DOI:
10.1039/C4RA15977B
(Paper)
RSC Adv., 2015,
5, 14359-14365
Two heteroleptic Ir(III)–bisthienylethene compounds: syntheses, structures and aggregation-induced luminescence†
Received
8th December 2014
, Accepted 20th January 2015
First published on 21st January 2015
Abstract
A new bisthienylethene 2-(2-hydroxynaphthyl)-4,5-bis(2,5-dimethyl(3-thienyl))-1H-imidazole (hnbdtiH) has been synthesized and applied for the coordination-assembly generation of two heteroleptic complexes [Ir(dfppy)2(hnbdti)]·2CH3OH (1) and [Ir(ppy)2(hnbdti)]·CH3OH (2) [dfppyH = 2-(2,4-difluorophenyl)-pyridine, ppyH = 2-phenyl-pyridine]. In the crystal structure of hnbdtiH, neighboring molecules are linked into a helical chain through N–H⋯π interactions between imidazole and naphthol rings. In 1 and 2, the {Ir(dfppy)2}+ or {Ir(ppy)2}+ unit is chelated by a hnbdti− ligand using both N and O binding sites, and the intramolecular aromatic stacking interactions are formed between the thiophene ring and the 2,4-difluorophenyl/phenyl group. The crystal structures of 1 and 2 indicate that neighboring molecules are connected by CH3OH molecules through hydrogen bond interactions, forming the [Ir⋯(CH3OH)4⋯Ir] dimer in the former, and the [Ir⋯(CH3OH)2⋯Ir] dimer in the latter. These dimers in 1 pack together through the intermolecular aromatic stacking interactions, while there are only van der Waals interactions in 2, resulting in their distinct luminescence behavior. At room temperature, 1 exhibits aggregation-induced phosphorescence emission. In contrast, 2 is almost non-luminescent both in solution and in the solid state. Compared with 1 and 2, free ligand hnbdtiH shows fluorescence both in CH2Cl2 solution and in the solid state. Moreover, no photochromism has been observed in hnbdtiH, 1 and 2.
Introduction
Metal–bisthienylethene complexes have attracted much attention because of their potential multifunctional properties, including bisthienylethene-centered photochromism, and metal-based properties, for example luminescence,1–4 nonlinear optical properties,5 redox properties,6 magnetic behavior,7 etc. In these complexes, only several Ir(III) compounds have been reported so far. Park, et al. synthesized three Ir(III) complexes where a picolinate ancillary ligand covalently links to a bisthienylethene (BTE) unit (Scheme S1,† left).2 Using a similar synthetic approach, Guerchais, et al. obtained Ir(C^N–ppy–Me)2(N^N–bpy–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–R) (R = BTE unit).3 In contrast to these Ir(III) compounds containing noncoordinated BTE unit, Tian, et al. reported compound (Py–BTE)2Ir(acac), in which Py–BTE serves as a cyclometalated ligand, using its pyridyl-thiophene moiety to chelate an Ir(III) ion (Scheme S1,† right).4 These Ir(III)–bisthienylethene compounds reveal the switching of Ir(III)-based luminescence by photochromism reaction. Their phosphorescence emissions become weaker upon the structural transition of the BTE unit from the open to the closed form. The number of these cases is quite limited, thus it is still a great challenge to synthesize more Ir(III)–bisthienylethene compounds to explore their structures, and related luminescences and photochromic behaviors.
CH–R) (R = BTE unit).3 In contrast to these Ir(III) compounds containing noncoordinated BTE unit, Tian, et al. reported compound (Py–BTE)2Ir(acac), in which Py–BTE serves as a cyclometalated ligand, using its pyridyl-thiophene moiety to chelate an Ir(III) ion (Scheme S1,† right).4 These Ir(III)–bisthienylethene compounds reveal the switching of Ir(III)-based luminescence by photochromism reaction. Their phosphorescence emissions become weaker upon the structural transition of the BTE unit from the open to the closed form. The number of these cases is quite limited, thus it is still a great challenge to synthesize more Ir(III)–bisthienylethene compounds to explore their structures, and related luminescences and photochromic behaviors.
Previously, we reported the photochromic behavior of hpbdtiH (Scheme 1).7b Upon irradiation with 321 nm light, its CH2Cl2–CH3CN solution changed color from colorless to pink, due to the structural transition from open to closed form. In this paper, we synthesize a derivative of hpbdtiH, namely hnbdtiH with a naphthol group (Scheme 1). Its coordination assemblies to [Ir(ppy)2Cl]2 and [Ir(dfppy)2Cl]2, respectively, generate two heteroleptic complexes [Ir(dfppy)2(hnbdti)]·2CH3OH (1) and [Ir(ppy)2(hnbdti)]·CH3OH (2) [dfppyH = 2-(2,4-difluorophenyl)-pyridine, ppyH = 2-phenyl-pyridine]. Herein, we report the crystal structures of hnbdtiH, 1 and 2, and discuss their luminescences and photochromisms.
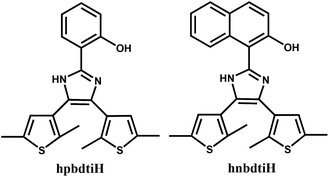 |
| | Scheme 1 | |
Experimental
Materials and methods
1,2-Bis[2,5-dimethyl(3-thienyl)]ethane-1,2-dione was prepared according to the literature method.8 [Ir(dfppy)2Cl]2 and [Ir(ppy)2Cl]2 were prepared following reported procedure.9 All other reagents were commercially available and used without further purification. Elemental analyses were performed on a Perkin Elmer 240C elemental analyzer. The IR spectra were obtained as KBr disks on a VECTOR 22 spectrometer. The 1H NMR spectra were recorded at room temperature with a 500 MHz BRUKER AM-500 spectrometer. The powder XRD patterns were recorded on a BRUKER D8 ADVANCE X-ray diffractometer. UV-vis absorption spectra were measured on a Cary 100 spectrophotometer. The luminescence spectra at room temperature were measured on a Perkin Elmer LS55 fluorescence spectrometer. The luminescence lifetimes were measured at room temperature on a HORIBA FluoroMax-4 TCSPC Spectrofluorometer with a 390 nm LED pulsed from a NanoLED resource. Quantum yield was obtained on a time-correlated single-photon counting lifetime spectroscopy system Edinburgh FL920 with a semiconductor laser as the excitation source (λex = 379 nm). Dynamic light scattering (DLS) measurement was conducted on a Brookhaven BI-200SM instrument. The transmission electron microscopy (TEM) study was performed using a JEM-2100 electron microscope. Photoirradiation was carried out using a 500 W Xe lamp and a monochromator.
Synthesis of 2-(2-hydroxynaphthyl)-4,5-bis(2,5-dimethyl(3-thienyl))-1H-imidazole (hnbdtiH)
A mixture of 2-hydroxy-1-naphthaldehyde (4 mmol, 0.6887 g), ammonium acetate (24 mmol, 1.8499 g), 1,2-bis[2,5-dimethyl(3-thienyl)]ethane-1,2-dione (3.5 mmol, 0.9743 g), and glacial acetic acid (30 mL) was refluxed for 24 hours. The solvent was removed under vacuum, and then the residue was mixed with H2O (20 mL). The resultant mixture was extracted with CH2Cl2 (20 mL × 3). The combined CH2Cl2 solution was dried with MgSO4, filtrated, and evaporated. The crude product was purified through preparative high-performance liquid chromatography, using a silicagel column and petroleum ether–ethyl acetate solution (v/v = 2/98), obtaining light yellow solid. Yield: 403 mg (27% based on 1,2-bis[2,5-dimethyl(3-thienyl)]ethane-1,2-dione). Anal. found (calcd) for C25H22ON2S2: C, 69.62 (69.74); H, 5.32 (5.15), N, 6.38 (6.51). IR (KBr, cm−1): 3407(s), 2914(w), 1618(w), 1595(w), 1547(w), 1466(m), 1410(w), 1334(w), 1277(m), 1245(s), 1207(w), 1144(m), 1127(m), 976(w), 832(s), 811(s), 783(w), 755(m), 729(w), 651(w), 608(m), 573(w), 540(w), 495(m), 442(w). 1H NMR (500 MHz, CDCl3), δ (ppm): 2.24 and 2.43 (12H, s, 4CH3), 6.66 (2H, s, thiophene-H), 7.30–8.18 (6H, m, naphthalene-H).
Synthesis of [Ir(dfppy)2(hnbdti)]·2CH3OH (1)
The mixture of hnbdtiH (0.18 mmol, 0.0775 g), [Ir(dfppy)2Cl]2 (0.08 mmol, 0.0973 g) and Na2CO3 (1.8 mmol, 0.1908 g) in methoxyethanol (25 mL) was stirred under argon at 136 °C for one day. After cooling down to room temperature, the mixture was filtered, and evaporated to dry under vacuum. The resultant brown solid was mixed with water (10 mL), and then the mixture was extracted with CH2Cl2 (25 mL × 3). The CH2Cl2 solution was dried with MgSO4, filtrated, and evaporated. The residue was crystallized with CH3OH, obtaining yellow blocky crystals as a monophasic material based on the powder XRD pattern. Yield: 111 mg (65% based on [Ir(dfppy)2Cl]2). Anal. found (calcd) for C49H41O3N4F4S2Ir: C, 55.13 (55.20); H, 4.06 (3.88); N, 5.03 (5.25). IR (KBr, cm−1): 3446(m), 2916(w), 1604(s), 1572(w), 1556(w), 1477(m), 1463(w), 1433(w), 1400(w), 1375(w), 1289(w), 1246(w), 1159(w), 1113(w), 1099(w), 987(m), 845(w), 827(m), 752(w). 1H NMR (500 MHz, CDCl3), δ (ppm): 1.56–2.28 (12H from 4CH3), 4.98–7.11, and 7.36–9.42 (2H from two thiophene rings, 6H from naphthalene ring, and 12H from two dfppy groups).
Synthesis of [Ir(ppy)2(hnbdti)]·CH3OH (2)
Compound 2 was synthesized by the method of preparing 1, using [Ir(ppy)2Cl]2 instead of [Ir(dfppy)2Cl]2. The yellow blocky crystals were obtained as a monophasic material based on the powder XRD pattern. Yield: 92 mg (60% based on [Ir(ppy)2Cl]2). Anal. found (calcd) for C48H41O2N4S2Ir: C, 59.84 (59.92); H, 4.12(4.29); N, 5.75 (5.82). IR (KBr, cm−1): 3433(w), 3052(w), 2913(w), 1605(m), 1584(m), 1559(w), 1477(s), 1463(m), 1433(m), 1419(w), 1289(w), 1267(w), 1155(w), 1138(w), 1058(w), 1030(w), 986(w), 826(w), 757(m), 728(m). 1H NMR (500 MHz, CDCl3), δ (ppm): 1.79–2.27 (12H, 4CH3), 5.71–7.08, and 7.27–9.43 (2H from two thiophene rings, 6H from naphthalene ring, and 16H from two ppy groups).
X-ray crystallographic studies. The colorless needlelike single crystal of hpbdtiH was obtained through the evaporation of its solution of petroleum ether and ethyl acetate. Single crystals of dimensions 0.30 × 0.20 × 0.15 mm3 for hnbdtiH, 0.28 × 0.18 × 0.15 mm3 for 1, and 0.20 × 0.18 × 0.15 mm3 for 2 were used for structural determinations on a Bruker SMART APEX CCD diffractometer using graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å) at 173 K for hnbdtiH, and at room temperature (296 K) for 1 and 2. A hemisphere of data were collected in the θ range of 2.58–27.01° for hnbdtiH, 2.43–25.31° for 1, and 2.51–22.47° for 2 using a narrow-frame method with scan widths of 0.30° in ω and an exposure time of 10 s per frame. Numbers of observed and unique [I > 2σ(I)] reflections are 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 948 and 4110 (Rint = 0.0486) for hnbdtiH, 12230 and 7873 (Rint = 0.0302) for 1, and 14085 and 9347 (Rint = 0.0385) for 2, respectively. The data were integrated using the Siemens SAINT program,10 with the intensities corrected for Lorentz factor, polarization, air absorption, and absorption due to variation in the path length through the detector faceplate. Multi-scan absorption corrections were applied. The structures were solved by direct methods and refined on F2 by full matrix least squares using SHELXTL.11 All the non-hydrogen atoms were located from the Fourier maps, and were refined anisotropically. All H atoms were put in calculated positions using riding model, and were refined isotropically, with the isotropic vibration parameters related to the non-H atom to which they are bonded. The crystallographic data for compounds hnbdtiH, 1 and 2 are listed in Table 1, and selected bond lengths are given in Tables 2–4.†
948 and 4110 (Rint = 0.0486) for hnbdtiH, 12230 and 7873 (Rint = 0.0302) for 1, and 14085 and 9347 (Rint = 0.0385) for 2, respectively. The data were integrated using the Siemens SAINT program,10 with the intensities corrected for Lorentz factor, polarization, air absorption, and absorption due to variation in the path length through the detector faceplate. Multi-scan absorption corrections were applied. The structures were solved by direct methods and refined on F2 by full matrix least squares using SHELXTL.11 All the non-hydrogen atoms were located from the Fourier maps, and were refined anisotropically. All H atoms were put in calculated positions using riding model, and were refined isotropically, with the isotropic vibration parameters related to the non-H atom to which they are bonded. The crystallographic data for compounds hnbdtiH, 1 and 2 are listed in Table 1, and selected bond lengths are given in Tables 2–4.†
Table 1 Crystallographic data and refinement for hnbdtiH, 1 and 2
| |
hnbdtiH |
1 |
2 |
| R1 = Σ‖Fo| − |Fc‖/Σ|Fo|. wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2. |
| Formula |
C25H22ON2S2 |
C49H41O3N4F4S2Ir |
C48H41O2N4S2Ir |
| M |
430.57 |
1066.18 |
962.17 |
| Crystal system |
Orthorhombic |
Triclinic |
Triclinic |
| Space group |
P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| T/K |
173 |
296 |
296 |
| a/Å |
7.2652(6) |
10.8322(15) |
11.8712(6) |
| b/Å |
8.0985(7) |
11.8007(16) |
12.7429(6) |
| c/Å |
35.690(3) |
18.990(3) |
15.5387(8) |
| α/° |
|
75.860(2) |
98.5550(10) |
| β/° |
|
73.658(2) |
102.6780(10) |
| γ/° |
|
83.633(2) |
111.6450(10) |
| V/Å3 |
2099.9(3) |
2256.5(5) |
2062.05(18) |
| Z |
4 |
2 |
2 |
| Dc/g cm−3 |
1.362 |
1.569 |
1.550 |
| F(000) |
904 |
1064 |
964 |
| GooF on F2 |
1.059 |
1.084 |
1.008 |
| R1, wR2 [I > 2σ(I)]a |
0.0393, 0.1115 |
0.0391, 0.1054 |
0.0477, 0.0741 |
| R1, wR2 (all data)a |
0.0427, 0.1139 |
0.0480, 0.1151 |
0.0738, 0.0813 |
| (Δρ)max, (Δρ)min (e Å−3) |
0.338, −0.292 |
1.608, −1.294 |
1.073, −1.266 |
Table 2 Selected bond lengths (Å) for hnbdtiH
| O1–C17 |
1.357(3) |
N2–C14 |
1.3790(3) |
| N1–C13 |
1.384(3) |
N2–C15 |
1.348(3) |
| N1–C15 |
1.326(3) |
|
|
Table 3 Selected bond lengths (Å) for 1
| Ir1–C1 |
1.983(6) |
O1–C39 |
1.327(7) |
| Ir1–C12 |
2.007(6) |
N1–C37 |
1.339(7) |
| Ir1–N3 |
2.041(5) |
N1–C36 |
1.421(7) |
| Ir1–N4 |
2.045(5) |
N2–C37 |
1.344(7) |
| Ir1–O1 |
2.186(4) |
N2–C35 |
1.388(7) |
| Ir1–N1 |
2.211(4) |
|
|
Table 4 Selected bond lengths (Å) for 2
| Ir1–C1 |
1.991(5) |
O1–C39 |
1.324(6) |
| Ir1–C12 |
2.004(5) |
N1–C37 |
1.338(6) |
| Ir1–N3 |
2.033(4) |
N1–C36 |
1.398(6) |
| Ir1–N4 |
2.026(4) |
N2–C37 |
1.350(6) |
| Ir1–O1 |
2.182(3) |
N2–C35 |
1.390(6) |
| Ir1–N1 |
2.194(4) |
|
|
Results and discussion
Crystal structure of hnbdtiH
Compound hnbdtiH crystallizes in orthorhombic non-centrosymmetric space group P212121. In its molecular structure (Fig. S7†), two thiophene groups adopt a non-photoactive parallel conformation, and naphthol and imidazole rings are not coplanar showing a dihedral angle of 31.9(1)°. Naphtholate oxygen O1 and imidazole nitrogen N1 form an intramolecular hydrogen bond [O1⋯N1 = 2.601(1) Å]. Neighboring molecules are linked into a left-handed 21 helical chain along the a axis through the N–H⋯π interactions [the N⋯π distance = 3.598(1) Å] (Fig. 1).12 It should be noted that no CD signal was observed for the bulk sample, indicating that hnbdtiH is a racemic mixture.
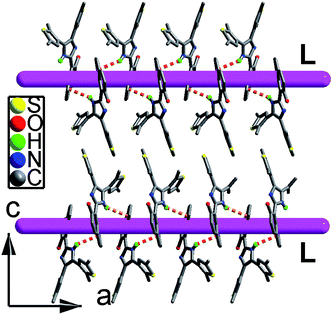 |
| | Fig. 1 Packing structure of hnbdtiH containing left-handed helical chains (denoted as L). | |
Crystal structures of 1 and 2
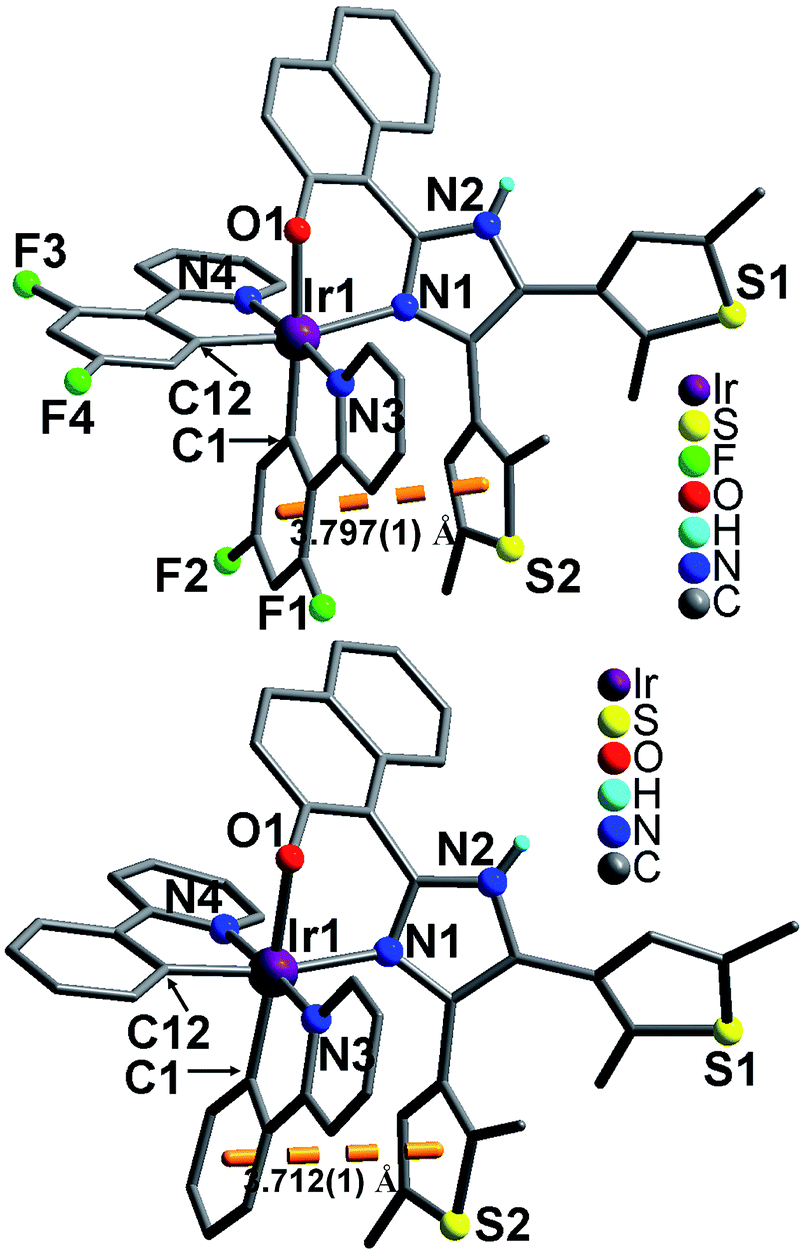
Both 1 and 2 crystallize in monoclinic space group P. As shown in Fig. 2, the Ir(III) ion in each compound adopts a distorted octahedral coordination geometry. Two of the six coordination sites are occupied by naphtholate oxygen O1 and imidazole nitrogen N1 from one hnbdti− ligand. The remaining four positions are occupied by N3, N4, C1 and C12 atoms from two nonequivalent cyclometalated C^N ligands (dfppy− in 1, and ppy− in 2). Both cases show similar Ir–C(N)dfppy−/ppy− distances, 1.983(6)–2.045(5) Å in 1 and 1.991(5)–2.033(4) Å in 2, which are comparable to those in the precursor compounds [Ir(dfppy)2Cl]2 [1.989(6)–2.053(3) Å] and [Ir(ppy)2Cl]2 [1.985(6)–2.061(5) Å].13 Compared with these Ir–C(N)dfppy/ppy bonds, the Ir–N(O)hnbdti− bonds in two cases show longer distances [2.182(3)–2.211(4) Å]. This could be due to the fact that O1 and N1 atoms in hnbdti− ligands are trans to corresponding σ-bound C1 and C12 atoms in the dfppy−/ppy− ligands.14
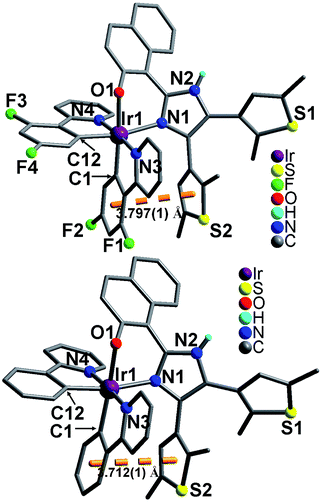 |
| | Fig. 2 Molecular structures of 1 (top) and 2 (bottom). Lattice CH3OH molecules and all H atoms attached to C atoms are omitted for clarity. | |
In the molecular structures of 1 and 2, two C^N ligands (dfppy− or ppy−) are arranged with the pyridine nitrogen atoms N3 and N4 lying in a trans position, which is consistent with the arrangement in [Ir(dfppy)2Cl]2 and [Ir(ppy)2Cl]2.13 The hnbdti− ligands in two cases adopt antiparallel conformation, which are different from free ligand showing parallel conformation. The dihedral angles between the imidazole and naphthol rings [51.4(1)° in 1, and 49.8(1)° in 2] are significantly larger than that in free ligand [31.9(1)°], which is assigned to the coordination of naphthol-imidazole moiety to Ir(III) ion, and the formation of a six-membered chelate ring [Ir1–O1–C–C–C–N1]. Both compounds show intra-molecule aromatic stacking interaction between a thiophene ring and a phenyl ring of ppy−/dfppy− (Fig. 2), showing the centroid–centroid distance of 3.797(1) Å in 1 and 3.712(1) Å in 2.15
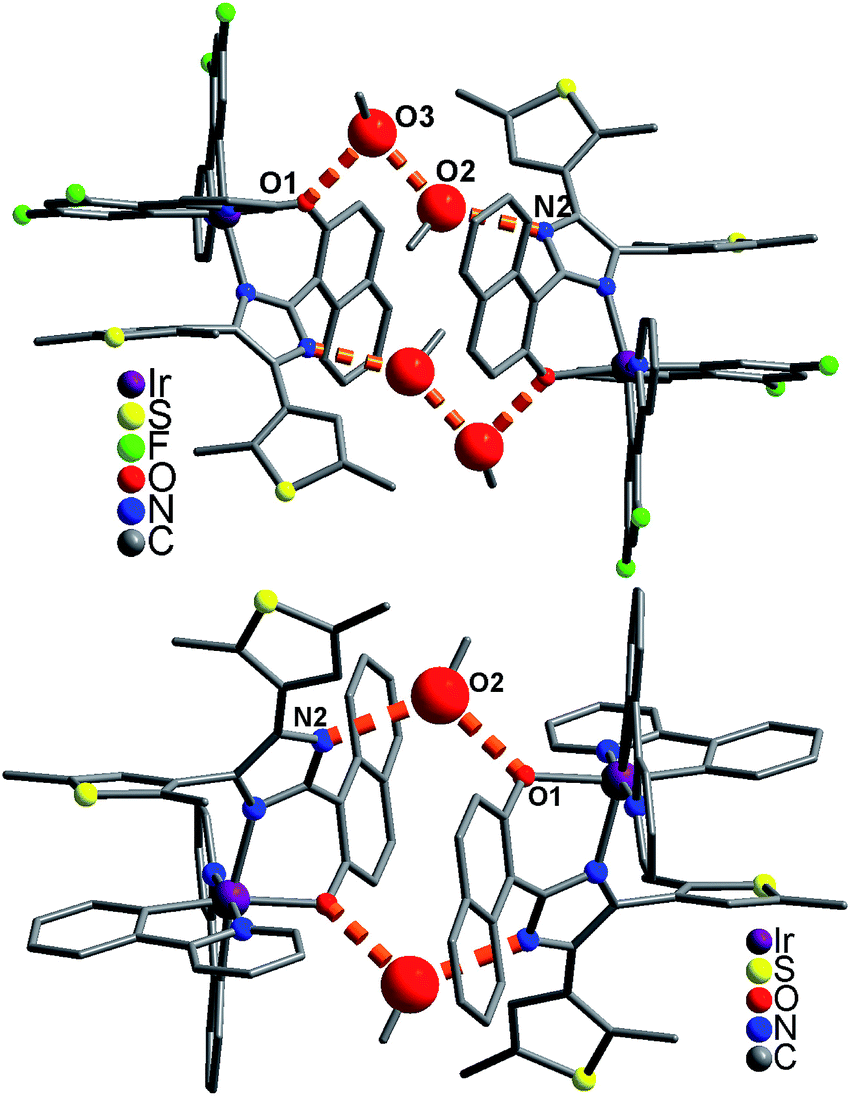
In the packing structure of 1, two neighboring molecules are connected by four CH3OH molecules with hydrogen bonds [O3⋯O1 = 2.727(1) Å, N2⋯O2a = 2.806(1) Å, O2⋯O3b = 2.750(1) Å, symmetry codes a = x, y, z + 1; b = −x + 1, −y + 1, −z + 1], forming a [Ir⋯(CH3OH)4⋯Ir] dimer (Fig. 3 top). These dimers pack together through aromatic stacking interactions, showing a plane–plane distance of 3.526(1) Å between two 2,4-difluorophenyl rings from adjacent dfppy− ligands (Fig. 4).15 In contrast, two neighboring molecules in 2 are linked by two CH3OH molecules with hydrogen bonds [O2⋯O1 = 2.671(1) Å, N2⋯O2a = 2.796(1) Å, symmetry code a = −x + 1, −y + 1, −z + 1], resulting in a [Ir⋯(CH3OH)2⋯Ir] dimer (Fig. 3 bottom). These dimers stack together only through van der Waals interactions (Fig. S8†). Therefore, 1 and 2 have significantly different packing structures, which is mainly due to their different cyclometalated ligand (dfppy− and ppy−, respectively).
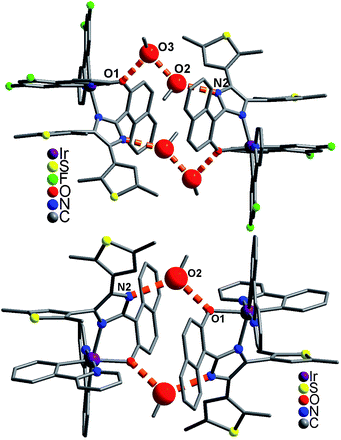 |
| | Fig. 3 Supramolecular dimer structure in 1 (top) and 2 (bottom). Big red balls are O atoms from CH3OH molecules. | |
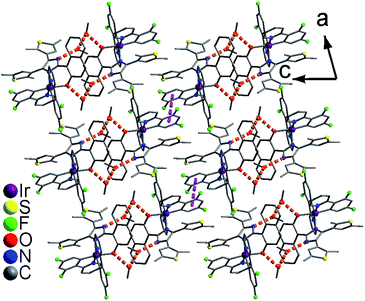 |
| | Fig. 4 Packing structure of 1 showing hydrogen bonds and aromatic stacking interactions. | |
Electronic absorption spectra
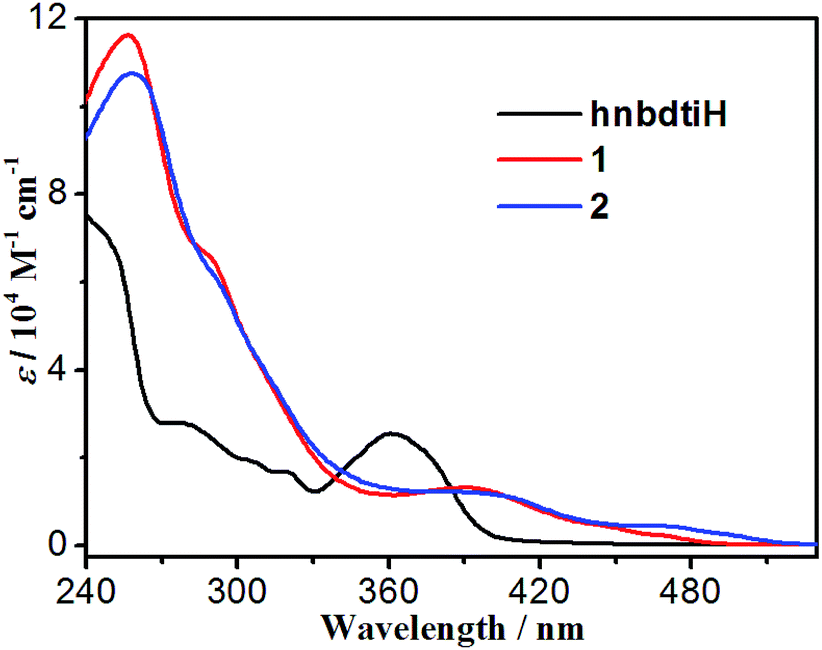
The UV-vis spectra of hnbdtiH, 1 and 2 were measured in CH2Cl2 at room temperature (Fig. 5, Table S1†). The hnbdtiH shows three absorption maxima at ca. 240, 276 and 361 nm. Compounds 1 and 2 reveal similar absorption patterns, and exhibit a wider absorption range than hnbdtiH (Fig. 5). Their strong absorptions in the range of 240–360 nm can be assigned to spin-allowed ligand-centered (1LC) transitions (dfppy−, ppy−, and hnbdti− ligands). The low-energy absorption bands around 390 nm are probably the combination of spin-allowed metal-to-ligand charge transfer (1MLCT) and ligand-centered (1LC) transitions, because of the high extinction coefficient (ε = 1.32 × 104 M−1 cm−1 for 1 and ε = 1.22 × 104 M−1 cm−1 for 2).3,14,16 The weak absorption tails towards 480 nm in 1 and 510 nm in 2 are mainly attributed to spin-forbidden 3MLCT absorptions.17
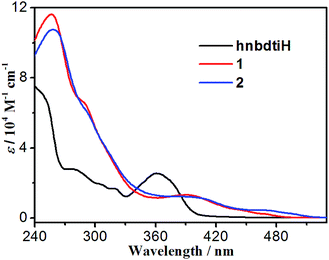 |
| | Fig. 5 UV-vis spectra of hnbdtiH, 1 and 2 in CH2Cl2. | |
Luminescence properties
The luminescence spectra of hnbdtiH, 1 and 2 in CH2Cl2 and in solid state were recorded at room temperature. In CH2Cl2, hnbdtiH shows an emission at 458 nm, but no obvious luminescence can be observed for 1 and 2, which is clear illustrated by the photographs under the irradiation with 365 nm light (Fig. 6). In the measurement of solid-state luminescence spectra (Fig. 7), hnbdtiH exhibits a slightly blue-shifted emission at 454 nm with respect to its emission at 458 nm in CH2Cl2, which could be due to the intermolecular N–H⋯π interactions in solid state. For the solid-state samples of 1 and 2, the former reveals strong luminescence with two main emissions at 537 and 567 nm, while the latter almost has no emission (Fig. 7). The emission lifetimes of hnbdtiH and 1 in CH2Cl2 and/or in solid state were measured at room temperature, with τ = 1.4 ns for hnbdtiH in CH2Cl2, τ = 1.0 ns for hnbdtiH in solid state, and τ = 240 ns for 1 in solid state. Compared to hnbdtiH, compound 1 shows relatively longer emission lifetime, which is in agreement with its characteristic phosphorescence emission.18 In addition, the absolute quantum yield of 1 in solid state was measured, with a value of 0.11.
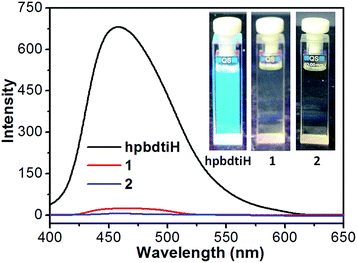 |
| | Fig. 6 Luminescence spectra of hnbdtiH, 1 and 2 (λex = 373 nm), and the photographs under the irradiation with 365 nm light. | |
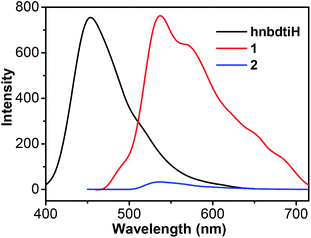 |
| | Fig. 7 Solid-state luminescence spectra of hnbdtiH, 1 and 2 (λex = 373 nm). | |
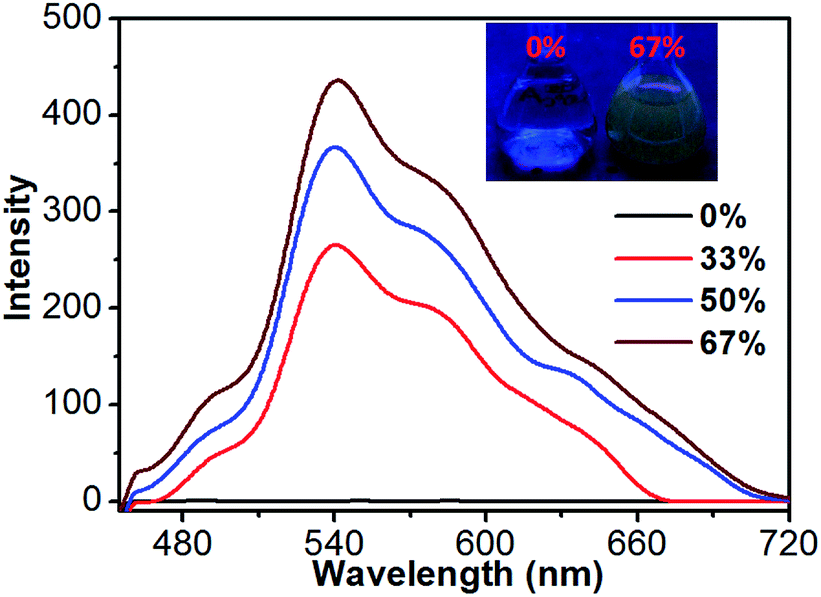
In order to clarify the luminescence behavior of 1, its luminescence spectra were further measured in CH3CN–H2O mixtures with different water fractions at room temperature. Since compound 1 is insoluble in water, its molecules can aggregate in the CH3CN–H2O mixture upon increasing the water fraction. As shown in Fig. 8, the solution of 1 is not luminescent when the water fraction is 0%. However, its luminescence intensity is gradually enhanced when such fraction increases from 0% to 67%. This indicates that compound 1 has an aggregation-induced emission (AIE) property.19 Moreover, the aggregation behavior of 1 in CH3CN–H2O mixture (v/v = 1/1) was studied by TEM and DLS measurements. As shown in the TEM image, the CH3CN–H2O mixture (c = 2 × 10−5 M) contains some particles with a size range of 200–300 nm (Fig. S9†), which is comparable to the DLS measurement showing an average diameters of 240.0 nm (Fig. S10†).
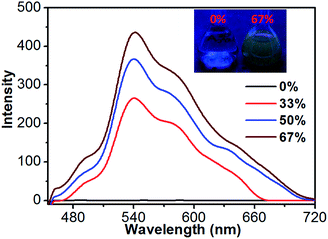 |
| | Fig. 8 Luminescence spectra of 1 in CH3CN–H2O mixtures with different water fractions (λex = 373 nm). | |
In CH2Cl2 solution, compound 1 has not shown obvious luminescence. This might be due to the lower energy level of 3(hpbdti−) triplet excited state than that of dfppy-centered 3MLCT state.19a In contrast, its solid state shows relatively strong luminescence resulted from AIE, which could be assigned to the formation of excimers by π⋯π stacking interactions between 2,4-difluorophenyl groups from adjacent dfppy− ligands (Fig. 4). The similar AIE behavior was observed in compounds Ir(ppy)2(DBM) and Ir(ppy)2(SB) [DBM = 1,3-diphenyl-1,3-propanedione, and SB = 2-(naphthalen-1-yliminomethyl)-phenol], which exhibit π⋯π stacking interactions involving the pyridine rings of adjacent ppy− ligands.19a In addition, compound 2 has not exhibited the similar AIE property to that in 1 (Fig. S11†), which is in agreement with its solid state structure without any intermolecular π⋯π stacking interactions. This could also support our conclusion that the π⋯π stacking interactions in 1 play a key role on its AIE behavior.
Lack of photochromism activity in hnbdtiH, 1 and 2
We previously reported the photochromic behavior of hpbdtiH in CH2Cl2–CH3CN solution,7b showing a color change from colorless to pink upon the irradiation with 321 nm light. In contrast, hnbdtiH does not show photochromic behavior in any solvent, such as CH2Cl2, CHCl3, CH3CN, THF, and CH3OH, under the irradiations with 300 nm and 360 nm lights. This should be due to its different molecular structure from that of hpbdtiH. As shown in Scheme 1, hnbdtiH contains a naphthol ring, and has a greater degree of π-electron conjugation compared with hpbdtiH. Considering the presence of such large π-conjugation system in hnbdtiH, the UV excitation energy is mainly used to generate luminescence, thus resulting in completely absent photocyclization process. The influence of luminescence on photochromism activity has also been observed in BTE containing a –NEt2 group (Scheme S2†).20 This compound has no luminescence in CH3CN, but exhibits photochromic behaviour. In contrast, its proton species with –HN+Et2 group shows luminescence in CH3CN, but no photocyclization reaction. In addition, neither 1 nor 2 exhibits photochromic behavior in any solution (such as CH2Cl2, CHCl3, CH3CN, THF and CH3OH), under the UV irradiations with 300 nm and 390 nm lights. This is mainly assigned to the non-photoactive hnbdti− molecules in two compounds.
Conclusions
We synthesized a new bisthienylethene hnbdtiH and its two heteroleptic complexes [Ir(dfppy)2(hnbdti)]·2CH3OH (1) and [Ir(ppy)2(hnbdti)]·CH3OH (2). In the packing structure of hnbdtiH, neighboring molecules are linked into a helical chain through N–H⋯π interactions. The crystal structures of 1 and 2 indicate that the C^N ligands (dfppy− and ppy−) could significantly influence their packing structures, in which two neighboring molecules are linked by CH3OH molecules with hydrogen bond interactions, forming the [Ir⋯(CH3OH)4⋯Ir] dimer in the former, and the [Ir⋯(CH3OH)2⋯Ir] dimer in the latter. These dimers are further packed through aromatic stacking interactions in 1, while only van der Waals interactions in 2. At room temperature, compound 1 shows aggregation-induced emission (AIE) property arising from the intermolecular π⋯π stacking interactions between 2,4-difluorophenyl groups from adjacent dfppy− ligands, exhibiting two main phosphorescence emissions at 537 and 567 nm. In contrast, compound 2 almost has no emission in solution and in solid state. Compared with 1 and 2, hnbdtiH is fluorescent in CH2Cl2 solution and in solid state, with the emission at 458 nm and 454 nm, respectively. Moreover, hnbdtiH does not exhibit photochromism in solution, which is different from previously reported photoactive hpbdtiH. This could be due to the relatively larger π-electron conjugation system in hnbdtiH which accepts the excitation energy, resulting in fluorescence instead of photochromism. Since hnbdtiH is non-photoactive, neither 1 nor 2 shows photochromism.
Acknowledgements
We thank financial supports by the NSF of Jiangsu province China (BK 20141314), the NSF of China (no. 21171090), and the National Science Fund for Talent Training in Basic Science (no. J1103310). We thank Mrs Zhi-Ping Zhou, Dr Zhen-Zhu Zhu, and Prof. Qiang Zhao for the measurements of TEM, DLS and luminescence properties.
References
-
(a) C.-C. Ko and V. W.-W. Yam, J. Mater. Chem., 2010, 20, 2063 RSC;
(b) J. C.-H. Chan, W. H. Lam, H.-L. Wong, N. Zhu, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12690 CrossRef CAS PubMed;
(c) H.-B. Cheng, H.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2013, 135, 10190 CrossRef CAS PubMed.
- I. Lee, Y. You, S. J. Lim and S. Y. Park, Chem. Lett., 2007, 36, 888 CrossRef CAS.
- V. Aubert, L. Ordronneau, M. Escadeillas, J. A. G. Williams, A. Boucekkine, E. Coulaud, C. Dragonetti, S. Righetto, D. Roberto, R. Ugo, A. Valore, A. Singh, J. Zyss, I. Ledoux-Rak, H. L. Bozec and V. Guerchais, Inorg. Chem., 2011, 50, 5027 CrossRef CAS PubMed.
-
(a) W. Tan, Q. Zhang, J. Zhang and H. Tian, Org. Lett., 2009, 11, 161 CrossRef CAS PubMed;
(b) S. Monaco, M. Semeraro, W. Tan, H. Tian, P. Ceroni and A. Credi, Chem. Commun., 2012, 48, 8652 RSC.
-
(a) V. Guerchais, L. Ordronneau and H. Le Bozec, Coord. Chem. Rev., 2010, 254, 2533 CrossRef CAS PubMed;
(b) L. Ordronneau, H. Nitadori, I. Ledoux, A. Singh, J. A. G. Williams, M. Akita, V. Guerchais and H. L. Bozec, Inorg. Chem., 2012, 51, 5627 CrossRef CAS PubMed;
(c) H. Nitadori, L. Ordronneau, J. Boixel, D. Jacquemin, A. Boucekkine, A. Singh, M. Akita, I. Ledoux, V. Guerchais and H. L. Bozec, Chem. Commun., 2012, 48, 10395 RSC;
(d) M.-Y. Zhang, C.-H. Wang, W.-Y. Wang, N.-N. Ma, S.-L. Sun and Y.-Q. Qiu, J. Phys. Chem. A, 2013, 117, 12497 CrossRef CAS PubMed.
-
(a) B. Li, J.-Y. Wang, H.-M. Wen, L.-X. Shi and Z.-N. Chen, J. Am. Chem. Soc., 2012, 134, 16059 CrossRef CAS PubMed;
(b) B. Li, J.-Y. Wang, H.-M. Wen, L.-X. Shi and Z.-N. Chen, J. Am. Chem. Soc., 2012, 134, 16059 CrossRef CAS PubMed;
(c) Y.-M. Hervault, C. M. Ndiaye, L. Norel, C. Lagrost and S. Rigaut, Org. Lett., 2012, 14, 4454 CrossRef CAS PubMed;
(d) H.-M. Wen, B. Li, J.-Y. Wang, L.-X. Shi, C.-N. Chen and Z.-N. Chen, Organometallics, 2013, 32, 1759 CrossRef CAS.
-
(a) M. Morimoto, H. Miyasaka, M. Yamashita and M. Irie, J. Am. Chem. Soc., 2009, 131, 9823 CrossRef CAS PubMed;
(b) D.-K. Cao, J.-Q. Feng, M. Ren, Y.-W. Gu, Y. Song and M. D. Ward, Chem. Commun., 2013, 49, 8863 RSC;
(c) D.-K. Cao, R.-H. Wei, Y.-W. Gu, Y. Zhao and M. D. Ward, RSC Adv., 2014, 4, 43064 RSC;
(d) M. Milek, F. W. Heinemann and M. M. Khusniyarov, Inorg. Chem., 2013, 52, 11585 CrossRef CAS PubMed.
- M. M. Krayushkin, S. N. Ivanov, A. Y. Martynkin, B. V. Lichitsky, A. A. Dudinov and B. M. Uzhinov, Russ. Chem. Bull., 2001, 50, 116 CrossRef CAS.
-
(a) P. Coppo, E. A. Plummer and L. De Cola, Chem. Commun., 2004, 1774 RSC;
(b) S. Kappaun, S. Eder, S. Sax, K. Mereiter, E. J. W. List and C. Slugovc, Eur. J. Inorg. Chem., 2007, 4207 CrossRef CAS.
- SAINT, Program for Data Extraction and Reduction, Siemens Analytical X-ray Instruments, Madison, WI, 1994–1996, SAINT program Search PubMed.
-
(a) SHELXTL, Reference Manual, version 5.0, Siemens Industrial Automation, Analytical Instruments, Madison, WI, 1997 Search PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- P. Sudhakar, S. Mukherjee and P. Thilagar, Organometallics, 2013, 32, 3129 CrossRef CAS and references therein.
-
(a) E. S. Andreiadis, D. Imbert, J. Pecaut, A. Calborean, I. Ciofini, C. Adamo, R. Demadrille and M. Mazzanti, Inorg. Chem., 2011, 50, 8197 CrossRef CAS PubMed;
(b) O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334 CrossRef CAS PubMed.
- E. Marchi, R. Sinisi, G. Bergamini, M. Tragni, M. Monari, M. Bandini and P. Ceroni, Chem.–Eur. J., 2012, 18, 8765 CrossRef CAS PubMed.
- C. J. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290 CrossRef CAS PubMed.
-
(a) M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. D. Cola, Adv. Funct. Mater., 2010, 20, 1812 CrossRef CAS;
(b) Y. You, S. Lee, T. Kim, K. Ohkubo, W.-S. Chae, S. Fukuzumi, G.-J. Jhon, W. Nam and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 18328 CrossRef CAS PubMed.
-
(a) D. R. Whang, Y. You, W.-S. Chae, J. Heo, S. Kim and S. Y. Park, Langmuir, 2012, 28, 15433 CrossRef CAS PubMed;
(b) G.-G. Shan, H.-B. Li, H.-Z. Sun, D.-X. Zhu, H.-T. Cao and Z.-M. Su, J. Mater. Chem. C, 2013, 1, 1440 RSC.
-
(a) Q. Zhao, L. Li, F. Li, M. Yu, Z. Liu, T. Yi and C. Huang, Chem. Commun., 2008, 685 RSC;
(b) N. Zhao, Y.-H. Wu, J. Luo, L.-X. Shi and Z.-N. Chen, Analyst, 2013, 138, 894 RSC;
(c) X.-G. Hou, Y. Wu, H.-T. Cao, H.-Z. Sun, H.-B. Li, G.-G. Shan and Z.-M. Su, Chem. Commun., 2014, 50, 6031 RSC.
- H.-H. Liu and Y. Chen, J. Phys. Org. Chem., 2012, 25, 142 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format, schemes of reported Ir(III)–bisthienylethene compounds, UV-vis absorption data, some figures including 1H NMR spectra, XRD patterns, structures, TEM image, DLS data, and luminescence spectra. CCDC 1037261–1037263. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra15977b |
|
| This journal is © The Royal Society of Chemistry 2015 |