
Recent advances in methanation catalysts for the production of synthetic natural gas
Jiajian Gaoab,
Qing Liua,
Fangna Gua,
Bin Liu*b,
Ziyi Zhongc and
Fabing Su*a
aState Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, China 100190. E-mail: fbsu@ipe.ac.cn; Fax: +86-10-82544851; Tel: +86-10-82544850
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459. E-mail: liubin@ntu.edu.sg; Fax: +65-67947553; Tel: +65-65137971
cInstitute of Chemical Engineering and Sciences, A*star, 1 Pesek Road, Jurong Island, Singapore 627833, Singapore
First published on 18th February 2015
Abstract
Methanation of coal- or biomass-derived carbon oxides for production of synthetic natural gas (SNG) is gaining considerable interest due to energy issues and the opportunity of reducing greenhouse gases by carbon dioxide conversion. The key component of the methanation process is the catalyst design. Ideally, the catalyst should show high activity at low temperatures (200–300 °C) and high stability at high temperatures (600–700 °C). In the past decades, various methanation catalysts have been investigated, among which transition metals including Ni, Fe, Co, Ru, Mo, etc. dispersed on metal oxide supports such as Al2O3, SiO2, TiO2, ZrO2, CeO2 etc. have received great attention due to their relatively high catalytic activity and selectivity. Furthermore, over the past few years, great efforts have been made both in methanation catalysts development and reaction mechanism investigation. Here we provide a comprehensive review to these most advancements, covering the reaction thermodynamics, mechanism and kinetics, the effects of catalyst active components, supports, promoters and preparation methods, hoping to outline the pathways for the future methanation catalysts design and development for SNG production.
1. Introduction
Among different forms of fossil fuels, natural gas that consists primarily of methane is ideal, owing to its ready availability, high energy density and conversion efficiency, and smoke- and slag-free composition.1 Additionally, natural gas can be transported efficiently at low cost using the existing natural gas pipelines and wide distribution network. In recent years, due to the rise of the natural gas price, the wish for less dependency on natural gas import, and replacement of oil products, synthetic or substitute natural gas (SNG) production from renewable biomass,2 coke oven gas (COG)3 or syngas from coal or wood4 is attracting increasing attention in some countries. Meanwhile, the actively investigated hydrogen production by photocatalytic or electrocatalytic water splitting powered by renewable energies (e.g., solar or wind) is regarded as future sources of hydrogen for carbon dioxide hydrogenation. Hence, the SNG production via carbon dioxide methanation process can not only produce fuels and chemicals, but also reduce carbon dioxide emission significantly to the atmosphere.5–9 Other applications of carbon monoxide methanation include removal of trace carbon monoxide present in H2-rich gases to produce high purity hydrogen for chemical industry such as NH3 synthesis and for fuel cells.The two key reactions of SNG production process are expressed as: CO + 3H2 → CH4 + H2O, CO2 + 4H2 → CH4 + 2H2O. Although the methanation reactions are thermodynamically favorable, catalyst is necessary to obtain an appropriate rate. It should be noted that methanation catalysts for the production of SNG deal with carbon monoxide and/or carbon dioxide at relatively high concentrations, making things quite different as compared with trace carbon monoxide (∼1 vol%) removal in hydrogen stream. In the earliest work by Sabatier and Senderens in 1902,10 nickel was found to be very active for methanation reaction. During the oil crisis in 1970s, the methanation catalysts and reactions were subjected to intensive investigations again because of the interest in producing SNG from naphtha and coal. In particular, Vannice conducted a systemic research on the catalytic synthesis of hydrocarbons from H2/CO mixtures over group VIII metals (Fe, Co, Ni, Ru, Rh, Pd, Ir, Pt).11–15 Meanwhile, Mills and Steffgen16 summarized and reviewed the catalytic methanation of carbon monoxide and carbon dioxide. However, these previous works focus on methanation at relatively low temperatures (about 200–400 °C) which are not optimum for energy recovery.17,18 Modern SNG processes dealing with high concentrations of carbon monoxide and/or carbon dioxide would result in large temperature increase (hot spots can reach 600–700 °C).18 It was reported that the methanation catalyst MCR-2X from Haldor Topsøe could be operated at high temperatures with high reaction rates to produce high-quality steam thus making the process more energy efficient.18 However, the high operating temperature would accelerate the sintering and coking of the catalyst. In short, the methanation catalysts should be stable at high temperatures and also be active at low temperatures to ignite the reaction in SNG production process.
Over the past ten years, methanation catalysts for the production of SNG have been investigated intensively again and some related reviews have been published, among which, Schildhauer and co-workers19 did a comprehensive technology review for SNG production from coal and dry biomass in the period from 1950 to 2009. Recently, Wang et al.20 summarized the catalytic hydrogenation of carbon dioxide. Considering the great progress achieved in this area, we feel it is necessary to contribute a new review paper, focusing on the recent advancements of methanation catalysts research and development. Also, the reaction thermodynamics, mechanism and kinetics, and the effects of catalyst active components, supports, promoters, and preparation methods will be reviewed and discussed. We hope this review will not only outline the achievements and technical problems, but also direct the future methanation catalysts design and development for SNG production.
2. Thermodynamics
During methanation, some side-reactions may occur which affects the purity of the SNG product. Table 1 lists the main possible reactions involved in the methanation process. Besides the normal methanation reactions (R1 and R2), carbon monoxide methanation reaction can also occur at lower H2/CO ratio (R3).21 The carbon monoxide disproportionation reaction (R4), also known as Boudouard reaction, is of great importance, since carbon on the catalyst surface is considered as a necessary intermediate during the methanation reaction.22 In addition, water plays an important role through the water–gas shift reaction (R5), which would modify the surface and catalytic chemistry of methanation catalysts.23 Among these reactions, it has to be noticed that R1, R2, and R4 can be regarded as three independent reactions. The other reactions can be described as a linear combination of these three reactions.| Reaction no. | Reaction formula | ΔH298![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K (kJ mol−1) K (kJ mol−1) |
ΔG298K (kJ mol−1) |
|---|---|---|---|
| R1 | CO + 3H2 ↔ CH4 + H2O | −206.1 | −141.8 |
| R2 | CO2 + 4H2 ↔ CH4 + 2H2O | −165.0 | −113.2 |
| R3 | 2CO + 2H2 ↔ CH4 + CO2 | −247.3 | −170.4 |
| R4 | 2CO ↔ C + CO2 | −172.4 | −119.7 |
| R5 | CO + H2O ↔ CO2 + H2 | −41.2 | −28.6 |
| R6 | 2H2 + C ↔ CH4 | −74.8 | −50.7 |
| R7 | CO + H2 ↔ C + H2O | −131.3 | −91.1 |
| R8 | CO2 + 2H2 ↔ C + 2H2O | −90.1 | −62.5 |
The equilibrium constants of the eight reactions involved in the methanation were calculated at different temperatures (Fig. 1).24 It can be seen that all the reactions are favorable at low temperatures (<∼400 °C) due to their exothermic characteristics. Obviously, low temperature and high pressure are preferred for carbon monoxide and carbon dioxide methanation. However, it is challenging to develop a catalyst that can achieve equilibration at low temperature for carbon monoxide and carbon dioxide methanation.17,18
 | ||
| Fig. 1 The calculated equilibrium constants (K) of the eight reactions involved in methanation process. Adapted from ref. 24 with permission of the Royal Society of Chemistry. | ||
Recently, we conducted a systematic thermodynamic analysis for methanation reactions of carbon oxides to produce SNG.24 The effects of temperature, pressure, ratios of H2/CO and H2/CO2, and the addition of other compounds in the feed gas on the conversion of carbon monoxide and carbon dioxide, methane selectivity and yield, as well as carbon deposition, were carefully investigated. The product composition distributions from stoichiometric carbon monoxide and carbon dioxide methanation reaction at equilibrium under 0.1 MPa are shown in Fig. 2a and b, respectively. For carbon monoxide methanation (Fig. 2a), the products mainly contain methane, water and little carbon dioxide by-product at low temperatures (200–300 °C) without deposition of carbon. With an increase in reaction temperature, the mole fraction of CH4 decreases, whereas the unreacted carbon monoxide, hydrogen, carbon dioxide, and deposited carbon increase simultaneously.
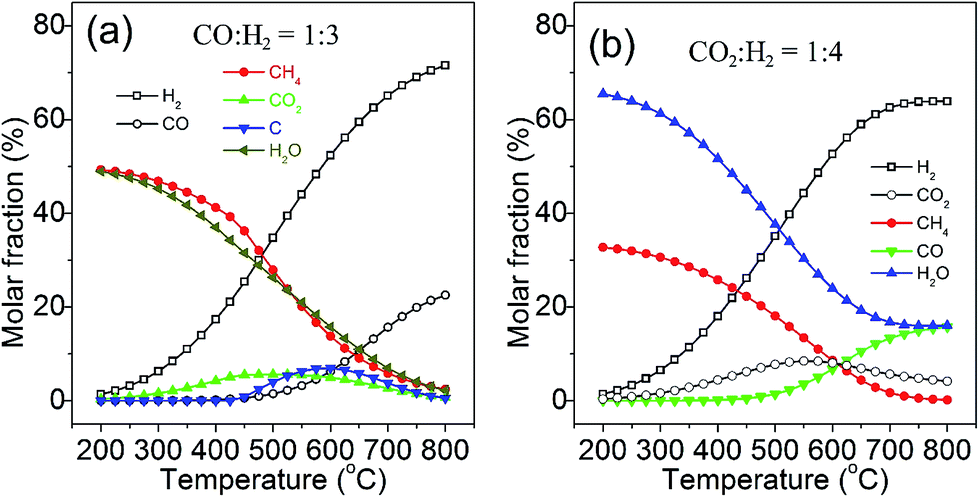 | ||
| Fig. 2 Product compositions for CO (a) and CO2 (b) methanation at equilibrium (0.1 MPa). Adapted from ref. 24 with permission of the Royal Society of Chemistry. | ||
Methane and water are the main products of carbon dioxide methanation (Fig. 2b) at low temperatures (200–250 °C). Noteworthily, the carbon dioxide methanation proceeds highly selectively as compared with the carbon monoxide methanation.25 Raising the reaction temperature above 450 °C results in the increase of the carbon monoxide by-product, due to the reverse water–gas shift reaction, and meanwhile, unreacted carbon dioxide and hydrogen also increase, along with a decrease in the methane yield. The reduction of the fully oxidized carbon to methane is an eight-electron process with significant kinetic barriers, which thus requires a highly active catalyst to achieve acceptable rate and selectivity.20 Through thermodynamic analysis, it is possible to obtain a useful guidance in the catalyst development and process control of methanation for the SNG production.
3. Methanation catalysts
Methanation catalysts are typically composed of active metal particles dispersed on metal oxide supports. Up to now, a number of active metals including Ni, Fe, Co, Ru, Rh, Pt, Pd, W, Mo and various oxide supports (Al2O3, SiO2, TiO2, SiC, ZrO2, CeO2, CexZr1−xO2) have been carefully investigated in both carbon monoxide and carbon dioxide methanation reactions for SNG production.3.1 Active components
In 1975, Vannice M. A.11 firstly compared the specific activity and product distributions of group VIII metals dispersed on Al2O3 in the synthesis of hydrocarbons from H2–CO mixtures, and found that carbon monoxide methanation reaction could occur readily over these metals. The specific activity follows the order of Ru ≫ Fe > Ni > Co > Rh > Pd > Pt > Ir. It is well known that methanation reactions involve hydrogen, carbon monoxide and/or carbon dioxide adsorption and dissociation. The reaction rate of carbon monoxide methanation is closely related to carbon monoxide dissociation. Bligaard et al.26 calculated the reaction energy for dissociative carbon monoxide adsorption at 550 K and compared it with the measured carbon monoxide methanation activities (Fig. 3). A clear volcano relationship was observed. The maximum of the volcano is approximately at a dissociative adsorption energy of −1.4 eV, which is in very good agreement with the prediction of Nørskov's results.27 Although the sequence of activity in Fig. 3 is a little different from that of Vannice's results, noble metal Ru is undoubtedly the most active one for the methanation reaction. However, Ni catalysts draw more attention for methanation due to their relatively high activity and low price.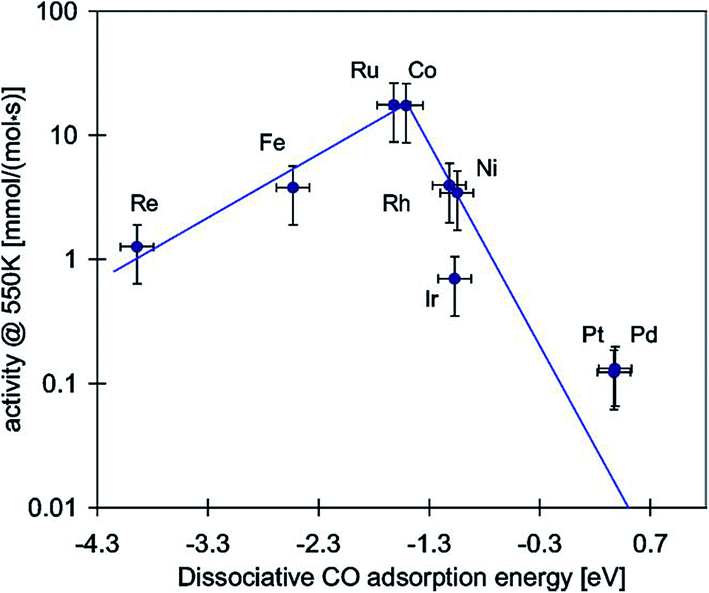 | ||
| Fig. 3 Activities of different supported transition metal catalysts as a function of the reaction energy for dissociative carbon monoxide chemisorption. Reprinted from ref. 26, Copyright (2004), with permission from Elsevier. | ||
Table 2 and 3 list some carbon monoxide and carbon dioxide methanation catalysts studied in recent years. It should be noted that it is difficult to directly compare the performance of these catalysts because different reaction conditions were used. However, some general results can be summarized. Ni with loading amounts of 10–40 wt% is the main active metal for carbon monoxide methanation. Nearly 100% conversion of carbon monoxide and about 90–100% selectivity of methane could be obtained over optimized Ni catalysts at 3.0 MPa with H2:CO feed ratio of 3. Some Ni catalysts could maintain good activity over 100 h.28–30 Even at 0.1 MPa, carbon dioxide methanation (Table 3) could still remain a high methane selectivity of nearly 100%, which is in accordance with the thermodynamic results.24 However, high carbon dioxide conversion is difficult to reach at low temperatures because of the high kinetic barriers of the reaction processes. During catalyst activity testing, space velocity deeply affects the carbon monoxide and carbon dioxide conversion and methane selectivity. High space velocity is recommended to test the activity of methanation catalysts far from thermodynamic equilibrium.
| Catalysts | Preparation methods | Active metal content wt% | Catalytic performance | Stability test time/h | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| P/MPa | WHSV/(mL g−1 h−1) | T/°C | XCO/% | SCH4/% | |||||
| a I: impregnation; P: precipitation; CP: co-precipitation; SC: solution combustion method; HT: hydrothermal synthesis; * deactivation was observed. | |||||||||
| Ni–Al2O3 | CP | 15 | 0.1 | 2500 h−1 | 400 | 98.2 | 84.7 | 120* | 1 |
| Ni–Al2O3 | Sol–gel | 40 | 1.0 | 8160 | 230 | 96.5 | 76.8 | — | 31 |
| Ni–Al2O3 | I | 10 | 0.1 | 240000 |
450 | 61 | 68 | 50* | 32 |
| Ni–SiC | I | 4.2 | 3.0 | 4006 | 500 | 96.7 | 100 | 120 | 28 |
| Ni–Mg–Al2O3 | I | 20 | 0.1 | 30000 |
400 | 100 | 80 | 196 | 29 |
| Ni–Al2O3 | I | 10 | 3.0 | 30000 |
400 | 97 | 90 | 10* | 33 |
| Ni–Mg–Al2O3 | I | 40 | 0.1 | 36000 |
300 | 64 | 58 | 50 | 34 |
| Ni–Mg–Al2O3 | CP + HT | 19 | 0.1 | 60000 |
427 | 85 | 84 | — | 35 |
| Ni–La2O3/Al2O3 | I | 15 | 1.5 | 10000 h−1 |
300 | 72 | 80 | 264 | 30 |
| Ni–TiO2 | Sonication | 23 | 0.1 | 38800 h−1 |
280 | 52 | 95 | — | 36 |
| Si–Ni/SiO2 | Silicification | 20 | 0.1 | 4800 | 350 | 50 | 28 | 42 | 37 |
| Ni/MCM-41 | HT | 10 | 0.1 | 12000 |
350 | 97.9 | 88.2 | 100 | 38 |
| Ni/CaTiO3 | I | 10 | 3.0 | 10000 |
350 | 95 | 72 | 50 | 39 |
| Ni/BaO·6Al2O3 | I | 40 | 3.0 | 30000 |
350 | 81 | 92 | 50 | 40 and 41 |
| Ni–Mo-MCM-41 | I | 10 | 0.1 | 12000 |
280 | 100 | 80 | 100 | 42 |
| Co3O4 | CP | — | 2.0 | 50000 h−1 |
300 | 99 | — | — | 43 |
| Ni/LaFeO3 | I | 30 | 1.0 | 3000 h−1 | 340 | 90 | 48 | 55* | 44 |
| Catalysts | Preparation methodsd | Active metal content wt% | Catalytic performance | Stability test time/h | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| P/MPa | WHSV/(mL g−1 h−1) | T/°C | XCO2/% | SCH4/% | |||||
| a MSN = mesostructured silica nanoparticles.b H-Al2O3 = hierarchical flowerlike Al2O3 matrix.c MC = mesoporous nanocrystalline γ-Al2O3.d I: impregnation; CP: co-precipitation; HT: hydrothermal synthesis; DP: deposition–precipitation. | |||||||||
| Ni–Ce0.5Zr0.5O2 | I | 10 | 3.0 | 30000 h−1 |
300 | 70 | — | — | 45 |
| Ni–TiO2 | DP | 15 | 0.1 | 2400 h−1 | 218 | 50 | 99 | — | 46 |
| Co/KIT-6 | I | 20 | 0.1 | 22000 |
260 | 45 | 99 | — | 47 |
| Ru–TiO2 | Barrel-sputtering | 0.8 | 0.1 | 864 | 180 | 100 | 100 | 170 | 48 |
| Ni–CeO2 | I | 10 | 0.1 | 10000 h−1 |
300 | ∼90 | 100 | — | 49 |
| Ni0.8Mg0.2O@SiO2 | CP | — | 0.1 | 60000 |
250 | 78 | 99 | 100 | 50 |
| Ni/MSNa | I | 5 | 0.1 | 50000 |
300 | 64.1 | 99.9 | 200 | 51 |
| Ni/H–Al2O3b | HT | 20 | 0.1 | 2400 | 234 | 50 | — | 252 | 52 |
| Ni–Ru/γ-Al2O3 | CP | Ni: 10; Ru: 1.0 | 0.1 | 9000 h−1 | 350 | 70 | — | 100 | 53 |
| Ru–CeO2/Al2O3 | I | 2 | 0.1 | 10000 h−1 |
300 | 60 | 99 | — | 54 |
| Ce0.95Ru0.05O2 | Combustion | — | 0.1 | 45000 |
450 | 55 | 99 | — | 55 |
| Ni/MCM-41 | HT | 3 | 0.1 | 5760 | 400 | 56 | 96.1 | — | 56 |
| Ni/MC γ-Al2O3c | I | 20 | 0.1 | 9000 | 300 | 74 | 100 | 10 | 57 |
| Co0.4Ni/SiO2 | I | 10 | 0.1 | 13200 |
300 | 58 | — | — | 58 |
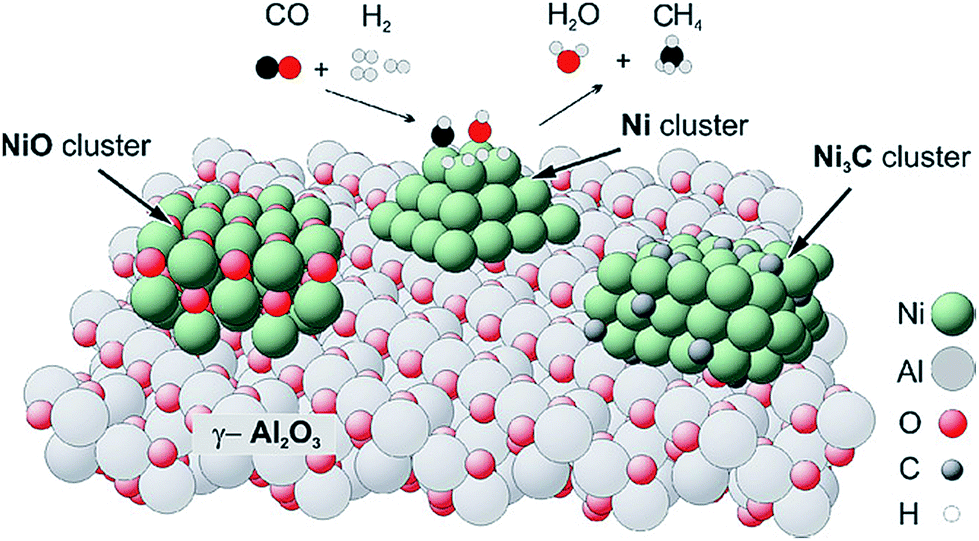 | ||
| Fig. 4 Sketch of the catalyst structure and selective reactions occurring during the synthesis of methane. Reprinted from ref. 75, Copyright (2007), with permission from Elsevier. | ||
 | ||
| Fig. 5 (a-1 and a-2) High-angle annular dark field (HAADF) STEM images of Ni nanoparticles in Ni/H–Al2O3. (b-1–b-6) HRTEM images of Ni nanoparticles selected from the Ni/H–Al2O3 sample (the scale bar is 2 nm). Reprinted with permission from ref. 52 Copyright (2013) American Chemical Society. | ||
Although Ni catalysts are preferred in catalytic methanation reaction, there still exist some problems, such as carbon deposition, sintering, Ni(CO)4 formation, and sulfur poisoning during SNG production.80,81 Therefore, the stability of methanation catalyst is of great importance besides the activity and selectivity. It is well known that the deactivation of supported metal catalysts by carbon or coke formation is a serious problem in methanation process.82 The typical causes are:83 (1) polluting the active metal surface, (2) blocking the voids and pores of catalysts, (3) physical disintegration of the catalyst support. Till now, three types of carbon have been identified on Ni catalysts: pyrolytic, encapsulating and whisker carbon.80 Thermodynamically, carbon formation in carbon monoxide methanation is more favorable as compared with carbon dioxide methanation under the same reaction conditions.24 In addition, higher hydrocarbons in biomass-derived syngas could dramatically enhance the formation of the carbon whiskers at the Ni surface.75 The nickel clusters were found to be associated with the formation of carbon whiskers, supporting a mechanism involving detachment of Ni from the catalyst support during whisker formation (Fig. 6).84
 | ||
| Fig. 6 Electron micrographs of the Ni catalyst and carbon nanofibres. (a) TEM image of a Ni nanocrystal supported on MgAl2O4. (b) TEM image showing a multi-walled tubular carbon nanofibre structure. (c) In situ TEM image showing a whisker-type carbon nanofibre. (d) In situ TEM image showing a Ni nanocrystal during carbon nanofibre growth. Scale bars, 5 nm. Reprinted by permission from Macmillan Publishers Ltd: [Nature] (ref. 84), Copyright (2004). | ||
Prevention of catalyst deactivation caused by coke formation in the methanation of carbon oxides is critical. In summary, there are two ways to prevent the carbon formation: (1) optimization of operating conditions, such as adjusting reaction temperature or pressure, increasing the H2/CO or H2/CO2 ratio, adding steam in the reactants.85 (2) modification of catalysts by formation of an alloy,86,87 and adding promoters like MgO,29 CeO2,88 etc.
Ni nanoparticle sintering during the highly exothermic methanation process is another challenge for Ni catalysts. Jens Sehested from Haldor Topsøe conducted a lot of studies on this aspect.89–92 Particle migration and coalescence, rather than Ostwald ripening was found to govern the sintering of Ni particles.92 The proposed coalescence mechanism89 explains the similar size of the sintered Ni particles independent of metal loading, and the rapid decrease in sintering rate with time. In addition, Sehested suggested a mathematical model that predicts well the experimental nickel surface area as a function of nickel loading, carrier surface area, temperature, and time.90 This model not only enhances our fundamental understanding of the sintering phenomena, but also provides a tool for predicting metal surface areas of the used supported catalysts. What's more, the established formulas assuming that Ni2–OH is dominating sintering91 is a tool for predicting the sintering data and the performance of industrial catalysts.
In methanation process, water as a product could further accelerate the sintering process. In fact, even hot liquid water could lead to the structural changes of γ-Al2O3-supported catalysts.93 Recently, Bai et al.94 investigated the sintering of Ni/Al2O3 methanation catalyst in SNG production. Primary encapsulation of metallic nickel due to the collapse of the support structure and sporadic agglomeration of nickel crystallites led to the reduction of nickel surface area. The steam ambience induced formation of a Ni2+ doped alumina phase, further accelerated the loss of surface nickel atoms. More importantly, it has been found that the sintering could decrease the specific activity of nickel due to the surface structure change.17
Ni sintering can be slowed down by increasing the metal-support interaction,38,56 adding promoters,29,75,88,95 adopting improved preparation methods,52,96 etc. recently, Lu et al. reported coking- and sintering-resistant palladium catalysts through atomic layer deposition of a thin Al2O3 protective layer,87 which gave some inspirations in developing highly stable methanation catalysts.
When Ni methanation catalysts are operated at low temperatures (<250 °C) and high pressures, the formation of nickel tetracarbonyl (Ni(CO)4) should be considered. Recently, Munnik et al.66 investigated the stability of silica gel supported Ni catalysts during the carbon monoxide methanation reaction. The deactivation caused by Ostwald ripening was limited not by diffusion but due to the formation and decomposition of Ni(CO)4 on metal surface, which were mainly determined by particle size. The supersaturation of Ni(CO)4 was low in catalysts with medium sized nanoparticles (Fig. 7a and b), which limited the possible extent of their growth to the pore diameter (Fig. 8a and b). By contrast, in the case of small particles (Fig. 7c and d), the Ni(CO)4 supersaturation was sufficiently high for the particles to break the pore walls, resulting in growth of very large nanoparticles (Fig. 8c and d).66 Therefore, it seems that larger Ni nanoparticles (∼10 nm) are more stable during low temperature carbon monoxide methanation process. Although the formation and decomposition of Ni(CO)4 is not favorable in methanation reaction and should be avoided, this process can be used to modify the size of nickel particles and increase the dispersion of the metallic nickel phase in the Ni/ZrO2 catalyst.97
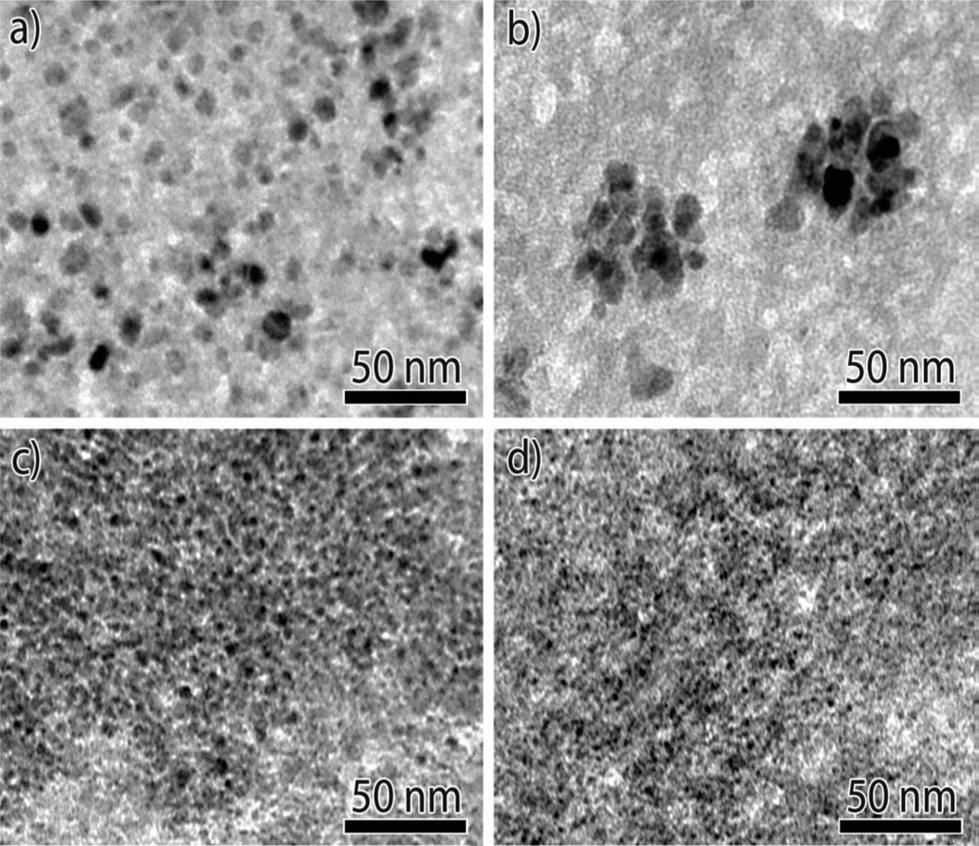 | ||
| Fig. 7 Bright-field TEM images of Ni/SiO2 catalysts with different Ni particle sizes after reduction: (a) ∼8 nm, (b) ∼9 nm, (c) ∼4 nm, and (d) ∼3 nm. Reproduced with permission from ref. 66 Copyright (c) [2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim]. | ||
 | ||
| Fig. 8 TEM images of Ni/SiO2 catalysts with different Ni particle sizes after 150 h on-stream (230 °C, H2:CO = 2:1, 0.1 MPa). (a) ∼8 nm, (b) ∼9 nm, (c) ∼4 nm, and (d) ∼3 nm. Reproduced with permission from ref. 66 Copyright (c) [2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim]. | ||
Sulfur compounds (H2S or thiophene (C4H4S)) are one of the major impurities in syngas which severely poison supported Ni catalysts.98–100 Trace ammonia was also found to decrease the methanation catalyst activity via ammonia adsorption on the active catalyst sites in dynamic biogas upgrading process.101 Even at ppm concentrations, sulfur compounds could still irreversibly affect the catalytic activity.102 The adsorption of S blocks the active sites as well as accelerates the sintering and the oxidation of Ni0 particles.103 Legras and co-workers99 studied the impact and detailed action of sulfur in syngas methanation on Ni/γ-Al2O3 catalyst. It was found that sulfur atoms preferentially adsorbed on the sites for reversible adsorption of molecular CO under the methanation conditions. The sites responsible for CO dissociation, which leads to CH4 production, are affected to a lesser extent by sulfur poisoning (Fig. 9).
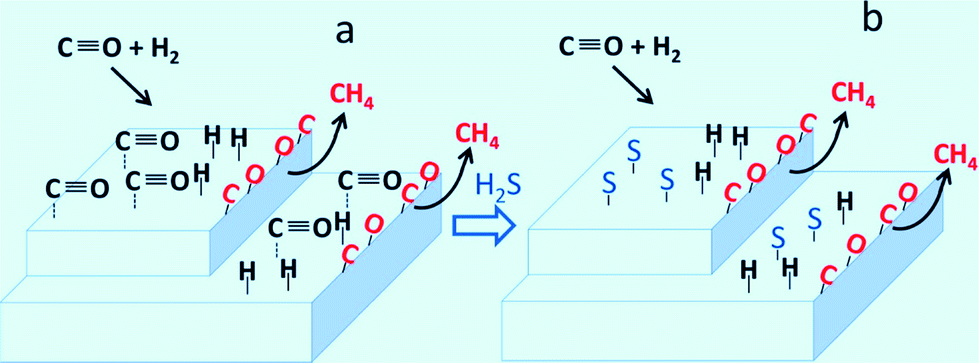 | ||
| Fig. 9 Carbon monoxide hydrogenation with sulfur-free syngas (a) and with syngas containing small amounts of sulfur (b). Reproduced with permission from ref. 99 Copyright (2014) American Chemical Society. | ||
Several strategies have been adopted to improve the sulfur resistance of Ni catalysts. For example, the application of plasma decomposition, which led to the obtained catalyst with less defect sites on Ni particles, could enhance H2S resistance for methanation of syngas.104 In addition, the S adsorption could be weakened when S bound to both Ni and Ru atoms simultaneously. Therefore, Ni–Ru/SiO2 catalyst with small bimetallic Ni–Ru particles showed enhanced sulfur tolerance.103 However, the applications of these methods are still limited. How to increase the sulfur resistance of Ni methanation catalysts still requires further investigation.
:Ni ratio has significant effects on activities of bimetallic catalysts over different supports, such as Al2O3 and CeZrOx.58,109,111,112 Alayoglu et al.113 studied the Co–Pt bimetallic nanoparticles supported on MCF-17 and found that Pt could promote the reduction of Co to its metallic state, resulting in different catalytic performance as compared with pure Co nanoparticles. Tuxen et al.114 demonstrated that the dissociation of carbon monoxide on Co nanoparticles could be facilitated by hydrogen, which was also size-dependent. It was suggested that the dissociation occurred through a –COH or –CHxO intermediate, however, such intermediates were not directly identified. A clear size-dependent dissociation of carbon monoxide on Co nanoparticles was also observed, with smaller nanoparticles favoring molecular adsorption of carbon monoxide and larger nanoparticles favoring carbon monoxide dissociation (Fig. 10). Importantly, the ability of the nanoparticles to dissociate hydrogen determines their ability to dissociate carbon monoxide via the hydrogen-assisted mechanism, which was also supported by the density functional theory (DFT) calculations.115
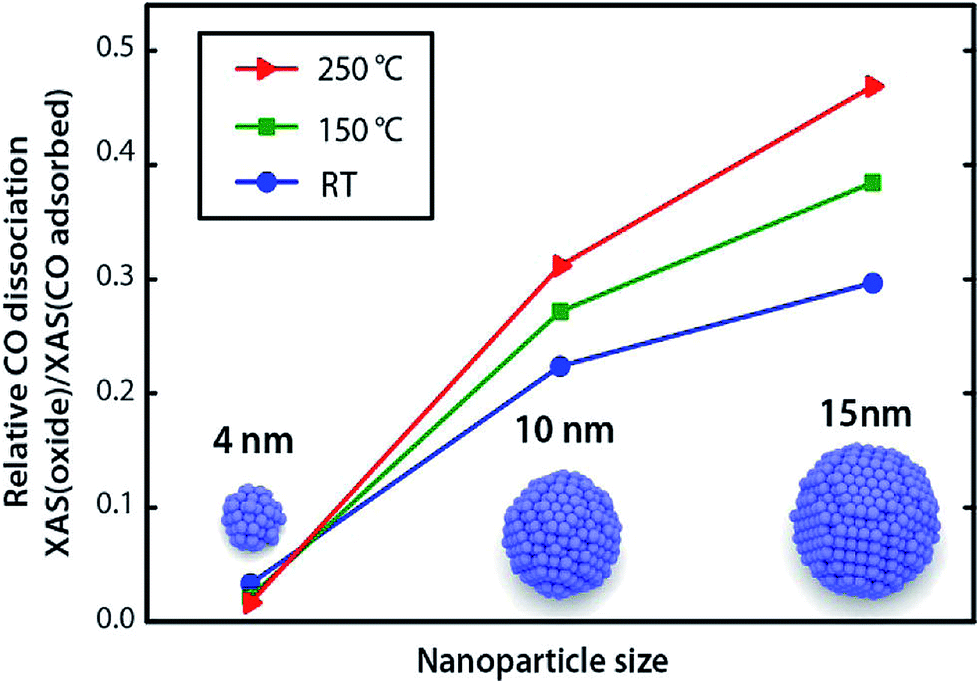 | ||
| Fig. 10 Relative concentrations of dissociated CO species on 4, 10, and 15 nm nanoparticles after exposure to CO/He at different temperatures. Reproduced with permission from ref. 114 Copyright (2013) American Chemical Society. | ||
In general, pure Fe has low methanation activity and selectivity to methane. However, when combined with Ni, in the form of Ni–Fe bimetal or alloy, it showed high activity for methanation44,63,116–118 and even surpassed monometallic Ni catalysts. Hwang and co-workers compared different second metal addition on the activity of nickel–M–Al2O3 (M = Fe, Co, Ce, La, Zr, Y, Mg) catalysts for carbon monoxide or carbon dioxide methanation. Among which, Fe exhibited the best promotional effect.110,119 Co and/or Fe can be combined with Ni to form enhanced methanation catalysts, however, their synergistic effect needs further study to understand the reaction mechanism.
Ru can also be combined with Ni to form a bimetallic methanation catalyst, which showed much enhanced performances.53,103 Zhen et al.53 found that the segregation of Ru on the Ni–Ru/γ-Al2O3 catalyst surface could provide more active Ru species and possess better sulfur-tolerance. Therefore, adding small amount of Ru in Ni catalyst is a promising way to promote methanation reaction.125 Long time stability, high activity, and low loading account for the crucial factors which lead to the use of the precious metal Ru in SNG production.
 | ||
| Fig. 11 TEM images of Pdimp/SiO2 (A), Pdcub@SiO2 (B) and Pdpol@SiO2 (C) catalysts before (1) and after (2) catalytic test. Reproduced from ref. 128 Copyright (2015), with permission from Elsevier. | ||
Pt dispersed on mesoporous silica showed poor activity towards carbon monoxide methanation.113,129 However, the catalytic activity of Pt could be significantly improved by depositing Pt on metal oxide supports through the well-known strong-metal-support-interaction effects,130 and TiO2 nanotube supported Pt shows high activity for carbon dioxide methanation.131 Similarly, CeO2 supported or promoted Rh catalysts132,133 also showed high activity for carbon dioxide methanation.134
Mo has relatively low activity for methanation and produces primarily non-methane hydrocarbons. Consequently, most studies of Mo catalysts have focused on MoOx and MoSx in sulfur-resistant methanation.135,136 The structure and resistance to carbon monoxide of Mo and MoO3 have been well reviewed.137 Unsupported MoS2 were used as sulfur-resistant carbon monoxide methanation catalyst and showed high activity.138 MoOx could promote Ni catalysts for carbon monoxide or carbon dioxide methanation both physically and electronically.42,139 In particular, the effects of CeO2–Al2O3 composite support73,140,141 and sulfidation process142–144 on MoO3/Al2O3 catalysts for sulfur-resistant methanation were intensively investigated. The main factors controlling SNG production by methanation of syngas in the presence of sulfur-resistant Mo-based catalysts were also determined.145 Mo based catalysts show high potential for sulfur-resistant methanation and more attention should be paid to further increase their activities.
3.2 Supports
Support plays an important role in the performance of a heterogeneous catalyst, which usually affects the metal-support interaction and metal dispersion, hence further influences the activity, selectivity, and stability of a catalyst.126 Till now, various metal oxides (Al2O3, SiO2, ZrO2, TiO2, CeO2 etc.), composite oxides (hexaaluminate, solid solution, perovskite) and SiC have been used as methanation catalyst supports. Of all these materials, Al2O3 is the most typical one for methanation reaction.TiO2 as a useful semiconductor material has been widely studied in photocatalysis.152 As the methanation catalyst support, TiO2 supported Ni catalysts have shown high activity for methanation reactions. Partial substitution of Ni in TiO2 lattice could be achieved through sonication, which creates oxide vacancies and facilitates hydrogen adsorption and spillover from nickel to support, further increases catalytic performance.36 In addition, different from Al2O3 support, electron transfer from TiOx could increase the electron cloud density of Ni atoms, which in turn could promote carbon monoxide dissociation on the catalyst surfaces, leading to a relatively high catalytic performance.153,154
ZrO2 support is similar to TiO2 which could improve the dispersion of Ni as well the hydrogen-promoted dissociation of carbon monoxide.155 High carbon monoxide or carbon dioxide methanation activities were obtained on MoS2/ZrO2,156 Yb2O3 doped Ni–ZrO2 catalysts157 and mesoporous ZrO2 supported Ni catalysts.158,159
CeO2 is a special catalyst support because of its partially reducing properties.160–163 Ni/CeO2 showed high carbon dioxide conversion and CH4 selectivity as compared with Ni/α-Al2O3, Ni/TiO2, and Ni/MgO in CO2 methanation reaction.49 It is well established that Ce has the ability to undergo rapid transformations between Ce4+ and Ce3+ states under oxidizing and reducing environments.161,164 The bulk vacancies created in CeO2 after reduction at high temperature could enhance the carbon dioxide methanation activity.134 Ni coverage on CeO2(111) surfaces also affects the carbon monoxide methanation activity.88,165 In addition, combining CeO2 with ZeO2 to form CexZr1−xO2 solid solution could lead to improved support properties such as high redox property, excellent thermal stability, resistance to sintering and suppressing coke formation.45,109,112,166,167
Hexaaluminate type materials possess a unique layered structure with alternative stacked spinel blocks separated by mirror planes. We investigated Ni catalysts supported on barium hexaaluminate (Ni/BHA) for carbon monoxide methanation.40,41 Compared with Ni catalysts supported on commercial Al2O3, the Ni/BHA catalysts exhibited much higher catalytic activity and thermal stability, as well as stronger resistance to carbon deposition.
Perovskite oxides have been widely used in high temperature reactions175 due to their high thermal stability, good reactivity of lattice oxygen, and low cost.176 CaTiO3 supported Ni catalyst and LaFeO3 supported Ni–Fe catalyst showed better catalytic performances than Ni/Al2O3 in carbon monoxide methanation.39,44
Other supports such as rice husk ash,177–179 mesoporous zirconia-modified clays,180 and carbon181 have also been used in methanation catalysts and showed high activity. Generally speaking, the ideal methanation catalyst support needs to be stable at high temperature under steam ambience, with high surface area to disperse active metal, and with appropriate surface properties to effectively anchor active metal nanoparticles.
3.3 Promoters
Promoters can be mainly classified as two types: (1) electron promoter to change the electron mobility of catalyst. (2) Structure promoter to improve the dispersion and thermal stability of catalyst by changing the chemical component, crystal texture, pore structure, dispersion state, and mechanical strength of catalyst. Some oxide promoters can serve with both functions.MgO is an effective promoter to improve resistance to carbon deposition and to minimize Ni particles sintering.29,182–185 MgO promoted Al2O3 (Mg2Al(O)) supported with Ni184 and Ni/Mg/Al hydrotalcite-like compounds186 showed excellent catalytic activity and thermal stability in methanation reaction, which demonstrates the effectiveness of adding promoters to tailor the properties of composite oxide catalyst support to improve the catalyst thermal stability for efficient SNG production.
La2O3 addition can restrain the growth of NiO particles, increase the H2 uptake and Ni dispersion, and therefore enhance the activity of catalysts.30,187,188 Tada et al.189 found La-electron-promoter could increase the electron density in Ru species, which enhanced the dissociation of the C–O bond on Ru due to back donation of electrons from Ru to carbon monoxide, hence led to high carbon monoxide methanation activity over Ru–La/TiO2. The addition of CeO2 can improve the reducibility of the methanation catalyst by altering the interaction between Ni and Al2O3.54,88,190,191 The Pt doping also can facilitate the reduction of Ni species.129 TiOx species153,192 and ZrO2 (ref. 155 and 193) were found to effectively restrict the formation of NiAl2O4 spinel phase and weaken the Ni–Al2O3 interaction, leading to a higher exposure of Ni species and thus enhancing the carbon monoxide adsorption capacity. In addition, electron transfer from TiOx could increase the electron cloud density of Ni, which facilitates the dissociation of CO.153 The same phenomenon was also found in MoO3 promoted Ni methanation catalysts.42 Zr doping improved the dispersion of Ni as well as hydrogen-promoted dissociation of carbon monoxide. Most recently, addition of V2O3 was found to enhance the activity and coking resistant of Ni–Al2O3 for carbon monoxide and carbon dioxide methanation due to the formation of Ni3V2O8.194 Na could also promote Ni/γ-Al2O3 catalysts for syngas methanation.195 Till today, a lot of research efforts have been made towards searching for new promoters to improve the performance of the methanation catalysts. However, a magic promoter seems not to be in sight yet.
3.4 Preparation methods
Various methods have been investigated for preparing methanation catalysts. Preparation methods affect the dispersion of active metal68 and metal-support interactions,196 which in turn further influences the catalytic performance.197 Traditional methods including impregnation,28,30,40,41,45,170,171,193,198,199 precipitation,43,95,109,157,200 and sol–gel methods31,63,110,201 have been widely applied in methanation catalysts synthesis. Some other methods such as deposition–precipitation,46,142 hydrothermal synthesis,38,202 ion exchange method,203 mechanical mixing,204 solution combustion method,96 and microwave heating77 were also used in preparing methanation catalysts.Previous studies205,206 showed that co-precipitated Ni–Al2O3 catalysts could obtain a high dispersion of active metal and strong metal-support interaction, which exhibited higher stability for methanation at high temperatures as compared with those obtained through the impregnation method. The precipitants (such as NH4OH, NaOH, (NH4)2CO3, and Na2CO3) used in co-precipitation strongly influenced the activity of Ni–Mg/Al2O3 catalysts in syngas methanation182 and NiFeAl catalysts in carbon dioxide methanation.117 In addition, the activities of Ni–Ru bimetallic catalysts for carbon dioxide methanation are highly dependent on the precipitation sequence during co-precipitation.53 Similarly, the Mg-modified Ni/SiO2 catalysts prepared by co-impregnation method showed better activity and stability than those prepared by sequential impregnation method.183 O'Brien and co-workers207 studied the active phase evolution in single Ni/Al2O3 methanation catalyst prepared by impregnation in real time using combined μ-XRD-CT and μ-absorption-CT. Both the oxidation procedure and the spatial distribution/concentration of the Ni just prior to oxidation affected the distribution of active Ni metal. Some new methods were also applied in methanation catalysts preparation to enhance the catalytic performance. For example, plasma treatment remarkably improved the dispersion of active components and enhanced the reactivity of Ni/SiO2 catalyst.149 The dielectric-barrier discharge plasma decomposition resulted in a higher dispersion of Ni, an enhanced interaction between Ni and the SiO2 support, as well as fewer defect sites on Ni particles that could enhance resistance to sintering and sulfur poisoning.104,150 It should be noted that the pretreatment/activation process can also affect the final performance of the methanation catalysts.159,166,208
4. Reaction mechanisms and kinetics
4.1 CO methanation
The mechanism of carbon monoxide hydrogenation has been examined under conditions of the FTS.209 Molecular level studies have revealed how the activated dissociation of hydrogen and carbon monoxide occurs.23 Generally, the dissociation of hydrogen on transition metal surfaces is facile. Carbon monoxide methanation process is initiated through carbon monoxide dissociation, and hence, a fundamental understanding of carbon monoxide activation is of utmost importance. Extensive experimental and theoretical studies78,210–212 have been carried out to investigate the reaction pathways of carbon monoxide dissociation and to identify the active sites responsible for the low carbon monoxide dissociation barrier. These studies have confirmed that the active sites present along stepped and corrugated metal surfaces could reduce the carbon monoxide dissociation barrier. In principle carbon monoxide dissociation may occur through one of three pathways:211,212| COads + * → Cads + Oads | (1) |
| COads + Hads ↔ HCOads + * → HCads + Oads | (2) |
| COads + Hads ↔ COHads + * → Cads + OHads | (3) |
Two mechanisms for carbon monoxide activation have been hypothesized. One suggests that the carbon monoxide dissociation directly (path a) follows hydrogenation to produce CHx species. The other proposed mechanism is that the carbon monoxide dissociation proceeds through hydrogen-assisted intermediates (path b and c). Shetty's results212 demonstrate that the direct carbon monoxide dissociation on corrugated Ru(1121) surface has a lower overall barrier than the hydrogen-assisted carbon monoxide pathways, which is due to the highly endothermic steps to form the intermediates during the hydrogenation paths. However, Ojeda et al.213 found that, by using theoretical analysis, the unassisted CO* activation was not competitive with the H-assisted route in Co catalysts, leading to oxygen rejection pathways exclusively. This study provided both experimental and theoretical evidences for hydrogen-assisted carbon monoxide activation as the predominant kinetically relevant step on Fe and Co catalysts. HCOad was also identified as reaction intermediate species in the dominant reaction pathway for carbon monoxide methanation on Ru/Al2O3 catalyst,214 but no such species was detected on Ru/zeolite. Panagiotopoulou et al.215 provided evidences that methanation of carbon monoxide on Ru/TiO2 occurs via two distinct reaction pathways: the first one involves hydrogenation of surface carbon produced by dissociative adsorption of carbon monoxide at low reaction temperatures, whereas the second one involves hydrogenation of Rux–CO species. Although debates about the mechanism of carbon monoxide methanation (direct dissociative adsorption vs. H-assist CO dissociation) still exit, it seems that the reaction pathway differs with different active metal sites and reaction conditions.
The kinetics of carbon monoxide methanation reaction has been extensively studied under different conditions, and various reaction rate expressions have been proposed.216–222 I. Alstrup216 proposed a kinetic model based on carbon monoxide dissociation and stepwise hydrogenation of surface carbon. Hydrogenation of surface methylidyne was considered as the rate-controlling step, which is consistent with the results of Goodman et al.217 and Polizzotti et al.218 later, Sehested et al.219 investigated the carbon monoxide methanation reaction over nickel, and the kinetics of this reaction could be well described by a first-order expression with carbon monoxide dissociation at the nickel surface. The first-order rate constant for CO* dissociation was 5 × 10−9 (s−1) exp[−96.7 (kJ mol−1)/RT] assuming that 5% of the nickel surface atoms are active. Kopyscinski et al.220 applied spatially resolved concentration and temperature measurements in a catalytic plate reactor for the kinetic study of carbon monoxide methanation. Three kinetics equations were obtained based on three different rate-determining steps (C* + H* → CH* + *, CH* + H* → CH2* + *, COH* + H* → CH* + OH*, * empty active site). Interestingly, all three models reflected the measured data equally well and the activation energy of the methanation reaction was 74 kJ mol−1. Transient experiments or spectroscopic methods are needed for further discrimination of the models. Zhang and co-workers investigated the kinetics of carbon monoxide hydrogenation under realistic conditions of methanation of biomass derived syngas.221 The reaction rates were fitted by two kinetic models with hydrogenation of adsorbed carbon species as the rate limiting steps. The validated models could be used for modeling the realistic methanation process of biomass-derived syngas to predict the catalyst performance and to better determine the operation conditions.
4.2 CO2 methanation
Even though the reaction mechanism of carbon dioxide methanation has been intensively investigated, there are still arguments on the nature of the intermediate compounds involved in the process and on the methane formation scheme.214,223–227 The main discrepancy is whether the reaction goes through the formation of carbon monoxide intermediate.214,223,225–227 Eckle and co-workers214 studied the reaction intermediates and side products in the methanation of carbon monoxide and carbon dioxide over supported Ru catalysts. It was found that carbon dioxide methanation proceeded via dissociation to COad, which was subsequently methanated. Formation and decomposition of surface formate only played a minor role in the carbon dioxide methanation reaction.Recently, Karelovic et al. studied low temperature carbon dioxide methanation over Rh/TiO2 catalysts.223 Two reaction mechanisms (Table 4) were proposed based on carbon monoxide intermediate. On the basis of a kinetic comparison between two proposed reaction paths, it was concluded that the dissociation of CO(ads) could proceed via a H-assisted path, probably by the formation of Rh carbonyl hydride species. These results reveal the similarities of the reaction path and the metal particle size dependence between carbon dioxide and carbon monoxide hydrogenations.
| (a) H-assisted carbon monoxide dissociation | (b) Unassisted carbon monoxide dissociation |
|---|---|
| 1. CO2 + 2* ↔ CO* + O* | 1. CO2 + 2* ↔ CO* + O* |
| 2. H2 + 2* ↔ 2H* | 2. H2 + 2* ↔ 2H* |
| 3. CO* + 2H* ↔ H2CO* + 2* | 3. CO* + * → C* + O* (RDS) |
| 4. H2CO* + * → CH* + OH* (RDS) | 4. C* + H* ↔ CH* + * |
| 5. CH* + 3H* ↔ CH4 + 4* | 5. CH* + 3H* ↔ CH4 + 4* |
| 6. O* + H* ↔ OH* + * | 6. O* + H* ↔ OH* + * |
| 7. OH* + H* → H2O + 2* | 7. OH* + H* → H2O + 2* |
Akamaru et al.224 carried out a DFT analysis of methanation reaction of carbon dioxide on Ru nanoparticle supported on TiO2 (101). The potential energy diagram is shown in Fig. 12. The adsorbed carbon dioxide on each site can transform into carbon monoxide through different reaction paths with nearly the same potential energy barriers.
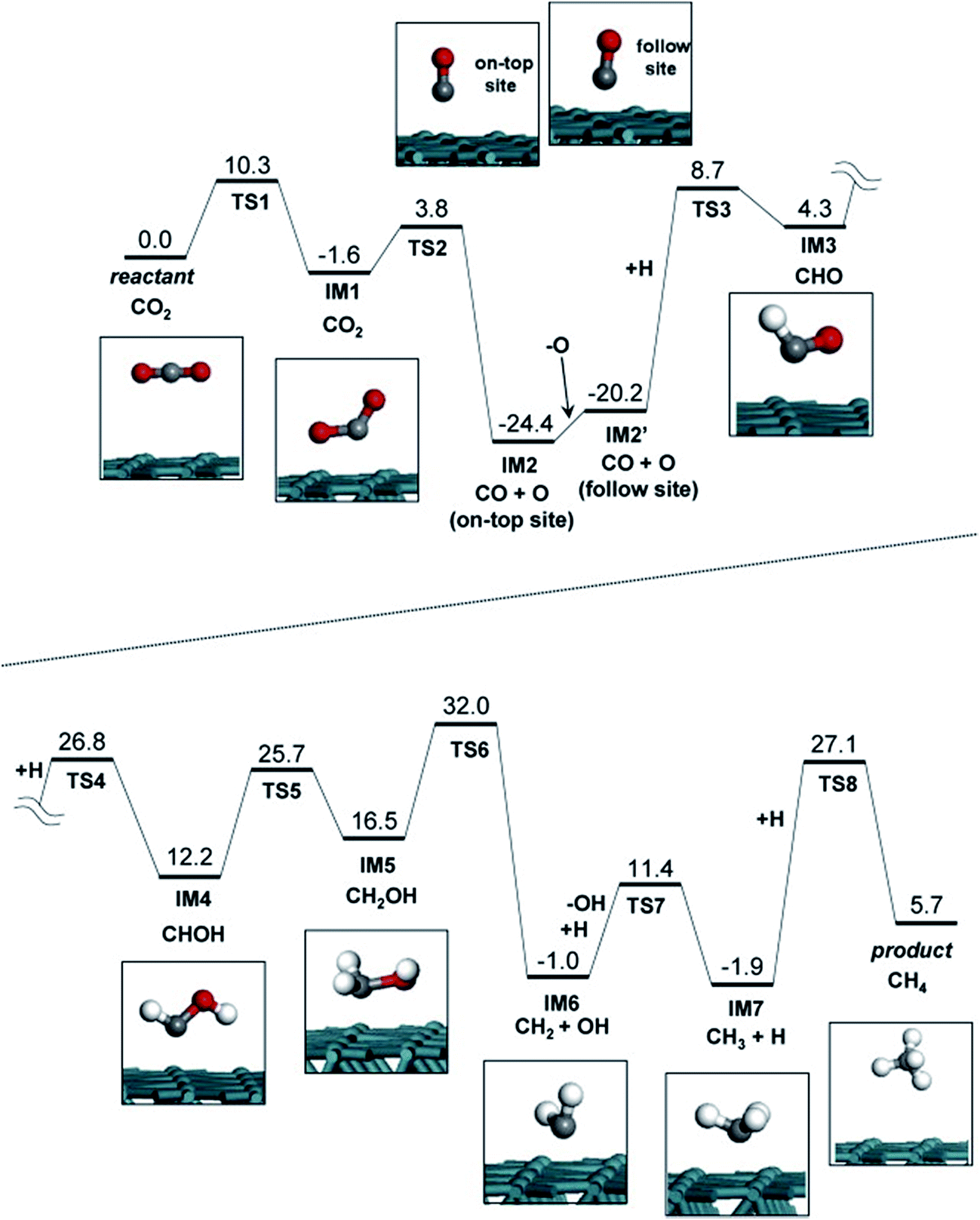 | ||
| Fig. 12 Potential energy diagram for carbon dioxide methanation on the Ru surface slab structure. Each reactant, product and intermediate structures are also shown in the inset of the figure. Reproduced with permission from ref. 224 Copyright (2014), with permission from Elsevier. | ||
Aldana et al.225 investigated carbon dioxide methanation mechanism over Ni-based ceria–zirconia catalysts. Different from the above results, H2 was found to dissociate on Ni0 sites while carbon dioxide was activated on the ceria–zirconia support to form carbonates which could be hydrogenated into formate and further into methoxy species (Fig. 13). This mechanism involves weak basic sites of the support for the adsorption of carbon dioxide and implies a stable metal-support interface, which explains the much better activity of these catalysts as compared to Ni–silica on which both carbon dioxide and hydrogen are activated on Ni0 particles. Pan et al.'s results also support this mechanism.226,227
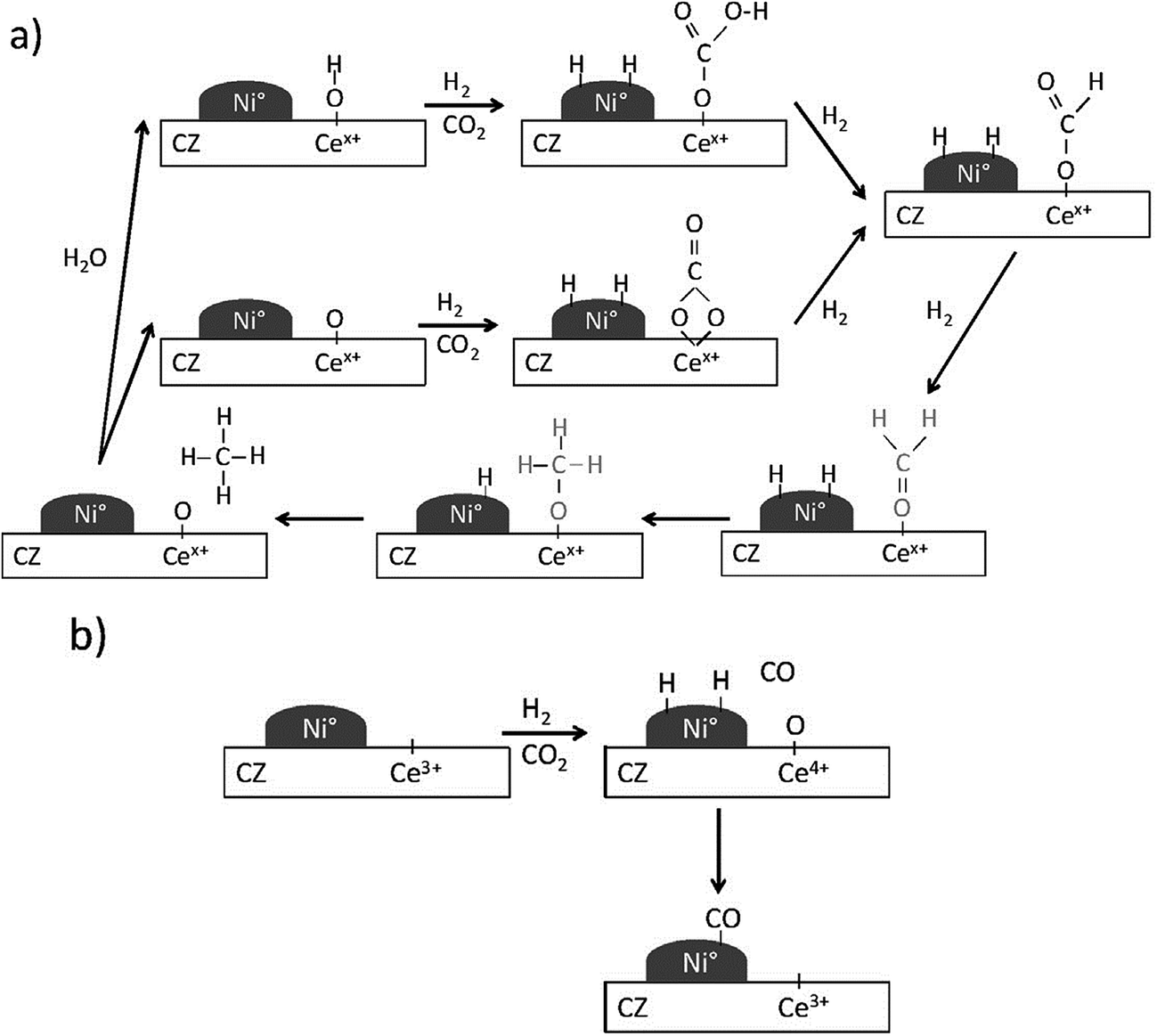 | ||
| Fig. 13 Reaction mechanism proposed on Ni–CZ sol–gel sample for: (a) carbon dioxide methanation and (b) carbon monoxide formation. Reproduced with permission from ref. 225 Copyright (2013), with permission from Elsevier. | ||
Park and co-workers proposed a bifunctional mechanism in Pd–Mg/SiO2 catalysts for CO2 methanation:228 carbon dioxide is stabilized by the magnesium-containing oxide as a surface carbonate and sequentially hydrogenated to form methane.
The methanation kinetics of carbon dioxide was also investigated in recent years.124,229,230 Lu and Kawamoto investigated CO2 methanation kinetics over mesoporous silica incorporated with Ni catalysts.230 A methanation rate equation of r = kCCO20.68CH23.31 was obtained with active energy of 73.1 kJ mol−1.230 Similarly, Kwak et al.124 measured the carbon dioxide methanation reaction rates over Ru/Al2O3 and the estimated apparent activation energies was 62 kJ mol−1. Carbon dioxide methanation kinetics was also used as a tool to reveal key insights into the role of platinum in promoted cobalt catalysis, H atom spillover and surface diffusion.231–233 It is found that increasing the spatial separation between discrete cobalt and platinum entities results in a dramatic ∼50% drop in apparent activation energy compared with that over pure cobalt (Ea = 80 kJ mol−1) in carbon dioxide methanation.
However, the arguments about the carbon dioxide methanation with or without the formation of carbon monoxide intermediate still exist. Further investigations are needed to determine the difference of carbon dioxide activation on different active metal surfaces, which should be the key to understand the mechanism in carbon dioxide conversion reactions.
5. Other aspects of methanation process
Over the past years, researchers have developed some new processes for methanation reaction, among which sorption assisted methanation is one of the most important ones.203,234,235 The sorption enhanced methanation reaction process showed unique performances for high grade SNG production at low methanation pressure. Photo-assisted catalytic methanation of carbon monoxide was also introduced over some semiconductor materials such as TiO2.236 Complete photocatalytic reduction of carbon dioxide to methane by hydrogen under solar light irradiation was also reported.237 Ternary diagrams were applied in the design of methanation systems238 and novel biochemical catalyzed methanation at mesophilic temperatures and ambient pressures have also been realized.239,240 Moreover, methanation reactor and operation conditions were thoroughly investigated.82,241–247 Fluidized bed reactor showed better performance than fixed bed in methanation process for SNG production in lab scale.4,34,35,200,248–254 But so far, no fluidized bed reactor for SNG production has been reported in industrial scale yet. Several problems including attrition of catalysts, stability of fluidized state, and difficulty of scaling-up still require further investigations. Methanation of syngas in slurry-phase bubble-column reactors was also investigated because they have good heat-removal capabilities and the catalysts can be exchanged on-line.155,255 The results show that the temperature of the catalyst bed is more stable and uniform in a slurry reactor as compared with that in a fixed bed reactor, therefore, using a slurry reactor is one potential route for syngas methanation. Magnetic fluidized bed,111 dielectric barrier discharge plasma reactor,256 micro-channel reactor257 were also studied recently, which may inspire some new ideas for the methanation reactor development.6. Conclusions and outlook
In conclusion, we have summarized the recent developments of carbon monoxide and carbon dioxide methanation catalysts for the production of SNG. Developing methanation catalysts with high activity at low temperature and high stability at high temperature are crucial for the methanation process because of the strong exothermic characteristic. How to increase the activity and stability of Ni catalysts and decrease/avoid the formation of Ni(CO)4 at low temperature still need further investigation. High mechanical strength and hydrothermal stability are also essential to maintain long catalyst stability under industrial conditions. Carbon deposition could be alleviated by controlling the reaction conditions, which should not be the major problem. Decreasing the rate of active metal sintering under high temperature is another big challenge to increase the stability of methanation catalysts. Mo based catalysts show high potential for sulphur-resistant methanation catalysis but the activity needs to be further enhanced. Theoretical calculations should be combined with experiments to further explore the methanation reaction mechanisms and to improve the methanation catalyst design. Surface characterization at high pressure would bridge the gap between surface science and heterogeneous catalysis under real reaction conditions. New methanation reactors and processes shall advance the methanation progress together with the catalyst design.Acknowledgements
The authors gratefully acknowledge the supports from the National Natural Science Foundation of China (no. 21476238), the National Basic Research Program (nos 2011CB200906 and 2014CB744306), and “Strategic Priority Research Program” of the Chinese Academy of Sciences (nos XDA07010100 and XDA07010200), and the National Research Foundation of Singapore (M4098015.121).References
- S. Ma, Y. Tan and Y. Han, J. Nat. Gas Chem., 2011, 20, 435–440 CrossRef CAS.
- I. Kiendl, M. Klemm, A. Clemens and A. Herrman, Fuel, 2014, 123, 211–217 CrossRef CAS PubMed.
- R. Razzaq, C. S. Li and S. J. Zhang, Fuel, 2013, 113, 287–299 CrossRef CAS PubMed.
- J. Kopyscinski, M. C. Seemann, R. Moergeli, S. M. A. Biollaz and T. J. Schildhauer, Appl. Catal., A, 2013, 462–463, 150–156 CrossRef CAS PubMed.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 CAS.
- K. Müller, M. Fleige, F. Rachow and D. Schmeißer, Energy Procedia, 2013, 40, 240–248 CrossRef PubMed.
- K. Müller, M. Städter, F. Rachow, D. Hoffmannbeck and D. Schmeißer, Environ. Earth Sci., 2013, 70, 3771–3778 CrossRef PubMed.
- P. Sabatier and J. B. Senderens, C. R. Acad. Sci., 1902, 134, 514–516 CAS.
- M. A. Vannice, J. Catal., 1975, 37, 449–461 CrossRef CAS.
- M. A. Vannice, J. Catal., 1975, 37, 462–473 CrossRef CAS.
- M. A. Vannice, J. Catal., 1975, 40, 129–134 CrossRef CAS.
- M. A. Vannice, J. Catal., 1976, 44, 152–162 CrossRef CAS.
- M. A. Vannice, J. Catal., 1977, 50, 228–236 CrossRef CAS.
- G. A. Mills and F. W. Steffgen, Catal. Rev.: Sci. Eng., 1974, 8, 159–210 Search PubMed.
- J. R. Rostrup Nielsen, K. Pedersen and J. Sehested, Appl. Catal., A, 2007, 330, 134–138 CrossRef CAS PubMed.
- T. T. M. Nguyen, L. Wissing and M. S. Skjoth-Rasmussen, Catal. Today, 2013, 215, 233–238 CrossRef CAS PubMed.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Fuel, 2010, 89, 1763–1783 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- B. Wang, G. Ding, Y. Shang, J. Lv, H. Wang, E. Wang, Z. Li, X. Ma, S. Qin and Q. Sun, Appl. Catal., A, 2012, 431–432, 144–150 CrossRef CAS PubMed.
- P. R. Wentrcek, B. J. Wood and H. Wise, J. Catal., 1976, 43, 363–366 CrossRef CAS.
- Y. Borodko and G. A. Somorjai, Appl. Catal., A, 1999, 186, 355–362 CrossRef CAS.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358–2368 RSC.
- S. I. Fujita and N. Takezawa, Chem. Eng. J., 1997, 68, 63–68 CrossRef CAS.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS PubMed.
- J. Nørskov, J. Catal., 2002, 209, 275–278 CrossRef.
- G. Zhang, T. Sun, J. Peng, S. Wang and S. Wang, Appl. Catal., A, 2013, 462–463, 75–81 CrossRef CAS PubMed.
- D. Hu, J. Gao, Y. Ping, L. Jia, P. Gunawan, Z. Zhong, G. Xu, F. Gu and F. Su, Ind. Eng. Chem. Res., 2012, 51, 4875–4886 CrossRef CAS.
- H. Qin, C. Guo, Y. Wu and J. Zhang, Korean J. Chem. Eng., 2014, 1–6 Search PubMed.
- S. Hwang, J. Lee, U. G. Hong, J. G. Seo, J. C. Jung, D. J. Koh, H. Lim, C. Byun and I. K. Song, J. Ind. Eng. Chem., 2011, 17, 154–157 CrossRef CAS PubMed.
- J. Gao, C. Jia, J. Li, M. Zhang, F. Gu, G. Xu, Z. Zhong and F. Su, Catal. Sci. Technol., 2013, 3, 2009–2015 CAS.
- J. Gao, C. Jia, J. Li, M. Zhang, F. Gu, G. Xu, Z. Zhong and F. Su, J. Energy Chem., 2013, 22, 919–927 CrossRef CAS.
- J. Li, L. Zhou, P. C. Li, Q. S. Zhu, J. J. Gao, F. N. Gu and F. B. Su, Chem. Eng. J., 2013, 219, 183–189 CrossRef CAS PubMed.
- J. Liu, W. Shen, D. Cui, J. Yu, F. Su and G. Xu, Catal. Commun., 2013, 38, 35–39 CrossRef CAS PubMed.
- V. M. Shinde and G. Madras, AIChE J., 2014, 60, 1027–1035 CrossRef CAS.
- X. Chen, J. Jin, G. Sha, C. Li, B. Zhang, D. Su, C. T. Williams and C. Liang, Catal. Sci. Technol., 2014, 4, 53–61 CAS.
- J. Zhang, Z. Xin, X. Meng and M. Tao, Fuel, 2013, 109, 693–701 CrossRef CAS PubMed.
- C. Jia, J. Gao, J. Li, F. Gu, G. Xu, Z. Zhong and F. Su, Catal. Sci. Technol., 2013, 3, 490–499 CAS.
- J. Gao, C. Jia, M. Zhang, F. Gu, G. Xu, Z. Zhong and F. Su, RSC Adv., 2013, 3, 18156–18163 RSC.
- J. Gao, C. Jia, J. Li, F. Gu, G. Xu, Z. Zhong and F. Su, Ind. Eng. Chem. Res., 2012, 51, 10345–10353 CrossRef CAS.
- J. Zhang, Z. Xin, X. Meng, Y. Lv and M. Tao, Fuel, 2014, 116, 25–33 CrossRef CAS PubMed.
- H. Zhu, R. Razzaq, L. Jiang and C. Li, Catal. Commun., 2012, 23, 43–47 CrossRef CAS PubMed.
- H. Wang, Y. Fang, Y. Liu and X. Bai, J. Nat. Gas Chem., 2012, 21, 745–752 CrossRef CAS.
- Q. Pan, J. Peng, T. Sun, D. Gao, S. Wang and S. Wang, Fuel Process. Technol., 2014, 123, 166–171 CrossRef CAS PubMed.
- J. Liu, C. Li, F. Wang, S. He, H. Chen, Y. Zhao, M. Wei, D. G. Evans and X. Duan, Catal. Sci. Technol., 2013, 3, 2627–2633 CAS.
- G. Zhou, T. Wu, H. Xie and X. Zheng, Int. J. Hydrogen Energy, 2013, 38, 10012–10018 CrossRef CAS PubMed.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315–321 CAS.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS PubMed.
- Y. R. Li, G. X. Lu and J. T. Ma, RSC Adv., 2014, 4, 17420–17428 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono, R. R. Mukti, Y. H. Taufiq-Yap and M. R. Sazegar, Appl. Catal., B, 2014, 147, 359–368 CrossRef CAS PubMed.
- S. He, C. Li, H. Chen, D. Su, B. Zhang, X. Cao, B. Wang, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2013, 25, 1040–1046 CrossRef CAS.
- W. Zhen, B. Li, G. Lu and J. Ma, RSC Adv., 2014, 4, 16472–16479 RSC.
- S. Tada, O. J. Ochieng, R. Kikuchi, T. Haneda and H. Kameyama, Int. J. Hydrogen Energy, 2014, 39, 10090–10100 CrossRef CAS PubMed.
- S. Sharma, Z. Hu, P. Zhang, E. W. McFarland and H. Metiu, J. Catal., 2011, 278, 297–309 CrossRef CAS PubMed.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS PubMed.
- S. Rahmani, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 1346–1352 CrossRef CAS PubMed.
- M. Guo and G. Lu, React. Kinet., Mech. Catal., 2014, 113, 101–113 CrossRef CAS.
- B. C. Enger and A. Holmen, Catal. Rev., 2012, 54, 437–488 CAS.
- P. Riani, G. Garbarino, M. A. Lucchini, F. Canepa and G. Busca, J. Mol. Catal. A: Chem., 2014, 383–384, 10–16 CrossRef CAS PubMed.
- S. K. Ryi, S. W. Lee, K. R. Hwang and J. S. Park, Fuel, 2012, 94, 64–69 CrossRef CAS PubMed.
- G. Lee, M. Moon, J. Park, S. Park and S. Hong, Korean J. Chem. Eng., 2005, 22, 541–546 CrossRef CAS.
- S. Hwang, J. Lee, U. G. Hong, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, J. Ind. Eng. Chem., 2013, 19, 698–703 CrossRef CAS PubMed.
- Y.-Z. Wang, F.-M. Li, H.-M. Cheng, L.-Y. Fan and Y.-X. Zhao, J. Fuel Chem. Technol., 2013, 41, 972–977 CrossRef CAS.
- C. W. Hu, J. Yao, H. Q. Yang, Y. Chen and A. M. Tian, J. Catal., 1997, 166, 1–7 CrossRef CAS.
- P. Munnik, M. E. Velthoen, P. E. de Jongh, K. P. de Jong and C. J. Gommes, Angew. Chem., Int. Ed., 2014, 126, 9647–9651 CrossRef.
- L. He, Q. Lin, Y. Liu and Y. Huang, J. Energy Chem., 2014, 23, 587–592 CrossRef.
- N. Yao, H. Ma, Y. Shao, C. Yuan, D. Lv and X. Li, J. Mater. Chem., 2011, 21, 17403–17412 RSC.
- B. Sen and M. A. Vannice, J. Catal., 1991, 130, 9–20 CrossRef CAS.
- C. Deleitenburg and A. Trovarelli, J. Catal., 1995, 156, 171–174 CrossRef CAS.
- M. A. Vannice, S. Y. Wang and S. H. Moon, J. Catal., 1981, 71, 152–166 CrossRef CAS.
- M. A. Vannice and R. L. Garten, J. Catal., 1979, 56, 236–248 CrossRef CAS.
- M. Jiang, B. Wang, Y. Yao, Z. Li, X. Ma, S. Qin and Q. Sun, Appl. Surf. Sci., 2013, 285(Part B), 267–277 CrossRef CAS PubMed.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and S. M. Sidik, Appl. Catal., A, 2014, 486, 115–122 CrossRef CAS PubMed.
- I. Czekaj, F. Loviat, F. Raimondi, J. Wambach, S. Biollaz and A. Wokaun, Appl. Catal., A, 2007, 329, 68–78 CrossRef CAS PubMed.
- G. Garbarino, I. Valsamakis, P. Riani and G. Busca, Catal. Commun., 2014, 51, 37–41 CrossRef CAS PubMed.
- Z. Qin, J. Ren, M. Miao, Z. Li, J. Lin and K. Xie, Appl. Catal., B, 2015, 164, 18–30 CrossRef CAS PubMed.
- M. P. Andersson, E. Abild Pedersen, I. N. Remediakis, T. Bligaard, G. Jones, J. Engbwk, O. Lytken, S. Horch, J. H. Nielsen, J. Sehested, J. R. Rostrup Nielsen, J. K. Norskov and I. Chorkendorff, J. Catal., 2008, 255, 6–19 CrossRef CAS PubMed.
- T. Shido, M. Lok and R. Prins, Top. Catal., 1999, 8, 223–236 CrossRef CAS.
- J. Sehested, Catal. Today, 2006, 111, 103–110 CrossRef CAS PubMed.
- H. H. Gierlich, M. Fremery, A. Skov and J. R. Rostrup-Nielsen, Deactivation Phenomena of a Ni-based Catalyst for High Temperature Methanation, in Studies in Surface Science and Catalysis, ed. B. Delmon and G. F. Froment, Elsevier, 1980, pp. 459–469 Search PubMed.
- X. Bai, S. Wang, T. Sun and S. Wang, Catal. Lett., 2014, 1–10 Search PubMed.
- C. H. Bartholomew, Catal. Rev.: Sci. Eng., 1982, 24, 67–112 CAS.
- S. Helveg, C. Lopez-Cartes, J. Sehested, P. L. Hansen, B. S. Clausen, J. R. Rostrup-Nielsen, F. Abild-Pedersen and J. K. Norskov, Nature, 2004, 427, 426–429 CrossRef CAS PubMed.
- T. Inui, T. Hagiwara and Y. Takegami, Fuel, 1982, 61, 537–541 CrossRef CAS.
- E. Nikolla, A. Holewinski, J. Schwank and S. Linic, J. Am. Chem. Soc., 2006, 128, 11354–11355 CrossRef CAS PubMed.
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS PubMed.
- Q. Liu, J. Gao, M. Zhang, H. Li, F. Gu, G. Xu, Z. Zhong and F. Su, RSC Adv., 2014, 4, 16094–16103 RSC.
- J. Sehested, A. Carlsson, T. V. W. Janssens, P. L. Hansen and A. K. Datyey, J. Catal., 2001, 197, 200–209 CrossRef CAS.
- J. Sehested, J. Catal., 2003, 217, 417–426 CrossRef CAS.
- J. Sehested, J. Catal., 2004, 223, 432–443 CrossRef CAS PubMed.
- J. Sehested, J. A. P. Gelten and S. Helveg, Appl. Catal., A, 2006, 309, 237–246 CrossRef CAS PubMed.
- R. M. Ravenelle, J. R. Copeland, W.-G. Kim, J. C. Crittenden and C. Sievers, ACS Catal., 2011, 1, 552–561 CrossRef CAS.
- X. Bai, S. Wang, T. Sun and S. Wang, React. Kinet., Mech. Catal., 2014, 112, 437–451 CrossRef CAS PubMed.
- A. M. Zhao, W. Y. Ying, H. T. Zhang, H. F. Ma and D. Y. Fang, J. Nat. Gas Chem., 2012, 21, 170–177 CrossRef CAS.
- A. M. Zhao, W. Y. Ying, H. T. Zhang, H. F. Ma and D. Y. Fang, Catal. Commun., 2012, 17, 34–38 CrossRef CAS PubMed.
- V. M. Gonzalez-Delacruz, R. Pereñiguez, F. Ternero, J. P. Holgado and A. Caballero, ACS Catal., 2011, 1, 82–88 CrossRef CAS.
- C. H. Bartholomew, G. D. Weatherbee and G. A. Jarvi, J. Catal., 1979, 60, 257–269 CrossRef CAS.
- B. Legras, V. V. Ordomsky, C. Dujardin, M. Virginie and A. Y. Khodakov, ACS Catal., 2014, 4, 2785–2791 CrossRef CAS.
- R. P. W. J. Struis, T. J. Schildhauer, I. Czekaj, M. Janousch, S. M. A. Biollaz and C. Ludwig, Appl. Catal., A, 2009, 362, 121–128 CrossRef CAS PubMed.
- L. Jurgensen, E. A. Ehimen, J. Born, J. B. Holm-Nielsen and D. Rooney, Bioresour. Technol., 2015, 178, 319–322 CrossRef CAS PubMed.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- C. Yuan, N. Yao, X. Wang, J. Wang, D. Lv and X. Li, Chem. Eng. J., 2015, 260, 1–10 CrossRef CAS PubMed.
- X. Yan, Y. Liu, B. Zhao, Y. Wang and C.-J. Liu, Phys. Chem. Chem. Phys., 2013, 15, 12132–12138 RSC.
- G. Prieto, A. Martínez, P. Concepción and R. Moreno-Tost, J. Catal., 2009, 266, 129–144 CrossRef CAS PubMed.
- P. Concepcion, C. Lopez, A. Martinez and V. Puntes, J. Catal., 2004, 228, 321–332 CrossRef CAS PubMed.
- S. Rojanapipatkul and B. Jongsomjit, Catal. Commun., 2008, 10, 232–236 CrossRef CAS PubMed.
- B. Wang, Y. Yao, M. Jiang, Z. Li, X. Ma, S. Qin and Q. Sun, J. Energy Chem., 2014, 23, 35–42 CrossRef CAS.
- H. Zhu, R. Razzaq, C. Li, Y. Muhmmad and S. Zhang, AIChE J., 2013, 59, 2567–2576 CrossRef CAS.
- S. Hwang, J. Lee, U. G. Hong, J. C. Jung, D. J. Koh, H. Lim, C. Byun and I. K. Song, J. Ind. Eng. Chem., 2012, 18, 243–248 CrossRef CAS PubMed.
- J. Li, L. Zhou, Q. Zhu and H. Li, Ind. Eng. Chem. Res., 2013, 52, 6647–6654 CrossRef CAS.
- R. Razzaq, H. Zhu, L. Jiang, U. Muhammad, C. Li and S. Zhang, Ind. Eng. Chem. Res., 2013, 52, 2247–2256 CrossRef CAS.
- S. Alayoglu, S. K. Beaumont, F. Zheng, V. V. Pushkarev, H. Zheng, V. Iablokov, Z. Liu, J. Guo, N. Kruse and G. A. Somorjai, Top. Catal., 2011, 54, 778–785 CrossRef CAS.
- A. Tuxen, S. Carenco, M. Chintapalli, C. H. Chuang, C. Escudero, E. Pach, P. Jiang, F. Borondics, B. Beberwyck, A. P. Alivisatos, G. Thornton, W. F. Pong, J. H. Guo, R. Perez, F. Besenbacher and M. Salmeron, J. Am. Chem. Soc., 2013, 135, 2273–2278 CrossRef CAS PubMed.
- J.-X. Liu, H.-Y. Su and W.-X. Li, Catal. Today, 2013, 215, 36–42 CrossRef CAS PubMed.
- D. Tian, Z. Liu, D. Li, H. Shi, W. Pan and Y. Cheng, Fuel, 2013, 104, 224–229 CrossRef CAS PubMed.
- S. Hwang, U. G. Hong, J. Lee, J. G. Seo, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, J. Ind. Eng. Chem., 2013, 19, 2016–2021 CrossRef CAS PubMed.
- M. P. Andersson, T. Bligaard, A. Kustov, K. E. Larsen, J. Greeley, T. Johannessen, C. H. Christensen and J. K. Norskov, J. Catal., 2006, 239, 501–506 CrossRef CAS PubMed.
- S. Hwang, U. Hong, J. Lee, J. Baik, D. Koh, H. Lim and I. Song, Catal. Lett., 2012, 142, 860–868 CrossRef CAS.
- S. Eckle, Y. Denkwitz and R. J. Behm, J. Catal., 2010, 269, 255–268 CrossRef CAS PubMed.
- C. Janke, M. S. Duyar, M. Hoskins and R. Farrauto, Appl. Catal., B, 2014, 152–153, 184–191 CrossRef CAS PubMed.
- F. Masini, C. E. Strebel, D. N. McCarthy, A. U. F. Nierhoff, J. Kehres, E. M. Fiordaliso, J. H. Nielsen and I. Chorkendorff, J. Catal., 2013, 308, 282–290 CrossRef CAS PubMed.
- B. T. Loveless, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2013, 135, 6107–6121 CrossRef CAS PubMed.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3, 2449–2455 CrossRef CAS.
- C. F. J. König, T. J. Schildhauer and M. Nachtegaal, J. Catal., 2013, 305, 92–100 CrossRef PubMed.
- W. J. Shen, M. Okumura, Y. Matsumura and M. Haruta, Appl. Catal., A, 2001, 213, 225–232 CrossRef CAS.
- S. Y. Wang, S. H. Moon and M. A. Vannice, J. Catal., 1981, 71, 167–174 CrossRef CAS.
- J. Martins, N. Batail, S. Silva, S. Rafik-Clement, A. Karelovic, D. P. Debecker, A. Chaumonnot and D. Uzio, Catal. Commun., 2015, 58, 11–15 CrossRef CAS PubMed.
- H. Yoshida, K. Watanabe, N. Iwasa, S.-I. Fujita and M. Arai, Appl. Catal., B, 2015, 162, 93–97 CrossRef CAS PubMed.
- M. A. Vannice and C. C. Twu, J. Catal., 1983, 82, 213–222 CrossRef CAS.
- K.-P. Yu, W.-Y. Yu, M.-C. Kuo, Y.-C. Liou and S.-H. Chien, Appl. Catal., B, 2008, 84, 112–118 CrossRef CAS PubMed.
- C. Swalus, M. Jacquemin, C. Poleunis, P. Bertrand and P. Ruiz, Appl. Catal., B, 2012, 125, 41–50 CrossRef CAS PubMed.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237–249 CrossRef CAS PubMed.
- A. Trovarelli, C. Deleitenburg, G. Dolcetti and J. L. Lorca, J. Catal., 1995, 151, 111–124 CrossRef CAS.
- S. Zaman and K. J. Smith, Catal. Rev., 2012, 54, 41–132 CAS.
- M. Y. Kim, S. B. Ha, D. J. Koh, C. Byun and E. D. Park, Catal. Commun., 2013, 35, 68–71 CrossRef CAS PubMed.
- L. Y. Novoselova, J. Alloys Compd., 2014, 615, 784–791 CrossRef CAS PubMed.
- J. Liu, E. Wang, J. Lv, Z. Li, B. Wang, X. Ma, S. Qin and Q. Sun, Fuel Process. Technol., 2013, 110, 249–257 CrossRef CAS PubMed.
- A. E. Aksoylu, Z. Mısırlı and Z. . İ. Önsan, Appl. Catal., A, 1998, 168, 385–397 CrossRef CAS.
- B. Wang, Y. Shang, G. Ding, J. Lv, H. Wang, E. Wang, Z. Li, X. Ma, S. Qin and Q. Sun, React. Kinet., Mech. Catal., 2012, 106, 495–506 CrossRef CAS.
- M. Jiang, B. Wang, Y. Yao, H. Wang, Z. Li, X. Ma, S. Qin and Q. Sun, Catal. Commun., 2013, 35, 32–35 CrossRef CAS PubMed.
- M. Jiang, B. Wang, Y. Yao, H. Wang, Z. Li, X. Ma, S. Qin and Q. Sun, Appl. Catal., A, 2014, 469, 89–97 CrossRef CAS PubMed.
- M. Jiang, B. Wang, J. Lv, H. Wang, Z. Li, X. Ma, S. Qin and Q. Sun, Appl. Catal., A, 2013, 466, 224–232 CrossRef CAS PubMed.
- M. Jiang, B. Wang, Y. Yao, Z. Li, X. Ma, S. Qin and Q. Sun, Catal. Sci. Technol., 2013, 3, 2793–2800 CAS.
- Z. Li, H. Wang, E. Wang, J. Lv, Y. Shang, G. Ding, B. Wang, X. Ma, S. Qin and Q. Su, Kinet. Catal., 2013, 54, 338–343 CrossRef CAS.
- I. Levin and D. Brandon, J. Am. Ceram. Soc., 1998, 81, 1995–2012 CrossRef CAS PubMed.
- G. Garbarino, P. Riani, L. Magistri and G. Busca, Int. J. Hydrogen Energy, 2014, 39, 11557–11565 CrossRef CAS PubMed.
- A. Tougerti, I. Llorens, F. D'Acapito, E. Fonda, J. L. Hazemann, Y. Joly, D. Thiaudiere, M. Che and X. Carrier, Angew. Chem., Int. Ed., 2012, 51, 7697–7701 CrossRef CAS PubMed.
- X. Zhang, W.-j. Sun and W. Chu, J. Fuel Chem. Technol., 2013, 41, 96–101 CrossRef.
- X. L. Yan, Y. Liu, B. R. Zhao, Z. Wang, Y. Wang and C. J. Liu, Int. J. Hydrogen Energy, 2013, 38, 2283–2291 CrossRef CAS PubMed.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and M. W. A. Saad, Chem. Eng. J., 2015, 260, 757–764 CrossRef CAS PubMed.
- S.-D. Mo and W. Ching, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13023–13032 CrossRef CAS.
- Y. Zeng, H. Ma, H. Zhang, W. Ying and D. Fang, Fuel, 2014, 137, 155–163 CrossRef CAS PubMed.
- J. Barrientos, M. Lualdi, M. Boutonnet and S. Järås, Appl. Catal., A, 2014, 486, 143–149 CrossRef CAS PubMed.
- J. Zhang, Y. Bai, Q. Zhang, X. Wang, T. Zhang, Y. Tan and Y. Han, Fuel, 2014, 132, 211–218 CrossRef CAS PubMed.
- Z. Li, Y. Tian, J. He, B. Wang and X. Ma, J. Energy Chem., 2014, 23, 625–632 CrossRef.
- Y.-H. Huang, J.-J. Wang, Z.-M. Liu, G.-D. Lin and H.-B. Zhang, Appl. Catal., A, 2013, 466, 300–306 CrossRef CAS PubMed.
- A. Chen, T. Miyao, K. Higashiyama and M. Watanabe, Catal. Sci. Technol., 2014, 4, 2508–2511 CAS.
- N. Perkas, G. Amirian, Z. Y. Zhong, J. Teo, Y. Gofer and A. Gedanken, Catal. Lett., 2009, 130, 455–462 CrossRef CAS.
- W. Q. Xu, Z. Y. Liu, A. C. Johnston-Peck, S. D. Senanayake, G. Zhou, D. Stacchiola, E. A. Stach and J. A. Rodriguez, ACS Catal., 2013, 3, 975–984 CrossRef CAS.
- I. I. Soykal, H. Sohn, D. Singh, J. T. Miller and U. S. Ozkan, ACS Catal., 2014, 4, 585–592 CrossRef CAS.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CAS.
- M. M. Zyryanova, P. V. Snytnikov, R. V. Gulyaev, Y. I. Amosov, A. I. Boronin and V. A. Sobyanin, Chem. Eng. J., 2014, 238, 189–197 CrossRef PubMed.
- G. Vilé, S. Colussi, F. Krumeich, A. Trovarelli and J. Pérez-Ramírez, Angew. Chem., 2014, 126, 12265–12268 CrossRef.
- S. D. Senanayake, J. Evans, S. Agnoli, L. Barrio, T.-L. Chen, J. Hrbek and J. A. Rodriguez, Top. Catal., 2011, 54, 34–41 CrossRef CAS.
- R. Razzaq, C. Li, N. Amin, S. Zhang and K. Suzuki, Energy Fuels, 2013, 27, 6955–6961 CrossRef CAS.
- W. Cai, Q. Zhong and Y. Zhao, Catal. Commun., 2013, 39, 30–34 CrossRef CAS PubMed.
- Z. Wang, Q. Fu and X. Bao, Progr. Chem., 2014, 26, 502–511 Search PubMed.
- M. Ledoux and C. Pham-Huu, CATTECH, 2001, 5, 226–246 CrossRef CAS.
- G. Zhang, J. Peng, T. Sun and S. Wang, Chin. J. Catal., 2013, 34, 1745–1755 CrossRef CAS.
- Y. Yu, G. Q. Jin, Y. Y. Wang and X. Y. Guo, Fuel Process. Technol., 2011, 92, 2293–2298 CrossRef CAS PubMed.
- Y. Yu, G. Jin, Y. Wang and X. Guo, Catal. Commun., 2013, 31, 5–10 CrossRef CAS PubMed.
- M. A. Vannice, Y. L. Chao and R. M. Friedman, Appl. Catal., 1986, 20, 91–107 CrossRef CAS.
- L. Song, Y. Yu, X. Wang, G. Jin, Y. Wang and X. Y. Guo, Korean Chem. Eng. Res., 2014, 52, 678–687 CrossRef CAS.
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS.
- M. A. Pena and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef CAS PubMed.
- F. W. Chang, T. J. Hsiao, S. W. Chung and J. J. Lo, Appl. Catal., A, 1997, 164, 225–236 CrossRef CAS.
- F. W. Chang, T. J. Hsiao and J. D. Shih, Ind. Eng. Chem. Res., 1998, 37, 3838–3845 CrossRef CAS.
- F.-W. Chang, M.-S. Kuo, M.-T. Tsay and M.-C. Hsieh, Appl. Catal., A, 2003, 247, 309–320 CrossRef CAS.
- H. Lu, X. Yang, G. Gao, K. Wang, Q. Shi, J. Wang, C. Han, J. Liu, M. Tong, X. Liang and C. Li, Int. J. Hydrogen Energy, 2014, 39, 18894–18907 CrossRef CAS PubMed.
- M. F. Variava, T. L. Church, N. Noorbehesht, A. T. Harris and A. I. Minett, Catal. Sci. Technol., 2015, 5, 515–524 CAS.
- J. Liu, J. Yu, F. Su and G. Xu, Catal. Sci. Technol., 2014, 4, 472–481 CAS.
- M. Guo and G. Lu, Catal. Commun., 2014, 54, 55–60 CrossRef CAS PubMed.
- M. T. Fan, K. P. Miao, J. D. Lin, H. B. Zhang and D. W. Liao, Appl. Surf. Sci., 2014, 307, 682–688 CrossRef CAS PubMed.
- H. Y. Kim, H. M. Lee and J.-N. Park, J. Phys. Chem. C, 2010, 114, 7128–7131 CAS.
- Z. Li, L. Bian, Q. Zhu and W. Wang, Kinet. Catal., 2014, 55, 217–223 CrossRef CAS.
- H. G. J. L. Rotgerink, R. P. A. M. Paalman, J. G. van Ommen and J. R. H. Ross, Appl. Catal., 1988, 45, 257–280 CrossRef.
- A. Zhao, W. Ying, H. Zhang, H. Ma and D. Fang, Proc. World Acad. Sci. Eng. Tech., 2011, 59, 1002–1006 Search PubMed.
- S. Tada, R. Kikuchi, A. Takagaki, T. Sugawara, S. Ted Oyama and S. Satokawa, Catal. Today, 2014, 232, 16–21 CrossRef CAS PubMed.
- H. Liu, X. Zou, X. Wang, X. Lu and W. Ding, J. Nat. Gas Chem., 2012, 21, 703–707 CrossRef CAS.
- H. G. J. Lansink Rotgerink, J. C. Slaa, J. G. van Ommen and J. R. H. Ross, Appl. Catal., 1988, 45, 281–290 CrossRef CAS.
- H. G. J. L. Rotgerink, P. D. L. Mercera, J. G. van Ommen and J. R. H. Ross, Appl. Catal., 1988, 45, 239–256 CrossRef.
- C. Guo, Y. Wu, H. Qin and J. Zhang, Fuel Process. Technol., 2014, 124, 61–69 CrossRef CAS PubMed.
- Q. Liu, F. Gu, X. Lu, Y. Liu, H. Li, Z. Zhong, G. Xu and F. Su, Appl. Catal., A, 2014, 488, 37–47 CrossRef CAS PubMed.
- Y. Zeng, H. Ma, H. Zhang and W. Ying, International Journal of Chemical, Nuclear, Metallurgical and Materials Engineering, 2014, 8, 582–586 Search PubMed.
- J. A. Schwarz, C. Contescu and A. Contescu, Chem. Rev., 1995, 95, 477–510 CrossRef CAS.
- E. C. Kruissink, E. B. M. Doesburg, L. L. van Reijen, L. E. Alzamora, S. Orr, J. R. H. Ross and G. van Veen, The Preparation and Pretreatment of Coprecipitated Nickel-Alumina Catalysts for Methanation at High Temperatures, in Studies in Surface Science and Catalysis, ed. P. G. P. J. B. Delmon and G. Poncelet, Elsevier, 1979, pp. 143–157 Search PubMed.
- X. Yang, X. Wang, G. Gao, Wendurima, E. Liu, Q. Shi, J. Zhang, C. Han, J. Wang, H. Lu, J. Liu and M. Tong, Int. J. Hydrogen Energy, 2013, 38, 13926–13937 CrossRef CAS PubMed.
- X. Yang, Wendurima, G. Gao, Q. Shi, X. Wang, J. Zhang, C. Han, J. Wang, H. Lu, J. Liu and M. Tong, Int. J. Hydrogen Energy, 2014, 39, 3231–3242 CrossRef CAS PubMed.
- B. Liu and S. Ji, J. Energy Chem., 2013, 22, 740–746 CrossRef CAS.
- S. Hwang, J. Lee, U. G. Hong, J. C. Jung, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, J. Nanosci. Nanotechnol., 2012, 12, 6051–6057 CrossRef CAS PubMed.
- J. Zhang, Z. Xin, X. Meng, Y. Lv and M. Tao, Ind. Eng. Chem. Res., 2013, 52, 14533–14544 CrossRef CAS.
- A. Borgschulte, N. Gallandat, B. Probst, R. Suter, E. Callini, D. Ferri, Y. Arroyo, R. Erni, H. Geerlings and A. Zuttel, Phys. Chem. Chem. Phys., 2013, 15, 9620–9625 RSC.
- A. Karelovic and P. Ruiz, ACS Catal., 2013, 3, 2799–2812 CrossRef CAS.
- L. E. Alzamora, J. R. H. Ross, E. C. Kruissink and L. L. Vanreijen, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 665–681 RSC.
- E. C. Kruissink, L. L. Vanreijen and J. R. H. Ross, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 649–663 RSC.
- M. G. O'Brien, S. D. M. Jacques, M. Di Michiel, P. Barnes, B. M. Weckhuysen and A. M. Beale, Chem. Sci., 2012, 3, 509–523 RSC.
- B. Vos, E. Poels and A. Bliek, J. Catal., 2001, 198, 77–88 CrossRef CAS.
- G. P. Van Der Laan and A. A. C. M. Beenackers, Catal. Rev., 1999, 41, 255–318 CAS.
- T. H. Pham, X. Z. Duan, G. Qian, X. G. Zhou and D. Chen, J. Phys. Chem. C, 2014, 118, 10170–10176 CAS.
- M. R. Elahifard, M. P. Jigato and J. W. Niemantsverdriet, ChemPhysChem, 2012, 13, 89–91 CrossRef CAS PubMed.
- S. Shetty, A. P. J. Jansen and R. A. van Santen, J. Am. Chem. Soc., 2009, 131, 12874–12875 CrossRef CAS PubMed.
- M. Ojeda, R. Nabar, A. U. Nilekar, A. Ishikawa, M. Mavrikakis and E. Iglesia, J. Catal., 2010, 272, 287–297 CrossRef CAS PubMed.
- S. Eckle, H.-G. Anfang and R. J. r. Behm, J. Phys. Chem. C, 2011, 115, 1361–1367 CAS.
- P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, J. Phys. Chem. C, 2011, 115, 1220–1230 CAS.
- I. Alstrup, J. Catal., 1995, 151, 216–225 CrossRef CAS.
- D. W. Goodman, R. D. Kelley, T. E. Madey and J. T. Yates, J. Catal., 1980, 63, 226–234 CrossRef CAS.
- R. S. Polizzotti and J. A. Schwarz, J. Catal., 1982, 77, 1–15 CrossRef CAS.
- J. Sehested, S. Dahl, J. Jacobsen and J. R. Rostrup-Nielsen, J. Phys. Chem. B, 2004, 109, 2432–2438 CrossRef PubMed.
- J. Kopyscinski, T. J. Schildhauer, F. Vogel, S. M. A. Biollaz and A. Wokaun, J. Catal., 2010, 271, 262–279 CrossRef CAS PubMed.
- J. Zhang, N. Fatah, S. Capela, Y. Kara, O. Guerrini and A. Y. Khodakov, Fuel, 2013, 111, 845–854 CrossRef CAS PubMed.
- A. C. Lausche, A. J. Medford, T. S. Khan, Y. Xu, T. Bligaard, F. Abild-Pedersen, J. K. Nørskov and F. Studt, J. Catal., 2013, 307, 275–282 CrossRef CAS PubMed.
- A. Karelovic and P. Ruiz, J. Catal., 2013, 301, 141–153 CrossRef CAS PubMed.
- S. Akamaru, T. Shimazaki, M. Kubo and T. Abe, Appl. Catal., A, 2014, 470, 405–411 CrossRef CAS PubMed.
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catal. Today, 2013, 215, 201–207 CrossRef CAS PubMed.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74–78 CrossRef CAS PubMed.
- Q. S. Pan, J. X. Peng, S. Wang and S. D. Wang, Catal. Sci. Technol., 2014, 4, 502–509 CAS.
- J.-N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS PubMed.
- M. Marwood, R. Doepper and A. Renken, Appl. Catal., A, 1997, 151, 223–246 CrossRef CAS.
- B. W. Lu and K. Kawamoto, Catal. Sci. Technol., 2014, 4, 4313–4321 CAS.
- S. K. Beaumont, S. Alayoglu, C. Specht, N. Kruse and G. A. Somorjai, Nano Lett., 2014, 14, 4792–4796 CrossRef CAS PubMed.
- S. K. Beaumont, S. Alayoglu, C. Specht, W. D. Michalak, V. V. Pushkarev, J. Guo, N. Kruse and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 9898–9901 CrossRef CAS PubMed.
- Z. A. Ibraeva, N. V. Nekrasov, B. S. Gudkov, V. I. Yakerson, Z. T. Beisembaeva, E. Z. Golosman and S. L. Kiperman, Theor. Exp. Chem., 1991, 26, 584–588 CrossRef.
- S. Walspurger, G. D. Elzinga, J. W. Dijkstra, M. Sarić and W. G. Haije, Chem. Eng. J., 2014, 242, 379–386 CrossRef CAS PubMed.
- V. M. Lebarbier, R. A. Dagle, L. Kovarik, K. O. Albrecht, X. Li, L. Li, C. E. Taylor, X. Bao and Y. Wang, Appl. Catal., B, 2014, 144, 223–232 CrossRef CAS PubMed.
- X. Lin, K. Yang, R. Si, X. Chen, W. Dai and X. Fu, Appl. Catal., B, 2014, 147, 585–591 CrossRef CAS PubMed.
- F. Sastre, A. V. Puga, L. Liu, A. Corma and H. Garcia, J. Am. Chem. Soc., 2014, 136, 6798–6801 CrossRef CAS PubMed.
- V. Frick, J. Brellochs and M. Specht, Fuel Process. Technol., 2014, 118, 156–160 CrossRef CAS PubMed.
- M. Burkhardt and G. Busch, Appl. Energy, 2013, 111, 74–79 CrossRef CAS PubMed.
- M. Burkhardt, T. Koschack and G. Busch, Bioresour. Technol., 2015, 178, 330–333 CrossRef CAS PubMed.
- W. Kang and K. Lee, Korean J. Chem. Eng., 2013, 30, 1386–1394 CrossRef CAS PubMed.
- R. Güttel, Chem. Eng. Technol., 2013, 36, 1675–1682 Search PubMed.
- H. Er-rbib and C. Bouallou, C. R. Chim., 2014, 17, 701–706 CrossRef CAS PubMed.
- N. R. Parlikkad, S. Chambrey, P. Fongarland, N. Fatah, A. Khodakov, S. Capela and O. Guerrini, Fuel, 2013, 107, 254–260 CrossRef CAS PubMed.
- L.-J. Zhao and Q. Sun, Int. J. Low-Carbon Technol., 2014, 1–6 CrossRef PubMed.
- X. Li, B. Yang and Y. Zhang, J. Process Control, 2013, 23, 1360–1370 CrossRef CAS PubMed.
- Y.-L. Kao, P.-H. Lee, Y.-T. Tseng, I. L. Chien and J. D. Ward, J. Taiwan Inst. Chem. Eng., 2014, 45, 2346–2357 CrossRef CAS PubMed.
- J. Liu, D. Cui, J. Yu, F. Su and G. Xu, Chin. J. Chem. Eng., 2015, 23, 86–92 CrossRef PubMed.
- Y. Liu and O. Hinrichsen, Ind. Eng. Chem. Res., 2014, 53, 9348–9356 CrossRef CAS.
- M. C. Seemann, T. J. Schildhauer and S. M. A. Biollaz, Ind. Eng. Chem. Res., 2010, 49, 7034–7038 CrossRef CAS.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Chem. Eng. Sci., 2011, 66, 924–934 CrossRef CAS PubMed.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Chem. Eng. Sci., 2011, 66, 1612–1621 CrossRef CAS PubMed.
- J. Kopyscinski, T. J. Schildhauer and S. M. A. Biollaz, Ind. Eng. Chem. Res., 2011, 50, 2781–2790 CrossRef CAS.
- M. C. Seemann, T. J. Schildhauer and S. M. A. Biollaz, Ind. Eng. Chem. Res., 2010, 49, 7034–7038 CrossRef CAS.
- J. Lefebvre, M. Götz, S. Bajohr, R. Reimert and T. Kolb, Fuel Process. Technol., 2015, 132, 83–90 CrossRef CAS PubMed.
- E. Jwa, S. B. Lee, H. W. Lee and Y. S. Mok, Fuel Process. Technol., 2013, 108, 89–93 CrossRef CAS PubMed.
- Z. H. Liu, B. Z. Chu, X. L. Zhai, Y. Jin and Y. Cheng, Fuel, 2012, 95, 599–605 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |