
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ternary polymer electrolytes incorporating pyrrolidinium-imide ionic liquids
Henrik
de Vries
abc,
Sangsik
Jeong
abc and
Stefano
Passerini
*abc
aInstitute of Physical Chemistry, University of Muenster, Corrensstrasse 28/30 & 46, 48149 Muenster, Germany. E-mail: Stefano.passerini@kit.edu
bHelmholtz Institute Ulm (HIU), Helmholtzstrasse 11, 89081 Ulm, Germany
cKarlsruhe Institute of Technology (KIT), P.O. Box 3640, 76021 Karlsruhe, Germany
First published on 16th January 2015
Abstract
Herein is reported the performance of ternary polymer electrolytes incorporating ionic liquids, showing higher ionic conductivity over a wide temperature range than binary polymer–salt systems, while guaranteeing higher safety compared to liquid, organic electrolytes or gel electrolytes. In particular, the electrochemical performance and the interactions between poly(ethylene oxide) (PEO), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and several pyrrolidinium-based ionic liquids is comparatively investigated. Eight different polymer electrolytes were produced to test the ionic conductivity and long-time (more than 1400 hours) cycling stability in symmetrical lithium cells. Thermal analysis was used to investigate the thermal stability and degree of crystallinity. Six of the eight investigated samples are found fully amorphous at room temperature. In general, the properties of the polymer electrolytes are influenced by both Ionic liquid ions. The ether function in the side chain of the pyrrolidinium increases the ionic conductivity but, in some cases, lowers the thermal and electrochemical stability.
Introduction
Polymer electrolytes are well known and established in the research field of lithium batteries.1 They are cheap and safe, and act as electrolyte, separator and binder in one. Furthermore, they guarantee a stable operation with lithium metal anodes, which is not true for liquid, carbonate-based electrolytes.2 The most popular polymer is poly(ethylene oxide), PEO, due to its high lithium ion conductivity and good electrochemical and chemical stability, especially with the lithium metal electrode.3 In industrial applications binary systems are already used, in which the polymer is combined with a conductive salt. While the salt cations (Li+ ions) supplies the electroactive species in the electrolyte, the anions might additionally exert a plasticizing effect on the polymer when they have low charge density, high flexibility and large hindrance (e.g., bis(trifluoromethanesulfonyl)imide a.k.a. TFSI).4 The main issue which prevents the use of these electrolyte systems in more applications is the low ionic conductivity at room temperature. This is caused by the limited mobility of the PEO chains, which support the cation mobility. In practice, PEO-based electrolytes are only conductive when the polymer is in the amorphous state.3 To solve this problem, cells are usually run at high temperatures over the melting point of PEO (e.g., at 80 °C). Although such a strict requirement, PEO-based cells are successfully used in the Bolloré Bluecar, which has been deployed as part of the Autolib' carsharing program in Paris, since December 2011.5In an attempt to address the high operative temperature issue of available polymer electrolytes, in 1996 Watanabe and colleagues proposed the incorporation of an ionic liquid mixture of two lithium salts (lithium acetate and LiTFSI) and triethylmethylammonium benzoate (TEMAB) into polyacrylonitrile or poly(vinyl butyrate), yielding to a polymer electrolyte with enhanced ionic conductivity.6 However, though complexation of the ionic liquid and polymer was reported, recrystallization and phase separation occurred after some time. No further investigation on the ionic liquid–polymer interaction was performed, however, it is very reasonable to assume that the liquid phase was just trapped within the polymer matrix, forming a self-standing polymer gel electrolyte, with the Li+ ions moving in the liquid phase.
In 2003, we have found that ternary mixtures of PEO, LiTFSI and N-alkyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide, a.k.a. Pyr1ATFSI, form electrolytes with high ionic conductivity even at room temperature.7 In following studies, we have demonstrated as such ternary electrolytes show excellent performance in terms of plating and stripping from lithium metal electrodes.8 Prototypes using these ternary electrolytes were developed and proved to operate at moderate rate (C/5 and C/3) at 40 °C and at low rate (C/20) even at 20 °C.9 The major hurdle limiting the room temperature performance was identified in the high value of the resistance at the lithium/polymer electrolyte interface.9 As an alternative approach poly-ionic liquids have been proposed,10–12 some of them including ethylene oxide units.11,12 However, their ionic conductivity is, at the best, only comparable to the cheaper and easier to make cross-linked ternary polymer electrolytes herein presented.
The conductivity improvement of PEO–LiTFSI–PYR1ATFSI at low temperatures is associated to the enhanced lithium mobility in the solid phase, resulting from the plasticisation of the polymer induced by the presence of the additional TFSI anions balancing the (rather non-interacting) pyrrolidinium cations.8 In a recent study we have presented a three-phase diagram outlining the compositions in which ternary polymer electrolytes exist in the fully amorphous state at room temperature.13
In this work the system is expanded to mixtures of PEO and LiTFSI with one of eight different ionic liquids synthesized combining different cations and fluorinated sulfonylimides (Fig. 1). The cation is either N-butyl-N-methylpyrrolidinium (Pyr14) or N-methoxy-ethyl-N-methylpyrrolidinium (Pyr12O1). In this latter one oxygen with two free electron pairs replace a CH2-group. In addition to TFSI, three different perfluorinated anions, FSI (bisfluorosulfonylimide; (FSO2)2N−), BETI (bis(pentafluoroethanesulfonyl)imide; (C2F5SO2)2N−), and IM14 ((nonafluorobutanesulfonyl) (trifluoromethanesulfonyl)imide; (C4F9SO2)(CF3SO2)N−)), were used to investigate their effect on the ternary polymer electrolytes. All ionic liquids used were in-house synthesized, purified and electrochemically characterized.14 However, with the exception of Pyr14TFSI, which is used as a baseline and is known to yield to the best performing polymer electrolyte system at the state of the art, they have never been used in polymer electrolytes.
 | ||
| Fig. 1 Pyrrolidinium cations (left) and perfluorinated imide anions (right) composing the ionic liquids investigated in this work. | ||
Experimental
Eight different polymer electrolytes were produced as described before.15 Briefly, PEO (Dow Chemicals, WSR 301, MW = 4 Mio) and LiTFSI (3 M, battery grade, vacuum dried [10−7 mbar] for 48 h at 110 °C) were dry-mixed. A mixture of benzophenone (Aldrich, 5 wt% based on PEO) and one of the eight ionic liquids was then added. After annealing the mixtures at 80 °C under vacuum, self-standing polymer electrolyte films were obtained by hot pressing the composites at 100 °C. The molar ratio of PEO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LiTFSI:ionic liquid was always 20:2:4, which is known to form stable, amorphous films with Pyr14TFSI.13 Finally, the polymer films were cross-linked by UV radiation for 10 minutes (Hönle UVA Cube 100).
:LiTFSI:ionic liquid was always 20:2:4, which is known to form stable, amorphous films with Pyr14TFSI.13 Finally, the polymer films were cross-linked by UV radiation for 10 minutes (Hönle UVA Cube 100).
Thermo-gravimetrical analyses was performed in oxygen and nitrogen gas. The stability of the different electrolytes was investigated using a thermogravimetric analyser (TGA; TA Instruments Model Q5000). The samples were hermetically sealed in aluminium pans inside a dry room (dew point: −60 °C). The TGA device punched the pans automatically prior to the measurement. After a purge step with the reaction gas, the temperature was ramped (5 K min−1) from room temperature to 600 °C. The sample weight change with time and temperature was recorded.
Differential scanning calorimetry (DSC; TA Instruments Model Q2000) was used to investigate melting and glass transition temperatures. Aluminium pans were filled in the dry room and remained sealed during the measurement. The samples were first quenched to 140 °C. Afterwards, three heating–cooling cycles were recorded between −140 and 200 °C with 5 K min−1.
Ionic conductivity was measured in symmetrical pouch cells. Polymer electrolytes films with an area of 4 cm2 were placed between two copper foils. The thickness of the polymer electrolyte films ranged between 80 and 120 μm. The cells were stored in a climatic chamber (Binder) and electrochemical impedance analysis (EIS) was performed with an impedance analyser (Solartron 1260). After every measurement the temperature was changed in 10 °C steps allowing the cells to stabilize for 24 h. A heating (20 °C to 80 °C) followed by a cooling (20 °C to −40 °C) test series were performed. To control the effect of the thermal history, a third measurement at 20 °C was always done afterwards.
Cycling tests were performed in symmetrical lithium pouch cells. Polymer electrolytes films with an area of 4 cm2 were placed between two lithium foils (Rockwood Lithium, 50 μm). The cycling tests were done in a climatic chamber (Binder) at 40 °C with a Maccor battery cycler. For limiting current tests, several potential steps were applied for 10 h each: 0.02; 0.05; 0.1 and 0.2mA cm−2; afterwards the potential was increased by 0.2 mA cm−2 in each step until the evolution of the cells potential indicated the decomposition of the electrolyte. The polarization of the cell was not changed during the test. However, after every current step a 10 h rest time was included for relaxation of the cell potential.
Results
Thermal analysis
The thermal stability was investigated in both oxidizing (O2) and inert (N2) atmospheres. Due to the very low vapour pressure and the high thermal stability of ionic liquids, only very small weight loss is detected at low temperature caused by the release of benzophenone and its photochemical reaction products.The thermal gravimetric analysis proves the high thermal stability of the ternary polymer electrolytes in inert atmosphere (Fig. 2). The TGA curves show as electrolytes with Pyr14 and Pyr12O1 cation have practically the same stability. However, while the samples containing TFSI anions and mixtures with BETI or IM14 anions are stable up to 300 °C those containing the FSI anion appear more reactive (i.e., less thermally stable). However, no weight loss was observed up to 200 °C even in the presence of the FSI anion.
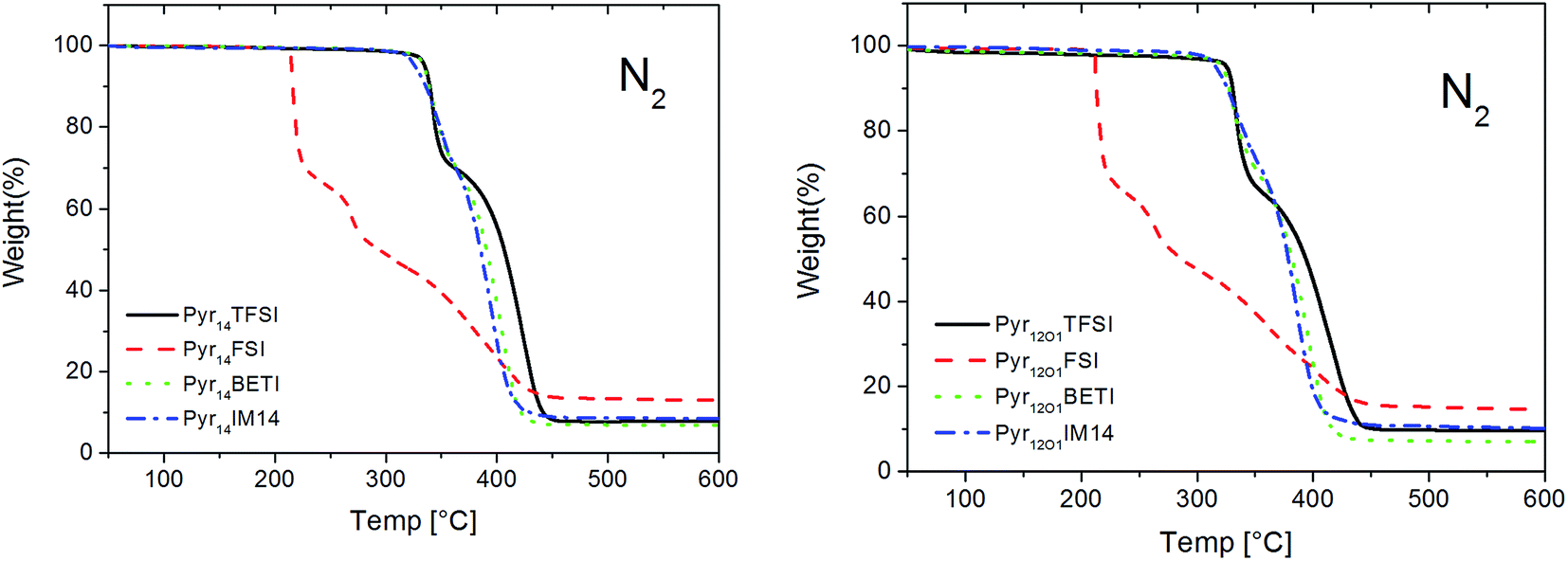 | ||
| Fig. 2 TGA of polymer electrolytes in inert atmosphere. Scan rate 5 °C min−1. | ||
The samples containing TFSI, BETI and IM14 anions show a two-step decomposition mechanism in nitrogen. The weight loss during the 1st step is most likely caused by PEO decomposition, followed by LiTFSI and ionic liquid decomposition.8,13 For the FSI-containing electrolytes three steps can be observed. The lower stability of these samples is caused by the ionic liquid anion which is much more reactive. It is known that in a mixture of Pyr14FSI and LiTFSI in situ formation of LiFSI takes place. While the pure ionic liquid Pyr14FSI shows only a small weight loss at 200 °C and a fast decomposition between 280 and 300 °C,16 LiFSI salt decomposes at 183 °C; Huang and Hollenkamp proposed a possible decomposition reaction under dry conditions.17 The decomposition products of FSI may catalyse the decomposition of PEO (31.7 wt% in electrolyte) in the 1st step, followed by decomposition of the remaining components in two steps.
The ternary polymer electrolytes show high thermal stability even in oxidizing atmosphere (Fig. 3). All samples are stable up to 200 °C with only small weight loss taking place below 100 °C. In this case, the FSI-based electrolytes do not decompose earlier compared to the other samples, indicating that the occurring decomposition at temperatures below 100 °C is related to a common component rather than the FSI anion. Although we did not pursue the identification of such a component, we fell reasonably to propose the oxidation of the reacted benzophenone (used as cross-linking initiator) as the cause of the weight loss at below 100 °C. Nevertheless, the FSI-containing samples own a lower thermal stability as evidenced by the fastest weight loss at higher temperature.
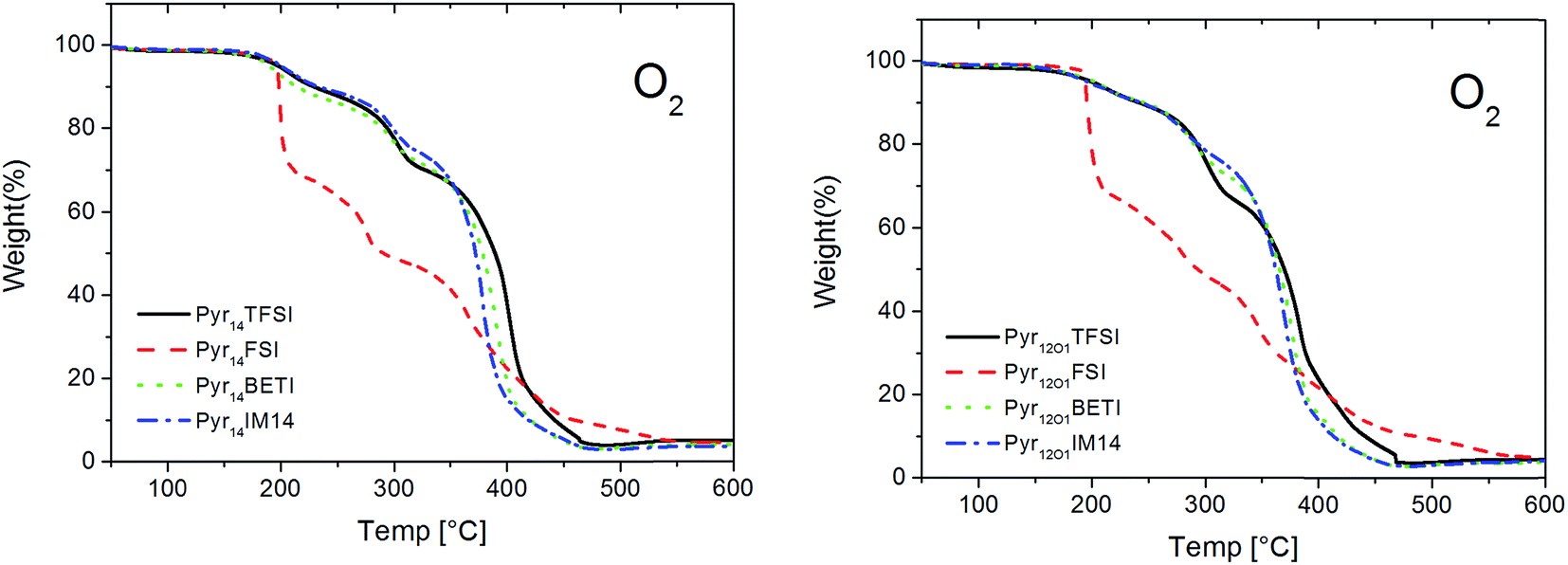 | ||
| Fig. 3 TGA of the polymer electrolytes in oxygen atmosphere. Scan rate 5 °C min−1. | ||
DSC investigation was performed to estimate the effect of the ionic liquid on the morphology of the polymer electrolytes. DSC measurements, in fact, can supply information about the amorphicity of the polymer electrolytes, which would improve the ionic conductivity of the system, and the effect the ionic liquid has on the glass transition temperature (Tg). The latter information is rather important since at temperatures below the Tg no movement in the polymer takes place. Thus, the lower the Tg, the higher is the ionic conductivity at room temperature expected to be.18
Fig. 4 reveals that the glass transition temperature of the polymer electrolytes depends on the anion. Additionally, the polymers including TFSI anions or mixtures with BETI or IM14 anions are fully amorphous while those containing FSI show the existence of crystalline phases indicated by the several peaks in the DSC curves between 0 °C and 100 °C. This is, indeed, in accordance with the appearance of the electrolytes, which look transparent when containing the TFSI, BETI, and IM14 anions, but exhibit opacity with the FSI anion. The glass transition temperatures measured during the 2nd heating and cooling scan are presented in Table 1.
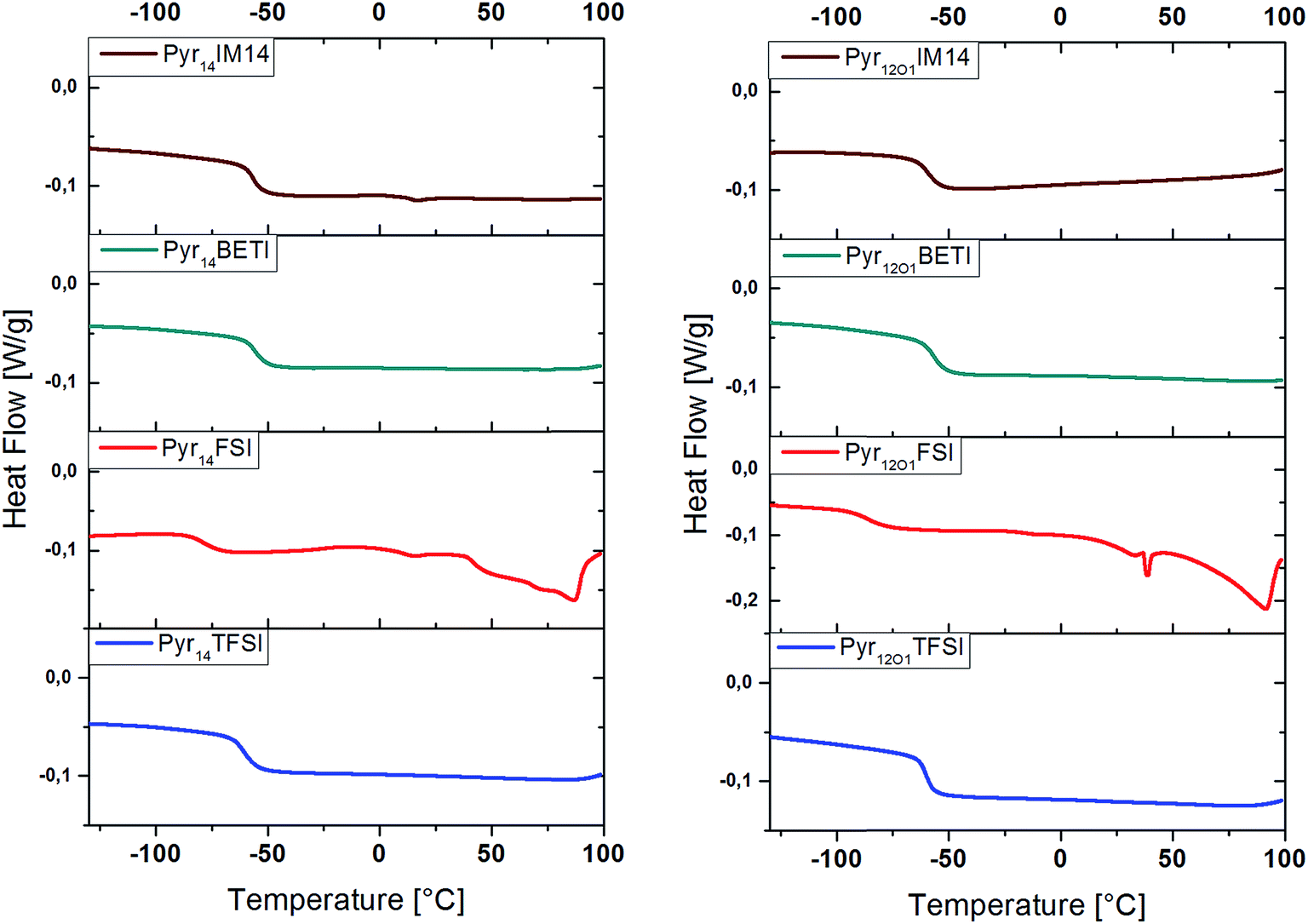 | ||
| Fig. 4 DSC of polymer electrolytes, 5 K min−1, 1st heating scan, exothermic heat up. | ||
| Ionic liquid | T g heating [°C] | T g cooling [°C] |
|---|---|---|
| Pyr14TFSI | −61.8 | −67.2 |
| Pyr14FSI | −79.8 | −88.9 |
| Pyr14BETI | −56.6 | −62.1 |
| Pyr14IM14 | −57.0 | −62.2 |
| 12O1_TFSI | −60.2 | −63.1 |
| 12O1_FSI | −85.3 | −93.2 |
| 12O1_BETI | −58.1 | −64.8 |
| 12O1_IM14 | −59.8 | −65.9 |
The comparison of the different anions shows as the Tg for FSI-containing samples are much lower than for the other samples, although the former ternary electrolytes have shown crystallization melting features above ambient temperature. Considering the effect of the ionic liquid cation, it is seen as Pyr14TFSI offers a slightly lower Tg than Pyr12O1TFSI while the contrary is observed for all other anions (FSI, BETI and IM14).
Ionic conductivity
The ionic conductivity was measured in symmetrical Cu cells using AC impedance spectroscopy in the temperature range between −40 °C and 80 °C. The following equation was used to calculate the overall ionic conductivity σEl from the electrolyte bulk resistance, which was taken as the high frequency intercept in the Nyquist plot of the impedance measurements (not shown):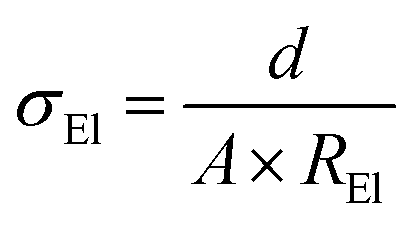
The thickness d of the polymer electrolyte films was around 100 μm (±20 μm) while the electrode area was 4 cm2.
The Arrhenius plots in Fig. 5 present the ionic conductivity (net charge transport of all ionic species) of the eight polymer electrolytes. All samples show a conductivity higher than 10−4 S cm−1 at room temperature and 10−3 S cm−1 at 60 °C.
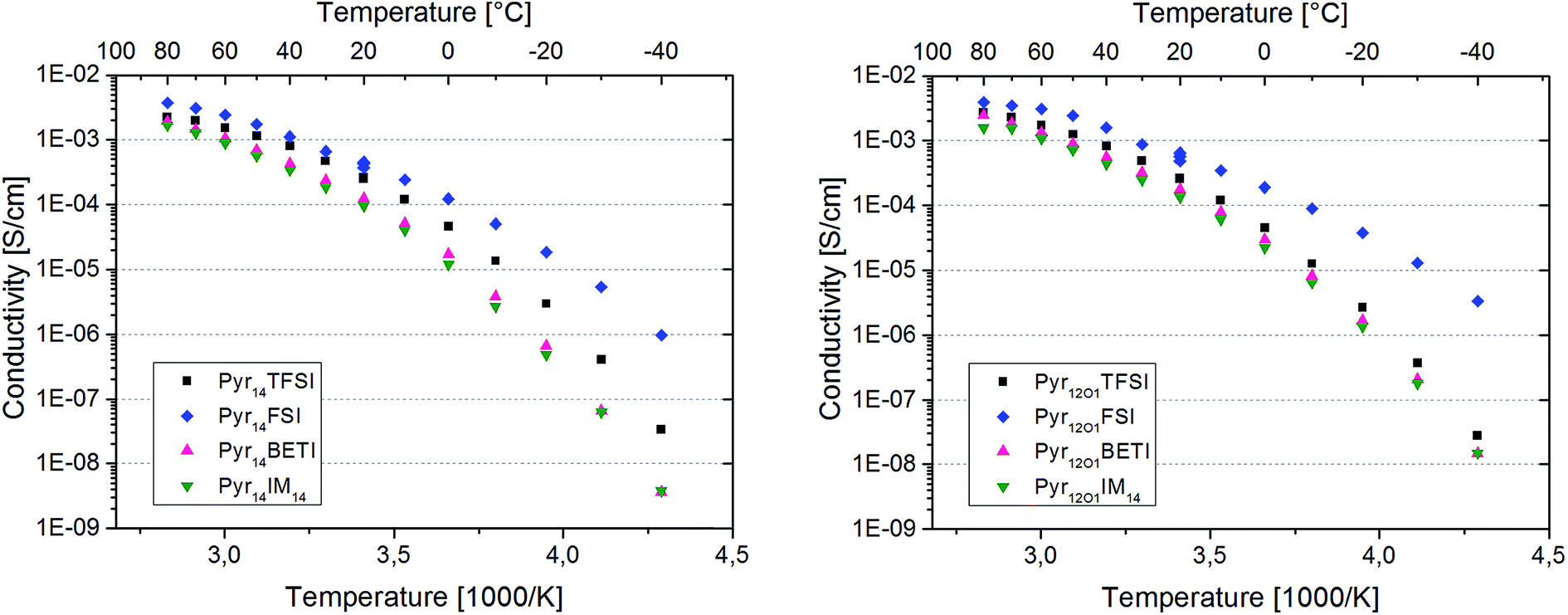 | ||
| Fig. 5 Arrhenius plot for Pyr14 – (left side) and Pyr12O1 – (right side) containing electrolytes. | ||
Binary PEO–LiTFSI polymer electrolytes show a jump in conductivity (sometimes including the switch from Arrhenius to VTF behaviour) when the melting of the crystalline PEO and PEO–Li salt phases takes place.8 However, this behaviour is not observed for the eight ternary electrolytes in this study because they are fully amorphous with the exception of the samples containing FSI. In fact, these samples are partly crystalline as indicated by the distinct melting features observed with DSC investigation. However, the melting of the crystalline fraction does not have a huge impact on the overall conductivity since no net transition is observed in the Arrhenius graphs.
The protocol used for the conductivity measurements allow us to control if the thermal history affects the polymer electrolyte conductivity. In fact, three data points were acquired at 20 °C for each sample, which are all reported in the graphs. They were recorded at the beginning of the heating scan, before the successive cooling scan (i.e., after cooling from the heating scan to 80 °C) and upon thermal equilibration after the cooling scan to −40 °C. For both FSI-containing samples the conductivity at 20 °C is seen to increase throughout the test series. The increase from the first to the second data point was somehow expected as a result of slow recrystallization kinetics after the heating scan to 80 °C. However, the increase detected with the third measurement is very surprising, since the crystallinity of these electrolytes should have increased after the cooling to −40 °C. In fact, DSC measurements show a few endothermic peaks starting from 0 °C up to almost 100 °C upon the heating cycle with corresponding crystallization features between 50 °C and 0 °C during cooling (Fig. 6). Therefore, a small crystalline fraction should be present at 20 °C, causing lower ionic conductivity. This is suggesting that the thermal history might affect the measured conductivity of those polymer electrolytes containing the FSI anion. However, no effect was observed for all other ternary electrolytes.
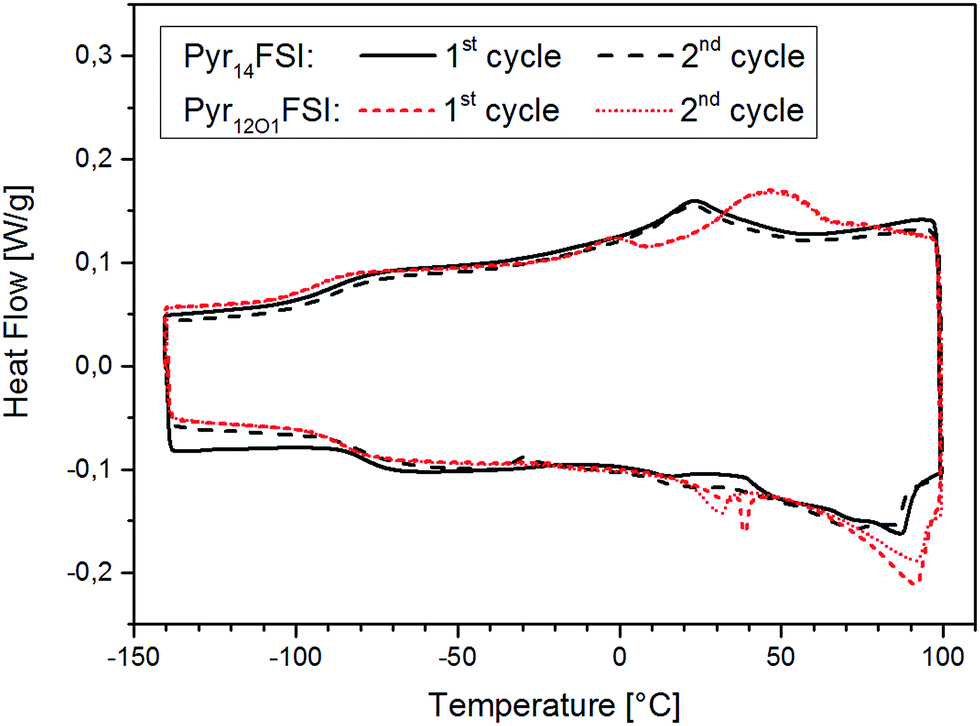 | ||
| Fig. 6 DSC curves of Pyr14FSI- and Pyr12O1FSI-containing electrolytes, 1st and 2nd heating/cooling cycle, exothermic heat up. | ||
The overall ionic conductivity results from the charge transport of all ionic species – in this case the Li+ and pyrrolidinium cations as well as the anions. To evaluate the specific lithium ion conductivity symmetrical lithium cells were assembled and cycled with two different test protocols each addressing the maximum steady-state current sustained by the electrolyte or the lithium plating/stripping performance.
The maximum steady-state current sustained by the polymer electrolytes was investigated by applying increasing potential steps, each for several hours. For each step the steady-state current was determined to depend on the applied potential. At some point, however, the steady-state current is expected not to rise upon increasing potential, this representing the limiting current of the electrolyte. This test also reveals relaxation processes and eventual decomposition reactions of the electrolyte (i.e., electrochemical stability vs. lithium).
In more details, symmetrical lithium cells were step-wise polarized (for 10 h) at increasing potentials, intermixed with 10 h relaxation steps. The voltage of the cell was recorded. With increasing potential the cells take longer to reach the steady-state current, i.e., the condition in which the current is sustained only by the electroactive ionic species (Li+) and is then limited by the lithium ions diffusion into the electrolyte.
Three of the potential steps are presented in Fig. 7. At 40 °C and 0.2 V polarization, the two samples containing FSI anions show the lowest current densities, 0.01 mA cm−2 for Pyr12O1FSI and 0.02 mA cm−2 for Pyr14FSI, although they exhibit the highest overall ionic conductivity, as evidenced by conductivity measurements. Such a poor performance indicates that the total ionic conductivity in FSI-based polymer electrolytes is mostly contributed by the small FSI anions rather than the strongly coordinated (by PEO) Li+ cations, although some effects due to the formation of a rather thick SEI layer on the lithium electrodes cannot be excluded. The two BETI-containing electrolytes show much better performance which was independent on the IL cation, with current density of more than 0.03 mA cm−2. Even better is the performance of IM14-based electrolytes with the one containing Pyr12O1IM14 approaching 0.05 mA cm−2. However, the highest steady-state current densities were achieved with those electrolytes containing TFSI only, especially that with Pyr14TFSI (0.06 mA cm−2).
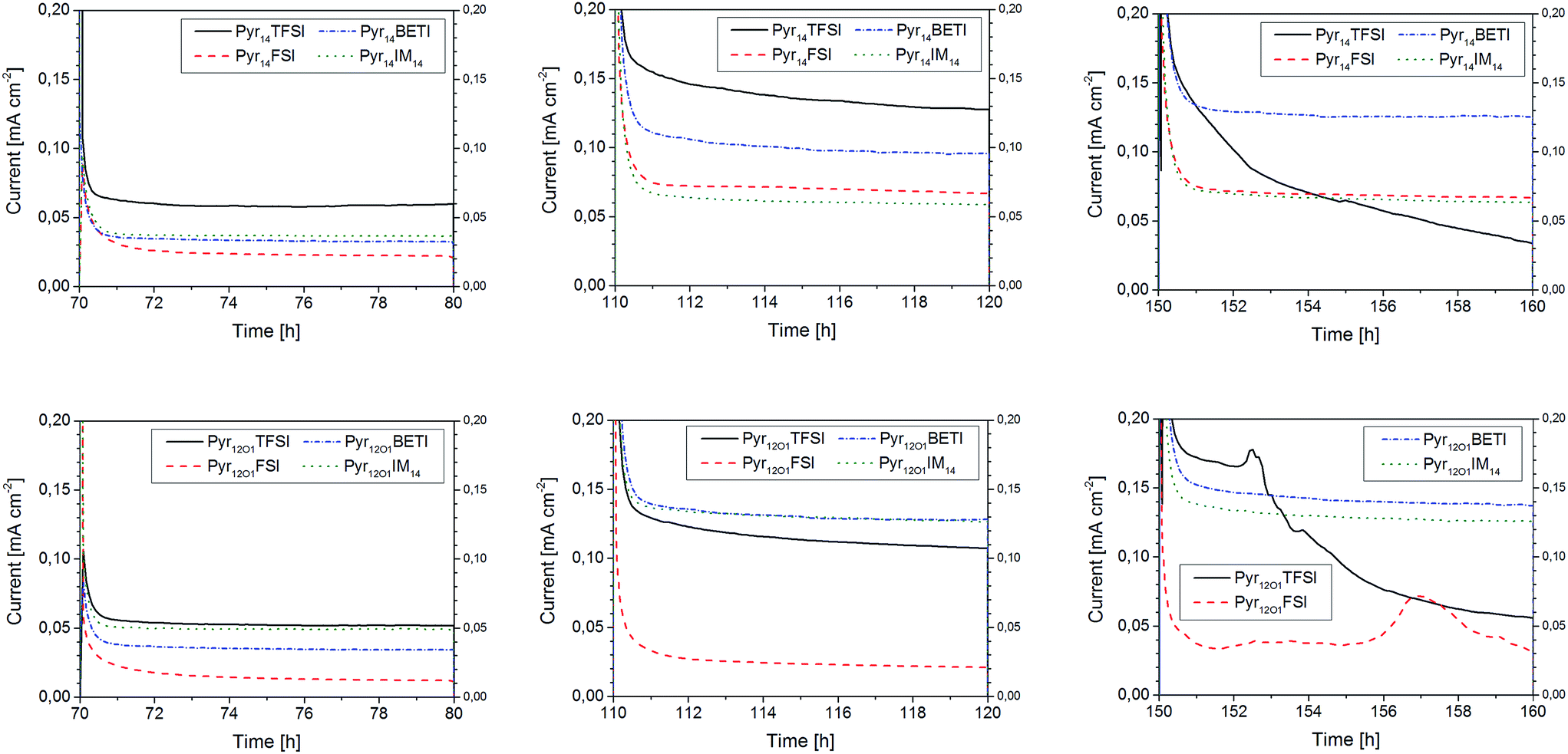 | ||
| Fig. 7 Chronoamperometry curves upon 10 h polarization steps (intermixed with 10 h relaxation steps) at 0.2 V (left), 0.6 V (center) and 1.0 V (right); Pyr14-containing electrolytes (top) and Pyr12O1-containing electrolytes (bottom). All measurements were performed at 40 °C. | ||
With increasing potential the current density increased for all samples. The electrolyte containing Pyr12O1FSI is still showing the worst performance though its steady-state current density doubled. For the one with Pyr14FSI a three-fold increase in current density, to more than 0.065 mA cm−2, is observed. For the Pyr14IM14-containing electrolyte, however, the polarization increase resulted in an increase of only about 75% (to 0.06 mA cm−2). A much higher value was reached with Pyr14BETI (increased by a factor of 3) as well as Pyr12O1TFSI (increased by a factor of 2) with around 0.1 mA cm−2. The highest current densities (0.125 mA cm−2) were achieved with Pyr14TFSI, Pyr12O1BETI and Pyr12O1IM14. It is noticeable, however, that the current flowing through the Pyr14TFSI-containing electrolyte did never reach the steady state, as indicated by the continuous current density decline.
With the applied potential of 1 V, the Pyr12O1 electrolytes with small anions (FSI and TFSI) show signs of electrochemical decomposition. This is somehow surprising since TFSI is known as a electrochemically very stable anion. However, the decomposition might very well involve the ionic liquid cation, instead, although the electrolytes incorporating Pyr12O1 and BETI or IM14 show no signs of decomposition. The highest current density – 0.14 mA cm−2 – in this test series was reached with Pyr12O1BETI, with Pyr12O1IM14 having the second highest value (0.125 mA cm−2). Also noticeable is the continuous current density decline observed for the electrolyte containing Pyr14TFSI, which supports degradation phenomena to occur at the polymer electrolyte–lithium interface resulting in a continuous increase of the interfacial resistance.
To better delimitate the range of stability of this latter polymer electrolyte, the time evolution of the current density curves, as well as the potential curves recorded during the relaxation steps are presented in Fig. 8. From the graphs is it observed that the continuous decline of the current density already starts at 0.6 V, although at a minor extent, to become very substantial at 0.8 V. Also, after the 1 V step the cell does not reach the 0 V during the following relaxation. This is a clear sign for the occurrence of decomposition reactions leading to the growth of a rather thick passivation layer at the lithium/electrolyte interface. This result indicates that Pyr14TFSI-containing polymer electrolytes should not be used at current densities higher than 0.12 mA cm−2 (at 40 °C) in order to avoid elevated overvoltages which would result in the electrolyte decomposition. Slightly higher current densities can be achieved using the Pyr12O1BETI-containing electrolyte.
 | ||
| Fig. 8 Chronoamperometry curves at various voltages (left panel) as detected upon 10 h polarization steps applied to the Pyr14TFSi-containing polymer electrolyte in symmetric lithium cells (T = 40 °C). The cell voltage relaxation recorded during the intermixed rest steps are also reported (right). | ||
Galvanostatic cycling tests were performed to investigate the long-term lithium stripping–plating performance. Freshly deposited lithium is extremely reactive, resulting in the occurrence of electrolyte decomposition reactions, in turns leading to lithium consumption (cell capacity loss) and SEI formation during cycling (increased internal cell resistance).2a,19 For these tests a specific current, which sign is reversed every hour, is applied to the cell and the overvoltage recorded. The evolution of overpotential can be used to monitor the SEI evolution (the increase of the internal resistance results in the increase of the overvoltage) while the overall number of cycles is limited by the lithium consumption during cycling or short-circuits caused by lithium dendrite growth. In order to perform this experiment in a reasonable amount of times (usually a few months) it is important to use rather thin lithium foils (here: 50 μm) to limit the lithium reservoir.
The stripping–plating tests were performed by reversing the polarization of the cells every hour. A current density of 0.1 mA cm−2 was chosen since this value was achieved with most of the electrolytes (except Pyr12O1FSI) at 40 °C. The cells were stopped after 10, 20, 50, 100, 200, 400, 600, 800 and 1000 cycles for impedance measurements (21 °C) in order to evaluate the changes of the electrolyte and interfacial resistances. The overvoltage evolution upon cycling (cell potential at the end of 1 h polarization) for all cells is shown in Fig. 9.
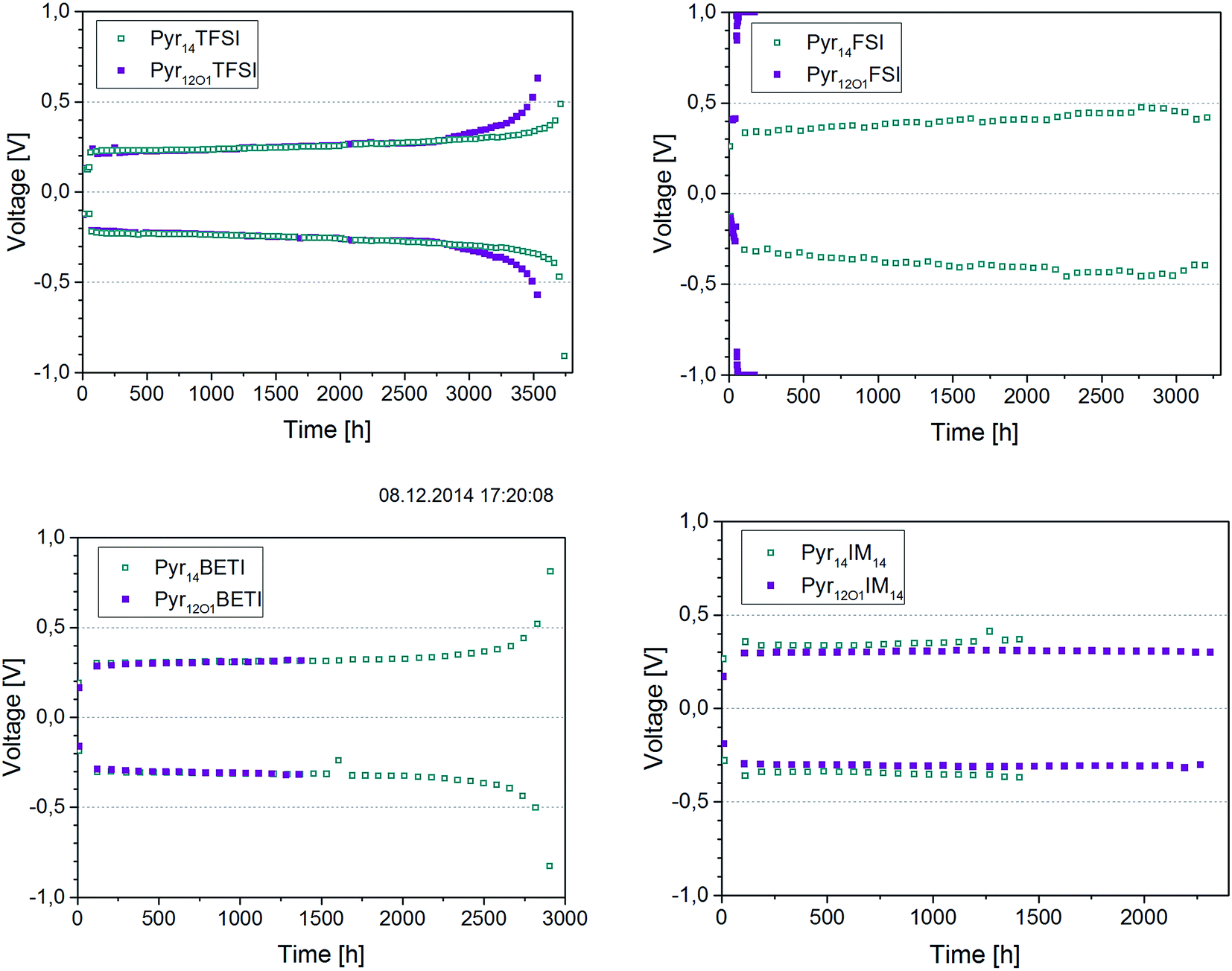 | ||
| Fig. 9 Stripping–plating in symmetrical lithium cells at 0.1 mA cm−2 and 40 °C. The ionic liquid present in the electrolytes is indicated in the legends. | ||
The expected behaviour for this test is exemplarily shown by the Pyr14BETI-containing electrolyte. In the first initial cycles the overvoltage increases resulting from the establishment of the interfaces between the electrolyte and both electrodes. In the following few thousand cycles the overvoltage is practically constant. However, a small fraction of freshly deposited lithium is consumed at each cycle, resulting, after 2500 hours, in the rapid increase of the overvoltage due to the scarcity of lithium available for stripping.
According to the results in Fig. 9 the best performing electrolyte in this test was that incorporating Pyr14TFSI (i.e., the TFSI anion only). This electrolyte, in fact, showed the best long-time stability (more than 3500 h) and the lowest overpotential. The sample containing Pyr12O1TFSI showed almost the same cycling stability, failing due to lithium consumption only slightly earlier. As expected, the Pyr12O1FSI electrolyte did not work since the applied current, which value was selected to perform the test in a reasonable time frame, was higher than the maximum steady-state current found in the previous experiment. Pyr14FSI shows a very high overpotential – around 0.4 V – but the cells was cycled for more than 3000 h. It is important to notice that, with the exception of the FSI-based ones, overvoltages lower than 0.4 V where observed for all other ternary electrolytes. This means that the plating–stripping tests were not affected by the massive electrochemical decomposition reactions previously discussed (see Fig. 7 and 8).
The two BETI-containing electrolytes showed performance very similar to the only TFSI-containing ones, however, with slightly higher overvoltages. In this case the Pyr12O1-containing electrolyte failed much earlier (1400 h vs. 2800 h). In the case of the IM14 anion, however, the opposite is observed with the Pyr12O1-containing electrolyte showing superior performance than that with Pyr14. As expected from the results illustrated in Fig. 7, these electrolytes showed slightly higher overvoltages than those containing BETI and TFSI.
Impedance measurements were performed on all cells upon the lithium plating–stripping tests. Fig. 10 shows a few, selected impedance spectra for the ternary electrolytes containing the ionic liquids composed by the two cations and either TFSI or BETI anions. Within the frequency range investigated only the polymer electrolyte impedance and the interfacial impedance are concerned, without any contribution from ion diffusion impedance. While the electrolyte impedance of each cell (given by the high frequency intercept with the real axis) was practically unaffected by the repeated plating–stripping cycles, the interfacial resistance (given by the difference between the high and low frequency intercepts with the real axis) changed substantially.9 It is also remarkable as the interfacial resistance is, at least, one order of magnitude larger than the electrolyte resistance, which is in a full agreement with previous reports.9 Also remarkable is the initial decrease of the interfacial resistance observed with all polymer electrolytes, which has been already associated with a substantial reconstruction of the native SEI caused by the lithium deposition and plating.9 However, upon long-term cycling, the SEI is seen to grow with all polymer electrolytes. After 800 cycles the interfacial impedance was equal if not higher than that measured for the pristine cells. A minor effect of the different cations and anions is observed on the evolution of the interfacial impedance, with the TFSI anion and the Pyr14 cation leading to lower values.
 | ||
| Fig. 10 Electrochemical impedance spectra of ternary electrolytes containing Pyr14TFSI (left, top) and Pyr12O1TFSI (right, top), Pyr14BETI (left, bottom) and Pyr12O1BETI (right, bottom) recorded at 21 °C during the plating–stripping cycle test at 40 °C. Frequency range: 100 kHz–1 Hz. | ||
Conclusion
Ternary polymer electrolytes were prepared with PEO, LiTFSI, and one of eight different ionic liquids, obtained from the combination of two cations (Pyr14 and Pyr12O1) and four anions (FSI, TFSI, BETI and IM14). For three of the four anions, the total ionic conductivity was increased with Pyr12O1 compared to the corresponding Pyr14 ionic liquid. The ionic conductivity increased with smaller anions (FSI > TFSI > BETI > IM14). Samples with TFSI, BETI and IM14 were found to be fully amorphous. Though both electrolytes with FSI show the highest ionic conductivity, they also showed a partial crystallinity and lower thermal stability. Additionally, these electrolytes showed a dependence of their room temperature conductivity from the prior thermal history. Further investigation would be necessary to explain the surprising increase of the room temperature ionic conductivity (i.e., 20 °C) after the cooling cycle (i.e., upon heating from −40 °C).Chronoamperometry tests at 40 °C show as the polymer electrolytes comprising only TFSI or TFSI in a mixture with BETI or IM14 anions are capable of sustaining steady-state currents, i.e., which are associated to the mobility of Li+ ions only, higher than 0.1 mA cm−2. This was not the case of FSI-containing electrolyte. A possible explanation for the poor performance of the latter might be the lower stability of the FSI anion resulting in the formation of thick SEI layers as well as the lower ability of the FSI anion to support the diffusion of Li+ cations.
Long-time stripping–plating (0.1 mA cm−1 and 40 °C) was shown feasible for seven of the eight electrolytes with cycling times ranging between 1400 and 3500 hour. In the impedance plots it can be observed that the resistance decreases during the first 100 (TFSI only) or 200 (BETI-containing) cycles. During further cycling the resistance increases again, reaching the initial resistance after around 800 cycles.
Summarizing, this comparative study shows as ternary polymer electrolytes can be made incorporating other pyrrolidinium cations than Pyr14, and imide anions than TFSI. However, no major improvements were achieved compared to the known systems composed of PEO, LiTFSI and Pyr14TFSI.
Acknowledgements
The authors kindly acknowledge the financial support from the EU under the grant agreements no. 314282 (LISSEN – Lithium Sulfur Superbattery Exploitating Nanotechnology) and no. 608502 (SIRBATT – Stable Interfaces for Rechargeable Batteries).References
- (a) M. Armand, Solid State Ionics, 1994, 69, 309–319 CrossRef CAS; (b) P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 172–172 CrossRef CAS.
- (a) E. Peled, D. Golodnitsky, G. Ardel and V. Eshkenazy, Electrochim. Acta, 1995, 40, 2197–2204 CrossRef CAS; (b) A. M. Christie, A. Lisowska-Oleksiak and C. A. Vincent, Electrochim. Acta, 1995, 40, 2405–2411 CrossRef CAS; (c) J. K. Stark, Y. Ding and P. A. Kohl, J. Electrochem. Soc., 2013, 160, D337–D342 CrossRef CAS PubMed.
- M. Armand, Adv. Mater., 1990, 2, 278–286 CrossRef CAS PubMed.
- W. Gorecki, M. Jeannin, E. Belorizky, C. Roux and M. Armand, J. Phys.: Condens. Matter, 1995, 7, 6823 CrossRef CAS.
- bluecar.fr, 2014.
- M. Watanabe and T. Mizumura, Solid State Ionics, 1996, 86–88(1), 353–356 CrossRef CAS.
- J.-H. Shin, W. A. Henderson and S. Passerini, Electrochem. Commun., 2003, 5, 1016–1020 CrossRef CAS PubMed.
- M. Joost, M. Kunze, S. Jeong, M. Schönhoff, M. Winter and S. Passerini, Electrochim. Acta, 2012, 86, 330–338 CrossRef CAS PubMed.
- G. B. Appetecchi, G. T. Kim, M. Montanino, F. Alessandrini and S. Passerini, J. Power Sources, 2011, 196, 6703–6709 CrossRef CAS PubMed.
- A. S. Shaplov, P. S. Vlasov, M. Armand, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, O. V. Okatova, G. M. Pavlov, C. Wandrey, I. A. Godovikov and Y. S. Vygodskii, Polym. Chem., 2011, 2, 2609–2618 RSC.
- H. Hu, W. Yuan, L. Lu, H. Zhao, Z. Jia and G. L. Baker, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2104–2110 CrossRef CAS PubMed.
- (a) Z. Jia, W. Yuan, H. Zhao, H. Hu and G. L. Baker, RSC Adv., 2014, 4, 41087–41098 RSC; (b) M. Döbbelin, I. Azcune, M. Bedu, A. Ruiz de Luzuriaga, A. Genua, V. Jovanovski, G. Cabañero and I. Odriozola, Chem. Mater., 2012, 24, 1583–1590 CrossRef.
- M. Joost, G. T. Kim, M. Winter and S. Passerini, Electrochim. Acta, 2013, 113, 181–185 CrossRef CAS PubMed.
- (a) M. Kunze, S. Jeong, E. Paillard, M. Winter and S. Passerini, J. Phys. Chem. C, 2010, 114, 12364–12369 CrossRef CAS; (b) M. Kunze, E. Paillard, S. Jeong, G. B. Appetecchi, M. Schönhoff, M. Winter and S. Passerini, J. Phys. Chem. C, 2011, 115, 19431–19436 CrossRef CAS; (c) M. Kunze, S. Jeong, E. Paillard, M. Schönhoff, M. Winter and S. Passerini, Adv. Energy Mater., 2011, 1, 274–281 CrossRef CAS PubMed; (d) J. Reiter, E. Paillard, L. Grande, M. Winter and S. Passerini, Electrochim. Acta, 2013, 91, 101–107 CrossRef CAS PubMed.
- J.-H. Shin, W. A. Henderson and S. Passerini, J. Electrochem. Soc., 2005, 152, A978–A983 CrossRef CAS PubMed.
- Q. Zhou, W. A. Henderson, G. B. Appetecchi, M. Montanino and S. Passerini, J. Phys. Chem. B, 2008, 112, 13577–13580 CrossRef CAS PubMed.
- J. Huang and A. F. Hollenkamp, J. Phys. Chem. C, 2010, 114, 21840–21847 CAS.
- M. B. Armand, P. G. Bruce, M. Forsyth, B. Scrosati and W. Wieczorek, in Energy Materials, ed. D. Bruce, D. O'Hare and R. I. Walton, Wiley, 2011, pp. 1–31 Search PubMed.
- (a) E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS PubMed; (b) Z. Li, J. Huang, B. Yann Liaw, V. Metzler and J. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS PubMed; (c) D. Lv, Y. Shao, T. Lozano, W. D. Bennett, G. L. Graff, B. Polzin, J. Zhang, M. H. Engelhard, N. T. Saenz, W. A. Henderson, P. Bhattacharya, J. Liu and J. Xiao, Adv. Energy Mater., 2014 DOI:10.1002/aenm.201400993.
| This journal is © The Royal Society of Chemistry 2015 |