surface patterning, deposition of contact killing and drug eluting coatings and for surface functionalization that can be subsequently used for drug immobilisation in non-release systems. Section 5 will concentrate on select examples of using plasma environment to process natural antimicrobial agents into bioactive coatings.
3. Antimicrobials of natural origin
The use of systemic antibiotics has been challenged on many levels, the key issue concerned with its contribution towards the development of microbial resistance. And while these agents remain among the most potent weapons in treating advanced infections, there has been an increasing interest in the use of alternative, nature-derived antimicrobials, whose physico-chemical structure and mechanism of bioactivity are sufficiently dissimilar to those of currently used synthetic antibiotics to eliminate the possibility of bacterial cross-resistance. Development of cross-resistance is an important problem, and methicillin resistant Staphylococcus aureus is one of the best known examples of microorganisms with multi-drug resistance against most currently available antibiotics, including recent cases of vancomycin-resistant S. aureus.Other notable clinically significant drug-resistant pathogens include Acinetobacter baumannii, P. aeruginosa, E. coli and Klebsiella pneumoniae resistant to β-lactamases, and Mycobacterium tuberculosis.32 Although some bacterial organisms are intrinsically resistant to some antimicrobials, excessive use and/or misadministration of antibiotics may select for pathogens that acquired resistance by either de novo mutations or via gene transfer, conjugation, transformation, and transduction.32 These newly acquired genes can complement and thus enhance the intrinsic resistance of the microorganism.
Phenotypically, the changes in genotype can manifest in many ways, including synthesis of enzymes capable of deactivating antibacterial agent, physico-chemical changes to the site targeted by the antimicrobial, activation of an alternative metabolic pathway to circumvent the activity of the drug, and to minimise the accessibility of internal drug targets via various efflux mechanisms. Amongst numerous alternative antimicrobials, metal ions, nitric oxide, antimicrobial peptides, and secondary metabolites derived from plant organisms provide a diverse range of antimicrobial agents.
3.1 Antimicrobial peptides
Antimicrobial peptides are produced by all complex organisms as well as some microbes as part of innate immune response, and display diverse and complex antimicrobial activities against a broad range of Gram negative and Gram positive bacteria, including those resistant to established antibiotic drug therapies, mycobacteria, enveloped viruses, parasites and fungi.35,36 Also known as host defense peptides, these are low molecular mass amphipathic molecules of 12–50 amino acids in length, and are secreted by many different cell types, either constitutively or in response to inflammatory stimuli.37 These molecules typically perform more than one function within the organism (Fig. 4). For instance, peptides produced by neurons, e.g. neurokinin-1, neuropeptide Y, orexins, function as both the neurotransmitters in the brain and the peripheral nervous system, and as immunomodulators, regulating immune function and neurogenic inflammatory responses through vasodilatation, plasma extravasation, and recruitment of immunocompetent cells.35,38–40 Orexin B has been reported to affect the function of peritoneal macrophages via activation of calcium-dependent potassium channels and to facilitate enhancement of phagocytosis in mouse peritoneal macrophages.38,41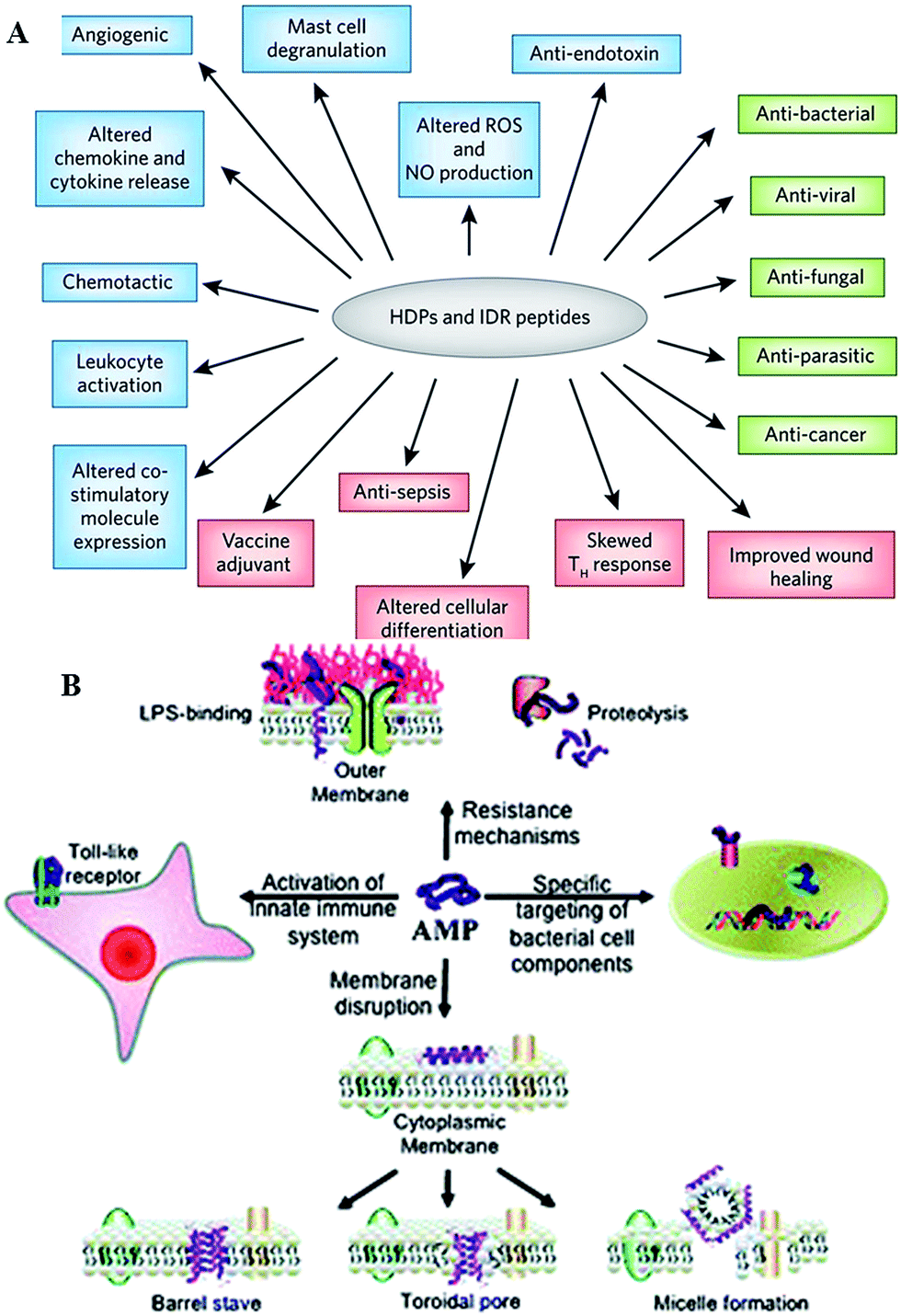 | ||
| Fig. 4 (A) Overview of the biological activities of host defense peptides (HDPs) and innate defense regulator (IDR) peptides. Direct cytotoxic activities are shown in green, direct and indirect immunomodulatory properties are in blue and pink, respectively. ROS, reactive oxygen species; NO, nitric oxide.33 (B) Overview of the broad spectrum of cellular interactions associated with antimicrobial peptides. Peptides exert antimicrobial activity by disrupting bacterial membranes, binding to specific target proteins within microbial cells and activating the innate immune system.34 Reproduced with permission from ref. 33 and 34. | ||
In plants, thionins, defensins, lipid transfer proteins, hevein- and knottin-like peptides, MBP1, IbAMP, and the recently reported snakins are the most commonly encountered antimicrobial peptides.42 Structurally, these are small cationic peptides with molecular masses of 2–10 kDa, with their structure stabilized via the formation of 2–6 disulfide bridges. The antibacterial mechanism of thionins is through the binding of phospholipids of the bacterial membrane which initiates a cascade of cytoplasmic events leading to cell death.43,44 High positive charge, which renders them extremely soluble (>300 mg ml−1), and the phospholipid-binding specificity of thionin allows the agent to bind areas of negatively charged phospholipids, either phosphatidic acid or phosphatidyl serine, and their subsequent withdrawal. The segregation of phospholipids destabilizes the membrane, causing its solubilisation and lysis.
Defensins, typically 45–54 amino acids long cationic peptides, display only modest antimicrobial activity, with sound efficacy against diverse fungi.45,46 Specific defensins have also been reported to inhibit protein synthesis, protease trypsin, or a-amylase activity.45 Lipid transfer proteins range in size between 7 and 10 kDa, and are typically of globular structure with a large hydrophobic cavity. The cavity serves as a binding site for mono- or diacylated lipids and other hydrophobic molecules, with a larger cavity of LTP2 allowing it to bind a planar sterol.
In terms of the use of antimicrobial peptides as antibacterial surface modification, numerous strategies have been tested to deliver these antimicrobials to the site of implantation. The ionic self-complementary of peptides allows for their use as building blocks for self-assembly of nanostructures. Each amphipathic molecule is comprised of distinct hydrophilic and hydrophobic regions. In aqueous environment, the hydrophobic region tries to minimise its exposure to water, resulting in folding of the molecule. The hydrophilic domain is composed of alternating positively charged (e.g. arginine, lysine) and negatively charged (e.g. aspartate, glutamate) amino acid residues, with various patterns of distribution of the charged residues. These residues will engage in ionic interactions with the oppositely charged residues of the complimentary molecule, driving the self-assembly. Although non-covalent in nature, the interactions are sufficiently strong to support highly stable structures.
Hydrogels of β-hairpin peptides rich in arginine displayed strong antibacterial activity against Gram-positive and Gram-negative bacteria, including multi-drug resistant P. aeruginosa.47 The fundamental and functional properties of the hydrogel, including killing efficacy, host cytocompatibility, bulk rheological properties and stimuli-responsiveness of this type of hydrogel can be controlled via selection of the specific peptide sequence at the monomer level.48–50 Pre-functionalisation of the implant surfaces, e.g. via plasma-assisted treatment, has been used for UV immobilisation of ε-poly-L-lysine-graft-methacrylamide hydrogel thin film.51 Coupled with excellent activity against bacteria and fungi, the low thickness of the coating makes it a good candidate for coating over medical devices and implants.
A mode of delivery via loading of antimicrobial peptides into a carrier platform has been trialled. Kazemzadeh-Narbat et al. used micro-porous octacalcium phosphate films to load broad spectrum antimicrobial peptides for orthopaedic applications.52 Shukla et al. used thin layers of polyionic polymer films to physically entrap the antimicrobial agent, varying layer number and composition for control over the amount of agent loaded into the structure.53 It has been suggested that the antimicrobial agent may not be able to diffuse through the layers of the polymer at a sufficient rate to ensure the steady level of the antimicrobial at the surface. In addition to the intrinsic properties of the layers, through which the peptide is to diffuse, its release may also be limited by the bacterial cells accumulated at the surface of the implant.54
Physical and chemical immobilisation of the peptide on the surface of the implant can circumvent the issues with diffusion, although accumulation of bacterial debris may still remain a challenge.56 Although reported as an effective approach for peptide surface immobilisation, non-specific physical adsorption may compromise the availability of physico-chemical particulates of the peptide present in the soluble analogue, potentially rendering the coating inactive.55 Even specific, covalent attachment of the peptide is likely to affect the structural exposure and flexibility characteristic of the peptide.29 For instance, even though notably larger amounts of peptide per unit area can be conjugated to a surface via polymer brush compared to the direct grafting of peptides, a significant portion of these peptides may not be available to interact with biomembranes due to steric restrictions exerted by the polymer brush structure.55 On the other hand, the surface-tethered peptides may be more effective in combating microbial colonisation due to higher concentration of appropriately structured peptides in one location (Fig. 5).
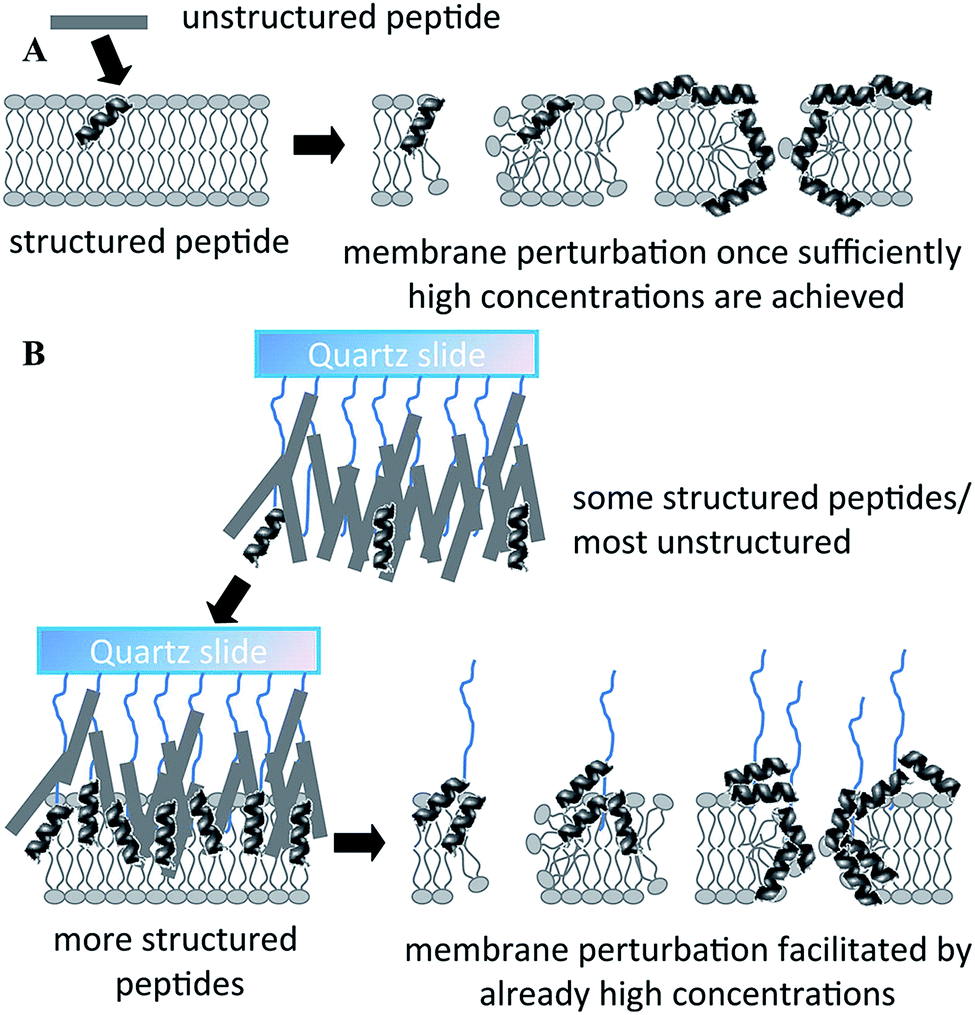 | ||
| Fig. 5 Proposed mechanism of action of free (A) and polymer brush-immobilized host defense peptide (B). In (B), more peptides adopt a structure before they interact with the membrane, and therefore there may be more structured peptides localized in one area when they bind/insert into the membrane. As a result, perturbation of the membrane may be more efficient. Reproduced with permission from ref. 55. | ||
To minimise the detrimental effects of the binding, many methods have been developed, employing a variety of chemical coupling strategies, length of spacers, and peptide orientation and concentration.57,58 Many of these strategies focus on minimising nonspecific interactions between the peptide and the substrate.59 Another important consideration in using peptide antimicrobials is their stability,60 as well as the stability of the coating system as a whole under physiological conditions.61
3.2 Antimicrobial secondary plant metabolites
Plants produce a broad assortment of secondary metabolites, including tannins, terpenoids, alkaloids, polyphenols and flavonoids, which have been found in vitro to have antimicrobial properties against both Gram positive and Gram negative bacteria. Furthermore, these phytochemicals have been shown to modulate or modify resistance mechanisms in bacteria.62 Yet, since the discovery of penicillin in the 1950s, the medical world has relied on antibiotics derived from bacterial and fungal sources, with the use of plant derivatives as antimicrobials being nearly non-existent.63 One of the possible reasons for this is that the relatively higher minimum inhibitory concentrations limited their utility as the sole agents, although certain combination of phytochemicals with conventional antimicrobial drugs demonstrated enhanced efficacy against methicillin resistant S. aureus.62 In that case, tannic acid was able to prolong and potentiate the bactericidal activity of fusidic acid, cefotaxime, minocycline and rifampicin, with a similar effect demonstrated for combinations of quercetin with fusidic acid, minocycline and rifampicin.Amongst the vast variety of phytochemicals, phenolics, terpenoids and other essential oils constituents, alkaloids, lectins and polypeptides, and polyacetylenes are most commonly associated with antimicrobial activity.32 These phytochemicals play other roles in plant physiological processes, e.g. flavonoids are the key pigments for plants that reproduce via biotic pollination; flavonoids are also involved in UV filtration and symbiotic nitrogen fixation; and as chemical messengers, physiological regulators, and cell cycle inhibitors. The use of these antimicrobial agents has been limited to traditional and alternative medical domains, yet to be recognised by the mainstream medical community as therapeutic agents. As mentioned above, one of the main reasons lies in the relatively weak and/or narrow spectrum of antimicrobial activity, and potentially high toxicity associated with the administration of sufficiently high antimicrobial doses. Indeed, the MICs typically reported for plant-derived antimicrobials are in the range of 100 to 1000 μg ml−1, orders of magnitude weaker than MICs of 0.01 to 10 μg ml−1 of antimicrobials synthesised by bacteria and fungi.64 It has been suggested that along with these antimicrobials, plants may produce a range of other chemicals, e.g. inhibitors of bacterial multidrug resistance pumps, which enhance permeation of the antimicrobials into the bacterial cells. Furthermore, there is a distinct lack of systematic description regarding the structure–property of antibacterial phytochemicals, potentially limiting their mainstream adoption.
Essential oils are abundant in nature, and most commonly associated with the distinctive flavours and aromas of many plants.65 Commonly used herbs and spices such as garlic, black cumin, cloves, cinnamon, thyme, bay leaves, mustard, and rosemary have essential oils with demonstrated antimicrobial properties.66 Garlic-derived allicin was found to exhibit antibacterial activity against a wide range of Gram-negative and Gram-positive bacteria, including multidrug-resistant enterotoxicogenic strains of Escherichia coli; antifungal activity, particularly against Candida albicans; antiparasitic activity, including some major human intestinal protozoan parasites such as Entamoeba histolytica and Giardia lamblia; and antiviral activity.67 The crude methanolic extracts of such spices and herbs as cumin (Cuminum cyminum), fennel seed (Nigella sativa), anise (Pimpinella anisum), ajowan (Trachyspermum copticum), and ginger (Zingiber officinale) were demonstrated to be effective against Gram-positive Bacillus amyloliquefaciens and S. aureus and Gram-negative E. coli and Pseudomonas aeruginosa bacteria.68 Importantly, the extracts demonstrated similar or higher broad-spectrum antimicrobial activity as compared with ampicillin, erythromycin, and tetracycline.
Extracts of Nigella sativa were also effective against pathogenic yeast, C. albicans, and its diethyl ether extract was revealed to be similar in antibacterial activity to that of streptomycin and gentamicin. Traditionally used to treat urinary tract infections, a combination of garlic and black cumin has been reported as being more effective than Cefalexin, Cotrimoxazole, and Nalidixic acid in the treatment of this infectious disease.66 Given the multicomponent nature of the extracts, which included carbohydrates, inulin, alkaloids, glycosides, flavonoids, terpenoids, tannins, reducing sugars, soluble phenols, and saponin glycosides, it is difficult to attribute the observed antimicrobial activity to a particular constituent (Table 1).68
| Plant name | Species | Test organism | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Acinetobacter baumanii | Aeromonas sobria | Candida albicans | Enterococcus faecalis | Escherichia coli | Klebsiella pneumoniae | Pseudomonas aeruginosa | Salmonella typhimurium | Serratia marcescens | Staphylococcus aureus | ||
| MIC, % v/v | |||||||||||
| Rosewood | Aniba rosaeodora | 0.1 | 0.1 | 0.3 | 0.5 | 0.1 | 0.5 | >2.0 | 0.3 | 0.5 | 0.3 |
| Celery seed | Apium graveolens | >2.0 | 1.0 | 1.0 | 2.0 | 2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 1.0 |
| Frankincense | Boswellia carterii | 1.0 | 1.0 | 1.0 | 2.0 | 1.0 | >2.0 | >2.0 | >2.0 | >2.0 | 1.0 |
| Ylang ylang | Cananga odorata | 1.0 | 0.5 | 1.0 | 2.0 | 2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 1.0 |
| Cedarwood | Cedrus atlantica | >2.0 | >2.0 | >2.0 | 0.5 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Cinnamon | Cinnamomum zeylanicum | 1.6 | >2.0 | 0.8 | >2.0 | ||||||
| Lime | Citrus aurantifolia | 1.0 | 1.0 | 2.0 | >2.0 | 1.0 | >2.0 | >2.0 | >2.0 | >2.0 | 2.0 |
| Orange | Citrus aurantium | >2.0 | 1.0 | 1.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 2.0 |
| Petitgrain | Citrus aurantium | 0.5 | 0.5 | 0.3 | 2.0 | 0.3 | >2.0 | >2.0 | >2.0 | >2.0 | 0.5 |
| Bergamot | Citrus aurantium var. bergamia | 2.0 | 2.0 | 1.0 | >2.0 | 1.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Lemon | Citrus limon | >2.0 | 1.0 | 2.0 | 2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 2.0 |
| Grapefruit | Citrus x paradisi | >2.0 | 1.0 | 1.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Mandarin | Citrus reticulata var. Madurensis | >2.0 | >2.0 | 2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Myrrh | Commiphora myrrha | >2.0 | >2.0 | >2.0 | 0.3 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 0.5 |
| Coriander | Coriandrum sativum | 0.3 | 0.3 | 0.3 | 1.0 | 0.3 | 0.5 | >2.0 | 1.0 | 0.5 | 0.3 |
| Pumpkin | Cucurbita pepo | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Cypress | Cupressus sempervirens | >2.0 | >2.0 | >2.0 | 1.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 2.0 |
| Turmeric | Curcuma longa L. | 0.02 | 0.02 | 0.01 | |||||||
| Lemongrass | Cymbopogon citratus | 0.0 | 0.1 | 0.1 | 0.1 | 0.1 | 0.3 | 1.0 | 0.3 | 0.3 | 0.1 |
| Palmarosa | Cymbopogon martinii | 0.1 | 0.1 | 0.1 | 0.3 | 0.1 | 0.3 | >2.0 | 0.5 | 0.3 | 0.1 |
| Citronella | Cymbopogon nardus | 0.3 | 0.1 | 1.0 | 0.5 | 1.0 | >2.0 | >2.0 | >2.0 | 0.3 | |
| Carrot seed | Daucus carota | >2.0 | >2.0 | 2.0 | 2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 1.0 |
| Eucalyptus | Eucalyptus polybractea | 1.0 | 0.5 | 1.0 | 2.0 | 1.0 | 2.0 | >2.0 | >2.0 | 1.0 | 2.0 |
| Clove | Eugenia caryophyllus | 1.6 | >2.0 | 1.6 | >2.0 | ||||||
| Fennel | Foeniculum vulgare | 1.0 | 0.5 | 0.5 | >2.0 | 0.5 | >2.0 | >2.0 | 1.0 | >2.0 | 0.3 |
| Wintergreen | Gaultheria procumbens | 0.3 | 0.3 | 0.3 | >2.0 | 0.5 | 1.0 | >2.0 | 0.5 | 0.5 | 2.0 |
| Juniper | Juniperus communis | >2.0 | 1.0 | 2.0 | 2.0 | >2.0 | >2.0 | >2.0 | 2.0 | >2.0 | 2.0 |
| French lavender | Lavandula angustifolia | 1.0 | 0.5 | >2.0 | 0.5 | 2.0 | >2.0 | >2.0 | >2.0 | 1.0 | |
| Tasmanian lavender | Lavandula angustifolia | 0.5 | 0.5 | 0.3 | 2.0 | 0.3 | >2.0 | >2.0 | >2.0 | 2.0 | 1.0 |
| Tea bush | Lippia chevalieri | 1.0 | >2.0 | 1.0 | |||||||
| Tea bush | Lippia multiflora | 0.3 | 0.3 | 0.1 | |||||||
| Macadamia | Macadamia integrifolia | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Tea tree | Melaleuca alternifolia | 0.3 | 0.5 | 0.5 | 2.0 | 0.3 | 0.5 | >2.0 | 0.5 | 0.5 | 0.5 |
| Cajuput | Melaleuca cajuputi | 1.0 | 1.0 | 1.0 | 2.0 | 1.0 | >2.0 | >2.0 | >2.0 | 2.0 | 1.0 |
| Niaouli | Melaleuca quinquenervia | 0.3 | 0.3 | 0.3 | 1.0 | 0.3 | 0.5 | >2.0 | 0.5 | 0.5 | 0.5 |
| Peppermint | Mentha x piperita | 0.5 | 0.5 | 2.0 | 0.5 | 1.0 | >2.0 | 1.0 | 2.0 | 1.0 | |
| Spearmint | Mentha spicata | 0.3 | 0.3 | 0.1 | 2.0 | 0.3 | 0.5 | >2.0 | 0.5 | 0.3 | 0.3 |
| Basil | Ocimum basilicum | 0.5 | 0.5 | 0.5 | >2.0 | 0.5 | 2.0 | >2.0 | 2.0 | >2.0 | 2.0 |
| Evening primrose | Oenothera biennis | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Marjoram | Origanum majorana | 0.3 | 0.3 | 0.3 | 2.0 | 0.3 | 0.5 | >2.0 | 0.5 | 0.5 | 0.5 |
| Oregano | Origanum vulgare | 0.1 | 0.1 | 0.1 | 0.3 | 0.1 | 0.1 | 2.0 | 0.1 | 0.3 | 0.1 |
| Geranium | Pelargonium graveolens | 0.3 | 0.3 | 0.1 | 0.5 | 0.3 | >2.0 | >2.0 | >2.0 | >2.0 | 0.3 |
| Aniseed | Pimpinella anisum | 0.5 | 0.3 | 0.5 | 2.0 | 0.5 | >2.0 | >2.0 | 2.0 | 1.0 | 0.3 |
| Bay | Pimenta racemosa | 0.1 | 0.1 | 0.1 | 0.5 | 0.1 | 0.3 | 1.0 | 0.3 | 0.3 | 0.3 |
| Pine | Pinus sylvestris | 2.0 | 2.0 | 2.0 | >2.0 | 2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Black pepper | Piper nigrum | >2.0 | >2.0 | >2.0 | 1.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Patchouli | Pogostemon patchouli | >2.0 | >2.0 | 0.5 | 0.1 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 0.3 |
| Apricot kernel | Prunus armeniaca | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Sweet almond | Prunus dulcis | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Rosemary | Rosmarinus officinalis | 1.0 | 0.5 | 1.0 | >2.0 | 1.0 | 2.0 | >2.0 | >2.0 | >2.0 | 1.0 |
| Sage | Salvia officinalis | 0.5 | 0.5 | 0.5 | 2.0 | 0.5 | 2.0 | >2.0 | 2.0 | 1.0 | 1.0 |
| Clary sage | Salvia sclarea | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 |
| Sandalwood | Santalum album | >2.0 | >2.0 | 0.1 | 0.3 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 0.1 |
| Clove | Syzygium aromaticum | 0.3 | 0.1 | 0.5 | 0.3 | 0.3 | >2.0 | >2.0 | 0.3 | 0.3 | |
| Thyme | Thymus vulgaris | 0.1 | 0.1 | 0.1 | 0.5 | 0.1 | 0.3 | >2.0 | >2.0 | 0.3 | 0.3 |
| Vetiver | Vetiveria zizanioides | >2.0 | >2.0 | 0.1 | 0.1 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 0.1 |
| Ginger | Zingiber officinale | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | >2.0 | 2.0 |
The antibacterial and antifungal potency of caraway (Carum carvi) oils are attributed to carvone, limonene and linalool, while antimicrobial activity of cumin is associated with the presence of limonene, eugenol, pinene and minor constituents, and the effect is likely to be synergistic.73 Cumin essential oil was found to the activity of the ciprofloxacin against biofilm-forming Klebsiella pneumoniae strains, although the oil on its own was not able to induce plasmid DNA degradation.74C. cyminum oil was also effective against Vibrio spp. strains.75 Essential oil from rosemary (Rosmarinus officinalis) was shown to be effective against E. coli, S. aureus and L. monocytogenes, although it was found less potent in comparison with Cu. Cyminum essential oil.76 Peppermint (Mentha piperita) oil was demonstrated to be more effective than chlorhexidine in preventing biofilm formation by Streptococcus mutans and Streptococcus pyogenes, with potential to be used in therapies against supragingival dental plaque.77
A survey of 35 different Indian spices showed clove, cinnamon, bishop's weed, chili, horse radish, cumin, tamarind, black cumin, pomegranate seeds, nutmeg, garlic, onion, tejpat, celery, have potent antimicrobial activities against the test organisms Bacillus subtilis, E. coli and Saccharomyces cerevisiae.78 Oils of chilli, cinnamon, cloves, ginger, nutmeg, oregano, rosemary, sage, thyme demonstrated a range of activities against psychrotrophic Aeromonas hydrophila, Listeria monocytogenes and Yersinia enterocolitica, from complete inhibition of growth in the case of cinnamon and cloves against A. hydrophila to no inhibition.79 The antimicrobial potency was also found to vary with the oil acquisition method, e.g. oil harvesting at different stages of plant development. Thyme (Thymus vulgaris) oil harvested at four ontogenetic stages had a significant bacteriostatic activity against nine strains of Gram-negative bacteria and six strains of Gram-positive bacteria. However, the activity was the highest for the oil harvested from the plants in full flower.80
Although oils and their individual components, such as terpenoids, carvacrol, thymol, have been recognised as potential antimicrobial agents, yet their exact mechanism of actions has not been fully elucidated. In part, this may be due to the numerous components that can potentially complement and/or enable the efficacy of the other component. For instance, Brophy et al. analysed over 800 samples of M. alternifolia essential oil by gas chromatography and mass spectrometry and found approximately 100 components in oil of M. alternifolia as well as significant batch to batch variation.81 The comparison may be complicated further by different methods used to quantify antibacterial activity (which also often differ from those used for evaluation of surface-immobilised antimicrobial agents).82–84
Oil from Australian native plant Melaleuca alternifolia has been reported to have the broad-spectrum activity against bacteria, including drug-resistant strains, fungi, viruses, and protozoa,85–88 but similar to other phytochemicals, in vivo and in vitro characterisation of tea tree oil thus far remains inadequate. Nonetheless, various preparations that include tea tree oil are readily available commercially in many countries, including in Australia, Europe, and North America. Tea tree oil is composed of terpene hydrocarbons based on an isoprene structure, mainly monoterpenes (C10H16), sesquiterpenes (C15H24), and their associated alcohols (terpenoids), with the antimicrobial activity of the oil is attributed mainly to terpinen-4-ol. The mechanism of action of terpenes is yet to be fully described but is believed to involve membrane disruption by the lipophilic compounds.63,89 In the case of tea tree oil, the ability of tea tree oil to disrupt the permeability barrier of cell membrane structures and the accompanying loss of chemiosmotic control were identified as the most likely source of its lethal action against E. coli, S. aureus, and Candida albicans.90 The predisposition to lysis, the loss of 260 nm-absorbing material, the loss of tolerance to NaCl, and the altered morphology by S. aureus cells suggest that tea tree oil and its components compromise the cytoplasmic membrane.91,92
Essential oils and their constituents are believed to interact with the bacterial membrane, causing disruption through lipophilic products (Fig. 6). These disruptions then lead to membrane expansion, increase of membrane fluidity and permeability, disturbance of membrane embedded proteins, inhibition of respiration, and alteration of ion transport processes in both Gram-positive and Gram-negative bacteria.32 An analysis of the chemical structure of these herbs and spices shows that the antimicrobial phytochemicals consist of phenols and oxygen-substituted phenolic rings,63 with the inhibitory action associated with the –OH groups in phenolic compounds.
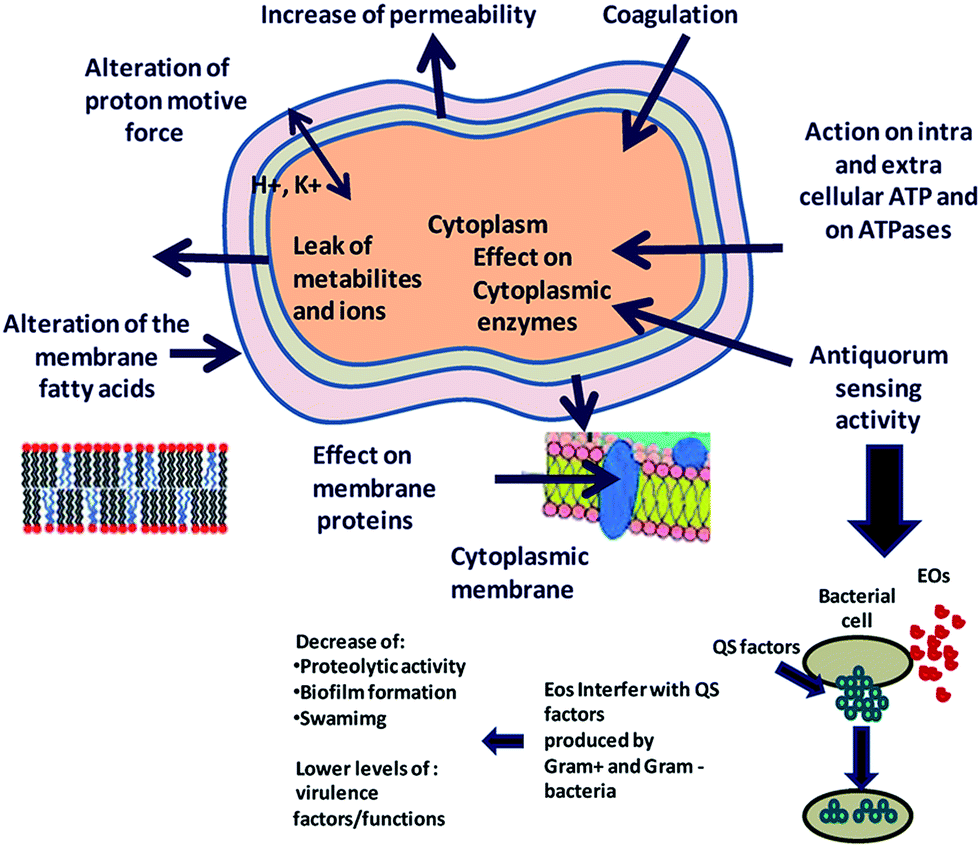 | ||
| Fig. 6 Proposed mechanism of action and target sites of secondary plant metabolites on microbial cells. Reproduced with permission from ref. 93. | ||
Garlic is different in such that it consists of non-aromatic sulfur compounds (thiosulfinates) that carry the antimicrobial properties. Diallyl thiosulfinate (allicin), the phytochemical agent found in garlic (Allium sativum) and believed to be responsible for the antibacterial and antifungal activity of extracts of this plant is thought to interact with intracellular thiols and thiol containing enzymes, including alcohol dehydrogenase, thioredoxin reductase and RNA polymerase.67 This can affect essential metabolism of cysteine proteinase activity involved in the virulence of E. histolytica. The effect of bacteriostatic concentrations of allicin (0.2 to 0.5 mM) on the growth of Salmonella typhimurium was characterised by a delayed and partial inhibition of DNA and protein syntheses and immediate and total inhibition of RNA synthesis, suggesting that the latter is the primary target of allicin.94 Garlic extract has also been shown to inhibit quorum sensing ability of biofilm-residing Pseudomonas aeruginosa, rendering the treated bacteria susceptible to the bactericidal activity of tobramycin and polymorphonuclear leukocytes.95,96
4. Nature-inspired catechol chemistry
In addition to nature-inspired antibiofouling and biocidal surfaces, e.g. lotus leaf, butterfly wing and shark skin-like biomimetic surfaces, bio-inspired chemistries can influence the manner in which biomaterials and biomaterial coatings are synthesized, functionalized and delivered in vivo.22,25,97,98 There are several methods of surface functionalisation that are inspired by the aquatic animals that are highly adept at fouling a variety of solid surfaces, both natural and man-made, in aqueous environments. Numerous sedentary marine organisms, including species of mussels, tubeworms, and barnacles attach to underwater surfaces by means of protein-rich adhesives.97,99Adhesive proteins form approximately 70 wt% of the cement of Belanus crenatus, where the proteinaceous cement is released from the pores to fill the space between the base of the attachment disk of the animal and the solid surface to which the organism is attaching. The cement cures within several hours, forming a high strength bond with attachment strength of up to 9.3 × 105 N m−2. Where cement produced by adult and cyprid acorn barnacles (order Sessilia) solidifies into a thin layer directly between the shell and the surface and is characterised by either solid or reticulate structure,100 the cement produced by Dosima fascicularis buoy barnacle is a gas-filled, foam-like structure.101 Whereas the barnacle uses the cement for attachment to surfaces, the Phragmatopoma californica marine worm uses its glue to build its mineralized shell from sand grains and fragments of seashell collected from its environment.102 Setting within 30 s under, the glue forms a microporous water-filled foam comprised of 50–80 nm spheres, and characterised by a sharp gradient in porosity.
Water-resistant, material-independent adhesive abilities of the mollusc (Mytilus edulis) byssus, a proteinaceous liquid from the phenol gland in the mussel foot that forms an adhesive holdfast, have been used to guide the development of substrate-independent adhesive hydrogels.31 Rapid solidification into a hardened adhesive and excellent adhesion to a variety of substrates, including tissues, is attributed to reactivity of catechol side chains on 3,4-dihydroxy-L-phenylalanine (DOPA).103 Readily oxidised, catechol side chains form reactive species that can undergo Michael-type addition, Schiff base formation with nucleophiles, and radical coupling with other catechols (Fig. 7). They can also form coordination bonds with diverse metals and inorganic surfaces, hydrogen bonds, and π–π aromatic interactions. The mechanism by which 3,4-dihydroxy-L-phenylalanine interacts with the wet surface depends on the state of the molecule.104 An atomic force microscopy (AFM) study of a single molecule immobilised on the scanning tip demonstrated high strength yet fully reversible, non-covalent interaction with a wet metal oxide surface, here titanium dioxide. Once 3,4-dihydroxy-L-phenylalanine was oxidised, the strength of this reversible interaction significantly decreased, although a new, high strength irreversible covalent bond was formed.
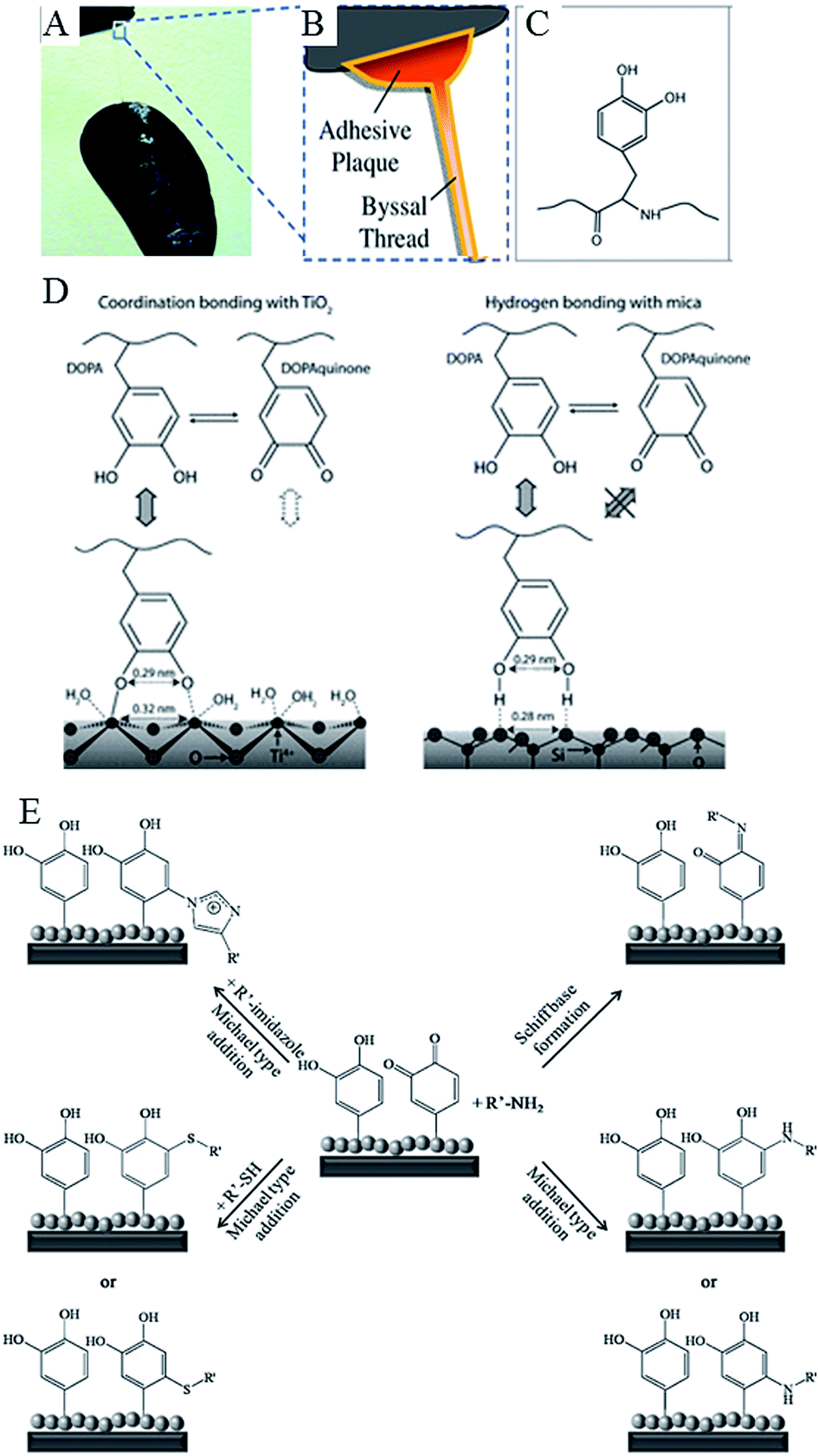 | ||
| Fig. 7 Catechols as versatile platforms in polymer chemistry. (A) A photograph of a mussel attached to the shell of another mussel. (B) Schematic representation of the adhesive plaque and byssal thread. (C) Chemical structure of the DOPA side chain found in mussel adhesive proteins.105 (D) Illustration of the proposed binding mechanism of DOPA to two types of surfaces, TiO2 and mica. DOPA and DOPAquinone, to a lesser extent, can form bidentate binuclear complexes with the TiO2 surface, whereas the interactions with mica are much less specific and may result from the hydrogen bonding of the phenolic OH groups to the oxygen atoms of the cleaved mica surface. DOPAquinone has no H to donate.106 (E) Possible reaction pathways of oxidized catechols with amines, thiols or imidazoles where R′ stands for a polymeric or peptidic backbone.107 Reproduced with permission from ref. 105–107. | ||
4.1 Catechol-based hydrogels
The incorporation of these catechol functionalities into water soluble hydrogels, such as polyethylene glycol, ensures rapid curing of these gels. The degradation properties of such adhesive hydrogel can be modified, by incorporating enzyme-degradable sites. For instance, a hydrogel based on polyethylene glycol functionalised by DOPA-mimetic catechol via biodegradable linker, e.g. an Ala–Ala dipeptide substrate of elastase, can be degraded by neutrophil elastase, the latter being a serine protease secreted by activated neutrophils as the result of their recruitment to a wound or site of local inflammation.108 The microstructure, composition and mechanical properties of the hydrogel can also be tuned by controlling the input catechol, linker, and polymer backbone.The hydrogel can be loaded with antibacterial agents for sustained release. For example, the use of silver nitrate to oxidize catechol-functionalised polyethylene glycol resulted in covalent cross-linking of the hydrogel and concomitant reduction of Ag(I).31 The resultant bulk hydrogels demonstrated inhibition of Staphylococcus epidermidis and Pseudomonas aeruginosa due to sustained release of silver, with minimal detriment to mammalian (3T3 fibroblast) cell viability.31 When used as a spin-cast 25 nm-thick coating over titanium dioxide substrate, the hydrogel resisted fouling by both bacterial and eukaryotic cells. Due to relatively low content of silver in the thin film hydrogel, the non-fouling by mammalian cells was attributed to the antifouling nature of the polyethylene glycol polymer, rather than cytotoxicity of released silver. However, the findings that the toxic effect of silver ions and silver nanoparticles occurs in a similar concentration range for Escherichia coli, Staphylococcus aureus, human mesenchymal stem cells, and peripheral blood mononuclear cells challenges this conclusion.109,110
4.2 Catechol adhesive layer
Dopamine has been demonstrated to possess the full adhesive properties of mussel adhesion protein, and can be used as a thin highly adherent coating on a range of biomaterial surfaces, organic and inorganic alike. Such dopamine-based surface coatings are resistant to hydrolysis and provide chemical activation on material surfaces for selective coupling of molecules and layers.111 The coating is deposited as poly(dihydroxyindole), but undergoes oxidation to polyorthoquinoneindole upon exposure to basic (pH 8.5) conditions.112 To this layer, biomolecules containing amine moieties can be covalently bonded via Schiff base type interactions, or Michael type reactions in the case of those molecules with amine and thiol functionalities.In addition to monolayers via self-assembly of long-chain molecular building blocks, secondary reactions on the dopamine-modified surfaces can be used for deposition of metal films by electroless metallization, and bioinert and bioactive surfaces via grafting of macromolecules.113 Silver nanoparticles were immobilised onto ferromagnetic Fe2O3 nanoparticles/fibrous bacterial cellulose nanocomposite by soaking dopamine-treated composite in silver nitrate solution.114 Dopamine coating was also shown to be a suitable platform for fabrication of polymer brushes via atom transfer radical polymerization.
The utility of barnacle cement for surface functionalisation has been demonstrated on stainless steel, where the adhesive was used as a surface anchor for coupling of functional polymer brushes via “click” reactions in both “grafting-to” and “grafting-from” processes.115 A surface rich in thiol, alkyne, and azide groups was obtained by first depositing a thin layer of the cement onto the metallic surface. The reactive amine and/or hydroxyl groups on the surface100 were then allowed to react with ethylene sulfide, propargyl carbonylimidazole, and azidoethyl carbonylimidazole, respectively, to introduce the desired functionality. Using these molecular anchors, a variety of stable functional polymer brush coatings were developed, including antifouling zwitterionic 2-methacryloyloxyethyl phosphorylcholine surfaces (via thiol photo polymerisation); protein-resistant hydrophilic poly(poly(ethylene glycol) methyl ether methacrylate) and protein-adsorbing hydrophobic poly(2,3,4,5,6-pentafluorostyrene) brushes (via azide–alkyne click reaction); antifouling poly(N-hydroxyethyl acrylamide) and antimicrobial poly(2-(methacryloyloxy)ethyl trimethylammonium chloride) surfaces (via alkynyl–azide click chemistry).115
Of the developed coatings, the zwitterionic and hydrophilic surfaces were most effective in reducing bovine serum albumin adsorption, with the zwitterionic, antifouling, and antimicrobial surfaces inhibiting the adhesion of Gram-negative E. coli and Gram-positive S. epidermidis. Similar to hydrophilic poly(ethylene glycol) and oligo(ethylene glycol), the antifouling properties of zwitterionic and polyampholyte polymer brushes rely on the formation of a strong hydrogen-bonded hydration layer, which limits protein interactions with the underlying surface.116 Surfaces rich in alkyl halide functionalities were also obtained by reacting the amine and hydroxyl moieties of barnacle cement with 2-bromoisobutyryl bromide.117 The initiator can be used for the surface-initiated atom transfer radical polymerization of 2-hydroxyethyl methacrylate, the hydroxyl groups of which can then be converted to carboxyl groups for coupling of chitosan. Thus functionalised stainless steel surfaces displayed antifouling property against bovine serum albumin and antibacterial activity against E. coli.
5. Plasma-assisted nanofabrication
Plasma-assisted technologies, especially those based on low-temperature, non-equilibrium plasmas, have found numerous applications in medicine, materials science, and biotechnology.118,119 The ability to remove biomolecules, such as proteins, pyrogens or peptides, and bacterial spores from biomaterial surfaces at high rates and low temperatures make plasma-assisted treatment an effective and practical tool for decontamination and sterilization of biomaterials and medically-relevant devices and surfaces.120–122 The neutral and reactive species, particularly reactive oxygen species (ROS) and reactive nitrogen species (RNS), electric fields, charges, and photons generated in low temperature ionized gas plasmas are responsible for the well-documented antimicrobial activity of these plasmas when applied directly to media.123,124 The low temperature (at or below physiological level) of such plasmas allow for their application onto living tissues, e.g. a wound, where they can be used to sterilise, suppress inflammation, and promote healing.125,126The unique chemistry of these plasmas also enables selective biomanipulation of the cells, where they can be used to increase cell proliferation, locally influence cell adhesion without causing necrosis or to initiate cell removal via induction of apoptosis, the result dependent on the dose.127,128 The selectivity whereby only one type of cells is affected, i.e. cancer cells and not healthy cells in co-culture, has a clear potential as a safer means for anti-cancer therapy.129–132
From biomaterials perspective, plasma-assisted techniques are widely used for lasting, highly controlled modification of a variety of medically relevant surfaces.136–138 Indeed, over the last 20 years, plasma-enabled nanoscale synthesis and modification have evolved from a relatively simple tool for materials science and microelectronics into a highly sophisticated instrument for development of a wide range of pure and hybrid nanoscale objects spanning across a vast number of materials systems and length scales (Fig. 8).139 At the present level of development, low-temperature plasmas afford chemists and material scientists the level of confidence comparable to, and in many cases superior to, conventional processing techniques, e.g. based on thermal chemical vapour deposition (CVD), wet chemistry-based synthesis and processing, laser-assisted microfabrication etc.139 Importantly, tailored plasmas enable the attainment of certain objectives conventional fabrication methodologies fail to achieve, such as providing the means for one-step green synthesis of functional materials from natural precursors.140–144
 | ||
| Fig. 8 Examples of plasma-treated surfaces. (A) Petal- and tree-like graphene networks. (B) Titanium pillars structured in bulk material using reactive ion etching with fluorine plasma. (C) Collagen-grafted titanium surface via allylamine-glow discharge treatment and collagen crosslinking. Reprinted with permission from ref. 133–135. | ||
5.1 Types of low-temperature plasma processing
Lower temperature processing suitable for temperature-sensitive biomaterials and implantable thin film structures and for production of polymer films where the functionality of the monomer is retained can be attained in low pressure, low energy plasma systems. In non-equilibrium plasma processing, the substrate is exposed to a reactive environment of a partially ionised gas comprising large concentrations of excited atomic, molecular, ionic, and free-radical species. The nature of the interactions between the excited species and the solid surface will determine the type and the degree of the chemical and physical modification that will take place, from film deposition, substitution, cross-linking to ablation. Generally, polymer deposition occurs when a monomer, either in vapour phase or at the surface, is fragmented into reactive species that then recombine and are deposited onto the surface of the substrate. As mentioned previously, even those monomers that do not contain functionalities required for conventional polymerisation, e.g. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or ring structures, can be deposited in this way.
C or ring structures, can be deposited in this way.
When film deposition is not desired, gases that do not fragment into polymerisable intermediates upon excitation are employed. Air, nitrogen, argon, oxygen, nitrous oxide, helium, tetrafluoromethane, water vapour, carbon dioxide, methane, and ammonia are amongst the most common gases used for surface modification. Exposure to these plasmas may result in the chemical functionalisation of the surface, with the degree and nature of the functionalities being highly dependent on the chemical composition of the biomaterial and the process gas. Plasma-assisted surface oxidation, nitration, hydrolysation, or amination are commonly used to increase the surface energy and hydrophilicity of the biomaterial. Surface ablation can also result from such plasma-exposure, whereby lower molecular weight species, such as volatile oligomers and monomers, are desorbed from the surface of the biomaterial. Cross-linking occurs when radicals from one chain on the surface of the polymer combine with radicals from another polymer chain to form a bond, thus changing the mechanical surface properties of the material. When plasma-generated radicals recombine with atoms or chemical groups that are different from those originally present at the surface of the biomaterial, surface activation takes place. Surface activation can also take place through opening of dangling bonds on the surface.
The surface functionalities that arise as a result of plasma deposition or functionalization can serve as a platform for further surface modification processes, such as the grafting of biomolecules and other functional structures,61,136 and to tune the properties of the biomaterial for a specific application.145 Surfaces coated with plasma polymers can be used to bind proteins specifically via covalent linkages, e.g. streptavidin conjugation to aldehyde groups where the binding ability of the protein is retained, or nonspecifically through other irreversible adsorption mechanisms, e.g. streptavidin binding to ethanol plasma polymer surfaces where protein denaturation occurred.146 Chemical gradients with different density of a specific functionality or with a changing concentration of two functional groups across the biomaterial surface can be fabricated via plasma polymerisation by using a mask.147 Such gradients can be highly useful for investigations of microbial and eukaryotic cell response to variations in surface chemistry, with each sample serving as a platform for high-throughput testing of a range of cell–surface interactions.148 Morphological gradients can also be obtained with the help of plasma polymerization, whereby a surface is first functionalised with a specific moiety, e.g. amine, and then subjected to controlled immersion into the solution of nanoparticles.149 The variation in nanoparticle density gives rise to differences in surface roughness, the effect of which on cell adhesion and metabolism can be investigated independently of surface chemistry with an addition of thin plasma polymer top layer. Furthermore, biomolecules (e.g. proteins) that display selective attachment to given nanoparticles can be immobilised on these surfaces. These surfaces can then be used to study the effect of biomolecule density on cell–surface interactions.
5.2 Controlling plasma-assisted surface modification
The processing conditions, such as power delivered to the reactor, pressure within the reactor, monomer molecular weight and flow rate, presence of feed gas, etc. will determine activation, fragmentation, rearrangement and recombination of the monomer units in plasma. The key determinant of the modification outcome is the amount of energy delivered into the chamber in relation to the building units (from which polymers and nanostructures are synthesised) or to substrate material (in the case of etching).Monomers do not always need to be fragmented; however, in a plasma environment, there are more options for monomer fragmentation. When fragmentation takes place, it typically involves the elimination of hydrogen atoms, and the scission of C–C bonds. Retention of the original chemical functionality within the resultant polymer deposit is highly dependent on the degree of monomer fragmentation. The technological challenge here lies in the ability to retain the desired chemistry and at the same time attain sound mechanical properties, desired density, stability, and adhesion to substrate in the material.150,151 To address this challenge, it is important to understand the mechanisms implicated in the plasma-assisted deposition of the polymer onto the surface.
While surface radical–plasma radical interactions were considered the primary route of polymer deposition for many years,152 recent findings have implicated ion adsorption and/or neutral grafting as potential drivers for plasma polymer formation at the surface.153–156 The mechanism to explain these phenomena centred on the energies at which depositing species arrive at the surface of the substrate. Under low pressure, low power conditions commonly used for fabrication of functional polymer coatings, neutral species such as radicals and unfragmented precursor molecules reach the surface at nearly ambient temperature (0.03–0.05 eV). On the other hand, ions are accelerated to the surface by the difference between the respective potentials of bulk plasma and the surface and thus arrive at the surface with much higher energies (15–20 eV).157 The higher energy of ions is sufficient to break chemical bonds at the biomaterial surface, leading to the formation of surface radicals. These radicals are then available for neutral grafting as per surface radical–plasma radical model, and can also promote cross-linking within the plasma polymer.
There is a clear link between the process parameters, the mechanism of film growth, and the resultant chemical and physical properties of the polymer structure.157 The chemical structure of the organic precursor was found to be critical, in particular, at low powers. The increased monomer fragmentation at high power reduces the ability of unsaturated monomers to grow via neutral grafting. For saturated monomers, there is a direct link between the deposition rate and ion flux to the substrate, whereas for unsaturated monomers, the neutral flux also plays a role.153 The material properties of these films also varied significantly. Polymers deposited from saturated monomers were characterised by higher moduli, lower solubility, and lower density compared to those grown from unsaturated precursors. As the utility of plasma coatings is reliant on the combination of desirable chemistry and morphology, as well as good substrate adhesion, controlled stability and suitable mechanical properties, understanding the relationship between the process parameters and material properties is crucial in the design of plasma polymer film processes to fully harness the unique plasma-specific chemistries and physical phenomena of non-equilibrium plasmas (Fig. 9).
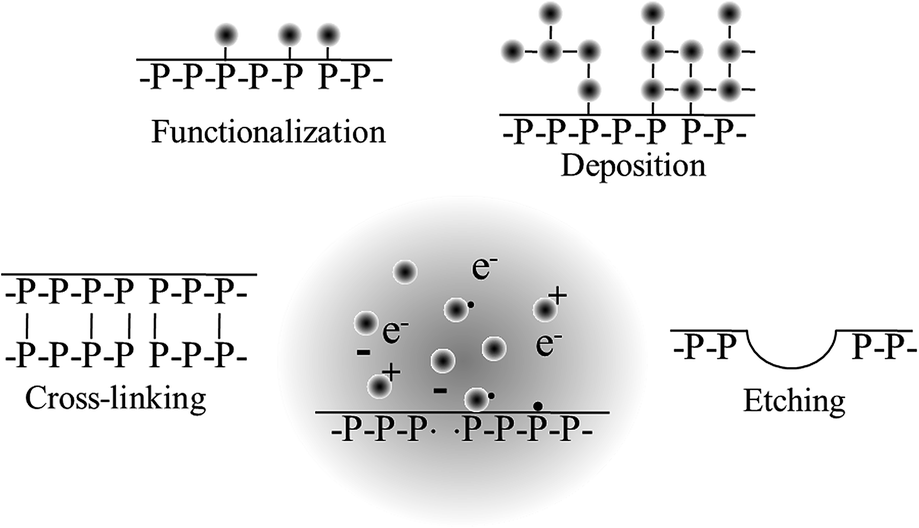 | ||
| Fig. 9 Processes that may take place during plasma treatment. | ||
5.3 Pulsed plasma deposition
Although low-power, low-pressure and low-temperature deposition is more conducive to the fabrication of plasma polymers with retained functionalities, the degree of fragmentation is still relatively high. As a result, polymers fabricated using this method retain only a fraction of the functionality present in the precursor, and are typically highly cross-linked and amorphous. Lowering power and temperature may reduce fragmentation even further, preserving more of the chemical structure present in the precursor. Yet, the utility of these coatings in vitro and in vivo is limited by their poor mechanical and chemical stability, and often unsatisfactory attachment to the substrate. These issues may be circumvented by pulsed plasma deposition, where two distinct regimes are employed (Fig. 10).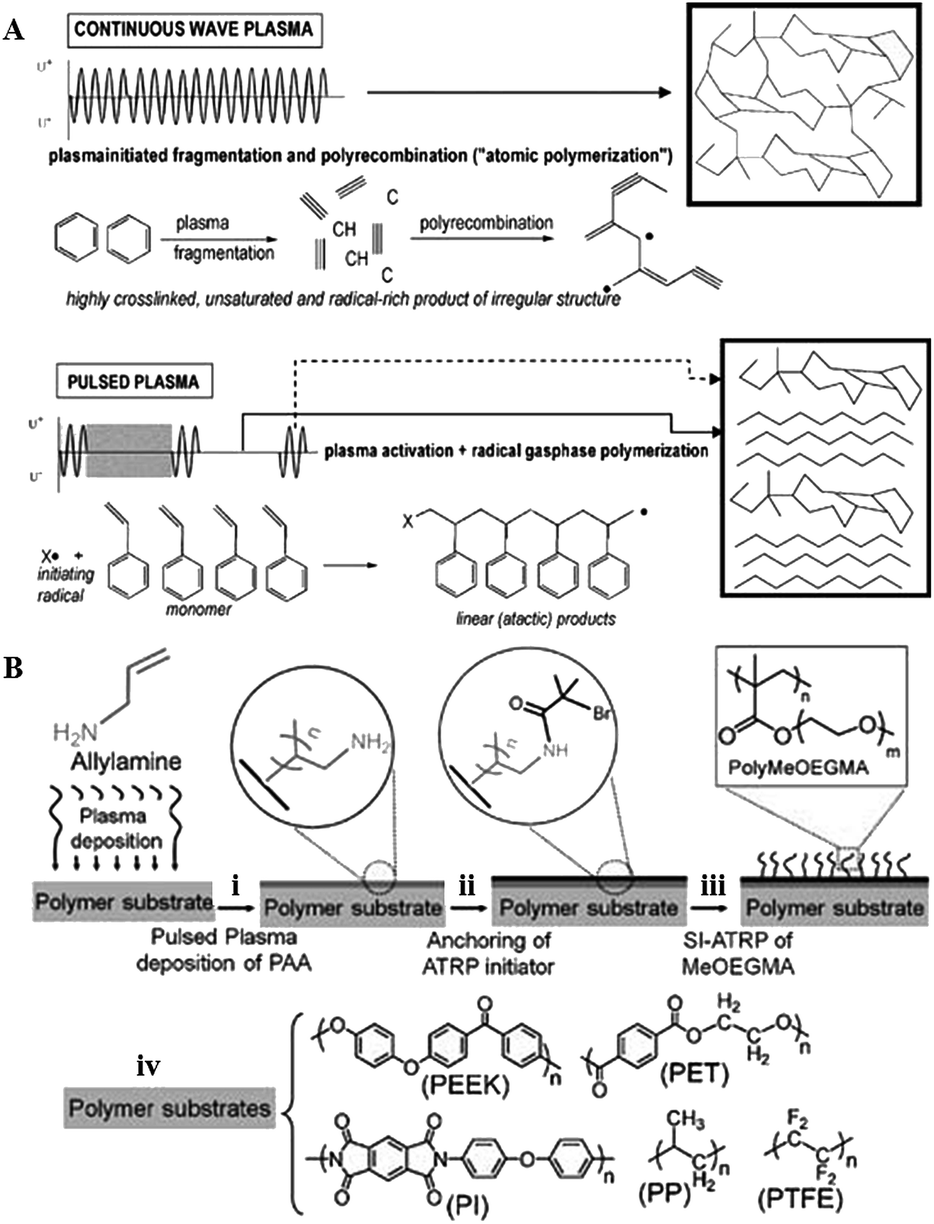 | ||
| Fig. 10 (A) Pulsed plasma deposition allows for fabrication of polymers that consist of more chemically-regular products than those fabricated by means of continuous wave plasma deposition, where predominantly random radical recombination occurs.159 (B) Abundant in functional groups, pulsed plasma treated surfaces can be used for covalent immobilisation of polymer brushes.160 Reproduced with permission from ref. 159 and 160. | ||
In contrast to continuous wave plasmas, monomer activation and generation of reactive site on the surface occur only during on-periods (typically microseconds) whereas polymerisation takes place during off-periods (usually milliseconds) in the absence of UV-, ion-, or electron-induced damage to the growing film.158
The resultant polymer is characterised by high retention of original chemistries, good stability and covalent attachment of the grown film to the substrate at the free radical sites generated during the on-period. By controlling the input power,161 pulsing frequency and the duration of the pulse it is possible to tune chemical functionality, surface morphology and density of desired chemical functionality at the polymer surface.162,163 The process is highly versatile in terms of resultant surface chemistry, with pyridine,158 anhydride,164 amine,165 ester,166 hydroxyl,167–169 sulfonic acid,170 carboxylic acid,171 cyano,172,173 epoxide,174 halide,175 thiol,137,176 and furan177 functionalised surfaces reported.
5.4 Plasma-assisted processing of essential oils
The limited understanding of the exact mechanism of antibacterial efficacy of the essential oils and their individual compounds significantly limited their potential clinical uses, especially as part of antimicrobial coatings for medical implants. Indeed, most in vitro and in vivo studies to date employed phytochemicals in their liquid or vapour, unbound form. Tea tree oil delivered into the cavities of prostatic abscesses in dogs in place of aspirated purulent matter resulted in the disappearance of the purulent matter in the cavities and a marked reduction in the volume of the cavities.178The ability of using these antimicrobials for site-specific applications, such as in release-based or non-leaching surfaces remains largely undiscovered. A range of polymer coatings based on ultra-high molecular weight polyethylene, very high molecular weight polyethylene and latex compounds and incorporating a wide range of biocidal phytochemical agents, alone and in combinations, have been proposed, with primary area of application being marine paints and coatings.179 In another patent, anti-fouling coating composition containing capsaicin were proposed, although these were not designed for medical implantation applications.180 A polymer system loaded with a variety of phytochemicals, phytonutrients, and chemical releasers has also been designed to inhibit the growth of pathogenic bacteria associated with packaged foodstuff.181 Recently, a number of antibacterial coatings containing curcumin have been developed. Sodium carboxymethyl cellulose silver nanocomposite films were loaded with curcumin by diffusion mechanism, with higher encapsulation of the agent observed in the films with higher cellulose content.182 Silver nanoparticles also enhanced the encapsulation of curcumin, suggesting a degree of interaction between these two antimicrobials. The synergistic effect between silver nanoparticles and curcumin was also observed in the antimicrobial activity against E. coli, with the activity being superior to either silver- or curcumin-only films. Sustained release and sound antibacterial efficacy was also observed for silver/curcumin-containing hydrogels based on poly(acrylamide)/poly(vinyl sulfonic acid sodium salt)183 and those based on chitosan–poly(vinyl alcohol) films.184
Although promising, the aforementioned strategies relied on the use of other polymers or chemical substances to produce a coating. Using non-equilibrium, low-temperature plasma polymerisation, Jacob and Bazaka and their colleagues demonstrated the possibility of producing solid polymer films exclusively from essential oils, including M. alternifolia and Lavandula angustifolia essential oils and their individual constituents.141,143 Fabricated over a wide range of processing parameters, these films varied in terms of chemical composition, surface morphology, stability and mechanical properties, while displaying uniform coverage and sound adhesion to a variety of substrates, including metals, ceramics, and polymers.185,186 Polymers fabricated from M. alternifolia oil and its major antimicrobial component terpinen-4-ol was demonstrated to be cytocompatible with a number of host cells. In combination with biological activity, their attractive optoelectronics properties make terpinen-4-ol films as potential candidates for inclusion in implantable electronics, where they can be used as both the device components and protective encapsulating layers.144,187–189
Films fabricated at conditions that favoured preservation of original functionalities of the monomer via limited fragmentation and incorporation of unfragmented species into the polymer matrix were able to retard attachment and colonisation by such bacteria as P. aeruginosa, S. aureus, S. epidermidis, and E. coli.141,142,192Fig. 11 shows the attachment preferences of two pathogens, S. aureus and S. epidermidis to polymers fabricated at three different input power levels.190 Surfaces rich in oxygen containing functional groups, particularly –OH, were characterised by higher antifouling and biocidal activity compared to more hydrocarbon dense coatings. In addition to availability of specific functionalities at the surface, it has been speculated that unfragmented monomer trapped within the polymer during deposition may be eluting over time, thus contributing to inhibition of biofilm formation at the polymer surface. It is believed that just as unmodified monomer, the eluted agent would interfere with the bacterial wall, leading to the suppression of cell biosynthesis, disruption of transport across the cell wall, loss of membrane integrity, and an increase in passive flux of protons across the membrane. However, the levels of the leached agent are sufficiently low to insure good cytocompatibility with mammalian cell lines.
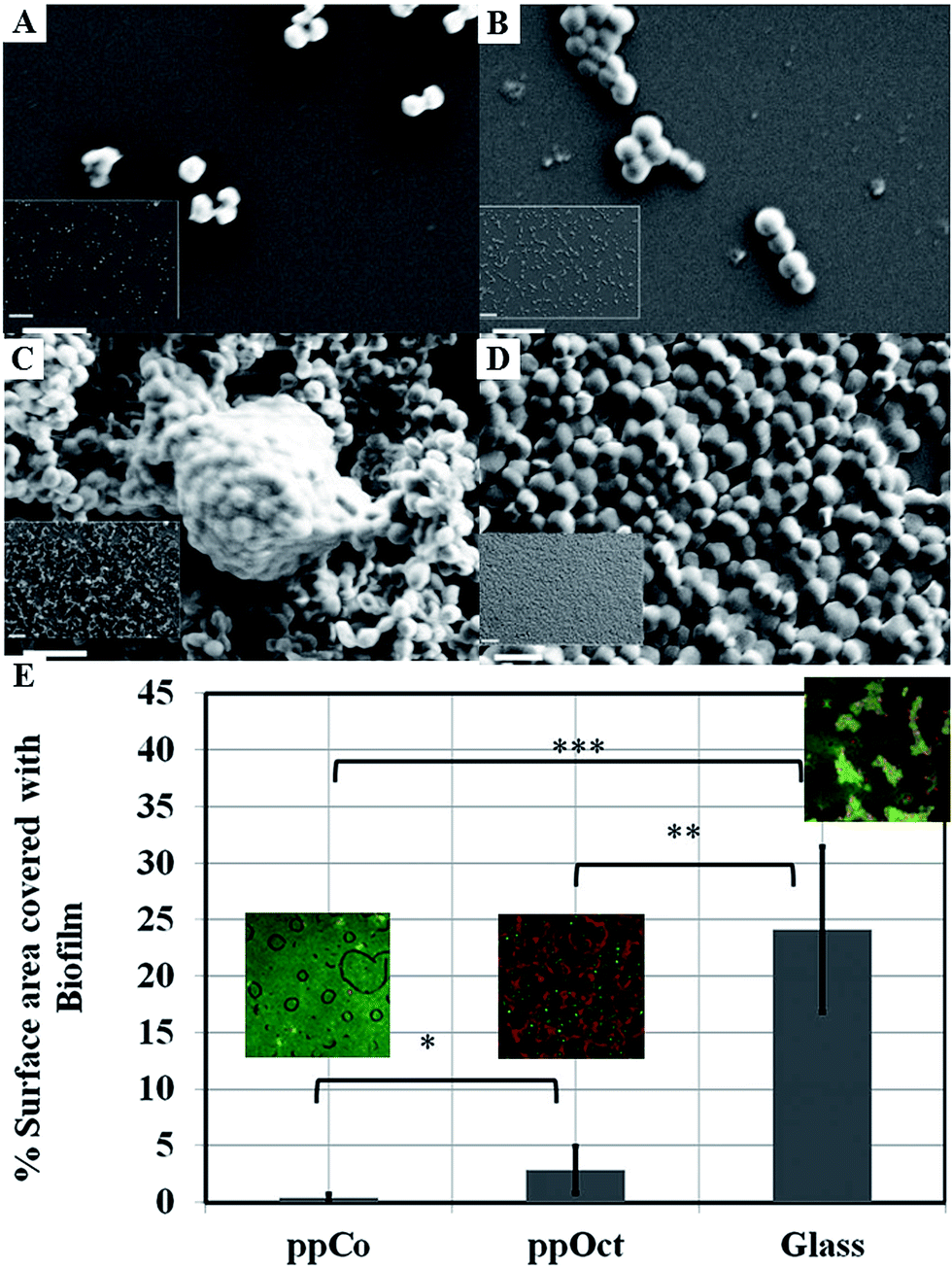 | ||
| Fig. 11 SEM images of attachment and proliferation of Streptococcus epidermidis (left panel) and Staphylococcus aureus (right panel) after 18 h incubation on surfaces subjected to monoterpene alcohol plasma deposition under varied input power conditions: (A and B) 10 W; (C and D) 50 W. Scale bar = 2 μm; 20 μm (inset).190 (E) Surface area covered by Escherichia coli biofilm formed on plasma polymerised 1,8-cineole (ppCo) and hydrophobic (ppOct) and hydrophilic (glass) controls. Samples were immersed in bacterial culture for 5 days (***p < 0.001, **p < 0.01, *p < 0.1).191 Reproduced with permission from ref. 190 and 191. | ||
Furthermore, the antimicrobial effect of these coatings is maintained over time under dry and wet conditions. Plasma polymer thin films fabricated from 1,8-cineole were able to successfully retard bacterial colonisation and biofilm formation by Escherichia coli for over 5 days, whereas plasma polymers of 1,7-octadiene, although hydrophobic and biofilm-retarding at early stages of bacterial exposure failed to maintain this activity. These results suggest that surface-immobilised and/or leachable constituents of cineol are primarily responsible for the observed activity.
The above examples relied on low-energy processing conditions to preserve the original chemistry of the film. However, increasing the processing energy can also produce useful structures fabricated from essential oils. Indeed, non-equilibrium and kinetics-driven phenomena of cold low-temperature plasma are often used for a simple, catalyst-free and highly-efficient production of self-organized nano-solids, e.g. graphenes nanowalls.193,194 While many other techniques exist for the fabrication of functional graphene sheets, most require the use of hazardous chemicals or toxic gases, complex, multistage processing, and are energy-, time-, and resource-consuming. In addition to being quick and efficient, the plasma-assisted processes allow for good adhesion to the substrate, and preservation of original minerals present in the precursor, resulting in varying chemistries and morphologies of the fabricated layers. It is worth noting that although plasma-assisted fabrication can substantially lower the overall processing temperature, the plasma heating effects limit the suitability of the technique for processing of polymers and other temperature-sensitive biomaterials.140
Graphene-based nanomaterials have been demonstrated to effectively inhibit the growth of bacteria, while showing minimal cytotoxicity to mammalian cell lines.197–201 Toxicity of graphene nanosheets in the form of graphene nanowalls on stainless steel towards E. coli and P. aeruginosa bacteria was attributed to the cell membrane damage from direct contact of the bacteria with the extremely sharp edges of the nanowalls,195 and a combination of membrane and oxidation stress, leading to DNA fragmentation and loss of cell viability.197–199 The sharper edges and more effective charge transfer between the bacteria and the nanowalls of reduced graphene oxide were responsible for its higher microbial toxicity compared to graphene oxide. The significance of direct interaction between bacterial membrane and the edge also meant that Gram-negative E. coli bacteria were more resistant compared to S. aureus which do not have an outer membrane.
Fig. 12(B) shows the structure of vertically-oriented graphenes prepared from Melaleuca alternifolia essential oil using plasma chemical vapour deposition. The graphene sheets had good adhesion to the substrate, low level of defects, and enhanced surface area with ultra-long edges. From the average circumference (about 600 nm) of the top edge of a well-like structure, the total length of the edges/folds in 1 cm2 area of a sample was estimated to be ∼2.6 km. Furthermore, plasmas can be used to chemically functionalise graphenes fabricated in this fashion during or post-synthesis, to thin the edges of graphene walls even further, or to decorate the surfaces with other nanostructures, creating more complex topographies.
 | ||
| Fig. 12 (A) Nanowalls of graphene oxide (GONWs) and reduced graphene (RGNWs) kill Gram-negative and Gram-positive bacteria upon direct contact, by causing membrane damage and loss of intracellular material (RNA). Sharper edges and a better charge transfer between the bacteria and the edge of the reduced nanowalls leads to more effective killing.195 (C–G) Mechanism of contact killing of graphenes. Respiratory proteins on the surface of bacteria behave as n-type semiconductors. The physical contact of bacteria with semimetal graphene results in Schottky barrier formation and Fermi level alignment. Electrons are then effectively transferred from microbial membranes to graphene acceptors and then to underlying conductor or semiconductor substrate (C and F). For graphene on insulating substrate (G), the electrons cannot be transferred to the underlying insulator, with bacteria cells remaining alive. Reproduced with permission from ref. 195 and 196. | ||
6. Conclusions
The continuous serious clinical complications related to multi-drug resistant pathogens have motivated researchers to explore alternative antibacterial agents whose mode of action differs sufficiently from the existing synthetic antibiotics, minimising the opportunity for bacteria to acquire resistance. Nature provides us with an abundant source of plant and animal-derived chemicals that have been specifically evolved by these organisms to ward off microbial invasion. Great physico-chemical diversity of these agents also brings about a significant challenge – only a very small fraction of the available agents have been identified. Their mechanism is even more elusive, as host organisms typically employ several defensive strategies that may complement, enhance and enable the activities of other contributors.Lack of consistency with regard to in vitro and in vivo testing reported in the literature often makes it difficult to compare these chemicals against each other and, importantly, against established antimicrobials. Limited understanding of the structure–property relationship of these antimicrobials makes it difficult to devise strategies for their controlled spatio-temporal delivery, thus hindering their potential application as coatings for implantable devices. In order to be used in practical applications, in addition to sound antimicrobial performance, the antimicrobials should be compatible with the processing, integration, implantation, and operation conditions. In turn, the processing environment should be flexible to accommodate a variety of surface modifications, allow for control of both physical and chemical surface characteristics, and should not compromise the biological performance of the bulk material with regard to its intended application nor diminish the efficacy of the antimicrobial agent. Plasma-enabled surface processing is a family of technologies that have the potential to amply address these requirements. Future research is ought to be directed towards development of plasma-enabled and other surface fabrication and modification methodologies with the capacity to independently control chemical, physical and biological properties of the material.
Acknowledgements
The authors acknowledge funding from ARC.Notes and references
- K. Bazaka, R. Crawford, E. Nazarenko and E. Ivanova, in Bacterial Adhesion, ed. D. Linke and A. Goldman, Springer Netherlands, 2011, vol. 715, pp. 213–226 Search PubMed.
- M. L. W. Knetsch and L. H. Koole, Polymers, 2011, 3, 340–366 CrossRef CAS.
- S. P. Valappil, D. Ready, E. A. Abou Neel, D. M. Pickup, W. Chrzanowski, L. A. O'Dell, R. J. Newport, M. E. Smith, M. Wilson and J. C. Knowles, Adv. Funct. Mater., 2008, 18, 732–741 CrossRef CAS.
- S. Valappil, D. Ready, E. Abou Neel, D. Pickup, L. O'Dell, W. Chrzanowski, J. Pratten, R. Newport, M. E. Smith and M. Wilson, Acta Biomater., 2009, 5, 1198–1210 CrossRef CAS PubMed.
- S. Forbes, A. J. McBain, S. Felton-Smith, T. A. Jowitt, H. L. Birchenough and C. B. Dobson, Biomaterials, 2013, 34, 5453–5464 CrossRef CAS PubMed.
- I. Ahmed, D. Ready, M. Wilson and J. C. Knowles, J. Biomed. Mater. Res., Part A, 2006, 79, 618–626 CrossRef CAS PubMed.
- S. P. Valappil, D. M. Pickup, D. L. Carroll, C. K. Hope, J. Pratten, R. J. Newport, M. E. Smith, M. Wilson and J. C. Knowles, Antimicrob. Agents Chemother., 2007, 51, 4453–4461 CrossRef CAS PubMed.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- K. Bazaka, M. Jacob, R. Crawford and E. Ivanova, Appl. Microbiol. Biotechnol., 2012, 95, 299–311 CrossRef CAS PubMed.
- W. Chrzanowski, S. P. Valappil, C. W. Dunnill, E. A. Abou Neel, K. Lee, I. P. Parkin, M. Wilson, D. A. Armitage and J. C. Knowles, Mater. Sci. Eng. C, 2010, 30, 225–234 CrossRef CAS.
- S. Mitragotri and J. Lahann, Nat. Mater., 2009, 8, 15–23 CrossRef CAS PubMed.
- Z. Wang, H. Wang, J. Liu and Y. Zhang, Desalination, 2014, 344, 313–320 CrossRef CAS.
- K. Bazaka, R. J. Crawford and E. P. Ivanova, Biotechnol. J., 2011, 6, 1103–1114 CrossRef CAS PubMed.
- J. Ma, Y. Sun, K. Gleichauf, J. Lou and Q. Li, Langmuir, 2011, 27, 10035–10040 CrossRef CAS PubMed.
- K. Koch, B. Bhushan and W. Barthlott, Soft Matter, 2008, 4, 1943–1963 RSC.
- S. H. Lee, H. W. Kim, J. O. Hwang, W. J. Lee, J. Kwon, C. W. Bielawski, R. S. Ruoff and S. O. Kim, Angew. Chem., Int. Ed., 2010, 49, 10084–10088 CrossRef CAS PubMed.
- S. Sethi, L. Ge, L. Ci, P. M. Ajayan and A. Dhinojwala, Nano Lett., 2008, 8, 822–825 CrossRef CAS PubMed.
- L. Zhang, Z. Zhou, B. Cheng, J. M. DeSimone and E. T. Samulski, Langmuir, 2006, 22, 8576–8580 CrossRef CAS PubMed.
- M. Psarski, J. Marczak, J. Grobelny and G. Celichowski, J. Nanomater., 2014, 2014, 547895 Search PubMed.
- K. Koch and W. Barthlott, Philos. Trans. R. Soc., A, 2009, 367, 1487–1509 CrossRef CAS PubMed.
- S. Pogodin, J. Hasan, V. A. Baulin, H. K. Webb, V. K. Truong, H. Phong Nguyen, V. Boshkovikj, C. J. Fluke, G. S. Watson, J. A. Watson, R. J. Crawford and E. P. Ivanova, Biophys. J., 2013, 104, 835–840 CrossRef CAS PubMed.
- E. P. Ivanova, J. Hasan, H. K. Webb, V. K. Truong, G. S. Watson, J. A. Watson, V. A. Baulin, S. Pogodin, J. Y. Wang, M. J. Tobin, C. Löbbe and R. J. Crawford, Small, 2012, 8, 2489–2494 CrossRef CAS PubMed.
- S. Mathews, M. Hans, F. Mücklich and M. Solioz, Appl. Environ. Microbiol., 2013, 79, 2605–2611 CrossRef CAS PubMed.
- P. Cronholm, H. L. Karlsson, J. Hedberg, T. A. Lowe, L. Winnberg, K. Elihn, I. O. Wallinder and L. Möller, Small, 2013, 9, 970–982 CrossRef CAS PubMed.
- J. Hasan, H. Webb, V. Truong, S. Pogodin, V. Baulin, G. Watson, J. Watson, R. Crawford and E. Ivanova, Appl. Microbiol. Biotechnol., 2013, 97, 9257–9262 CrossRef CAS PubMed.
- P. Kurt, L. Wood, D. E. Ohman and K. J. Wynne, Langmuir, 2007, 23, 4719–4723 CrossRef CAS PubMed.
- M. L. Eva, K. Yvonne, D. Joerg, E. Matthias and D. Ralf, Nanotechnology, 2011, 22, 375101 CrossRef PubMed.
- L. Ferreira and A. Zumbuehl, J. Mater. Chem., 2009, 19, 7796–7806 RSC.
- K. Hilpert, M. Elliott, H. Jenssen, J. Kindrachuk, C. D. Fjell, J. Körner, D. F. H. Winkler, L. L. Weaver, P. Henklein, A. S. Ulrich, S. H. Y. Chiang, S. W. Farmer, N. Pante, R. Volkmer and R. E. W. Hancock, Chem. Biol., 2009, 16, 58–69 CrossRef CAS PubMed.
- G. W. Charville, E. M. Hetrick, C. B. Geer and M. H. Schoenfisch, Biomaterials, 2008, 29, 4039–4044 CrossRef CAS PubMed.
- D. E. Fullenkamp, J. G. Rivera, Y.-K. Gong, K. H. A. Lau, L. He, R. Varshney and P. B. Messersmith, Biomaterials, 2012, 33, 3783–3791 CrossRef CAS PubMed.
- M. Simões, M. Lemos and L. Simões, in Dietary Phytochemicals and Microbes, ed. A. K. Patra, Springer Netherlands, 2012, pp. 185–205 Search PubMed.
- A. L. Hilchie, K. Wuerth and R. E. W. Hancock, Nat. Chem. Biol., 2013, 9, 761–768 CrossRef CAS PubMed.
- E. N. G. Marsh, B. C. Buer and A. Ramamoorthy, Mol. BioSyst., 2009, 5, 1143–1147 RSC.
- B. S. Holub, I. Rauch, S. Radner, W. Sperl, M. Hell and B. Kofler, Int. J. Antimicrob. Agents, 2011, 38, 76–80 CrossRef CAS PubMed.
- E. Gonzalez-Rey, A. Chorny and M. Delgado, Ann. N. Y. Acad. Sci., 2006, 1070, 303–308 CrossRef CAS PubMed.
- N. Afacan, L. Janot and R. W. Hancock, in Antimicrobial Peptides and Innate Immunity, ed. P. S. Hiemstra and S. A. J. Zaat, Springer Basel, 2013, pp. 321–358 Search PubMed.
- K. Ohta, M. Kajiya, T. Zhu, H. Nishi, H. Mawardi, J. Shin, L. Elbadawi, N. Kamata, H. Komatsuzawa and T. Kawai, J. Neuroimmunol., 2011, 233, 37–45 CrossRef CAS PubMed.
- I. M. Chiu, C. A. von Hehn and C. J. Woolf, Nat. Neurosci., 2012, 15, 1063–1067 CrossRef CAS PubMed.
- I. Vouldoukis, Y. Shai, P. Nicolas and A. Mor, FEBS Lett., 1996, 380, 237–240 CrossRef CAS PubMed.
- D. Augustyniak, J. Nowak and F. T. Lundy, Curr. Protein Pept. Sci., 2012, 13, 723–738 CrossRef CAS PubMed.
- F. García-Olmedo, A. Molina, J. M. Alamillo and P. Rodríguez-Palenzuéla, Pept. Sci., 1998, 47, 479–491 CrossRef.
- M. J. Carmona, A. Molina, J. A. Fernández, J. J. López-Fando and F. García-Olmedo, Plant J., 1993, 3, 457–462 CrossRef CAS PubMed.
- B. Stec, O. Markman, U. Rao, G. Heffron, S. Henderson, L. P. Vernon, V. Brumfeld and M. M. Teeter, J. Pept. Res., 2004, 64, 210–224 CrossRef CAS PubMed.
- H. U. Stotz, F. Waller and K. Wang, in Antimicrobial Peptides and Innate Immunity, ed. P. S. Hiemstra and S. A. J. Zaat, Springer Basel, 2013, pp. 29–51 Search PubMed.
- S. Yokoyama, K. Kato, A. Koba, Y. Minami, K. Watanabe and F. Yagi, Peptides, 2008, 29, 2110–2117 CrossRef CAS PubMed.
- A. S. Veiga, C. Sinthuvanich, D. Gaspar, H. G. Franquelim, M. A. R. B. Castanho and J. P. Schneider, Biomaterials, 2012, 33, 8907–8916 CrossRef CAS PubMed.
- D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS PubMed.
- D. A. Salick, D. J. Pochan and J. P. Schneider, Adv. Mater., 2009, 21, 4120–4123 CrossRef CAS.
- Y. Liu, Y. Yang, C. Wang and X. Zhao, Nanoscale, 2013, 5, 6413–6421 RSC.
- C. Zhou, P. Li, X. Qi, A. R. M. Sharif, Y. F. Poon, Y. Cao, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Biomaterials, 2011, 32, 2704–2712 CrossRef CAS PubMed.
- M. Kazemzadeh-Narbat, J. Kindrachuk, K. Duan, H. Jenssen, R. E. W. Hancock and R. Wang, Biomaterials, 2010, 31, 9519–9526 CrossRef CAS PubMed.
- A. Shukla, K. E. Fleming, H. F. Chuang, T. M. Chau, C. R. Loose, G. N. Stephanopoulos and P. T. Hammond, Biomaterials, 2010, 31, 2348–2357 CrossRef CAS PubMed.
- S. A. Onaizi and S. S. J. Leong, Biotechnol. Adv., 2011, 29, 67–74 CrossRef CAS PubMed.
- G. Gao, J. T. J. Cheng, J. Kindrachuk, R. E. W. Hancock, S. K. Straus and J. N. Kizhakkedathu, Chem. Biol., 2012, 19, 199–209 CrossRef CAS PubMed.
- K. Glinel, P. Thebault, V. Humblot, C. M. Pradier and T. Jouenne, Acta Biomater., 2012, 8, 1670–1684 CrossRef CAS PubMed.
- F. Costa, I. F. Carvalho, R. C. Montelaro, P. Gomes and M. C. L. Martins, Acta Biomater., 2011, 7, 1431–1440 CrossRef CAS PubMed.
- G. Gao, D. Lange, K. Hilpert, J. Kindrachuk, Y. Zou, J. T. J. Cheng, M. Kazemzadeh-Narbat, K. Yu, R. Wang, S. K. Straus, D. E. Brooks, B. H. Chew, R. E. W. Hancock and J. N. Kizhakkedathu, Biomaterials, 2011, 32, 3899–3909 CrossRef CAS PubMed.
- M. Gabriel, K. Nazmi, E. C. Veerman, A. V. Nieuw Amerongen and A. Zentner, Bioconjugate Chem., 2006, 17, 548–550 CrossRef CAS PubMed.
- N. K. Brogden and K. A. Brogden, Int. J. Antimicrob. Agents, 2011, 38, 217–225 CAS.
- C. Vreuls, G. Zocchi, B. Thierry, G. Garitte, S. S. Griesser, C. Archambeau, C. Van de Weerdt, J. Martial and H. Griesser, J. Mater. Chem., 2010, 20, 8092–8098 RSC.
- B. M. Kyaw, S. arora and C. S. Lim, Braz. J. Microbiol., 2012, 43, 938–945 CrossRef CAS PubMed.
- M. M. Cowan, Clin. Microbiol. Rev., 1999, 12, 564–582 CAS.
- G. Tegos, F. R. Stermitz, O. Lomovskaya and K. Lewis, Antimicrob. Agents Chemother., 2002, 46, 3133–3141 CrossRef CAS PubMed.
- N. S. Iacobellis, P. Lo Cantore, F. Capasso and F. Senatore, J. Agric. Food Chem., 2004, 53, 57–61 CrossRef PubMed.
- P. K. Lai and J. Roy, Curr. Med. Chem., 2004, 11, 1451–1460 CrossRef CAS PubMed.
- S. Ankri and D. Mirelman, Microbes Infect., 1999, 1, 125–129 CrossRef CAS PubMed.
- M. Singh and N. Singh, Comparison of antimicrobial activity of herbs & spices and their phytochemical determination, 2011 Search PubMed.
- K. A. Hammer, C. F. Carson and T. V. Riley, J. Appl. Microbiol., 1999, 86, 985–990 CrossRef CAS PubMed.
- P. S. Negi, G. K. Jayaprakasha, L. Jagan Mohan Rao and K. K. Sakariah, J. Agric. Food Chem., 1999, 47, 4297–4300 CrossRef CAS PubMed.
- I. H. N. Bassole, A. S. Ouattara, R. Nebie, C. A. T. Ouattara, Z. I. Kabore and S. A. Traore, Phytochemistry, 2003, 62, 209–212 CrossRef CAS PubMed.
- S. Prabuseenivasan, M. Jayakumar and S. Ignacimuthu, BMC Complementary Altern. Med., 2006, 6, 39 CrossRef PubMed.
- R. K. Johri, Pharmacogn. Rev., 2011, 5, 63–72 CrossRef CAS PubMed.
- M. Bigdeli, M. Sattari and S. Derakhshan, Effect of cumin Cuminum cyminum seed essential oil on biofilm formation and plasmid integrity of Klebsiella pneumoniae, 2010 Search PubMed.
- H. Hajlaoui, H. Mighri, E. Noumi, M. Snoussi, N. Trabelsi, R. Ksouri and A. Bakhrouf, Food Chem. Toxicol., 2010, 48, 2186–2192 CrossRef CAS PubMed.
- L. Gachkar, D. Yadegari, M. B. Rezaei, M. Taghizadeh, S. A. Astaneh and I. Rasooli, Food Chem., 2007, 102, 898–904 CrossRef CAS.
- S. Shayegh, I. Rasooli, M. Taghizadeh and S. D. Alipoor Astaneh, Nat. Prod. Res., 2008, 22, 428–439 CrossRef CAS PubMed.
- M. De, A. Krishna De and A. B. Banerjee, Phytother. Res., 1999, 13, 616–618 CrossRef CAS PubMed.
- A. Fabio, A. Corona, E. Forte and P. Quaglio, New Microbiol., 2003, 26, 115–120 CAS.
- M. Marino, C. Bersani and G. Comi, J. Food Prot., 1999, 62, 1017–1023 CAS.
- J. J. Brophy, N. W. Davies, I. A. Southwell, I. A. Stiff and L. R. Williams, J. Agric. Food Chem., 1989, 37, 1330–1335 CrossRef CAS.
- J. M. Wilkinson, in Modern Phytomedicine, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 157–171 Search PubMed.
- C. Valgas, S. M. D. Souza, E. F. A. Smânia and A. Smânia Jr, Braz. J. Microbiol., 2007, 38, 369–380 CrossRef.
- A. Klančnik, S. Piskernik, B. Jeršek and S. S. Možina, J. Microbiol. Methods, 2010, 81, 121–126 CrossRef PubMed.
- C. F. Carson, K. A. Hammer and T. V. Riley, Clin. Microbiol. Rev., 2006, 19, 50–62 CrossRef CAS PubMed.
- C. J. Papadopoulos, C. F. Carson, K. A. Hammer and T. V. Riley, J. Antimicrob. Chemother., 2006, 58, 449–451 CrossRef CAS PubMed.
- A. Brady, R. Loughlin, D. Gilpin, P. Kearney and M. Tunney, J. Med. Microbiol., 2006, 55, 1375–1380 CrossRef CAS PubMed.
- B. Oliva, E. Piccirilli, T. Ceddia, E. Pontieri, P. Aureli and A. M. Ferrini, Lett. Appl. Microbiol., 2003, 37, 185–187 CrossRef CAS PubMed.
- J. Sikkema, J. A. de Bont and B. Poolman, Microbiol. Rev., 1995, 59, 201–222 CAS.
- S. D. Cox, C. M. Mann, J. L. Markham, H. C. Bell, J. E. Gustafson, J. R. Warmington and S. G. Wyllie, J. Appl. Microbiol., 2000, 88, 170–175 CrossRef CAS PubMed.
- T. Hada, Y. Inoue, A. Shiraishi and H. Hamashima, J. Microbiol. Methods, 2003, 53, 309–312 CrossRef CAS PubMed.
- C. F. Carson, B. J. Mee and T. V. Riley, Antimicrob. Agents Chemother., 2002, 46, 1914–1920 CrossRef CAS PubMed.
- F. Nazzaro, F. Fratianni, L. De Martino, R. Coppola and V. De Feo, Pharmaceuticals, 2013, 6, 1451–1474 CrossRef PubMed.
- R. S. Feldberg, S. C. Chang, A. N. Kotik, M. Nadler, Z. Neuwirth, D. C. Sundstrom and N. H. Thompson, Antimicrob. Agents Chemother., 1988, 32, 1763–1768 CrossRef CAS PubMed.
- T. Bjarnsholt, P. Ø. Jensen, T. B. Rasmussen, L. Christophersen, H. Calum, M. Hentzer, H.-P. Hougen, J. Rygaard, C. Moser, L. Eberl, N. Høiby and M. Givskov, Microbiology, 2005, 151, 3873–3880 CrossRef CAS PubMed.
- T. B. Rasmussen, T. Bjarnsholt, M. E. Skindersoe, M. Hentzer, P. Kristoffersen, M. Köte, J. Nielsen, L. Eberl and M. Givskov, J. Bacteriol., 2005, 187, 1799–1814 CrossRef CAS PubMed.
- K. Kamino, Mar. Biotechnol., 2008, 10, 111–121 CrossRef CAS PubMed.
- E. Fadeeva, V. K. Truong, M. Stiesch, B. N. Chichkov, R. J. Crawford, J. Wang and E. P. Ivanova, Langmuir, 2011, 27, 3012–3019 CrossRef CAS PubMed.
- R. J. Stewart, T. C. Ransom and V. Hlady, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 757–771 CrossRef CAS PubMed.
- K. Kamino, J. Adhes., 2010, 86, 96–110 CrossRef CAS.
- V. Zheden, J. Von Byern, A. Kerbl, N. Leisch, Y. Staedler, I. Grunwald, A. M. Power and W. Klepal, Biol. Bull., 2012, 223, 192–204 Search PubMed.
- H. Shao, K. N. Bachus and R. J. Stewart, Macromol. Biosci., 2009, 9, 464–471 CrossRef CAS PubMed.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- B. P. Lee and S. Konst, Adv. Mater., 2014, 26, 3415–3419 CrossRef CAS PubMed.
- T. H. Anderson, J. Yu, A. Estrada, M. U. Hammer, J. H. Waite and J. N. Israelachvili, Adv. Funct. Mater., 2010, 20, 4196–4205 CrossRef CAS PubMed.
- E. Faure, C. Falentin-Daudré, C. Jérôme, J. Lyskawa, D. Fournier, P. Woisel and C. Detrembleur, Prog. Polym. Sci., 2013, 38, 236–270 CrossRef CAS.
- C. E. Brubaker and P. B. Messersmith, Biomacromolecules, 2011, 12, 4326–4334 CrossRef CAS PubMed.
- C. Greulich, D. Braun, A. Peetsch, J. Diendorf, B. Siebers, M. Epple and M. Koller, RSC Adv., 2012, 2, 6981–6987 RSC.
- A. Peetsch, C. Greulich, D. Braun, C. Stroetges, H. Rehage, B. Siebers, M. Köller and M. Epple, Colloids Surf., B, 2013, 102, 724–729 CrossRef CAS PubMed.
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed.
- T. Shalev, A. Gopin, M. Bauer, R. W. Stark and S. Rahimipour, J. Mater. Chem., 2012, 22, 2026–2032 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. Sureshkumar, D. Y. Siswanto and C.-K. Lee, J. Mater. Chem., 2010, 20, 6948–6955 RSC.
- W. J. Yang, T. Cai, K.-G. Neoh, E.-T. Kang, S. L.-M. Teo and D. Rittschof, Biomacromolecules, 2013, 14, 2041–2051 CrossRef CAS PubMed.
- T. Tah and M. T. Bernards, Colloids Surf., B, 2012, 93, 195–201 CrossRef CAS PubMed.
- W. J. Yang, T. Cai, K.-G. Neoh, E.-T. Kang, G. H. Dickinson, S. L.-M. Teo and D. Rittschof, Langmuir, 2011, 27, 7065–7076 CrossRef CAS PubMed.
- M. G. Kong, M. Keidar and K. Ostrikov, J. Phys. D: Appl. Phys., 2011, 44, 174018 CrossRef.
- D. Mariotti and K. Ostrikov, J. Phys. D: Appl. Phys., 2009, 42, 092002 CrossRef.
- M. J. Pavlovich, Z. Chen, Y. Sakiyama, D. S. Clark and D. B. Graves, Plasma Processes Polym., 2013, 10, 69–76 CrossRef CAS.
- F. Rossi, O. Kylián, H. Rauscher, M. Hasiwa and D. Gilliland, New J. Phys., 2009, 11, 115017 CrossRef.
- A. Mai-Prochnow, A. B. Murphy, K. M. McLean, M. G. Kong and K. Ostrikov, Int. J. Antimicrob. Agents, 2014, 43, 508–517 CrossRef CAS PubMed.
- D. B. Graves, J. Phys. D: Appl. Phys., 2012, 45, 263001 CrossRef.
- M. Laroussi, IEEE Trans. Plasma Sci., 2002, 30, 1409–1415 CrossRef CAS.
- R. E. J. Sladek, E. Stoffels, R. Walraven, P. J. A. Tielbeek and R. A. Koolhoven, IEEE Trans. Plasma Sci., 2004, 32, 1540–1543 CrossRef.
- T. Nosenko, T. Shimizu and G. E. Morfill, New J. Phys., 2009, 11, 115013 CrossRef.
- I. E. Kieft, M. Kurdi and E. Stoffels, IEEE Trans. Plasma Sci., 2006, 34, 1331–1336 CrossRef CAS.
- S. Kalghatgi, C. M. Kelly, E. Cerchar, B. Torabi, O. Alekseev, A. Fridman, G. Friedman and J. Azizkhan-Clifford, PLoS One, 2011, 6, e16270 CAS.
- M. Keidar, R. Walk, A. Shashurin, P. Srinivasan, A. Sandler, S. Dasgupta, R. Ravi, R. Guerrero-Preston and B. Trink, Br. J. Cancer, 2011, 105, 1295–1301 CrossRef CAS PubMed.
- M. Ishaq, M. Evans and K. Ostrikov, Int. J. Cancer, 2014, 134, 1517–1528 CrossRef CAS PubMed.
- M. Vandamme, E. Robert, S. Lerondel, V. Sarron, D. Ries, S. Dozias, J. Sobilo, D. Gosset, C. Kieda, B. Legrain, J.-M. Pouvesle and A. L. Pape, Int. J. Cancer, 2012, 130, 2185–2194 CrossRef CAS PubMed.
- M. Ishaq, M. D. M. Evans and K. Ostrikov, Biochim. Biophys. Acta, Mol. Cell Res., 2014, 1843, 2827–2837 CrossRef CAS PubMed.
- R. Löffler, M. Fleischer and D. P. Kern, Microelectron. Eng., 2012, 97, 361–364 CrossRef.
- Z. Yue, I. Levchenko, S. Kumar, D. Seo, X. Wang, S. Dou and K. Ostrikov, Nanoscale, 2013, 5, 9283–9288 RSC.
- W.-J. Chang, K.-L. Ou, S.-Y. Lee, J.-Y. Chen, Y. Abiko, C.-T. Lin and H.-M. Huang, Dent. Mater. J., 2008, 27, 340–346 CrossRef CAS.
- L. Russo, S. Zanini, P. Giannoni, E. Landi, A. Villa, M. Sandri, C. Riccardi, R. Quarto, S. Doglia, F. Nicotra and L. Cipolla, J. Mater. Sci.: Mater. Med., 2012, 23, 2727–2738 CrossRef CAS PubMed.
- D. Çökeliler, H. Gökta, P. D. Tosun and S. Mutlu, J. Nanosci. Nanotechnol., 2010, 10, 2583–2589 CrossRef.
- M. A. Surmeneva, A. Kovtun, A. Peetsch, S. N. Goroja, A. A. Sharonova, V. F. Pichugin, I. Y. Grubova, A. A. Ivanova, A. D. Teresov, N. N. Koval, V. Buck, A. Wittmar, M. Ulbricht, O. Prymak, M. Epple and R. A. Surmenev, RSC Adv., 2013, 3, 11240–11246 RSC.
- K. Ostrikov, E. C. Neyts and M. Meyyappan, Adv. Phys., 2013, 62, 113–224 CrossRef CAS.
- D. H. Seo, A. E. Rider, Z. J. Han, S. Kumar and K. Ostrikov, Adv. Mater., 2013, 25, 5638–5642 CrossRef CAS PubMed.
- K. Bazaka, M. V. Jacob, V. K. Truong, F. Wang, W. A. A. Pushpamali, J. Y. Wang, A. V. Ellis, C. C. Berndt, R. J. Crawford and E. P. Ivanova, Biomacromolecules, 2010, 11, 2016–2026 CrossRef CAS PubMed.
- K. Bazaka, M. V. Jacob and E. P. Ivanova, Mater. Sci. Forum, 2010, 654–656, 2261–2264 CrossRef CAS.
- M. V. Jacob, N. S. Olsen, L. Anderson, K. Bazaka and R. A. Shanks, Thin Solid Films, 2013, 546, 167–170 CrossRef CAS.
- K. Bazaka, M. V. Jacob, D. Taguchi, T. Manaka and M. Iwamoto, Chem. Phys. Lett., 2011, 503, 105–111 CrossRef CAS.
- B. R. Coad, M. Jasieniak, S. S. Griesser and H. J. Griesser, Surf. Coat. Technol., 2013, 233, 169–177 CrossRef CAS.
- M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS PubMed.
- A. Mierczynska, A. Michelmore, A. Tripathi, R. V. Goreham, R. Sedev and K. Vasilev, Soft Matter, 2012, 8, 8399–8404 RSC.
- J. Yang, F. R. A. J. Rose, N. Gadegaard and M. R. Alexander, Adv. Mater., 2009, 21, 300–304 CrossRef CAS.
- R. V. Goreham, A. Mierczynska, M. Pierce, R. D. Short, S. Taheri, A. Bachhuka, A. Cavallaro, L. E. Smith and K. Vasilev, Thin Solid Films, 2013, 528, 106–110 CrossRef CAS.
- A. Michelmore, D. A. Steele, J. D. Whittle, J. W. Bradley and R. D. Short, RSC Adv., 2013, 3, 13540–13557 RSC.
- R. A. Surmenev, M. A. Surmeneva, K. E. Evdokimov, V. F. Pichugin, T. Peitsch and M. Epple, Surf. Coat. Technol., 2011, 205, 3600–3606 CrossRef CAS.
- H. Yasuda, Polym. Prepr., 1975, 16, 57–59 CrossRef.
- A. Michelmore, P. Gross-Kosche, S. A. Al-Bataineh, J. D. Whittle and R. D. Short, Langmuir, 2013, 29, 2595–2601 CrossRef CAS PubMed.
- A. Michelmore, P. M. Bryant, D. A. Steele, K. Vasilev, J. W. Bradley and R. D. Short, Langmuir, 2011, 27, 11943–11950 CrossRef CAS PubMed.
- P. N. Brookes, S. Fraser, R. D. Short, L. Hanley, E. Fuoco, A. Roberts and S. Hutton, J. Electron Spectrosc. Relat. Phenom., 2001, 121, 281–297 CrossRef CAS.
- D. Barton, R. D. Short, S. Fraser and J. W. Bradley, Chem. Commun., 2003, 348–349 RSC.
- A. Michelmore, D. A. Steele, D. E. Robinson, J. D. Whittle and R. D. Short, Soft Matter, 2013, 9, 6167–6175 RSC.
- T. J. Wood, G. A. Hurst, W. C. E. Schofield, R. L. Thompson, G. Oswald, J. S. O. Evans, G. J. Sharples, C. Pearson, M. C. Petty and J. P. S. Badyal, J. Mater. Chem., 2012, 22, 3859–3867 RSC.
- J. Friedrich, Plasma Processes Polym., 2011, 8, 783–802 CrossRef CAS.
- B. Yameen, H. U. Khan, W. Knoll, R. Förch and U. Jonas, Macromol. Rapid Commun., 2011, 32, 1735–1740 CrossRef CAS PubMed.
- G. P. Wells, I. C. Estrada-Raygoza, P. L. S. Thamban, C. T. Nelson, C.-W. Chung, L. J. Overzet and M. J. Goeckner, Plasma Processes Polym., 2013, 10, 119–135 CrossRef CAS.
- D. T. K. Kwok, L. Tong, C. Y. Yeung, C. G. D. Remedios and P. K. Chu, Surf. Coat. Technol., 2010, 204, 2892–2897 CrossRef CAS.
- L. Denis, P. Marsal, Y. Olivier, T. Godfroid, R. Lazzaroni, M. Hecq, J. Cornil and R. Snyders, Plasma Processes Polym., 2010, 7, 172–181 CAS.
- A. Manakhov, M. Moreno-Couranjou, N. D. Boscher, V. Rogé, P. Choquet and J.-J. Pireaux, Plasma Processes Polym., 2012, 9, 435–445 CrossRef CAS.
- H. Duran, B. Yameen, H. U. Khan, R. Förch and W. Knoll, React. Funct. Polym., 2013, 73, 606–612 CrossRef CAS.
- L. Duque, B. Menges, S. Borros and R. Förch, Biomacromolecules, 2010, 11, 2818–2823 CrossRef CAS PubMed.
- A. Fahmy, A. Schönhals and J. Friedrich, J. Phys. Chem. B, 2013, 117, 10603–10611 CrossRef CAS PubMed.
- J. C. Shearer and E. R. Fisher, Nanosci. Nanotechnol. Lett., 2012, 4, 358–363 CrossRef CAS.
- C. Tarducci, W. C. E. Schofield, J. P. S. Badyal, S. A. Brewer and C. Willis, Chem. Mater., 2002, 14, 2541–2545 CrossRef CAS.
- T. J. Wood and J. P. S. Badyal, ACS Appl. Mater. Interfaces, 2012, 4, 1675–1682 CAS.
- J.-P. Chen, C.-Y. Kuo and W.-L. Lee, Appl. Surf. Sci., 2012, 262, 95–101 CrossRef CAS.
- K. D. Anderson, S. L. Young, H. Jiang, R. Jakubiak, T. J. Bunning, R. R. Naik and V. V. Tsukruk, Langmuir, 2011, 28, 1833–1845 CrossRef PubMed.
- C. Tarducci, W. C. E. Schofield, J. P. S. Badyal, S. A. Brewer and C. Willis, Chem. Mater., 2001, 13, 1800–1803 CrossRef CAS.
- C. Tarducci, E. J. Kinmond, J. P. S. Badyal, S. A. Brewer and C. Willis, Chem. Mater., 2000, 12, 1884–1889 CrossRef CAS.
- D. O. H. Teare, D. C. Barwick, W. C. E. Schofield, R. P. Garrod, L. J. Ward and J. P. S. Badyal, Langmuir, 2005, 21, 11425–11430 CrossRef CAS PubMed.
- D. Thiry, N. Britun, S. Konstantinidis, J.-P. Dauchot, M. Guillaume, J. Cornil and R. Snyders, J. Phys. Chem. C, 2013, 117, 9843–9851 CAS.
- C. Tarducci, J. P. S. Badyal, S. A. Brewer and C. Willis, Chem. Commun., 2005, 406–408 RSC.
- E. Kawakami, M. Washizu, T. Hirano, M. Sakuma, M. Takano, T. Hori and T. Tsutsui, J. Vet. Med. Sci., 2006, 68, 1215–1217 CrossRef PubMed.
- S. G. Seabrook, Google Patents, EP1940431 A1, 2008.
- J. L. Watts, Google Patents, US5397385 A, 2008.
- W. E. Craver and S. G. Seabrook, Google Patents, WO1999020258 A1, 1999.
- K. Varaprasad, K. Vimala, S. Ravindra, N. Narayana Reddy, G. Venkata Subba Reddy and K. Mohana Raju, J. Mater. Sci.: Mater. Med., 2011, 22, 1863–1872 CrossRef CAS PubMed.
- K. Varaprasad, Y. M. Mohan, K. Vimala and K. Mohana Raju, J. Appl. Polym. Sci., 2011, 121, 784–796 CrossRef CAS.
- K. Vimala, M. Mohan Yallapu, K. Varaprasad, N. Narayana Reddy, S. Ravindra, N. Sudhakar Naidu and K. Mohana Raju, J. Biomater. Nanobiotechnol., 2011, 2, 55–64 CrossRef CAS.
- K. Bazaka and M. V. Jacob, Polym. Degrad. Stab., 2010, 95, 1123–1128 CrossRef CAS.
- K. Bazaka, M. V. Jacob and B. F. Bowden, J. Mater. Res., 2011, 26, 1018–1025 CrossRef CAS.
- M. V. Jacob, K. Bazaka, M. Weis, D. Taguchi, T. Manaka and M. Iwamoto, Thin Solid Films, 2010, 518, 6123–6129 CrossRef CAS.
- K. Bazaka and M. Jacob, Electronics, 2012, 2, 1–34 CrossRef.
- M. V. Jacob, K. Bazaka, D. Taguchi, T. Manaka and M. Iwamoto, Chem. Phys. Lett., 2012, 528, 26–28 CrossRef CAS.
- K. Bazaka, M. V. Jacob, R. J. Crawford and E. P. Ivanova, Acta Biomater., 2011, 7, 2015–2028 CrossRef CAS PubMed.
- A. Pegalajar-Jurado, C. D. Easton, K. E. Styan and S. L. McArthur, J. Mater. Chem. B, 2014, 2, 4993–5002 RSC.
- K. Bazaka, M. Jacob, V. K. Truong, R. J. Crawford and E. P. Ivanova, Polymers, 2011, 3, 388–404 CrossRef CAS.
- D. H. Seo, A. E. Rider, S. Kumar, L. K. Randeniya and K. Ostrikov, Carbon, 2013, 60, 221–228 CrossRef CAS.
- D. H. Seo, Z. J. Han, S. Kumar and K. Ostrikov, Adv. Energy Mater., 2013, 3, 1316–1323 CrossRef CAS.
- O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS PubMed.
- J. Li, G. Wang, H. Zhu, M. Zhang, X. Zheng, Z. Di, X. Liu and X. Wang, Sci. Rep., 2014, 4, 4359 Search PubMed.
- W. Hu, C. Peng, W. Luo, M. Lv, X. Li, D. Li, Q. Huang and C. Fan, ACS Nano, 2010, 4, 4317–4323 CrossRef CAS PubMed.
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5, 6971–6980 CrossRef CAS PubMed.
- S. Gurunathan, J. Han, A. Dayem, V. Eppakayala and J. Kim, Int. J. Nanomed., 2012, 2012, 5901–5914 CrossRef PubMed.
- L. Yu, Y. Zhang, B. Zhang, J. Liu, H. Zhang and C. Song, J. Membr. Sci., 2013, 447, 452–462 CrossRef CAS.
- L. Yu, Y. Zhang, B. Zhang and J. Liu, Sci. Rep., 2014, 4, 4551 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |