
Electrospun composite nanofibres of PVA loaded with nanoencapsulated n-octadecane†
Abstract
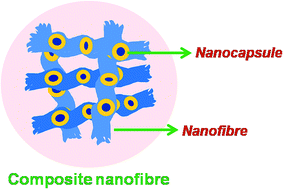
In this study composite nanofibres of PVA consisting of polystyrene/n-octadecane nanocapsules were prepared. The n-octadecane present as a core of the nanocapsules acts as a phase change material (PCM) for thermal storage properties. Controlled size nanoencapsulated PCMs were prepared by surfactant free RAFT miniemulsion polymerization of styrene. These nanocapsules were incorporated into nanofibres by electrospinning a mixture of nanocapsule latex containing octadecane as a core and polystyrene (PS) as a shell material with an aqueous solution of poly(vinyl alcohol). The morphology and thermal properties of the resultant PVA nanofibres were studied by field emission scanning microscopy (FE-SEM), scanning transmission electron microscopy (STEM), high resolution transmission electron microscopy (HRTEM) and differential scanning calorimetry (DSC). The composite nanofibres could show a latent heat storage capacity of approx. 4–5 J g−1. This method offers the opportunity to produce low diameter composite nanofibres with uniform dispersion of encapsulated material for various applications.
 Please wait while we load your content...
Please wait while we load your content...

